

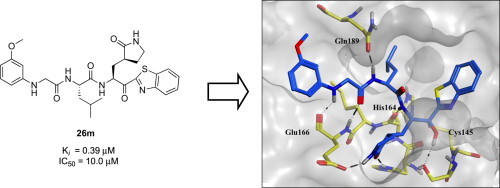
Abstract
This work describes the design, synthesis, and evaluation of low-molecular weight peptidic SARS-CoV 3CL protease inhibitors. The inhibitors were designed based on the potent tripeptidic Z-Val-Leu-Ala(pyrrolidone-3-yl)-2-benzothiazole (8; Ki = 4.1 nM), in which the P3 valine unit was substituted with a variety of distinct moieties. The resulting series of dipeptide-type inhibitors displayed moderate to good inhibitory activities against 3CLpro. In particular, compounds 26m and 26n exhibited good inhibitory activities with Ki values of 0.39 and 0.33 μM, respectively. These low-molecular weight compounds are attractive leads for the further development of potent peptidomimetic inhibitors with pharmaceutical profiles. Docking studies were performed to model the binding interaction of the compound 26m with the SARS-CoV 3CL protease. The preliminary SAR study of the peptidomimetic compounds with potent inhibitory activities revealed several structural features that boosted the inhibitory activity: (i) a benzothiazole warhead at the S1′ position, (ii) a γ-lactam unit at the S1-position, (iii) an appropriately hydrophobic leucine moiety at the S2-position, and (iv) a hydrogen bond between the N-arylglycine unit and a backbone hydrogen bond donor at the S3-position.
Keywords: SARS, SARS-CoV 3CL protease, Peptidomimetics, Dipeptide, Cysteine protease inhibitors, Docking study
Graphical abstract
1. Introduction
Since its first appearance in Southern China in late 2002, severe acute respiratory syndrome (SARS) has been recognized as a global threat [1], [2]. Its rapid and unexpected spread to 32 countries has affected more than 8000 individuals and caused nearly 800 (∼10%) fatalities worldwide within a few months [1], [2], [3]. The causative SARS pathogen is a novel coronavirus, SARS-CoV [4], [5]. SARS-CoV is a positive-strand RNA virus with a genome sequence that is only moderately homologous to other known coronaviruses [6], [7]. SARS-CoV encodes a chymotrypsin-like protease (3CLpro), also referred to as the main protease (Mpro), which plays a pivotal role in processing viral polyproteins and controlling replicase complex activity [8]. This enzyme is indispensable for viral replication and infection processes, making it an ideal target for the design of antiviral therapies. The 3CLpro active site contains a catalytic dyad in which a cysteine residue (Cys145) acts as a nucleophile and a histidine (His41) residue acts as a general acid or base [9], [10]. The SARS epidemic was successfully controlled in 2003; however, the potential reemergence of pandemic SARS-CoV continues to pose a risk, and new strains of SARS could potentially be more virulent than the strains that contributed to the 2003 outbreak. Since 2003, two additional human coronaviruses, NL63 and HKU1, have been identified in patients around the world [11], [12]. Recently, a new SARS-like virus, HCoV-EMC, was identified in at least two individuals, one of whom died [13]. Very recently, the first case of a fatal respiratory illness similar to the deadly SARS was confirmed in Britain [14]. The World Health Organization (WHO) has announced that it is closely monitoring the situation and is working to “ensure a high degree of preparedness, should the new virus be found to be sufficiently transmissible to cause a community outbreak”. There is a significant need to develop anti-SARS agents that are capable of treating this potentially fatal respiratory illness. Several reports of crystalline forms of the SARS-CoV 3CLpro protein bound to hexapeptidyl chloromethyl ketone inhibitors have been reported [6], [9], and numerous peptidic structures have been reported in the context of targeted antiviral drug design [15], [16], [17], [18], [19], [20], [21], [22]. The reported protease inhibitors are generally peptidic in nature, often five to three residues in length, and bear a reactive warhead group at the C-terminus which forms an interaction with the protease catalytic Cys145 (Fig. 1 , 1–9). Two of these compounds (8 and 9), recently described in a separate report from our group [22], exhibited excellent potent inhibitory activities with K i values of 4.1 and 3.1 nM, respectively. These peptidic inhibitors provided valuable insight into the design constraints for this system and quickly led to the development of non-peptidic small molecule inhibitors (Fig. 1, 10–17) [23], [24], [25], [26], [27], [28], [29], [30]. These small molecular inhibitors generally showed moderate to good activities.
Fig. 1.
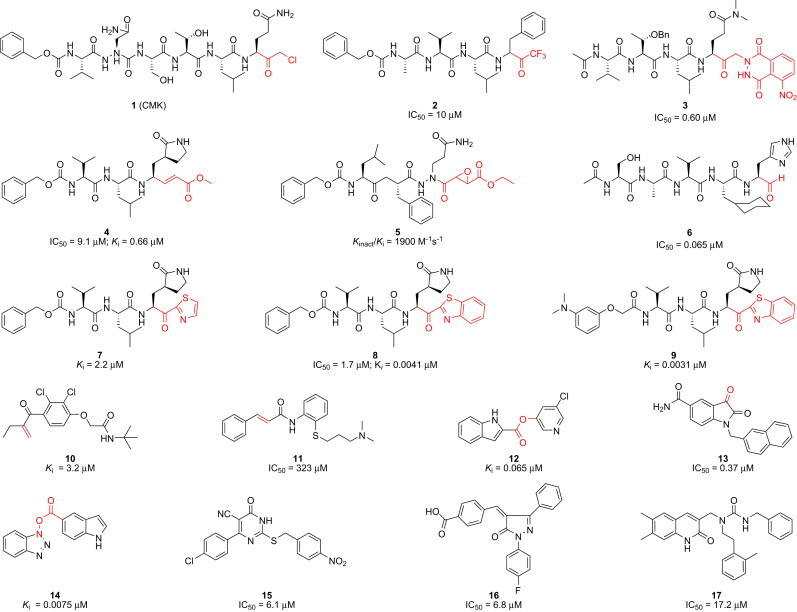
Representative peptidomimetics (1–9) and small molecular (10–17) 3CLpro inhibitors highlighting reactive warhead groups (red).
Recently, we performed a structure–activity relationship study based on the lead compound, Z-Val-Leu-Ala(pyrrolidone-3-yl)-2-thiazole (7) [21]. This study led to the discovery of the potent compounds 8 and 9, with K i values in the low nanomolar range [22].
Extending our studies toward the development of new anti-SARS agents, we now report the design, synthesis, and evaluation of a series of low-molecular weight dipeptide-type compounds in which the P3 valine unit is removed from the previous lead Z-Val-Leu-Ala(pyrrolidone-3-yl)-2-benzothiazole compound (8, Fig. 1). A preliminary SAR study led to the identification of inhibitors with moderate to good inhibitory activities. In particular, compounds 26m and 26n exhibited potent inhibitory activities with K i values of 0.39 and 0.33 μM, respectively. The binding interactions of 26m were predicted using molecular modeling studies. We describe the results of these extensive studies in detail, including the design, synthesis, molecular modeling, and biological evaluation of a series of SARS-CoV 3CLpro inhibitors.
2. Results and discussion
2.1. Synthesis
The synthesis of the title inhibitors was achieved through a coupling reaction involving two key fragments, as shown in Scheme 1 . One of the key fragment intermediates (19) was synthesized from the amino acid esters 18 with either corresponding carboxylic acids via 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride–1-hydroxybenzotriazole (EDC·HCl–HOBt) mediated coupling in the presence of triethylamine (TEA) in DMF or acid chlorides in the presence of TEA in dichloromethane (CH2Cl2). The resulting N-protected amino acid esters (19) were consequently deprotected in the presence of TFA or lithium hydroxide·water (LiOH·H2O) to afford the N-protected amino acids (20), which were used directly in the subsequent step.
Scheme 1.
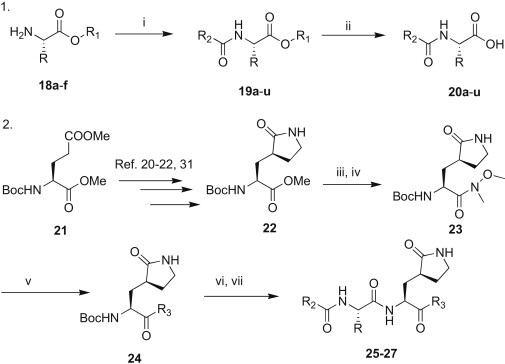
Reagents and conditions: i) EDC·HCl, HOBt·H2O, TEA/DMF (if reactant is a carboxylic acid) or Et3N/CH2Cl2 (if reactant is a benzyloxycarbonyl chloride), 0 °C–rt; ii) TFA/H2O, CH2Cl2 (if R1 = tert-butyl unit), or LiOH·H2O, THF/H2O, (if R1 = methyl unit); iii) 4M NaOH, MeOH; iv) HN(OMe)Me·HCl, EDC·HCl, HOBt·H2O, Et3N/DMF, 0 °C–rt; v) n-BuLi, THF, −78 °C (if R3 = benzothiazole) or LDA, THF, −78 °C (if R3 = 5-arylated thiazoles); vi) TFA/H2O (10:1), CH2Cl2; vii) 20, HBTU, DIPEA/DMF, 0 °C–rt followed by HPLC purification. Note: The substituents R–R3 are indicated in Table 1, Table 2, Table 3
The γ-lactam-thiazoles (24) were synthesized using an approach similar to the syntheses reported previously [21], [22], [31]. Accordingly, the optically pure l-glutamic acid ester 21 was converted to the γ-lactam ester 22 by treatment with bromoacetonitrile, followed by reduction with PtO2 (5%) and cyclization. The resulting ester 22 was hydrolyzed in the presence of 4 M NaOH in methanol to yield the corresponding acid, which was coupled to N,O-dimethylhydroxylamine via the EDC–HOBt method to afford the Weinreb amide 23. The Weinreb amide 23 was then coupled to the appropriate thiazoles in the presence of n-butyl lithium (n-BuLi) or lithium diisopropylamide (LDA) at −78 °C to afford the γ-lactam-thiazoles 24. The Boc protecting group was then removed, and the resulting intermediate was subsequently reacted with the above-mentioned N-protected amino acids 20 in the presence of O-benzotriazole-N,N,N′,N′-tetramethyluroniumhexafluoro phosphate (HBTU) and DIPEA in DMF to afford the title compounds 25–27. All compounds were purified by reverse phase HPLC and characterized by 1H and 13C NMR, and mass spectrometry analysis. The purity of each compound exceeded 90–95%.
2.2. Biological activity
The compounds were subjected to a fluorometric protease inhibitory assay against SARS-CoV 3CLpro, as described previously [32], [33]. Briefly, the kinetic parameters were determined at a constant substrate concentration, and the inhibitor concentrations were varied to assess the K i values [22]. The IC50 values were determined only for certain potent inhibitors, based on the apparent decrease in the substrate concentration (H-Thr-Ser-Ala-Val-Leu-Gln-Ser-Gly-Phe-Arg-Lys-NH2) upon digestion by R188I SARS 3CLpro, as described previously [19], [34]. The cleavage reaction was monitored by analytical HPLC, and the cleavage rates were calculated from the decrease in the substrate peak area. Table 1, Table 2, Table 3, Table 4 report the K i or IC50 values as the mean of 3 independent experiments.
Table 1.
SARS-CoV 3CLpro inhibitory activities (Ki) of 25a–h.
| Entry no. | Inhibitors | Ki (μM) | Entry no. | Inhibitors | Ki (μM) |
|---|---|---|---|---|---|
| 25a |  |
5.90 | 25e | 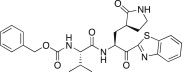 |
1.71 |
| 25b | 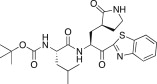 |
23.0 | 25f | 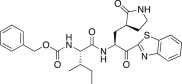 |
29.0 |
| 25c | 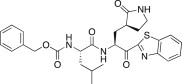 |
0.46 | 25g | 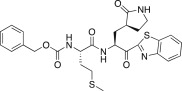 |
9.40 |
| 25d | 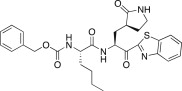 |
1.60 | 25h | 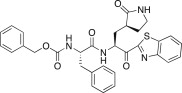 |
1.20 |
Table 2.
SARS-CoV 3CLpro inhibitory activities (Ki) of 26a–n.
| Entry No. | Inhibitors | Ki (μM) | Entry No. | Inhibitors | Ki (μM) |
|---|---|---|---|---|---|
| 26a | 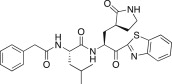 |
3.20 | 26h | 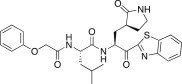 |
0.56 |
| 26b | 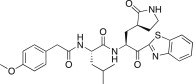 |
0.42 | 26i | 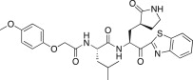 |
1.56 |
| 26c | 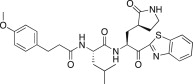 |
0.61 | 26j | 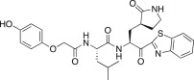 |
8.4 |
| 26d | 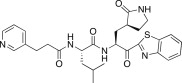 |
7.4 | 26k | 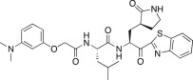 |
0.84 |
| 26e | 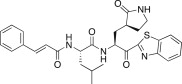 |
0.69 | 26l | 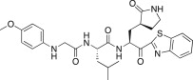 |
3.20 |
| 26f | 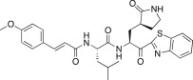 |
0.70 | 26m | 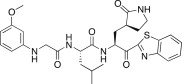 |
0.39 |
| 26g | 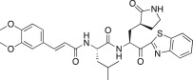 |
1.30 | 26n | 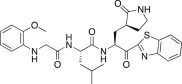 |
0.33 |
Table 3.
SARS-CoV 3CLpro inhibitory activities (Ki) of 27a–d.
| Entry no. | Inhibitors | Ki (μM) |
|---|---|---|
| 27a | 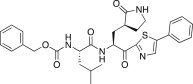 |
0.66 |
| 27b | 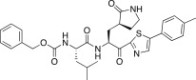 |
37.0 |
| 27c | 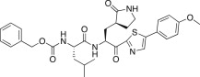 |
52.0 |
| 27d |  |
2.50 |
Table 4.
Inhibitory activities (IC50) of selected inhibitors.
| Entry no. | IC50 (μM) |
|---|---|
| 25c | 21.0 |
| 26b | 43.0 |
| 26h | 24.0 |
| 26m | 10.0 |
| 26n | 14.0 |
In an effort to develop low-molecular weight peptidic SARS-CoV 3CLpro inhibitors, we designed a series of dipeptide-type inhibitors based on the previous potent lead compound 8 (see Fig. 1). In a first attempt, the P3 valine region was removed and the P4-benzyloxycarbonyl (Cbz) in 8 was replaced with a series of small moieties with sizes similar to that of the P3 valine group in 8. The inhibitory activities of the resulting dipeptidic compounds bearing a valine mimic, isopentanoyl (25a; K i = 5.9 μM) or tert-butoxy carbonyl (25b; K i = 23 μM), were dramatically reduced compared to the activity of 8 (K i = 0.0041 μM) [22]; however, the introduction of Cbz (25c; K i and IC50 = 0.46 and 21.0 μM) as a P3 moiety resulted in a 12-fold or 50-fold activity increase for 25a or 25b, respectively, although the potency was reduced relative to the value for the tripeptidic lead 8. This result suggested that the Cbz group, which was introduced in place of the P3 scaffold in the dipeptidic 25c, conveyed appreciable activity; therefore, compound 25c could serve as a lead for further optimization steps. By retaining the P3 Cbz moiety in 25c, we examined the relevance of the leucine residue (or isobutyl unit) for P2 substrate selectivity in comparison with a variety of its congeners. Accordingly, a series of isosteres was introduced, including n-butyl (25d; K i = 1.60 μM), isopropyl (25e; K i = 1.71 μM), sec-butyl (25f; K i = 29.0 μM), 3-methyl(thio)ethyl (25g; K i = 9.40 μM), and benzyl (25h; K i = 1.20 μM). The compounds bearing n-butyl (25d), isopropyl (25e), or benzyl (25h) groups exhibited reasonable inhibitory activities, although the potencies were reduced by a factor of 3–4 relative to the value for 25c. The inhibitory activity of the compound bearing sec-butyl (25f) or 3-methyl(thio)ethyl (25g) was severely reduced compared to the activity of 25c. Therefore, the leucine residue (or isobutyl group) was more selective and appropriate as a P2 group in 25c, providing enhanced inhibitory activity.
The P3 moiety of the lead compound 25c was examined by introducing a wide variety of substituents, such as aryl (or heteroaryl) acetyls and propionyls, arylacrylyls, aryloxyacetyls, and N-arylglycyls. Initially, the aryl (or heteroaryl) acetyls and propionyls were introduced, including phenylacetyl (26a; K i = 3.20 μM), 4-methoxyphenylacetyl (26b; K i and IC50 = 0.42 and 43 μM), 4-methoxyphenylpropionyl (26c; K i = 0.62 μM), and pyridine-3-propionyl (26d; K i = 7.4 μM). Among the compounds, a 4-methoxyphenylacetyl derivative (26b) exhibited potent inhibitory activity; the phenylacetyl (26a) and pyridine-3-propionyl (26d) derivatives in particular displayed low inhibitory activities. Therefore, the 4-methoxyphenylacetyl moiety provided a good alternative to the P3 Cbz group in 25c. We next introduced the arylacrylyls, including cinnamoyl (26e; K i = 0.69 μM), 4-methoxycinnamoyl (26f; K i = 0.70 μM), and 3,4-dimethoxycinnamoyl (26g; K i = 1.30 μM). These compounds showed moderate inhibitory activities relative to the lead 25c; thus, the acrylyls were not considered further as P3 moieties in the context of 25c.
A previous report describing a SAR study of the tripeptidomimetic 7 (see Fig. 1) as a SARS inhibitor revealed that the introduction of an aryloxyacetyl at the N-terminal position (the P4 position) appeared to significantly enhance the inhibitory activity against SARS-CoV 3CLpro [22]. Therefore, we introduced a series of aryloxyacetyls, including phenoxyacetyl (26h; K i and IC50 = 0.56 and 24 μM), 4-methoxyphenoxyacetyl (26i; K i = 1.56 μM), 4-hydroxyphenoxyacetyl (26j; K i = 8.4 μM), and 3-dimethylaminophenoxyacetyl (26k; K i = 0.84 μM) as P3 groups in place of the Cbz group in 25c. Compounds 26h and 26k exhibited comparable activities to 25c; however, the other derivatives (26i and 26j) displayed reduced inhibitory activities relative to the lead compound 25c. These results suggested that the introduction of an aryloxyacetyl P3 group in the dipeptidic 25c did not appreciably improve the inhibitory activity against 3CLpro. We also introduced several N-arylglycyls, including N-(4-methoxyphenyl)glycyl (26l; K i = 3.20 μM), N-(3-methoxyphenyl)glycyl (26m; K i and IC50 = 0.39 and 10.0 μM), and N-(2-methoxyphenyl)glycyl (26n; K i and IC50 = 0.33 and 14.0 μM). The results of these studies revealed that compounds 26m and 26n displayed relatively potent inhibitory activities compared to the lead 25c. The compound bearing an N-(2-methoxyphenyl)glycyl (26n) was identified as the most potent inhibitor in the present study. This result suggested that the hydrogen bonding properties of the amino group on the N-arylglycyl moiety might have contributed to the improvement in activity (see Fig. 2 B).
Fig. 2.
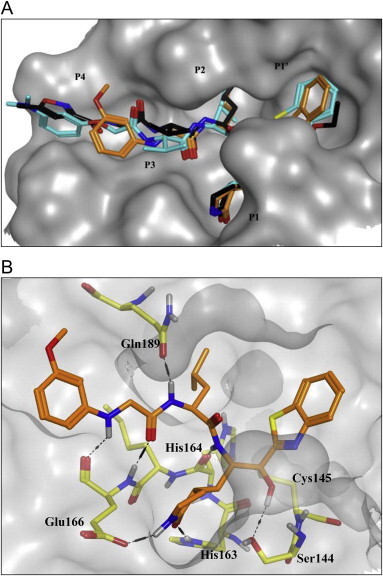
Molecular dynamics stimulated pose of compound 26m (orange stick) bound to SARS-CoV 3CLpro (PDB ID: 1WOF (black stick)). (A) Overlapped view of 26m with an original vinyl ester (black stick) and lead 9 (light blue stick); (B) compound 26m was shown as an orange stick. Dotted black lines represent the hydrogen bonding interaction. A notable hydrogen bonding interaction between the amino group (–NH) of N-(3-methoxyphenyl)glycyl of 26m with a backbone amino acid residue Glu166 of 3CLpro.
The P1′ moiety was examined next by varying the 5-substituted thiazoles (27a–d). The inhibitory activities of compounds 27a–d are illustrated in Table 3. Inhibitor 27a exhibited an inhibitory activity comparable to that of 25c. The other 5-arlylated thiazoles (27b–d) generally exhibited very low inhibitory activities compared to 25c. These studies confirmed that the benzothiazole unit was more suitable as a warhead group on the P1′ moiety in 25c.
2.3. Molecular docking study
The binding mode of compound 26m with 3CLpro was simulated using a molecular docking program as described previously [22]. We examined the molecular docking of the potent active compound 26m in comparison with docking of the tripeptidic 9 and a structurally similar ligand, the docking structure of which has been elucidated previously by X-ray crystallography (PDB ID: 1WOF, K i = 10.7 μM) (Fig. 2) [35]. Several minimization processes were performed using the MMFF94X force field to model the solvation environment surrounding the inhibitor. A molecular simulation was then performed. As shown in Fig. 2A, the P1′–P2 moieties in 26m, in the tripeptidic compound 9, and in the original ligand interacted with the same region of the protease. Interestingly, the aminoacetyl group on the N-(3-methoxyphenyl)glycyl group in 26m mimicked the P3 valine moieties of the tripeptidomimetic 9 and the reported ligand. The interactions of compound 26m with the protein are described in detail in Fig. 2B. Particularly, a notable hydrogen bonding involved between the amino group (–NH) of P3 N-(3-methoxyphenyl)glycine with a backbone amino acid residue Glu166 of 3CLpro was observed to have relatively potent inhibition than other inhibitors presented in the study against SARS-CoV 3CLpro. The essential nature of this hydrogen bond will be explored in further optimization studies of dipeptide-type SARS-CoV 3CLpro inhibitors.
3. Conclusion
In an effort to develop low-molecular weight peptidic anti-SARS agents, we designed, synthesized, and evaluated a series of dipeptide-type inhibitors in which the P3 valine unit in our potent tripeptidomimetic lead compound 8 was replaced with a number of distinct functionalities. In a preliminary study, the compound 25c, bearing a benzyloxycarbonyl (Cbz) P3 moiety, was identified as a lead compound. The P3 moiety in 25c was systematically modified with a view to improve the biological activity. This study led to the identification of a compound bearing an N-arylglycyl as a P3 moiety as having higher inhibitory activity due to the presence of a hydrogen bond with the backbone amino acid residue Glu166 of 3CLpro. Leucine and benzothiazole units were identified as appropriate P2 and P1′ moieties, respectively, in 25c. Accordingly, compounds 26m and 26n were recognized as potent inhibitors in the present study. These potent dipeptidic inhibitors provide attractive leads, which are undergoing further structural modifications in an effort to improve the pharmaceutical profiles. These processes are underway and will be reported in the near future.
4. Experimental section
4.1. Materials and methods
Reagents and solvents were purchased from Wako Pure Chemical Ind., Ltd. (Osaka, Japan), and Aldrich Chemical Co. Inc. (Milwaukee, WI) and were used without further purification. Analytical thin-layer chromatography (TLC) was performed on Merck Silica Gel 60F254 pre-coated plates. Preparative HPLC was performed using a C18 reverse-phase column (19 × 150 mm; Sun-Fire Prep C18 OBD™, 5 μm) with a binary solvent system: a linear gradient of CH3CN in 0.1% aqueous TFA at a flow rate of 6 mL/min, detected at UV 254 and 230 nm. All solvents used for HPLC were HPLC-grade. All other chemicals were of analytical grade or better. 1H and 13C NMR spectra were obtained using a JEOL 400 MHz spectrometer, a Varian Mercury 300 spectrometer (300 MHz), or a BRUKER AV600 spectrometer (600 MHz) with tetramethylsilane as an internal standard. High-resolution mass spectra (ESI or EI) were recorded on a micromass Q-Tof Ultima API or a JEOL JMS-GCmate BU-20 spectrometer. Mass spectra (ESI) were recorded on an LCMS-2010EV (SHIMADZU).
4.2. Synthetic procedures
4.2.1. Synthetic procedures for the preparation of (S)-tert-butyl 2-(3-methylbutanamido)-4-methylpentanoate (19a)
To a solution of the commercially available l-leucine tert-butyl ester 18a (0.200 g, 0.89 mmol) in DMF (15 mL) was added an isovaleric acid (0.100 g, 0.98 mmol), HOBt·H2O (0.151 g, 0.98 mmol), and EDC·HCl (0.189 g, 0.98 mmol). The resulting solution was cooled to 0 °C under ice bath conditions, and TEA was then added dropwise. After 5 min, the ice bath was removed and the mixture was allowed to stir for 2 h at ambient temperature. DMF was removed under high vacuum, and the resulting residue was dissolved in EtOAc (30 mL). The organic layer was washed with 5% citric acid (20 mL × 2), 5% NaHCO3 (20 mL × 2), and brine (20 mL). The solution was dried over Na2SO4, filtered, and evaporated under reduced pressure to give compound 19a. The resulting crude compound was purified by silica gel column chromatography using hexane–EtOAc as eluents. Yield 65%; white solid; 1H NMR (400 MHz, CD3OD): δ 4.20 (t, J = 7.6 Hz, 1H), 2.03–1.90 (m, 3H), 1.57–1.42 (m, 3H), 1.34 (s, 9H), 0.86–0.79 (m, 12H). HRMS (ESI): m/z calcd for C15H30NO3 [M + H]+ 272.2226, found 272.2230.
The intermediates 19h–u were prepared from l-leucine tert-butyl ester 18a and various commercially available carboxylic acids according to the procedure described for the synthesis of 19a.
4.2.2. Synthesis of benzyl (S)-1-(tert-butoxycarbonyl)-3-methylbutylcarbamate (19b)
To a solution of the commercially available l-leucine tert-butyl ester 18a (0.500 g, 2.20 mmol) in CH2Cl2 (20 mL) was added benzyloxycarbonyl chloride (0.410 mL, 2.5 mmol). The resulting solution was cooled to 0 °C and TEA (0.380 mL, 2.5 mmol) was then added dropwise. After 5 min, the ice bath was removed and the mixture was allowed to stir for 2 h at room temperature. The mixture was washed with H2O (20 mL) and brine (10 mL). The resulting solution was dried over Na2SO4, filtered, and evaporated under reduced pressure to give compound 19b, which was purified by silica gel column chromatography using hexane–EtOAc as eluents. Yield 85%; colorless oil; 1H NMR (400 MHz, CDCl3): δ 7.35–7.28 (m, 5H, merged with CDCl3), 5.10 (s, 2H), 4.29–4.23 (m, 1H), 1.74–1.67 (m, 2H), 1.62–1.58 (m, 1H), 1.44 (s, 9H), 0.95–0.93 (m, 6H). HRMS (ESI): m/z calcd for C18H27NO4Na [M + Na]+ 344.1838, found 344.1848.
The intermediates 19c–g were prepared from benzyloxycarbonyl chloride and various commercially available amino acid esters 18b–f according to the procedure described for the synthesis of 19c.
4.2.3. Benzyl (S)-1-(methoxycarbonyl)pentylcarbamate (19c)
Yield 67% from l-norleucine methyl ester (18b); colorless oil; 1H NMR (400 MHz, CDCl3): δ 7.36–7.29 (m, 5H), 5.11 (s, 2H), 4.39–4.34 (q, J = 5.6 Hz, 1H), 3.73 (s, 3H), 1.88–1.79 (m, 1H), 1.66–1.63 (m, 1H), 1.38–1.33 (m, 4H), 0.90–0.87 (t, J = 6.4 Hz, 3H). HRMS (ESI): m/z calcd for C15H22NO4 [M + H]+ 280.1549, found 280.1545.
4.2.4. Benzyl (S)-1-(tert-butoxycarbonyl)-2-methylpropylcarbamate (19d)
Yield 71% from l-valine tert-butyl ester (18c); colorless oil; 1H NMR (400 MHz, CDCl3): δ 7.39–7.31 (m, 5H), 5.10 (s, 2H), 4.20–4.17 (m, 1H), 2.15–2.12 (m, 1H), 1.46 (s, 9H), 0.96–0.87 (m, 6H). HRMS (ESI): m/z calcd for C17H25NO4Na [M + Na]+ 330.1681, found 330.1683.
4.2.5. Benzyl (1S,2R)-1-(tert-butoxycarbonyl)-2-methylbutylcarbamate (19e)
Yield 66% from l-isoleucine tert-butyl ester (18d); colorless oil; 1H NMR (400 MHz, CDCl3): δ 7.36–7.29 (m, 5H), 5.30–5.28 (m, NH, 1H), 5.17 (s, 2H), 4.25–4.20 (m, 1H), 1.85–1.80 (m, 1H), 1.44 (s, 9H), 1.21–1.01 (m, 2H), 0.94–0.91 (m, 6H). HRMS (ESI): m/z calcd for C18H28NO4 [M + H]+ 322.2018, found 322.2010.
4.2.6. Benzyl (S)-1-(tert-butoxycarbonyl)-3-(methylthio)propylcarbamate (19f)
Yield 72% from l-methionine tert-butyl ester (18e); colorless oil; δ 7.36–7.28 (m, 5H), 5.10 (s, 2H), 4.36 (q, J = 6.0 Hz, 1H), 2.54–2.46 (m, 2H), 2.08 (m, 5H), 1.94–1.92 (m, 2H), 1.46 (s, 9H). HRMS (ESI): m/z calcd for C17H26NO4S [M + H]+ 340.1583, found 340.1580.
4.2.7. Benzyl (S)-1-(methoxycarbonyl)-2-phenylethylcarbamate (19g)
Yield 82% from l-phenylalanine methyl ester (18f): colorless solid; 1H NMR (400 MHz, CDCl3): δ 7.36–7.31 (m, 4H), 7.29–7.21 (m, 3H), 7.10–7.07 (m, 3H), 5.09 (s, 2H), 4.68–4.63 (m, 1H), 3.70 (s, 3H), 3.16–3.04 (m, 2H). HRMS (ESI): m/z calcd for C18H19NO4Na [M + Na]+ 336.1212, found 336.1224.
4.2.8. (S)-tert-Butyl 2-(2-phenylacetamido)-4-methylpentanoate (19h)
Yield 59% from l-leucine tert-butyl ester (18a); colorless solid; 1H NMR (400 MHz, CDCl3): δ 7.35 (t, J = 7.6 Hz, 2H), 7.31–7.28 (m, 3H, merged with CDCl3), 4.52–4.47 (m, 1H), 3.59 (s, 2H), 1.57–1.52 (m, 3H), 1.42 (s, 9H), 0.88 (d, J = 4.8 Hz, 6H). HRMS (ESI): m/z calcd for C18H28NO3 [M + H]+ 306.2069, found 306.2065.
4.2.9. (S)-tert-Butyl 2-(2-(4-methoxyphenyl)acetamido)-4-methylpentanoate (19i)
Yield 69% from l-leucine tert-butyl ester (18a); colorless solid; 1H NMR (400 MHz, CDCl3): δ 7.17 (d, J = 8.4 Hz, 2H), 6.87 (d, J = 8.0 Hz, 2H), 4.51–4.46 (m, 1H), 3.79 (s, 3H), 3.53 (s, 2H), 1.57–1.52 (m, 3H), 1.42 (s, 9H), 0.88–0.87 (m, 6H). HRMS (ESI): m/z calcd for C19H27NO4 [M + H]+ 336.2175, found 336.2173.
4.2.10. (S)-tert-Butyl 2-(3-(4-methoxyphenyl)propanamido)-4-methylpentanoate (19j)
Yield 79% from l-leucine tert-butyl ester (18a); colorless oil; 1H NMR (400 MHz, CDCl3): δ 7.13 (d, J = 8.8 Hz, 2H), 6.83 (d, J = 8.0 Hz, 2H), 4.54–4.48 (m, 1H), 3.79 (s, 3H), 2.94–2.90 (m, 2H), 2.55–2.44 (m, 2H), 1.56–1.44 (m, 3H), 1.44 (s, 9H), 0.90–0.88 (m, 6H). HRMS (ESI): m/z calcd for C20H31NO4Na [M + Na]+ 372.2151, found 372.2145.
4.2.11. (S)-tert-Butyl 2-(3-(pyridin-3-yl)propanamido)-4-methylpentanoate (19k)
Yield 56% from l-leucine tert-butyl ester (18a); yellow oil; 1H NMR (400 MHz, CDCl3): δ 8.52 (s, 1H), 8.46 (d, J = 3.6 Hz, 1H), 7.74–7.69 (t, J = 8.0 Hz, 1H), 7.32–7.28 (m, 1H), 4.52–4.47 (m, 1H), 3.05–3.01 (m, 2H), 2.62–2.52 (m, 2H), 1.58–1.48 (m, 3H), 1.46 (s, 9H), 0.91–0.89 (m, 6H). HRMS (ESI): m/z calcd for C18H29N2O3 [M + H]+ 321.2178, found 321.2188.
4.2.12. (S)-tert-Butyl 2-(cinnamamido)-4-methylpentanoate (19l)
Yield 79% from l-leucine tert-butyl ester (18a); colorless oil; 1H NMR (400 MHz, CD3OD): δ 7.44 (d, J = 21.0 Hz, 1H), 7.41 (d, J = 7.4 Hz, 2H), 7.29–7.26 (m, 5H), 6.58 (d, J = 21.0 Hz, 1H), 4.40–4.30 (m, 1H), 1.86–1.50 (m, 3H), 1.36 (s, 9H), 0.95–0.79 (m, 6H). HRMS (ESI): m/z calcd for C19H28NO3 [M + H]+ 318.2069, found 318.2077.
4.2.13. (S)-tert-Butyl 2-((E)-3-(4-methoxyphenyl)acrylamido)-4-methylpentanoate (19m)
Yield 76% from l-leucine tert-butyl ester (18a); colorless oil; 1H NMR (400 MHz, CDCl3): δ 7.58 (d, J = 15.6 Hz, 1H), 7.44 (d, J = 8.6 Hz, 2H), 6.88 (d, J = 8.6 Hz, 1H), 6.29 (d, J = 15.6 Hz, 1H), 4.70–4.65 (m, 1H), 3.89 (s, 3H), 1.73–1.48 (m, 3H), 1.43 (s, 9H), 0.98–0.95 (m, 6H). HRMS (ESI): m/z calcd for C20H31NO4 [M + H]+ 348.2175, found 348.2183.
4.2.14. (S)-tert-Butyl 2-((E)-3-(3,4-dimethoxyphenyl)acrylamido)-4-methylpentanoate (19n)
Yield 69% from l-leucine tert-butyl ester (18a); colorless oil; 1H NMR (400 MHz, CDCl3): δ 7.53 (d, J = 15.6 Hz, 1H), 7.04 (d, J = 8.0 Hz, 1H), 7.00 (s, 1H), 6.84 (d, J = 8.0 Hz, 1H), 6.30 (d, J = 15.6 Hz, 1H), 4.71–4.65 (m, 1H), 3.90 (s, 6H), 1.74–1.49 (m, 3H), 1.43 (s, 9H), 0.98–0.95 (m, 6H). HRMS (ESI): m/z calcd for C21H33NO5 [M + H]+ 378.2280, found 378.2282.
4.2.15. (S)-tert-Butyl 2-(2-phenoxyacetamido)-4-methylpentanoate (19o)
Yield 77% from l-leucine tert-butyl ester (18a); colorless oil; 1H NMR (400 MHz, CDCl3): δ 7.33 (t, J = 8.4 Hz, 2H), 7.03 (t, J = 8.0 Hz, 1H), 6.96 (d, J = 8.0 Hz, 2H), 4.63–4.60 (m, 1H), 4.52 (s, 2H), 1.68–1.54 (m, 3H), 1.47 (s, 9H), 0.96–0.94 (m, 6H). HRMS (ESI): m/z calcd for C18H27NO4Na [M + Na]+ 344.1838, found 344.1835.
4.2.16. (S)-tert-Butyl 2-(2-(4-methoxyphenoxy)acetamido)-4-methylpentanoate (19p)
Yield 79% from l-leucine tert-butyl ester (18a); yellow oil; 1H NMR (400 MHz, CDCl3): δ 6.90–6.84 (m, 4H), 4.62–4.59 (m, 1H), 4.46 (s, 2H), 3.78 (s, 3H), 1.68–1.54 (m, 3H), 1.47 (s, 9H), 0.96–0.94 (m, 6H). HRMS (ESI): m/z calcd for C18H29NO4Na [M + Na]+ 374.1943, found 374.1953.
4.2.17. (S)-tert-Butyl 2-(2-(4-hydroxyphenoxy)acetamido)-4-methylpentanoate (19q)
Yield 63% from l-leucine tert-butyl ester (18a); colorless solid; 1H NMR (400 MHz, CD3OD): δ 6.75–6.72 (m, 2H), 6.63–6.60 (m, 2H), 4.36 (s, 2H), 4.32–4.29 (m, 1H), 1.56–1.50 (m, 3H), 1.35 (s, 9H), 0.84–0.78 (m, 6H). HRMS (ESI): m/z calcd for C18H27NO5 [M + H]+ 338.1967, found 338.1961.
4.2.18. (S)-tert-Butyl 2-(2-(3-(dimethylamino)phenoxy)acetamido)-4-methylpentanoate (19r)
Yield 62% from l-leucine tert-butyl ester (18a); yellow solid; 1H NMR (400 MHz, CDCl3): δ 7.17 (t, J = 8.0 Hz, 1H), 6.97 (d, J = 8.0 Hz, 1H), 6.43 (d, J = 8.0 Hz, 1H), 6.32 (s, 1H), 4.66–4.61 (m, 1H), 4.52 (s, 2H), 2.85 (s, 6H), 1.68–1.55 (m, 3H), 1.46 (s, 9H), 0.97–0.94 (m, 6H). HRMS (ESI): m/z calcd for C20H33N2O4 [M + H]+ 365.2440, found 365.2430.
4.2.19. (S)-tert-Butyl 2-(2-((4-methoxyphenyl)amino)acetamido)-4-methylpentanoate (19s)
Yield 54% from l-leucine tert-butyl ester (18a); brown viscous oil; 1H NMR (400 MHz, CDCl3): δ 6.78 (d, J = 8.0 Hz, 2H), 6.58 (d, J = 8.0 Hz, 1H), 4.56–4.49 (m, 1H), 3.77–3.71 (m, 5H), 1.55–1.38 (m, 12H), 0.96–0.86 (m, 6H). HRMS (ESI): m/z calcd for C19H30N2O4 [M + H]+ 351.2284, found 351.2279.
4.2.20. (S)-tert-Butyl 2-(2-((3-methoxyphenyl)amino)acetamido)-4-methylpentanoate (19t)
Yield 57% from l-leucine tert-butyl ester (18a); brown viscous oil; 1H NMR (400 MHz, CDCl3): δ 7.00 (d, J = 8.0 Hz, 1H), 6.87–6.85 (m, 2H), 6.77 (s, 1H), 4.58–4.53 (m, 1H), 3.89–3.71 (m, 5H), 1.57–1.51 (m, 2H), 1.48–1.46 (m, 1H), 1.43 (s, 9H), 0.95–0.91 (m, 6H). HRMS (ESI): m/z calcd for C19H30N2O4 [M + H]+ 351.2284, found 351.2297.
4.2.21. (S)-tert-Butyl 2-(2-((2-methoxyphenyl)amino)acetamido)-4-methylpentanoate (19u)
Yield 60% from l-leucine tert-butyl ester (18a); light pink solid; 1H NMR (400 MHz, CDCl3): δ 6.87–6.74 (m, 3H), 6.51 (d, J = 8.0 Hz, 1H), 4.58–4.53 (m, 1H), 3.87–3.75 (s, 5H), 1.57–1.52 (m, 2H), 1.48–1.46 (m, 1H), 1.43 (s, 9H), 0.92–0.87 (m, 6H). HRMS (ESI): m/z calcd for C19H30N2O4 [M + H]+ 351.2284, found 351.2277.
4.3. Synthetic procedure for the preparation of 20
Procedure A: To a solution of the corresponding tert-butyl ester 19 (0.110 g, 0.31 mmol) in CH2Cl2 (2 mL) at 0 °C was added TFA/H2O (10:1, 3 mL). After 5 min stirring, the reaction mixture was allowed to stir at room temperature for 1 h. The solvent was completely evaporated under reduced pressure to give the corresponding acids 20, which were used directly in the subsequent step without further characterization.
Procedure B: To a solution of the corresponding methyl ester 19 (0.100 g, 0.35 mmol) in THF (3 mL) at room temperature was added LiOH in water (0.102 g, 2.45 mmol). After 3 h stirring, the solvent was completely evaporated under reduced pressure and the resulting residue was neutralized with 2 M HCl. The solution was extracted with EtOAc (20 mL × 2), dried over Na2SO4, filtered and evaporated under reduced pressure to give the corresponding acids 20, which were directly used for next step without further characterizations.
4.4. Synthesis of tert-butyl (S)-1-(N-methoxy-N-methylcarbamoyl)-2-((S)-2-oxopyrrolidin-3-yl)ethylcarbamate (23)
Compound 22 was prepared through sequential reactions from the well-known intermediate 21, as reported previously [20], [21], [22], [31].
To a solution of 22 (3.27 g, 8.0 mmol) in methanol (5 mL) was added a 4 M NaOH solution (0.04 mol) at room temperature. The mixture was stirred for 4 h and the methanol was evaporated. The residue was neutralized with 2 M HCl and extracted with EtOAc (20 mL × 4). The organic layer was washed with brine, dried over Na2SO4, and evaporated under reduced pressure to yield the corresponding acid. To a solution of the acid (12.0 mmol) in DMF (30 mL) were added EDC·HCl (0.540 g, 10.0 mmol), HOBt·H2O (0.330 g, 11.0 mmol), and N,O-dimethylhydroxylamine (0.213 g, 11.0 mmol) at ambient temperature. The solution was cooled to 0 °C, and TEA (0.240 mL, 11.0 mmol) was added slowly. After 2 h, the DMF was evaporated under high vacuum, and the resulting residue was dissolved in EtOAc (100 mL). The resulting solution was subsequently washed with 5% citric acid (20 mL × 2), 5% NaHCO3 (20 mL × 2), and brine (50 mL). The organic layer was then dried over Na2SO4 and concentrated under reduced pressure to yield the Weinreb amide derivative 23, which was purified by column chromatography (EtOAc/MeOH = 9.5:0.5).
The data for compounds 22 and 23 were reported previously [20], [21].
4.4.1. tert-Butyl ((S)-1-(benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)carbamate (24a)
To a solution of benzothiazole (1.37 g, 10.0 mmol) in THF at −78 °C was added n-BuLi (2.0 M in THF, 1.67 mL) dropwise over 20 min. After 1 h stirring, the Weinreb amide 23 (0.064 g, 2.0 mmol) in THF was slowly added dropwise over 20 min and then the solution was stirred for 3 h at same temperature. The reaction was quenched with sat. NH4Cl and allowed to stir at 0 °C for 20 min. The mixture was evaporated and dissolved in EtOAc. This solution was washed with water (100 mL) and brine (50 mL), and then dried over Na2SO4. After filtration, the organic layer was concentrated under reduced pressure and the resulting residue was subjected to flash chromatography (EtOAc/MeOH = 9:1) to obtain the pure compound 24a. The data for the compound 24a has been reported in a previous article [21].
4.4.2. tert-Butyl ((S)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)-1-(5-phenylthiazol-2-yl)propan-2-yl)carbamate (24b)
To a cooled solution of the commercially available 5-phenylthiazole (0.175 g, 10 mmol) in dry THF at −78 °C was slowly added a solution of LDA (1.0 mL, 14 mmol). After 1 h, a pre-cooled solution containing the Weinreb amide 23 (0.329 g, 10 mmol) in anhydrous THF was slowly added, and the reaction was stirred at −78 °C over 2 h. The solution was allowed to warm to room temperature, was quenched by the addition of water (35 mL), and then extracted with EtOAc (3 × 50 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and evaporated in vacuo. The crude mixture was then purified using flash chromatography (n-hexane/EtOAc = 3:7) to furnish 24b. The data for the compound 24b were reported previously [22].
Compounds 24c–e were prepared from 23 according to the procedure described for the synthesis of 24b.
4.4.3. tert-Butyl ((S)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)-1-(5-(p-tolyl)thiazol-2-yl)propan-2-yl)carbamate (24c)
The data for the compound 24c has been reported in a previous article [22].
4.4.4. tert-Butyl ((S)-1-(5-(4-methoxyphenyl)thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)carbamate (24d)
The data for the compound 24d has been reported in a previous article [22].
4.4.5. tert-Butyl ((S)-1-(5-(4-methoxyphenyl)thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)carbamate (24e)
The data for the compound 24e has been reported in a previous article [22].
4.5. Synthesis of (S)-N-((S)-1-(benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)-4-methyl-2-(3-methylbutanamido)pentanamide (25a)
To a solution of 24a (0.200 g, 0.5 mmol) in CH2Cl2 (3 mL) at 0 °C was added TFA/H2O (10:1, 5 mL), and the solution was stirred for 1 h. After evaporating the solvent under reduced pressure, the corresponding deprotected lactam residue (0.100 g, 0.53 mmol) was coupled to the carboxylic acid 20a (0.136 g, 0.38 mmol) using the coupling agent HBTU (0.147 g, 0.38 mmol) in the presence of diisopropylethylamine (0.050 mL, 0.38 mmol) in DMF (3 mL) at 0 °C. The reaction mixture was allowed to stir for 2–3 h under ambient conditions. The solvent was then evaporated under a high vacuum, and the residue was dissolved in EtOAc (50 mL). The organic layer was washed with 5% citric acid (20 mL × 2), 5% NaHCO3 (20 mL × 2), and brine (25 mL). This solution was dried over Na2SO4, filtered, and evaporated under reduced pressure to give compound 25a. Yield 35%; light yellow solid; 1H NMR (400 MHz, CD3OD): δ 8.21 (d, J = 8.0 Hz, 1H), 8.11 (d, J = 8.0 Hz, 1H), 7.65–7.51 (m, 2H), 5.72–5.69 (m, 1H), 4.61–4.42 (m, 1H), 3.38–3.29 (m, 4H, merged with CD3OD), 2.73–2.66 (m, 1H), 2.46–2.40 (m, 1H), 2.22–2.00 (m, 4H), 1.67–1.53 (m, 3H), 1.28–1.21 (m, 2H), 0.95–0.86 (m, 12H). 13C NMR (400 MHz, CD3OD): δ 193.3, 181.8, 175.8, 165.5, 154.8, 138.4, 129.3, 126.5, 123.7, 55.3, 53.1, 52.9, 46.1, 45.9, 41.8, 41.5, 40.0, 39.8, 29.4, 28.8, 27.5, 25.8, 23.4, 22.8, 21.9. HRMS (ESI): m/z calcd for C25H35N4O4S [M + H]+ 487.2379 found 487.2373.
Compounds 25b–h were prepared from 24a with 20a–h using a procedure similar to that described for the synthesis of 25a.
4.5.1. tert-Butyl ((S)-1-(((S)-1-(benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (25b)
Yield 37% from 24a; colorless solid; 1H NMR (400 MHz, CDCl3): δ 8.18 (d, J = 8.0 Hz, 1H), 7.99 (d, J = 7.6 Hz, 1H), 7.60–7.52 (m, 2H), 5.75–5.66 (m, 1H), 4.24–4.04 (m, 1H), 3.40–3.37 (m, 2H), 2.66–2.45 (m, 2H), 2.17–1.95 (m, 2H), 1.94–1.72 (m, 1H), 1.70–1.53 (m, 3H), 1.44 (s, 9H), 0.96–0.88 (m, 6H). HRMS (ESI): m/z calcd for C25H35N4O5SNa [M + Na]+ 525.2148, found 525.2145.
4.5.2. Benzyl ((S)-1-(((S)-1-(benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (25c)
Yield 43% from 24a; colorless solid; 1H NMR (400 MHz, CDCl3): δ 8.16 (d, J = 8.4 Hz, 1H), 7.97 (d, J = 8.4 Hz, 1H), 7.58–7.51 (m, 2H), 7.32–7.27 (m, 5H, merged with CDCl3), 5.70–5.58 (m, 1H), 5.12–5.08 (m, 2H), 4.39–4.14 (m, 1H), 3.30–3.27 (m, 2H), 2.64–2.49 (m, 2H), 2.24–2.10 (m, 2H), 2.09–1.89 (m, 1H), 1.86–1.67 (m, 2H), 1.66–1.48 (m, 1H), 0.93–0.85 (m, 6H). 13C NMR (400 MHz, CD3OD): δ 193.5, 181.8, 175.8, 165.5, 158.4, 154.8, 138.4, 129.4, 128.7, 127.3, 126.5, 123.7, 67.7, 67.5, 56.0, 55.1, 54.9, 42.0, 41.9, 39.9, 33.9, 32.7, 29.3, 28.7, 25.8, 23.7, 22.0, 21.8. HRMS (ESI): m/z calcd for C28H33N4O5S [M + H]+ 537.2172 found 537.2153.
4.5.3. Benzyl ((S)-1-(((S)-1-(benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-1-oxohexan-2-yl)carbamate (25d)
Yield 45% from 24a; light yellow solid; 1H NMR (400 MHz, CD3OD): δ 8.37–8.28 (m, 2H), 7.69–7.64 (m, 2H), 7.40–7.30 (m, 5H), 5.56–5.47 (m, 1H), 5.04–4.99 (m, 2H), 4.07–4.01 (m, 1H), 3.22–3.11 (m, 2H, merged with CD3OD), 2.51–2.11 (m, 3H), 1.97–1.75 (m, 2H), 1.67–1.49 (m, 2H), 1.21–1.08 (m, 4H), 0.85–0.72 (m, 3H). 13C NMR (400 MHz, CD3OD): δ 202.2, 192.8, 178.0, 172.4, 164.3, 155.8, 152.8, 137.0, 136.3, 77.2, 65.2, 64.2, 42.1, 41.9, 41.5, 37.8, 36.6, 32.0, 31.6, 31.5, 28.1, 27.4, 25.9, 23.6, 21.7, 21.5, 13.6. HRMS (ESI): m/z calcd for C28H33N4O5S [M + H]+ 537.2172 found 537.2166.
4.5.4. Benzyl ((S)-1-(((S)-1-(benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-3-methyl-1-oxobutan-2-yl)carbamate (25e)
Yield 39% from 24a; yellow solid; 1H NMR (400 MHz, CDCl3): δ 8.18 (d, J = 8.0 Hz, 1H), 7.98 (d, J = 8.0 Hz, 1H), 7.59–7.51 (m, 2H), 7.39–7.30 (m, 5H), 5.74–5.65 (m, 1H), 5.15–5.09 (m, 2H), 4.40–4.20 (m, 1H), 3.39–3.36 (m, 2H), 2.65–2.52 (m, 2H), 2.30–2.16 (m, 3H), 2.03–1.96 (m, 1H), 1.10–0.89 (m, 6H). 13C NMR (400 MHz, CD3OD): δ 193.5, 181.8, 174.6, 165.4, 158.5, 154.7, 138.4, 129.5, 129.0, 128.7, 126.6, 123.7, 79.5, 78.9, 67.6, 61.1, 55.3, 41.6, 40.0, 38.2, 38.1, 33.9, 33.2, 32.7, 28.7, 25.9, 23.7. HRMS (ESI): m/z calcd for C27H31N4O5S [M + H]+ 523.2015 found 523.2003.
4.5.5. Benzyl ((2S,3R)-1-(((S)-1-(benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-3-methyl-1-oxopentan-2-yl)carbamate (25f)
Yield 42% from 24a; light yellow solid; 1H NMR (400 MHz, CD3OD): δ 8.19 (d, J = 8.4 Hz, 1H), 8.08 (d, J = 8.4 Hz, 1H), 7.63–7.56 (m, 2H), 7.33–7.27 (m, 5H), 5.75–5.73 (m, 1H), 5.11–5.03 (s, 2H), 4.12–4.05 (m, 1H), 3.35–3.25 (m, 2H, merged with CD3OD), 2.70–2.50 (m, 1H), 2.49–2.39 (m, 1H), 2.38–2.01 (m, 2H), 2.00–1.85 (m, 1H), 1.80–1.75 (m, 1H), 1.67–1.49 (m, 2H), 1.08–0.86 (m, 6H). HRMS (ESI): m/z calcd for C28H33N4O5S [M + H]+ 537.2178 found 537.2178.
4.5.6. Benzyl ((S)-1-(((S)-1-(benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-(methylthio)-1-oxobutan-2-yl)carbamate (25g)
Yield 45% from 24a; white solid; 1H NMR (400 MHz, CDCl3): δ 8.19 (d, J = 8.4 Hz, 1H), 8.00 (d, J = 8.0 Hz, 1H), 7.59–7.52 (m, 2H), 7.35–7.25 (m, 5H), 5.78–5.61 (m, 1H), 5.20–5.10 (m, 2H), 4.60–4.44 (m, 1H), 3.40–3.34 (m, 2H), 2.69–2.50 (m, 1H), 2.49–2.38 (m, 2H), 2.38–2.09 (m, 2H), 2.08–2.00 (m, 5H), 1.99–1.89 (m, 2H). 13C NMR (400 MHz, CD3OD): δ 193.5, 181.8, 174.8, 165.7, 165.4, 158.4, 154.7, 138.4, 129.4, 129.3, 128.8, 127.3, 126.5, 123.7, 67.8, 56.0, 41.5, 40.0, 33.8, 32.8, 31.7, 31.0, 30.7, 29.3, 28.7, 24.1, 15.2. HRMS (ESI): m/z calcd for C27H31N4O5S2 [M + H]+ 555.1736 found 555.1743.
4.5.7. Benzyl ((S)-1-(((S)-1-(benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate (25h)
Yield 43% from 24a; colorless solid; 1H NMR (400 MHz, CDCl3): δ 8.19 (d, J = 7.6 Hz, 1H), 7.99 (d, J = 7.6 Hz, 1H), 7.61–7.55 (m, 2H), 7.51–7.17 (m, 10H, merged with CDCl3), 5.82–5.79 (m, 1H), 5.11–5.08 (m, 2H), 4.43–4.30 (m, 1H), 3.39–3.31 (m, 2H), 3.20–3.15 (m, 2H), 2.65–2.45 (m, 2H), 2.39–2.14 (m, 3H). 13C NMR (400 MHz, CDCl3): δ 192.9, 181.2, 174.3, 165.6, 163.8, 161.8, 154.8, 141.9, 139.2, 129.3, 128.9, 127.4, 127.0, 126.2, 125.9, 125.5, 123.3, 117.9, 114.1, 63.1, 55.2, 54.4, 41.6, 44.3, 41.4, 39.7, 38.2, 33.0, 27.8, 24.7, 22.8. HRMS (ESI): m/z calcd for C31H32N4O5S [M + H]+ 571.2015 found 571.2003.
4.6. Synthesis of 26
Compounds 26a–r were prepared from 24a with 20i–u using a procedure similar to that described for the synthesis of 25a.
4.6.1. (S)-N-((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)-4-methyl-2-(2-phenylacetamido)pentanamide (26a)
Yield 47% from 24a; white solid; 1H NMR (400 MHz, CD3OD): δ 8.09 (d, J = 8.0 Hz, 1H), 8.01 (d, J = 8.0 Hz, 1H), 7.55–7.47 (m, 2H), 7.19–7.11 (m, 5H), 5.61–5.57 (m, 1H), 4.35–4.32 (m, 1H), 3.44 (s, 2H), 3.40–3.36 (m, 1H), 3.19–3.07 (m, 1H), 2.57–2.52 (m, 1H), 2.39–2.20 (m, 2H), 2.12–1.88 (m, 2H), 1.79–1.48 (m, 3H), 0.88–0.76 (m, 6H). 13C NMR (400 MHz, CD3OD): δ 193.5, 181.8, 175.1, 165.5, 154.8, 138.4, 136.9, 130.2, 129.5, 128.4, 127.8, 126.5, 124.2, 123.7, 55.9, 55.2, 53.3, 43.7, 41.8, 39.9, 39.2, 33.7, 32.8, 29.4, 28.7, 25.8, 23.7, 22.1. HRMS (ESI): m/z calcd for C28H33N4O4S [M + H]+ 521.2223 found 521.2216.
4.6.2. (S)-N-((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)-2-(2-(4-methoxyphenyl)acetamido)-4-methylpentanamide (26b)
Yield 37% from 24a; yellow solid; 1H NMR (400 MHz, CD3OD): δ 8.20 (d, J = 8.0 Hz, 1H), 8.11 (d, J = 8.0 Hz, 1H), 7.64–7.58 (m, 2H), 7.20 (d, J = 8.0 Hz, 2H), 6.84 (d, J = 8.4 Hz, 2H), 5.69–5.67 (m, 1H), 4.45–4.41 (m, 1H), 3.73 (s, 3H), 3.51–3.21 (m, 4H, merged with CD3OD), 2.63–2.52 (m, 1H), 2.51–2.21 (m, 2H), 2.10–1.88 (m, 2H), 1.86–1.55 (m, 3H), 0.92–0.81 (m, 6H). 13C NMR (400 MHz, CD3OD): δ 193.5, 181.8, 175.1, 174.4, 165.5, 160.1, 154.8, 138.4, 131.4, 129.3, 128.4, 126.5, 123.7, 114.9, 55.6, 55.2, 42.8, 42.6, 41.6, 41.5, 39.9, 33.7, 29.4, 28.7, 25.8, 25.4, 24.2, 23.2, 22.1. HRMS (ESI): m/z calcd for C29H34N4O5SNa [M + Na]+ 573.2148 found 573.2148.
4.6.3. (S)-N-((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)-2-(3-(4-methoxyphenyl)propanamido)-4-methylpentanamide (26c)
Yield 43% from 24a; colorless solid; 1H NMR (400 MHz, CDCl3): δ 8.17 (d, J = 8.4 Hz, 1H), 7.98 (d, J = 8.0 Hz, 1H), 7.60–7.51 (m, 2H), 7.10 (d, J = 8.4 Hz, 2H), 6.81 (d, J = 8.4 Hz, 2H), 5.67–5.55 (m, 1H), 4.59–4.50 (m, 1H), 3.79 (s, 3H), 3.40–3.37 (m, 2H), 2.93–2.89 (m, 2H), 2.66–2.43 (m, 4H), 2.22–1.99 (m, 3H), 1.77–1.57 (m, 2H), 1.46–1.40 (m, 1H), 0.91–0.86 (m, 6H). 13C NMR (400 MHz, CDCl3): δ 192.2, 180.3, 172.2, 162.9, 162.5, 158.0, 155.7, 155.4, 147.6, 140.6, 132.7, 129.7, 129.3, 127.3, 113.8, 55.2, 54.8, 50.0, 41.5, 40.6, 38.6, 38.4, 34.8, 33.4, 30.0, 28.7, 27.9, 24.7, 22.8, 21.9. HRMS (ESI): m/z calcd for C30H36N4O5SNa [M + Na]+ 587.2034 found 587.2032.
4.6.4. (S)-N-((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)-4-methyl-2-(3-(pyridin-3-yl)propanamido)pentanamide (26d)
Yield 27% from 24a; brown solid; 1H NMR (400 MHz, CDCl3): δ 8.30–8.28 (m, 2H), 7.88–7.86 (d, J = 8.0 Hz, 1H), 7.66–7.64 (m, 2H), 7.51–7.26 (m, 2H), 7.20–7.16 (m, 1H), 5.61–5.57 (m, 1H), 4.49–4.45 (m, 1H), 3.35–3.29 (m, 2H), 3.01–2.97 (m, 2H), 2.83–2.80 (m, 2H), 2.71–2.69 (m, 2H), 2.58–2.42 (m, 3H), 2.41–2.25 (m, 2H), 1.90–1.84 (m, 1H), 1.73–1.53 (m, 2H), 0.96–0.85 (m, 6H). HRMS (ESI): m/z calcd for C28H34N5O4S [M + H]+ 536.2332 found 536.2327.
4.6.5. (S)-N-((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)-2-cinnamamido-4-methylpentanamide (26e)
Yield 41% from 24a; yellow solid; 1H NMR (400 MHz, CDCl3): δ 8.17–8.15 (m, J = 8.0 Hz, 1H), 7.97–7.93 (m, 1H), 7.70–7.50 (m, 5H), 7.49–7.45 (m, 2H), 6.47–6.43 (m, 1H), 5.70–5.60 (m, 1H), 4.90–4.72 (m, 1H), 3.40–3.27 (m, 2H), 2.60–2.45 (m, 2H), 2.40–1.99 (m, 2H), 1.95–1.80 (m, 1H), 1.76–1.42 (m, 3H), 0.96–0.83 (m, 6H). 13C NMR (400 MHz, CD3OD): δ 193.5, 181.2, 176.3, 165.7, 154.7, 142.4, 138.3, 136.2, 130.9, 129.9, 128.4, 126.5, 123.7, 121.3, 56.1, 55.6, 54.4, 53.7, 42.1, 41.6, 40.1, 33.8, 30.7, 29.52, 28.9, 26.0, 25.9, 23.3, 22.1. HRMS (ESI): m/z calcd for C29H33N4O4S [M + H]+ 533.2223 found 533.2243.
4.6.6. (S)-N-((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)-2-((E)-3-(4-methoxyphenyl)acrylamido)-4-methylpentanamide (26f)
Yield 33% from 24a; yellow solid; 1H NMR (400 MHz, CD3OD): δ 8.26 (d, J = 7.0 Hz, 1H), 8.18–8.10 (m, 1H), 7.70–7.55 (m, 2H), 7.54–7.42 (m, 3H), 6.95 (d, J = 8.8 Hz, 2H), 6.90–6.88 (m, 1H), 6.61–6.50 (m, 2H), 5.80–5.62 (m, 1H), 4.62–4.50 (m, 1H), 3.84 (s, 3H), 3.40–3.29 (m, 2H, merged with CD3OD), 2.80–2.65 (m, 1H), 2.63–2.45 (m, 1H), 2.43–2.20 (m, 1H), 2.19–1.99 (m, 1H), 1.92–1.78 (m, 1H), 1.77–1.50 (m, 3H), 1.05–0.88 (m, 6H). 13C NMR (400 MHz, CD3OD): δ 191.9, 180.2, 173.3, 166.6, 163.7, 160.8, 153.2, 140.9, 137.1, 129.3, 127.9, 127.4, 127.0, 125.5, 122.3, 117.9, 117.7, 114.1, 55.2, 54.4, 51.6, 51.4, 41.6, 40.4, 38.7, 33.0, 27.6, 24.6, 22.7, 21.6. HRMS (ESI): m/z calcd for C30H34N4O5S [M + H]+ 563.2328 found 563.2328.
4.6.7. (S)-N-((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)-2-((E)-3-(3,4-dimethoxyphenyl)acrylamido)-4-methylpentanamide (26g)
Yield 42% from 24a; yellow solid; 1H NMR (400 MHz, CDCl3): δ 8.20–8.14 (m, 1H), 8.00–7.94 (m, 1H), 7.60–7.50 (m, 3H), 7.10–6.99 (m, 2H), 6.84 (d, J = 8.0 Hz, 1H), 6.30 (d, J = 16.0 Hz, 1H), 5.85–5.60 (m, 1H), 4.78–4.66 (m, 1H), 3.90 (s, 6H), 3.42–3.30 (m, 2H), 2.70–2.45 (m, 2H), 2.40–2.25 (m, 2H), 2.20–1.85 (m, 2H), 1.80–1.64 (m, 2H), 1.10–0.88 (m, 6H). 13C NMR (400 MHz, CD3OD): δ 193.5, 182.4, 176.2, 169.7, 155.5, 152.3, 149.5, 142.8, 138.3, 135.3, 131.9, 129.3, 127.3, 125.7, 123.5, 119.3, 112.7, 117.7, 56.5, 54.2, 53.6, 42.3, 41.6, 40.0, 33.8, 30.7, 29.3, 28.8, 25.8, 23.6, 22.1. HRMS (ESI): m/z calcd for C31H36N4O6SNa [M + Na]+ 615.2253 found 615.2253.
4.6.8. (S)-N-((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)-4-methyl-2-(2-phenoxyacetamido)pentanamide (26h)
Yield 45% from 24a; colorless solid; 1H NMR (400 MHz, CDCl3): δ 8.17 (d, J = 8.0 Hz, 1H), 8.00–7.97 (m, 1H), 7.60–7.51 (m, 2H), 7.31–7.27 (m, 2H, merged with CDCl3), 7.10–6.85 (m, 3H), 5.70–5.65 (m, 1H), 4.76–4.66 (m, 1H), 4.55–4.50 (m, 2H), 3.40–3.21 (m, 2H), 2.70–2.50 (m, 2H), 2.35–2.19 (m, 1H), 2.34–2.20 (m, 2H), 2.19–1.85 (m, 1H), 1.80–1.65 (m, 2H), 1.00–0.88 (m, 6H). 13C NMR (400 MHz, CD3OD): δ 193.6, 181.8, 174.8, 174.5, 171.5, 165.5, 159.2, 154.7, 138.4, 130.6, 129.3, 126.7, 123.7, 115.8, 68.1, 55.9, 52.9, 41.9, 40.7, 39.8, 33.7, 29.2, 28.8, 25.7, 23.5, 22.0, 21.7, 21.2. HRMS (ESI): m/z calcd for C28H33N4O5S [M + H]+ 537.2174 found 537.2174.
4.6.9. (S)-N-((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)-2-(2-(4-methoxyphenoxy)acetamido)-4-methylpentanamide (26i)
Yield 45% from 24a; yellow solid; 1H NMR (400 MHz, CDCl3): δ 8.18 (d, J = 7.6 Hz, 1H), 7.99 (d, J = 7.6 Hz, 1H), 7.60–7.52 (m, 2H), 6.90–6.80 (m, 4H), 5.81–5.65 (m, 1H), 4.70–4.61 (m, 1H), 4.60–4.45 (m, 2H), 3.76 (s, 3H), 3.40–3.28 (m, 2H), 2.75–2.40 (m, 2H), 2.39–1.90 (m, 3H), 1.89–1.58 (m, 3H), 1.00–0.89 (m, 6H). 13C NMR (400 MHz, CD3OD): δ 192.5, 180.2, 172.4, 169.4, 168.7, 163.8, 154.7, 151.5, 145.3, 137.2, 128.0, 127.1, 125.6, 122.4, 115.9, 114.7, 68.1, 55.6, 53.8, 41.7, 40.6, 38.0, 37.8, 32.9, 28.8, 25.8, 23.5, 22.0, 21.8. HRMS (ESI): m/z calcd for C29H35N4O6S [M + H]+ 567.2277, found 567.2268.
4.6.10. (S)-N-((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)-2-(2-(4-hydroxyphenoxy)acetamido)-4-methylpentanamide (26j)
Yield 40% from 24a; yellow solid; 1H NMR (400 MHz, CD3OD): δ 8.21 (d, J = 8.1 Hz, 1H), 8.11 (d, J = 8.1 Hz, 1H), 7.65–7.58 (m, 2H), 6.82–6.78 (m, 2H), 6.70–6.65 (m, 2H), 5.78–5.66 (m, 1H), 4.55–4.50 (m, 1H), 4.60–4.45 (m, 2H), 3.35–3.18 (m, 2H, merged with CD3OD) 2.75–2.45 (m, 1H), 2.44–2.20 (m, 2H), 2.19–1.89 (m, 2H), 1.88–1.61 (m, 1H), 1.60–1.35 (m, 2H), 0.99–0.97 (m, 6H). 13C NMR (400 MHz, CD3OD): δ 192.9, 181.5, 173.2, 165.3, 154.7, 138.4, 132.3, 128.5, 127.2, 126.5, 124.3, 123.7, 116.8, 113.1, 69.2, 56.0, 54.2, 42.0, 40.0, 39.8, 34.0, 32.6, 29.2, 28.8, 25.7, 23.4, 22.0, 21.7. HRMS (ESI): m/z calcd for C28H32N4O6SNa [M + Na]+ 575.1940 found 575.1926.
4.6.11. (S)-N-((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)-2-(2-(3-(dimethylamino)phenoxy)acetamido)-4-methylpentanamide (26k)
Yield 35% from 24a; light green solid; 1H NMR (400 MHz, CDCl3): δ 8.18 (d, J = 7.6 Hz, 1H), 7.99–7.97 (d, J = 8.0 Hz, 1H), 7.60–7.52 (m, 2H), 7.29–7.22 (m, 1H, merged with CDCl3), 7.11 (d, J = 8.4 Hz, 1H), 6.77–6.70 (m, 1H), 6.56 (dd, 8.1, 8.4 Hz, 2H), 5.70–5.66 (m, 1H), 4.79–4.67 (m, 1H), 4.66–4.50 (m, 2H), 3.41–3.28 (m, 2H), 3.04 (s, 3H), 3.01 (s, 3H), 2.75–2.60 (m, 1H), 2.59–2.49 (m, 1H), 2.48–2.35 (m, 1H), 2.34–2.10 (m, 2H), 2.05–1.85 (m, 1H), 1.79–1.57 (m, 2H), 0.99–0.90 (m, 6H). 13C NMR (400 MHz, CD3OD): δ 193.6, 181.9, 178.0, 172.3, 171.6, 171.3, 165.7, 142.6, 138.4, 131.2, 129.3, 128.5, 126.8, 123.7, 121.9, 90.7, 77.2, 66.7, 57.4, 52.8, 41.5, 40.0, 33.7, 28.8, 25.7, 23.4, 22.0, 21.7. HRMS (ESI): m/z calcd for C30H38N5O5S [M + H]+ 580.2594 found 580.2563.
4.6.12. (S)-N-((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)-2-(2-((4-methoxyphenyl)amino)acetamido)-4-methylpentanamide (26l)
Yield 45% from 24a; light yellow solid; 1H NMR (400 MHz, CD3OD): δ 8.24 (d, J = 8.0 Hz, 1H), 8.14 (d, J = 8.0 Hz, 1H), 7.68–7.60 (m, 2H), 7.06 (t, J = 8.4 Hz, 1H), 6.30–6.17 (m, 3H), 5.72–5.68 (m, 1H), 4.53–4.50 (m, 1H), 3.84–3.65 (m, 2H), 3.82 (s, 3H), 3.37–3.28 (m, 2H, merged with CD3OD), 2.72–2.55 (m, 1H), 2.54–2.29 (m, 1H), 2.28–1.83 (m, 3H), 1.62–1.41 (m, 3H), 0.90–0.83 (m, 6H). 13C NMR (400 MHz, CD3OD): δ 193.4, 181.8, 175.4, 165.6, 164.0, 155.8, 154.7, 138.3, 132.0, 129.3, 129.2, 128.4, 127.1, 126.4, 124.2, 122.9, 116.7, 113.9, 105.2, 103.2, 56.2, 53.4, 41.8, 40.0, 39.4, 33.9, 29.3, 28.8, 26.1, 22.1, 21.9. HRMS (ESI): m/z calcd for C29H36N5O5S [M + H]+ 566.2437 found 566.2428.
4.6.13. (S)-N-((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)-2-(2-((3-methoxyphenyl)amino)acetamido)-4-methylpentanamide (26m)
Yield 41% from 24a; light yellow solid; 1H NMR (400 MHz, CD3OD): δ 8.21 (d, J = 7.6 Hz, 1H), 8.12 (d, J = 7.6 Hz, 1H), 7.66–7.58 (m, 2H), 7.10–6.89 (m, 4H), 5.80–5.65 (m, 1H), 4.60–4.45 (m, 1H), 3.94–3.72 (m, 2H), 3.76 (s, 3H), 3.36–3.29 (m, 2H, merged with CD3OD), 2.75–2.55 (m, 1H), 2.49–2.25 (m, 1H), 2.20–1.80 (m, 3H), 1.70–1.45 (m, 3H), 0.99–0.84 (m, 6H). 13C NMR (400 MHz, CD3OD): δ 193.4, 181.8, 177.4, 162.3, 160.2, 157.8, 141.2, 138.4, 133.9, 129.3, 128.4, 126.5, 123.7, 119.3, 116.4, 109.9, 66.0, 55.6, 55.4, 53.0, 42.0, 41.7, 41.5, 34.3, 28.8, 25.7, 23.3, 21.9, 21.5. HRMS (ESI): m/z calcd for C29H36N5O5S [M + H]+ 566.2437 found 566.2415.
4.6.14. (S)-N-((S)-1-(Benzo[d]thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)-2-(2-((2-methoxyphenyl)amino)acetamido)-4-methylpentanamide (26n)
Yield 45% from 24a; light yellow solid; 1H NMR (400 MHz, CD3OD): δ 8.11 (d, J = 7.6 Hz, 1H), 8.02 (d, J = 7.6 Hz, 1H), 7.56–7.48 (m, 2H), 6.85–6.82 (m, 1H), 6.79–6.87 (m, 2H), 6.56–6.54 (m, 1H), 5.65–5.56 (m, 1H), 4.43–4.39 (m, 1H), 3.81–3.73 (m, 5H), 3.31–3.25 (m, 2H, merged with CD3OD), 2.58–2.45 (m, 2H), 2.44–2.10 (m, 1H), 2.09–1.68 (m, 3H), 1.40–1.38 (m, 3H), 0.80–0.71 (m, 6H). 13C NMR (400 MHz, CD3OD): δ 193.4, 181.8, 174.9, 173.3, 165.5, 159.8, 154.4, 138.4, 133.3, 130.2, 129.3, 128.0, 126.9, 123.7, 123.2, 116.3, 65.4, 56.1, 55.4, 53.0, 42.0, 41.6, 40.1, 34.3, 33.7, 28.8, 25.7, 23.3, 21.9, 21.2. HRMS (ESI): m/z calcd for C29H36N5O5S [M + H]+ 566.2437 found 566.2426.
4.7. Synthesis of 27
Compounds 27a–d were prepared from 24b–d with 20a using a similar procedure to that described for the synthesis of 25a.
4.7.1. Benzyl ((S)-4-methyl-1-oxo-1-(((S)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)-1-(5-phenylthiazol-2-yl)propan-2-yl)amino)pentan-2-yl)carbamate (27a)
Yield 55% from 24b; colorless solid; 1H NMR (400 MHz, CD3OD): δ 8.32–8.30 (m, 1H), 7.72 (d, J = 7.20 Hz, 2H), 7.48–7.40 (m, 3H), 7.37–7.24 (m, 5H), 5.65–5.61 (m, 1H), 5.15–5.03 (m, 2H), 4.30–4.20 (m, 1H), 3.40–3.23 (m, 2H, merged with CD3OD), 2.75–2.65 (m, 1H), 2.64–2.06 (m, 2H), 2.05–1.80 (m, 2H), 1.79–1.60 (m, 1H), 1.59–1.52 (m, 2H), 0.99–0.89 (m, 6H). 13C NMR (400 MHz, CD3OD): δ 192.0, 181.8, 175.8, 164.2, 158.4, 148.9, 141.9, 131.5, 130.9, 130.7, 129.4, 128.9, 128.7, 128.3, 67.7, 61.5, 55.7, 54.8, 42.4, 41.5, 40.1, 38.9, 34.2, 29.3, 28.6, 25.9, 24.2, 23.4, 22.1, 21.8. HRMS (ESI): m/z calcd for C30H35N4O5S [M + H]+ 563.2328 found 563.2308.
4.7.2. Benzyl ((S)-4-methyl-1-oxo-1-(((S)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)-1-(5-(p-tolyl)thiazol-2-yl)propan-2-yl)amino)pentan-2-yl)carbamate (27b)
Yield 51% from 24c; colorless solid; 1H NMR (400 MHz, CD3OD): δ 8.32–8.29 (m, 1H), 7.62 (d, J = 8.4 Hz, 2H), 7.40–7.20 (m, 7H), 5.70–5.57 (m, 1H), 5.15–5.01 (m, 2H), 4.30–4.18 (m, 1H), 3.40–3.25 (m, 2H, merged with CD3OD), 2.75–2.40 (m, 1H), 2.50–2.30 (m, 2H), 2.38 (s, 3H), 2.29–2.09 (m, 1H), 2.08–1.75 (m, 2H), 1.74–1.65 (m, 1H), 1.63–1.49 (m, 2H), 0.97–0.90 (m, 6H). HRMS (ESI): m/z calcd for C31H37N4O5S [M + H]+ 577.2485 found 577.2484.
4.7.3. Benzyl ((S)-1-(((S)-1-(5-(4-methoxyphenyl)thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (27c)
Yield 47% from 24d; light yellow solid; 1H NMR (400 MHz, CDCl3): δ 8.05–8.03 (m, 1H), 7.60–7.52 (m, 2H), 7.60–7.27 (m, 5H), 7.00–6.95 (m, 2H), 5.64–5.56 (m, 1H), 5.20–5.08 (m, 1H), 4.40–4.25 (m, 1H), 3.86 (s, 3H), 3.35–3.34 (m, 2H), 2.67–2.42 (m, 2H), 2.22–2.04 (m, 2H), 2.02–1.96 (m, 1H), 1.80–1.52 (m, 3H), 0.97–0.86 (m, 6H). HRMS (ESI): m/z calcd for C31H37N4O6S [M + H]+ 593.2434 found 593.2414.
4.7.4. Benzyl ((S)-1-(((S)-1-(5-(2-methoxyphenyl)thiazol-2-yl)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (27d)
Yield 51% from 24e; light yellow solid; 1H NMR (400 MHz, CDCl3): δ 8.12–8.10 (m, 1H), 7.37–7.27 (m, 6H), 7.19 (d, J = 7.6 Hz, 1H), 7.13–7.11 (m, 1H), 6.95 (dd, 1.8, 8.2 Hz, 1H), 5.70–5.53 (m, 1H), 5.15–5.08 (m, 2H), 4.40–4.30 (m, 1H), 3.85 (s, 3H), 3.50–3.25 (m, 2H), 2.70–2.37 (m, 2H), 2.35–2.05 (m, 2H), 2.04–1.80 (m, 1H), 1.79–1.60 (m, 2H), 1.59–1.45 (m, 1H), 0.99–0.80 (m, 6H). 13C NMR (400 MHz, CDCl3): δ 190.8, 180.3, 172.8, 162.9, 160.1, 156.1, 147.4, 140.6, 136.4, 131.5, 130.4, 128.4, 127.9, 119.7, 115.1, 113.1, 66.8, 55.4, 53.4, 42.2, 41.7, 40.7, 38.9, 33.5, 31.9, 28.2, 24.7, 24.6, 23.0, 22.9, 22.0. HRMS (ESI): m/z calcd for C31H37N4O6S [M + H]+ 593.2434 found 593.2427.
4.8. Molecular docking studies
The crystal structure of the SARS-CoV 3CL protease in complex with a substrate analog inhibitor (coded 1WOF) [35] was obtained from the Protein Data Bank (PDB; http://www.rcsb.org/pdb/home/home.do). Initially, a binding model of 26m with 3CLpro was simulated to form the basis of a comparison with our previous potent lead tripeptidomimetic 9 and a substrate analog, using molecular operating environment (MOE) software. Several minimization processes were performed using the MMFF94X force field to model the solvation environment surrounding the inhibitor. Structures having a relatively low binding free energy and a high number of cluster members were selected for the subsequent docking conformation optimization step. The minimized energies of 26m, obtained from the docking study, were −40.67 and −35.52 kcal/mol.
Acknowledgments
This research was supported by Grants from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, including a Grant-in-aid for Young scientist (Tokubetsu Kenkyuin Shorei-hi) 23·01104 and a Grant-in-aid for Scientific Research 23659059 and 23390029. E.F. acknowledges support from the National Institutes of Health (grant GM57144).
Footnotes
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ejmech.2013.05.005.
Appendix A. Supplementary data
References
- 1.Drosten C., Gunther S., Preiser W., Ven der Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolensnikova L., Fouchier R.A.M., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D.M.E., Schmitz H., Doerr H.W. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 2.Lee N., Hui D., Wu A., Chan P., Cameron P., Joynt F.M., Ahuja A., Yung M.Y., Leung C.B., To K.F., Leu M.D., Szeto C.C., Chung S., Sung J.J.Y. N. Engl. J. Med. 2003;348:1986–1994. doi: 10.1056/NEJMoa030685. [DOI] [PubMed] [Google Scholar]
- 3.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 4.Anand K., Ziebuhr J., Wadhwani P., Mesturs J.R., Hilgenfeld R. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 5.Peiris J.S., Lai S.T., Poon L.L., Guan Y., Yam L.Y., Lim W., Nicholls J., Yee W.K., Yan W.W., Cheung M.T., Cheng V.C., Chan K.H., Tsang D.N., Yung R.W., Ng T.K., Yuen K.Y. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. (and references therein) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chem M.H., Tong W., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 7.Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Sott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bermard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 8.Thiel V., Ivanov K.A., Putics A.A., Hertzig T., Schelle B., Bayer S., Weiabrich B., Snijder E.J., Rabenau H., Doerr H.W., Ziebuhr J. J. Gen. Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 9.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. Proc. Natl. Acad. Sci. U. S. A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chou K., Wei D., Zhong W. Biochem. Biophys. Res. Commun. 2003;308:148–151. doi: 10.1016/S0006-291X(03)01342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pyrc K., Berkhout B., van der Hoek L. J. Virol. 2007;81:3051–3057. doi: 10.1128/JVI.01466-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fielding B.C. Future Microbiol. 2011;6:153–159. doi: 10.2217/fmb.10.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zaki A.M., van Boheemen S., Bestebroer T.M., Osterhaus A.D.M.E., Fouchier R.A.M. N. Engl. J. Med. 2012;367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 14.http://www.independent.co.uk/life-style/health-and-families/health-news/sarslike-virus-spreads-persontoperson-in-the-uk-8492750.html.
- 15.Shao Y.M., Yang W.B., Kuo T.H., Tsai K.C., Lin C.H., Yang A.S., Liang P.H., Wong C.H. Bioorg. Med. Chem. 2008;16:4652–4660. doi: 10.1016/j.bmc.2008.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jain R.P., Pettersson H.I., Zhang J., Aull K.D., Fortin P.D., Huitema C., Eltis L.D., Parrish J.C., James M.N., Wishart D.S., Vederas J.C. J. Med. Chem. 2004;47:6113–6116. doi: 10.1021/jm0494873. [DOI] [PubMed] [Google Scholar]
- 17.Yang S., Chen S.J., Hsu M.F., Wu J.D., Tseng C.T., Liu Y.F., Chen H.C., Kuo C.W., Wu C.S., Chang L.W., Chen W.C., Liao S.Y., Chang T.Y., Hung H.H., Shr H.L., Liu C.Y., Huang Y.A., Chang L.Y., Hsu J.C., Peters C.J., Wang A.H., Hsu M.C. J. Med. Chem. 2006;49:4971–4980. doi: 10.1021/jm0603926. [DOI] [PubMed] [Google Scholar]
- 18.Lee T.W., Cherney M.M., Huitema C., Liu J., James K.E., Powers J.C., Eltis L.D., James M.N.G. J. Mol. Biol. 2005;353:1137–1151. doi: 10.1016/j.jmb.2005.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akaji K., Konno H., Mitsui H., Teruya K., Shimamoto Y., Hattori Y., Ozaki T., Kusunoki M., Sanjoh A. J. Med. Chem. 2011;54:7962–7973. doi: 10.1021/jm200870n. [DOI] [PubMed] [Google Scholar]
- 20.Sydnes M.O., Hayashi Y., Sharma V.K., Hamada T., Bacha U., Barrila J., Freire E., Kiso Y. Tetrahedron. 2006;62:8601–8609. doi: 10.1016/j.tet.2006.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Regnier T., Sharma D., Hidaka K., Bacha U., Freire E., Hayashi Y., Kiso Y. Bioorg. Med. Chem. Lett. 2009;19:2722–2727. doi: 10.1016/j.bmcl.2009.03.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Konno S., Thanigaimalai P., Nakada K., Yamamoto T., Yamazaki Y., Yakushiji F., Akaji K., Kiso Y., Kawasaki Y., Freire E., Hayashi Y. Bioorg. Med. Chem. 2013;21:412–424. doi: 10.1016/j.bmc.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaeppler U., Stiefl N., Schiller M., Vicik R., Breuning A., Schmitz W., Rupprecht D., Schmuck C., Baumann K., Ziebuhr J., Schirmeister T. J. Med. Chem. 2005;48:6832–6842. doi: 10.1021/jm0501782. [DOI] [PubMed] [Google Scholar]
- 24.Chen L., Gui C., Luo X., Yang Q., Gunther S., Scandella E., Drosten C., Bai D., He X., Ludewig B., Chen J., Luo H., Yang Y., Yang Y., Zou J., Thiel V., Chen K., Shen J., Shen X., Jiang H. J. Virol. 2005;79:7095–7103. doi: 10.1128/JVI.79.11.7095-7103.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blanchard J.E., Elowe N.H., Huitema C., Fortin P.D., Cechetto J.D., Eltis L.D., Brown E.D. Chem. Biol. 2004;11:1445–1453. doi: 10.1016/j.chembiol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou L., Liu Y., Zhang W., Wei P., Huang C., Pei J., Yuan Y., Lai L. J. Med. Chem. 2006;49:3440–3443. doi: 10.1021/jm0602357. [DOI] [PubMed] [Google Scholar]
- 27.Wu C.Y., King K.Y., Kuo C.J., Fang J.M., Wu Y.T., Ho M.Y., Liao C.L., Shie J.J., Liang P.H., Wong C.H. Chem. Biol. 2006;13:261–268. doi: 10.1016/j.chembiol.2005.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramajayam R., Tan K.P., Liu H.G., Liang P.H. Bioorg. Med. Chem. Lett. 2010;20:3569–3572. doi: 10.1016/j.bmcl.2010.04.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramajayam R., Tan K.P., Liu H.G., Liang P.H. Bioorg. Med. Chem. 2010;18:7849–7854. doi: 10.1016/j.bmc.2010.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mukherjee P., Desai P., Ross L., White E.L., Avery M.A. Bioorg. Med. Chem. 2008;7:4138–4149. doi: 10.1016/j.bmc.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tian Q., Nayyar N.K., Babu S., Chen L., Tao J., Lee S., Tibbetts A., Moran T., Liou J., Guo M., Kennedy T.P. Tetrahedron Lett. 2001;42:6807–6809. [Google Scholar]
- 32.Bacha U., Barrila J., Gabelli B., Kiso Y., Amzel L.M., Freire E. Chem. Biol. Drug Des. 2008;72:34–39. doi: 10.1111/j.1747-0285.2008.00679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barrila J., Bacha U., Freire E. Biochemistry. 2006;45:14908–14916. doi: 10.1021/bi0616302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akaji K., Konno H., Onozuka M., Makino A., Saito H., Nosaka K. Bioorg. Med. Chem. 2008;16:9400–9408. doi: 10.1016/j.bmc.2008.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang H., Xie W., Xue X., Yang K., Ma J., Liang W., Zhao Q., Zhou Z., Pei D., Ziebuhr J., Hilgenfeld R., Yuen K.Y., Wong L., Gao G., Chen S., Chen Z., Ma D., Bartlam M., Rao Z. PLoS Biol. 2005;3:1742–1752. doi: 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.