

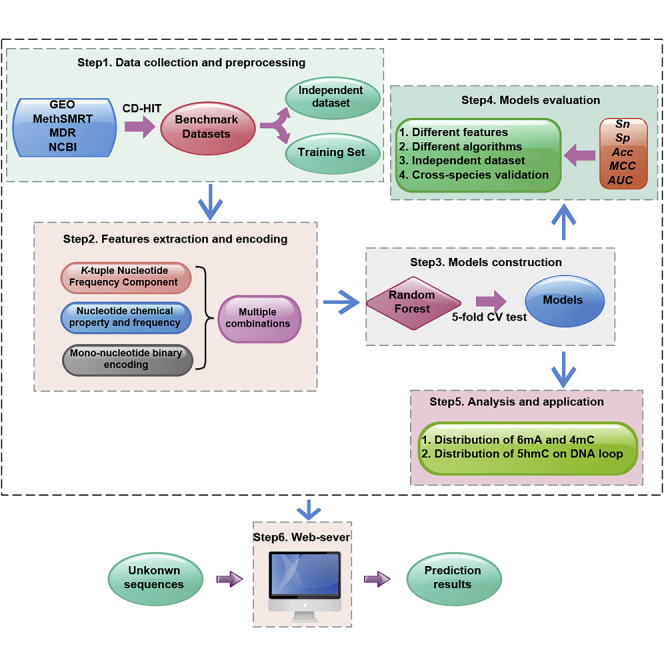
Summary
5hmC, 6mA, and 4mC are three common DNA modifications and are involved in various of biological processes. Accurate genome-wide identification of these sites is invaluable for better understanding their biological functions. Owing to the labor-intensive and expensive nature of experimental methods, it is urgent to develop computational methods for the genome-wide detection of these sites. Keeping this in mind, the current study was devoted to construct a computational method to identify 5hmC, 6mA, and 4mC. We initially used K-tuple nucleotide component, nucleotide chemical property and nucleotide frequency, and mono-nucleotide binary encoding scheme to formulate samples. Subsequently, random forest was utilized to identify 5hmC, 6mA, and 4mC sites. Cross-validated results showed that the proposed method could produce the excellent generalization ability in the identification of the three modification sites. Based on the proposed model, a web-server called iDNA-MS was established and is freely accessible at http://lin-group.cn/server/iDNA-MS.
Subject Areas: Genetics, Quantitative Genetics, Bioinformatics
Graphical Abstract
Highlights
-
•
A computational tool was developed for identification of 5hmC, 6mA, and 4mC
-
•
6mA and 4mC mark similar regions in the C. equisetifolia and F. vesca genomes
-
•
5hmC enriches in the initial and middle of the DNA loops
-
•
A user-friendly webserver was available at http://lin-group.cn/server/iDNA-MS
Genetics; Quantitative Genetics; Bioinformatics
Introduction
The common kinds of DNA modifications, namely, 5-methylcytosine (5mC), 5-hydroxymethylcytosine (5hmC), N6-methyladenine (6mA), and N4-methylcytosine (4mC), have been identified in diverse species (Hu et al., 2019, Ratel et al., 2006). All these modifications are essential in a number of key biological reactions (Fu and He, 2012). As an epigenetic mechanism, DNA methylation is introduced into a DNA molecule by adding methyl or hydroxymethyl groups to nucleotides (see also Figure S1), which plays an essential role for the normal development of organism, including genomic imprinting, X chromosome inactivation, repression of transposable elements, aging, and carcinogenesis (Bergman and Cedar, 2013, Smith and Meissner, 2013).
The 5hmC is generated from 5mC by Ten-eleven translocation (Tet) family proteins. Various studies indicate that 5hmC not only acts as an intermediate during 5mC demethylation, but also plays a potential active role during maintenance of pluripotency in embryo stem cells (ESCs), neural system development, and tumorigenesis (Thomson and Meehan, 2017). Moreover, 5hmC may be involved in regulating gene expression by association with different regulatory elements and processes (Szulwach et al., 2011). The 6mA is formed by transferring methyl groups to the sixth position of the adenine ring catalyzed by methyltransferases. It is a non-canonical DNA modification that is present at low levels in different eukaryotes (Greer et al., 2015, Mondo et al., 2017). 6mA possesses similar characteristics between prokaryotes and eukaryotes (Heyn and Esteller, 2015) and has a variety of roles, such as guiding the discrimination between original and newly synthesized DNA strand after replication (Wion and Casadesus, 2006), regulating gene transcription and repressing transposable elements, and reducing the stability of base pairings for opening DNA duplexes during the cell cycle (Fang et al., 2012). Strikingly, the protection from methylation is an inherited state that, however, can be modified by environmental conditions (Wion and Casadesus, 2006). The 4mC is catalyzed by the N-4 cytosine-specific DNA methyltransferase (DNMT) that specifically methylates the amino group at the fourth position of cytosine in DNA (Timinskas et al., 1995). It is a member of the restriction modification (RM) systems and can protect the host DNA against degradation by restriction enzymes (Schweizer, 2008). In prokaryotes, 4mC is primarily used for distinguishing self from foreign DNA and correcting DNA replication errors (Iyer et al., 2011, Modrich, 1991).
Recently, several studies have revealed the genome-wide distribution of 5hmC and 6mA in Homo sapiens and Mus musculus (Hu et al., 2019) (Wu et al., 2016, Xiao et al., 2018) and the 4mC in Tolypocladium sp SUP5-1 (Stamps et al., 2015), Casuarina equisetifolia (Ye et al., 2019), and so on. Although these studies testified the presence of 5hmC, 6mA, and 4mC in various genomes based on experiments and indeed achieved encouraging results, the short-read sequencing and long-read sequencing techniques still have drawbacks. For example, bisulfite sequencing with short-read techniques is widely used to call methylated cytosines by converting unmethylated cytosines to uracil (Liu et al., 2019), but its positioning efficiency is low and genome coverage is uneven, resulting in poor sequencing quality; PacBio followed by long-read sequencing can identify DNA modifications in genome-wide scale, but it lacks high signal-to-noise ratio for DNA modifications. Moreover, in the organism kingdom, the levels of 5hmC, 6mA, and 4mC were suggested to be low and to be detectable by highly sensitive technologies. Thus, computationally prediction of 5hmC, 6mA, and 4mC is a good choice to reduce the experimental costs and provide informational guidance for the experimental study on epigenetic modification research.
In fact, several machine learning-based methods have emerged as an attractive approach for DNA methylation sites identification (Basith et al., 2019, Manavalan et al., 2019b). Recently, Ni et al. (Ni et al., 2019) extracted sequence information from Nanopore sequencing reads and proposed a deep learning-based classifier called DeepSignal to identify 6mA and 5mC sites in H. sapiens and Escherichia coli. Later, Liu et al. (2019) designed DeepMod, a bidirectional recurrent neural network with long short-term memory to identify 6mA and 5mC sites in E. coli, Chlamydomonas reinhardtii, and H. sapiens. Subsequently, Chen et al. (2019) developed a webserver named i6mA-Pred based on nucleotide chemical properties and support vector machine (SVM) to predict 6mA sites in rice genome. Similarly, Yu et al. (Yu and Dai, 2019) proposed another computational tool-based deep learning, called SNNRice6mA, for 6mA identification in rice genome. By using manually crafted DNA sequence features, Kong et al. (Kong and Zhang, 2019) built a bagging classifier to identify 6mA sites in rice genome. In addition, Basith et al. (2019) developed a novel computational predictor, called the Sequence-based DNA N6-methyladenine predictor (SDM6A), which is a two-layer ensemble approach for identifying 6mA sites in the rice genome. Based on the experimentally confirmed 4mC sites data, Chen et al. (2017) first built a novel predictor called iDNA4mC to identify 4mC sites in multiple species. Later on, based on Chen et al.’s dataset (Chen et al., 2017), Wei et al. (2019) proposed an iterative feature representation algorithm that enables one to learn informative features from several sequential models in a supervised iterative mode to identify 4mC sites. Manavalan et al. (2019a) developed Meta-4mCpred and 4mCpred-EL for the identification of 4mC sites in the mouse genome and six other species, respectively, using meta and ensemble approaches. Hasan et al. (2019) developed a tool called i4mC-ROSE for identifying 4mC sites in the genomes of Fragaria vesca and Rosa chinensis based on random forest classifier with various aspects of DNA sequence information. However, the above-mentioned predictors are trained by using species-specific data and thus may produce a low true-positive rate with a high false-positive rate when generalized to other species. Thus, it is urgent to develop novel 5hmC, 6mA, and 4mC site predictors that will be applicable for different species.
In view of the aforementioned description, this study was devoted to developing new methods for identifying 5hmC, 6mA, and 4mC sites in different species. We first collected and constructed 17 objective benchmark datasets including experimentally confirmed 5hmC/6mA/4mC sequences and non-5hmC/non-6mA/non-4mC sequences from 17 genomes. Subsequently, three kinds of sequence encoding features were proposed to formulate samples and then used as the input of the Random Forest algorithm (RF) to discriminate 5hmC/6mA/4mC from non-5hmC/non-6mA/non-4mC, respectively. Several experiments were performed to investigate the performance of the proposed methods. Finally, based on the proposed methods, a predictor called iDNA-MS was established.
Results and Discussion
Sequence Analysis
In genome, some DNA consensus sequences are widespread and are conjectured to have biological functions, such as serving as transcription factor binding sites (Xiao et al., 2018). Finding the potential oligonucleotide distribution patterns of sequences around modification site will be an effective step in understanding why the site is modified and in revealing the biological functions of modifications (Smith and Meissner, 2013). To investigate the nucleotide distribution surrounding modification sites, the Two Sample Logos (Crooks et al., 2004) was plotted to observe the statistical difference of nucleotide occurrence between positive and negative samples. As shown in Figures 1 and S2, the modification sequences are significantly different (t test, p value<0.05) from non-modification samples in terms of nucleotide distribution. For 5hmC shown in Figure 1A, the nucleotide distribution patterns between H. sapiens and M. musculus are extremely similar. A highly conserved guanine appears at position 22 (+1 site related to 5hmC site) of the 5hmC-containing sequence; in contrast, the corresponding position in non-5hmC-containing sequence prefers to adenine, cytosine, and thymine. Moreover, we also observed the enrichment of cytosine in upstream regions of 5hmC site. These results imply that these special nucleotide distribution patterns could provide functional signal to MTases. Thus, it is reasonable to extract the positional information of the sequences to construct prediction model.
Figure 1.

The Nucleotide Distribution
(A) The nucleotide distribution around 5hmC and non-5hmC sites in (a) H. sapiens and (b) M. musculus, respectively. In each figure, the top panel of the x axis is for 5hmC site-containing sequences, whereas the bottom panel of the x axis is for non-5hmC site-containing sequences.
(B) The nucleotide distribution around 6mA and non-6mA sites in (a) A. thaliana, (b) C. elegans, (c) C. equisetifolia, (d) D. melanogaster, (e) H. sapiens, (f) S. cerevisiae, and (g) Ts. SUP5-1. In each figure, the top panel of the x axis is for 6mA site-containing sequences, whereas the bottom panel of the x axis is for non-6mA site-containing sequences.
(C) The nucleotide distribution around 4mC and non-4mC sites in (a) C. equisetifolia, (b) F. vesca, (c) S. cerevisiae, and (d) Ts. SUP5-1. In each figure, the top panel of the x axis is for 4mC site-containing sequences, whereas the bottom panel of the x axis is for non-4mC site-containing sequences.
As shown in Figure 1B, we found some similar patterns of the nucleotide distributions around 6mA among seven species. First, a consensus motif, namely, [G/A]AGG, was observed in 6mA samples, which has been also reported in publications (Lv et al., 2019, Zhou et al., 2018). Second, there are high-frequency repeat A-containing segments in both upstream and downstream of 6mA sites, which is consistent with the periodic pattern of deposition in zebrafish (Liu et al., 2016). Especially, the upstream of 6mA sites have a consensus sequence of AAAAA from position 15 to 19 (from −6 to −2 upstream 6mA site). Finally, we further investigated the sequence motif of the 6mA sites in other species (See also Figure S2). We found that the nucleotide composition bias regions exist from position 22 to 23 (GG) in F. vesca, R. chinensis, and Xoc. BLS256.
As shown in Figure 1C (a) and (b), the consensus motif, namely, CCC[C/G]G[G/C], was observed over the range of position 17–23 (from −4 to +2 around 4mC site) in C. equisetifolia and F. vesca. For S. cerevisiae and Ts. SUP5-1, we found that two motifs, namely, CAA and AAC, were located at positions 15–17 (from −6 to −4 upstream 4mC site) and 25–27 (from +4 to +6 downstream 4mC site), respectively. These results indicated that the 4mCs have different nucleotide-enriched regions. Their nucleotide distribution patterns are species specific; however, within plants (C. equisetifolia and F. vesca) or fungi (S. cerevisiae and Ts. SUP5-1), some conserved motifs can still be observed.
Performance Evaluation on Different Features
In this section, we attempt to answer the following question: which features are the most important for identifying 5hmC/6mA/4mC in each species? For this, we first investigated the prediction performances of different kinds of features, namely, K-tuple nucleotide frequency component (KNFC), nucleotide chemical property and nucleotide frequency (NCPNF), and mono-nucleotide binary encoding (MNBE) and their four combinations for identifying three types of modification in 17 genomes. The prediction results of the RF-based models were recorded in Figure 2 and Table S1. We noticed that the best prediction performances for the 5hmC recognition are obtained by NCPNF and NCPNF-MNBE in the H. sapiens and M. musculus, respectively. However, for all 6mA and 4mC identifications, the best prediction performances are always generated by MNBE.
Figure 2.

The AUC of Proposed Features with RF for Identifying 5hmC/6mA/4mC Sites in 17 Genomes
(A) The AUC of proposed features with RF for identifying 5hmC sites in H. sapiens and M. musculus. (B) The AUC of proposed features with RF for identifying 6mA sites in A. thaliana, C. elegans, C. equisetifolia, D. melanogaster, H. sapiens, S. cerevisiae, and Ts. SUP5-1. (C) The AUC of proposed features with RF for identifying 4mC sites in C. equisetifolia, F. vesca, S. cerevisiae, and Ts. SUP5-1.Figure 3: The Performance Evaluation of the RF and Other Three Machine Learning Algorithms on 17 Genomes in Terms of AUC(A) The AUC of RF and other three machine learning algorithms for identifying 5hmC inH. sapiens and M. musculus. (B) The AUC of RF and other three machine learning algorithms for identifying 6mA in A. thaliana, C. elegans, C. equisetifolia, D. melanogaster, H. sapiens, S. cerevisiae, and Ts. SUP5-1. (C) The AUC of RF and other three machine learning algorithms for identifying 4mC in C. equisetifolia, F. vesca, S. cerevisiae, and Ts. SUP5-1.Figure 4: Illustration to show the prediction indexes for identifying 5hmC, 6mA, and 4mC sites by using independent datasets. (A) The prediction indexes for identifying 5hmC by using independent datasets on H. sapiens and M. musculus. (B) The prediction indexes for identifying 6mA by using independent datasets on A. thaliana, C. elegans, C. equisetifolia, D. melanogaster, H. sapiens, S. cerevisiae, and Ts. SUP5-1. (C) The prediction indexes for identifying 4mC by using independent datasets on C. equisetifolia, F. vesca, S. cerevisiae, and Ts. SUP5-1.
Although NCPNF is not better than MNBE, its area under the curves (AUCs) for all species are acceptable. The reason is that, although the core ideas of the two feature extraction methods are different, when they are used to transform the sequence into a feature matrix, some same features are produced, whereas MNBE can capture more position-specific information of one single nucleotide in positive and negative samples so as to obtain higher prediction performance. In fact, the KNFC is the worst descriptor among all features. This is because an active methyltransferase methylates nearly all (often >95%) target sequence motifs in a prokaryotic genome (Blow et al., 2016, Fang et al., 2012). In contrast, 6mA is a much less motif-driven modification in eukaryotes (Luo et al., 2016, Wu et al., 2016), likely owing to their involvement in functional regulation rather than RM systems. Moreover, we speculated that the 5hmC and 4mC modifications have a similar motif-driven rule as the 6mA modification. Taken together, the final models of 17 genomes were established based on their best features. For instance, the optimal features for identifying 5hmC in H. sapiens and M. musculus are NCPNF and NCPNF-MNBE, respectively. However, the MNBE is the best feature for identifying 6mA and 4mC sites in the other 15 genomes.
Performance Evaluation on Different Algorithms
To further testify the superiority of our proposed method, we investigated the discriminant capabilities of three classic algorithms, i.e., Naive Bayes, Bayes Net, and Decision Tree, on the benchmark dataset by using 5-fold cross-validation. All algorithms were implemented in WEKA (Frank et al., 2004). The results are shown in Figure 3 and Table S2. It was found that RF was the best one among the four compared algorithms in some cases for identifying modification sites. But in some species, Naive Bayes and Bayes Net can also produce the best predictive performances. Thus, the final model was built based on the best classification algorithm. For instance, all models that identify 5hmC sites were constructed based on Bayes Net, whereas models that recognize 6mA and 4mC sites were built almost exclusively on RF.
Figure 3.

The Performance Evaluation of the RF and Other Three Machine Learning Algorithms on 17 Genomes in Terms of AUC
Performance Evaluation on Independent Dataset
In order to further evaluate the performance of our proposed method, the independent datasets were used to measure the stability and generality of the proposed model (Basith et al., 2020). The predictive results thus obtained were listed in Figure 4 and Table S3. We observed that all models produced satisfactory results regardless of whether the modification type was 5hmC or 6mA, suggesting that our method is robust and reliable. However, the AUC obtained by the 4mC site recognition models was around 0.8, indicating that it is difficult to identify 4mC sites in fungi (S. cerevisiae) and plants (C. equisetifolia, Ts. SUP5-1). But these models are still the state-of-the-art tools for methylation sites analysis.
Figure 4.

Illustration to show the prediction indexes for identifying 5hmC, 6mA, and 4mC sites by using independent datasets.
Cross-Species Validation
To explore whether knowledge transfer information can be applied to study the relationships of interacting species, it is necessary to demonstrate whether a model trained with the data from one species could recognize the modification sites in other species. To this end, we trained 17 species-specific models using the species-specific 5hmC/6mA/4mC data and validated these models on the 5hmC/6mA/4mC data from other species. The predictive accuracies thus obtained were shown in Figure 5 and Table S4.
Figure 5.

The Heatmap of Cross-Species Validation
The heatmap showing the cross-species prediction accuracies for identifying 5hmC (A), 6mA (B), and 4mC (C). Once a species-specific model was established on its own training dataset, it was tested on the data from the other species.
As shown in Figure 5A, when H. sapiens and M. musculus data were regarded as independent testing data, we evaluated the H. sapiens- and M. musculus-based models and obtained satisfactory results. The accuracies for H. sapiens and M. musculus are 97.09% and 94.92%, respectively, suggesting that the knowledge based on a transfer learning method from the source domain (H. sapiens or M. musculus data) was well applied in the target domain (H. sapiens or M. musculus data). As shown in Figure 5B and Table S4, we can clearly find the poor prediction accuracies (<71%) for C. elegans, T. thermophile, and Xoc. BLS256 obtained by using other species-based models. This indicates a huge difference between these species in the 6mA modification pattern. Moreover, as shown in Figure 5C, the best accuracy is always obtained by model built based on the data from itself. However, the result based on the S. cerevisiae model is unsatisfactory with the accuracy of 74.24%. The reason is perhaps that the S. cerevisiae-based model was trained on less samples than other models. In summary, our proposed models display high accuracy, robustness, and generality for identifying the modification sites.
6mA and 4mC Mark Similar Regions in the C. equisetifolia and F. vesca Genomes
There is evidence that 6mA and 4mC have similar functions in prokaryotes and are primarily used for distinguishing self from foreign DNA (Iyer et al., 2011). These modifications are considered to be signaling or epigenetic modifications because they are predicted not to disrupt DNA base pairing (Iyer et al., 2011). These suggest that 6mA and 4mC may have synergistic effects through co-localization during the development of the organism. Therefore, we calculated the distant distribution between 6mA sites and 4mC sites to investigate the co-localization of the two modifications in the genome. The results showed that 6mA and 4mC had significant co-localization in C. equisetifolia and F. vesca, and slight co-localization in S. cerevisiae, whereas 6mA generally does not co-localize with 4mC in Ts. SUP5-1 (Figure 6). This indicates that 6mA and 4mC are two similar marks in plant genome (C. equisetifolia and F. vesca) rather than fungal genome (S. cerevisiae and Ts. SUP5-1) (Quandt et al., 2014).
Figure 6.

The Distance Between 6mA and 4mC
Distance between 6mA and 4mC was plotted to show the correlation between the two modifications in C. equisetifolia (A), F. vesca (B), S. cerevisiae (C), and Ts. SUP5-1 (D).
Distribution of 5hmC on DNA Loop
Genome-wide 3D chromosome organization mapping technologies have revealed important insights on genome folding that the spatial organization of genome plays a significant role in the transcriptional control of genes (Dekker and Mirny, 2016, Dixon et al., 2012). The DNA loop, as the smallest spatial structure that can be recognized by chromosome conformation capture technology, is thought to reflect the activities of transcription factors (TFs), cohesion, and CCCTC-binding factor (CTCF) (Dowen et al., 2014, Gorkin et al., 2014). Although the role of histone marks in DNA loops has been extensively explored, we do not yet know the relationship between the DNA modifications and loops (Bonev and Cavalli, 2016, Dowen et al., 2014). To address this question, we examined the distribution of 5hmC in loops from M. musculus embryonic stem cells (ESs) (Salameh et al., 2019). The results in Figure 7 showed that 5hmC modification sites enrich in the initial and middle of the loops. The reason might be that ESs need a large number of promoter-associated loops to maintain the active transcriptional state of the genes. These promoters locate at the 5′ region of the loop and function independently of CTCF (Bonev and Cavalli, 2016). More experiments showed that 5hmC is enriched at both extended promoter regions of Polycomb-repressed developmental regulators and gene bodies of actively transcribed genes (Wu et al., 2011).
Figure 7.

The Distribution of 5hmC in DNA Loop Shows that the 5hmC Sites Enrich in the 5′ and Middle Regions of the Loop
Web-Server
More and more researches have demonstrated that the database and the web-server can provide scholars with more convenient services. Thus, for convenience of researchers, we established a user-friendly web-server called iDNA-MS to identify 5hmC, 6mA, and 4mC modification sites in 17 genomes, which can be freely accessed at http://lin-group.cn/server/iDNA-MS.
Below, we give researchers a step-by-step guideline on how to use the web-server. Users can open the homepage as shown in Figure 8 to see a short introduction about iDNA-MS. One may first click the “Web-server” button, then type or copy/paste the DNA sequence in the input box, or upload the FASTA format file. Note that the length of each sequence should be greater than 41 nt long. Subsequently, after clicking the “submit” button, the predicted results will appear on a new page. We hope that it will provide a convenient way for users concerned on DNA modifications.
Figure 8.

A Semi-Screenshot for the Web-Server Page of the iDNA-MS Web-Server at http://lin-group.cn/server/iDNA-MS
Limitations of the Study
Novel feature description methods should be used. In addition, the positional distribution of epigenetic modification sites and different DNA elements can be further studied, for example, the relationship between DNA modification and nucleosome in whole genome.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work has been supported by the National Natural Science Foundation of China (61772119, 31771471), Sichuan Provincial Science Fund for Distinguished Young Scholars (20JCQN0262), the Natural Science Foundation for Distinguished Young Scholar of Hebei Province (No. C2017209244), and the Science Strength Promotion Programme of UESTC.
Author Contributions
Conceptualization, H.D., W.C., and H. Lin.; Investigation, W.S. and M.-L.L.; Coding, D.Z., Z.-X.G., and H.Y.; Writing - Original Draft, H. Lv. and F.-Y.D.; Writing – Review & Editing, W.C. and H. Lin.; Funding Acquisition, W.C. and H. Lin.
Declaration of Interests
The authors declare that they have no competing interests.
Published: April 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.100991.
Data and Code Availability
The datasets are publicly available at http://lin-group.cn/server/iDNA-MS. All software used are published and/or in the public domain. All pipelines and Python scripts used in the study are available at http://lin-group.cn/server/iDNA-MS/download.html.
Supplemental Information
References
- Basith S., Manavalan B., Shin T.H., Lee G. SDM6A: a web-based integrative machine-learning framework for predicting 6mA sites in the rice genome. Mol. Ther. Nucleic Acids. 2019;18:131–141. doi: 10.1016/j.omtn.2019.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basith S., Manavalan B., Shin T.H., Lee G. Machine intelligence in peptide therapeutics: a next-generation tool for rapid disease screening. Med. Res. Rev. 2020 doi: 10.1002/med.21658. [DOI] [PubMed] [Google Scholar]
- Bergman Y., Cedar H. DNA methylation dynamics in health and disease. Nat. Struct. Mol. Biol. 2013;20:274–281. doi: 10.1038/nsmb.2518. [DOI] [PubMed] [Google Scholar]
- Blow M.J., Clark T.A., Daum C.G., Deutschbauer A.M., Fomenkov A., Fries R., Froula J., Kang D.D., Malmstrom R.R., Morgan R.D. The epigenomic landscape of prokaryotes. PLoS Genet. 2016;12:e1005854. doi: 10.1371/journal.pgen.1005854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonev B., Cavalli G. Organization and function of the 3D genome. Nat. Rev. Genet. 2016;17:661–678. doi: 10.1038/nrg.2016.112. [DOI] [PubMed] [Google Scholar]
- Chen W., Lv H., Nie F., Lin H. i6mA-Pred: identifying DNA N6-methyladenine sites in the rice genome. Bioinformatics. 2019;35:2796–2800. doi: 10.1093/bioinformatics/btz015. [DOI] [PubMed] [Google Scholar]
- Chen W., Yang H., Feng P., Ding H., Lin H. iDNA4mC: identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics. 2017;33:3518–3523. doi: 10.1093/bioinformatics/btx479. [DOI] [PubMed] [Google Scholar]
- Crooks G.E., Hon G., Chandonia J.M., Brenner S.E. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J., Mirny L. The 3D genome as moderator of chromosomal communication. Cell. 2016;164:1110–1121. doi: 10.1016/j.cell.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowen J.M., Fan Z.P., Hnisz D., Ren G., Abraham B.J., Zhang L.N., Weintraub A.S., Schujiers J., Lee T.I., Zhao K. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell. 2014;159:374–387. doi: 10.1016/j.cell.2014.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G., Munera D., Friedman D.I., Mandlik A., Chao M.C., Banerjee O., Feng Z., Losic B., Mahajan M.C., Jabado O.J. Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat. Biotechnol. 2012;30:1232–1239. doi: 10.1038/nbt.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank E., Hall M., Trigg L., Holmes G., Witten I.H.J.B. Data mining in bioinformatics using Weka. Bioinformatics. 2004;20:2479–2481. doi: 10.1093/bioinformatics/bth261. [DOI] [PubMed] [Google Scholar]
- Fu Y., He C. Nucleic acid modifications with epigenetic significance. Curr. Opin. Chem. Biol. 2012;16:516–524. doi: 10.1016/j.cbpa.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorkin D.U., Leung D., Ren B. The 3D genome in transcriptional regulation and pluripotency. Cell Stem Cell. 2014;14:762–775. doi: 10.1016/j.stem.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer E.L., Blanco M.A., Gu L., Sendinc E., Liu J., Aristizabal-Corrales D., Hsu C.H., Aravind L., He C., Shi Y. DNA methylation on N6-adenine in C. elegans. Cell. 2015;161:868–878. doi: 10.1016/j.cell.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan M.M., Manavalan B., Khatun M.S., Kurata H. i4mC-ROSE, a bioinformatics tool for the identification of DNA N4-methylcytosine sites in the Rosaceae genome. Int. J. Biol. Macromol. 2019 doi: 10.1016/j.ijbiomac.2019.12.009. [DOI] [PubMed] [Google Scholar]
- Heyn H., Esteller M. An adenine code for DNA: a second life for N6-methyladenine. Cell. 2015;161:710–713. doi: 10.1016/j.cell.2015.04.021. [DOI] [PubMed] [Google Scholar]
- Hu L., Liu Y., Han S., Yang L., Cui X., Gao Y., Dai Q., Lu X., Kou X., Zhao Y. Jump-seq: genome-wide capture and amplification of 5-hydroxymethylcytosine sites. J. Am. Chem. Soc. 2019;141:8694–8697. doi: 10.1021/jacs.9b02512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer L.M., Abhiman S., Aravind L. Natural history of eukaryotic DNA methylation systems. Prog. Mol. Biol. Transl Sci. 2011;101:25–104. doi: 10.1016/B978-0-12-387685-0.00002-0. [DOI] [PubMed] [Google Scholar]
- Kong L., Zhang L. i6mA-DNCP: computational identification of DNA N(6)-methyladenine sites in the rice genome using optimized dinucleotide-based features. Genes (Basel) 2019;10 doi: 10.3390/genes10100828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Zhu Y., Luo G.Z., Wang X., Yue Y., Wang X., Zong X., Chen K., Yin H., Fu Y. Abundant DNA 6mA methylation during early embryogenesis of zebrafish and pig. Nat. Commun. 2016;7:13052. doi: 10.1038/ncomms13052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Fang L., Yu G., Wang D., Xiao C.L., Wang K. Detection of DNA base modifications by deep recurrent neural network on Oxford Nanopore sequencing data. Nat. Commun. 2019;10:2449. doi: 10.1038/s41467-019-10168-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo G.Z., Wang F., Weng X., Chen K., Hao Z., Yu M., Deng X., Liu J., He C. Characterization of eukaryotic DNA N(6)-methyladenine by a highly sensitive restriction enzyme-assisted sequencing. Nat. Commun. 2016;7:11301. doi: 10.1038/ncomms11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv H., Dao F.Y., Guan Z.X., Zhang D., Tan J.X., Zhang Y., Chen W., Lin H. iDNA6mA-Rice: a computational tool for detecting N6-methyladenine sites in rice. Front. Genet. 2019;10:793. doi: 10.3389/fgene.2019.00793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manavalan B., Basith S., Shin T.H., Lee D.Y., Wei L., Lee G. 4mCpred-EL: an ensemble learning framework for identification of DNA N(4)-methylcytosine sites in the mouse genome. Cells. 2019;8 doi: 10.3390/cells8111332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manavalan B., Basith S., Shin T.H., Wei L., Lee G. Meta-4mCpred: a sequence-based meta-predictor for accurate DNA 4mC site prediction using effective feature representation. Mol. Ther. Nucleic Acids. 2019;16:733–744. doi: 10.1016/j.omtn.2019.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrich P. Mechanisms and biological effects of mismatch repair. Annu. Rev. Genet. 1991;25:229–253. doi: 10.1146/annurev.ge.25.120191.001305. [DOI] [PubMed] [Google Scholar]
- Mondo S.J., Dannebaum R.O., Kuo R.C., Louie K.B., Bewick A.J., LaButti K., Haridas S., Kuo A., Salamov A., Ahrendt S.R. Widespread adenine N6-methylation of active genes in fungi. Nat. Genet. 2017;49:964–968. doi: 10.1038/ng.3859. [DOI] [PubMed] [Google Scholar]
- Ni P., Huang N., Zhang Z., Wang D.P., Liang F., Miao Y., Xiao C.L., Luo F., Wang J. DeepSignal: detecting DNA methylation state from Nanopore sequencing reads using deep-learning. Bioinformatics. 2019;35:4586–4595. doi: 10.1093/bioinformatics/btz276. [DOI] [PubMed] [Google Scholar]
- Quandt C.A., Kepler R.M., Gams W., Araujo J.P., Ban S., Evans H.C., Hughes D., Humber R., Hywel-Jones N., Li Z. Phylogenetic-based nomenclatural proposals for Ophiocordycipitaceae (Hypocreales) with new combinations in Tolypocladium. IMA Fungus. 2014;5:121–134. doi: 10.5598/imafungus.2014.05.01.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratel D., Ravanat J.L., Berger F., Wion D. N6-methyladenine: the other methylated base of DNA. BioEssays. 2006;28:309–315. doi: 10.1002/bies.20342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salameh T.J., Wang X., Song F., Zhang B., Wright S.M., Khunsriraksakul C., Yue F.J.b. A supervised learning framework for chromatin loop detection in genome-wide contact maps. bioRxiv. 2019:739698. doi: 10.1038/s41467-020-17239-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer H. Bacterial genetics: past achievements, present state of the field, and future challenges. Biotechniques. 2008;44:633–634. doi: 10.2144/000112807. 636–641. [DOI] [PubMed] [Google Scholar]
- Smith Z.D., Meissner A. DNA methylation: roles in mammalian development. Nat. Rev. Genet. 2013;14:204–220. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- Stamps B.W., Du L., Mitchell C.A., Cichecwicz R.H., Stevenson B.S. Draft genomes of two sordariomycete fungi that produce novel secondary metabolites. Genome Announc. 2015;3 doi: 10.1128/genomeA.00291-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulwach K.E., Li X., Li Y., Song C.X., Wu H., Dai Q., Irier H., Upadhyay A.K., Gearing M., Levey A.I. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 2011;14:1607–1616. doi: 10.1038/nn.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson J.P., Meehan R.R. The application of genome-wide 5-hydroxymethylcytosine studies in cancer research. Epigenomics. 2017;9:77–91. doi: 10.2217/epi-2016-0122. [DOI] [PubMed] [Google Scholar]
- Timinskas A., Butkus V., Janulaitis A. Sequence motifs characteristic for DNA [cytosine-N4] and DNA [adenine-N6] methyltransferases. classification of all DNA methyltransferases. Gene. 1995;157:3–11. doi: 10.1016/0378-1119(94)00783-o. [DOI] [PubMed] [Google Scholar]
- Wei L., Su R., Luan S., Liao Z., Manavalan B., Zou Q., Shi X. Iterative feature representations improve N4-methylcytosine site prediction. Bioinformatics. 2019;35:4930–4937. doi: 10.1093/bioinformatics/btz408. [DOI] [PubMed] [Google Scholar]
- Wion D., Casadesus J. N6-methyl-adenine: an epigenetic signal for DNA-protein interactions. Nat. Rev. Microbiol. 2006;4:183–192. doi: 10.1038/nrmicro1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., D'Alessio A.C., Ito S., Wang Z., Cui K., Zhao K., Sun Y.E., Zhang Y. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 2011;25:679–684. doi: 10.1101/gad.2036011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T.P., Wang T., Seetin M.G., Lai Y., Zhu S., Lin K., Liu Y., Byrum S.D., Mackintosh S.G., Zhong M. DNA methylation on N(6)-adenine in mammalian embryonic stem cells. Nature. 2016;532:329–333. doi: 10.1038/nature17640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C.L., Zhu S., He M., Chen, Zhang Q., Chen Y., Yu G., Liu J., Xie S.Q., Luo F. N(6)-methyladenine DNA modification in the human genome. Mol. Cell. 2018;71:306–318 e307. doi: 10.1016/j.molcel.2018.06.015. [DOI] [PubMed] [Google Scholar]
- Ye G., Zhang H., Chen B., Nie S., Liu H., Gao W., Wang H., Gao Y., Gu L. De novo genome assembly of the stress tolerant forest species Casuarina equisetifolia provides insight into secondary growth. Plant J. 2019;97:779–794. doi: 10.1111/tpj.14159. [DOI] [PubMed] [Google Scholar]
- Yu H., Dai Z. SNNRice6mA: a deep learning method for predicting DNA N6-methyladenine sites in rice genome. Front. Genet. 2019;10:1071. doi: 10.3389/fgene.2019.01071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C., Wang C., Liu H., Zhou Q., Liu Q., Guo Y., Peng T., Song J., Zhang J., Chen L. Identification and analysis of adenine N(6)-methylation sites in the rice genome. Nat. Plants. 2018;4:554–563. doi: 10.1038/s41477-018-0214-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets are publicly available at http://lin-group.cn/server/iDNA-MS. All software used are published and/or in the public domain. All pipelines and Python scripts used in the study are available at http://lin-group.cn/server/iDNA-MS/download.html.