

Abstract
Six series of semisynthetic lipophilic glycopeptide antibiotic derivatives were evaluated for in vitro activity against influenza A and B viruses. The new teicoplanin pseudoaglycon-derived lipoglycopeptides were prepared by coupling one or two side chains to the N-terminus of the glycopeptide core, using various conjugation methods. Three series of derivatives bearing two lipophilic groups were synthesized by attaching bis-alkylthio maleimides directly or through linkers of different lengths to the glycopeptide. Access to the fourth and fifth series of compounds was achieved by click chemistry, introducing single alkyl/aryl chains directly or through a tetraethylene glycol linker to the same position. A sixth group of semisynthetic derivatives was obtained by sulfonylation of the N-terminus. Of the 42 lipophilic teicoplanin pseudoaglycon derivatives tested, about half showed broad activity against influenza A and B viruses, with some of them having reasonable or no cytotoxicity. Minor differences in the side chain length as well as lipophilicity appeared to have significant impact on antiviral activity and cytotoxicity. Several lipoglycopeptides were also found to be active against human coronavirus.
Keywords: Teicoplanin, Lipoglycopeptide, Maleimide, Sulfonamide, Influenza virus inhibitor, Coronavirus
Abbreviations: DCM, dichloromethane; DMF, dimethylformamide; CPE, cytopathic effect; Et3N, triethylamine; Galp, galactopyranose; HA, hemagglutinin; HIV, human immunodeficiency virus; logP, logarithm of the partition coefficient; MCC, minimum cytotoxic concentration; MDCK, Madin−Darby Canine Kidney; M2, Matrix-2; MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium; NA, neuraminidase; Ph, phenyl; PMB, p-methoxybenzyl; SARS-CoV, severe acute respiratory syndrome coronavirus; SI, selectivity index; SEM, standard error of the mean; TEG, tetraethylene glycol; TLC, thin layer chromatography; tosyl, p-toluenesulfonyl
Graphical abstract

Highlights
-
•
Multiple series of lipophilic teicoplanin pseudoaglycon derivatives were prepared.
-
•
Alkyl or aryl chains were coupled to the N-terminus by various conjugation methods.
-
•
The activity of new antibiotic derivatives was evaluated against influenza viruses.
-
•
Half of the 42 derivatives showed high activity against influenza A and B viruses.
-
•
The length and lipophilicity of the side chains influence the antiviral activity.
1. Introduction
Human influenza A and B viruses cause the annual influenza epidemics and sporadic pandemics associated with high fatality rate [1]. The viral envelope contains two glycoproteins with a crucial role in virus replication. The hemagglutinin (HA) is responsible for initial attachment of the virus to sialylated cell surface glycans, and fusion of the viral and endosomal membranes after endocytosis of the virus particle [2]. The influenza virus neuraminidase (NA) catalyzes release of newly formed virions at the end of the viral life cycle. Currently available influenza virus blockers are the NA inhibitors oseltamivir and zanamivir, and the M2 ion channel blockers amantadine and rimantadine [3]. The latter two are rarely used nowadays because of global viral resistance against them [4]. The increasing awareness of potential oseltamivir resistance [5,6] advocates the need for new anti-influenza medications. Clinical trials are ongoing for a few HA-targeting approaches, i.e. the fusion inhibitor arbidol and diverse broadly neutralizing anti-HA antibodies [3]. In addition, the possibility to target a host factor involved in HA functioning is tested with the HA maturation inhibitor nitazoxanide and a receptor-destroying sialidase enzyme, besides various concepts in the preclinical stage [7].
Antiviral drugs are also required for pandemic preparedness against zoonotic and highly virulent influenza viruses [8]. In this context, antiviral glycopeptide analogues seem particularly relevant since they often display broad activity against influenza plus some other emerging viruses. Derivatives of teicoplanin or related antibiotics were reported to inhibit, among others, influenza virus [9]; coronaviruses [10] including SARS-CoV (severe acute respiratory syndrome coronavirus) [11]; Ebola pseudovirus [12]; HIV [13]; or hepatitis C virus [14]. Our focus of the last years was to investigate the structure-activity relationship and mechanism of action for influenza virus. We reported lipophilic derivatives of ristocetin aglycon modified on the N-terminal part of the molecule, which proved to be strong inhibitors of influenza virus replication in cell culture [15]. Mechanistic studies with the lead compound demonstrated that it interferes with influenza virus endocytosis [16]. Its favorable selectivity index encouraged us to prepare a series of analogues to gain further insight into structure activity relationships [17,18]. The outstanding antiviral properties were lost in analogues containing teicoplanin aglycon or pseudoaglycon, meaning that minor structural differences between the aglycon of teicoplanin and that of ristocetin, have major impact on antiviral activity [19].
Next, we found that a special lipophilic modification of teicoplanin pseudoaglycon also resulted in derivatives with high anti-influenza virus activity [20]. These derivatives contain a sugar unit carrying two n-octyl chains, and this lipophilic auxiliary is attached to teicoplanin pseudoaglycon through a tetraethylene glycol chain and a triazole ring (1a and 1b, Fig. 1 ). Although the antiviral mode of action of 1a and 1b remains to be elucidated, we observed that these dually octylated teicoplanin pseudoaglycon derivatives inhibit influenza virus-induced hemagglutination, suggesting that they interfere with the binding interaction between the viral HA and sialylated host cell receptors. We also found that the lipophilic side chains in these molecules are essential for anti-influenza virus activity, since changing these octyl chains to methyl groups (1c) completely abolished the antiviral effect [20]. Unfortunately, the strong activity of 1a and 1b was accompanied by high cytotoxicity, while the inactive and less amphiphilic 1c was only moderately toxic.
Fig. 1.

Structure of previously synthesized teicoplanin derivatives. Compounds 1a, 1b, 2a and 2d showed promising anti-influenza virus activity; 1c, 2b, and 2c were inactive; and 2e showed modest activity [20,21]. (PMB = p-methoxybenzyl).
Surprisingly, we recently found that teicoplanin pseudoaglycon achieves anti-influenza virus properties by a simple lipophilic modification based on the well-known azide-alkyne cycloaddition click reaction (2a-j) [21]. The activity oddly correlated to the structure, since it disappeared and then reappeared by increasing the length of the lipophilic alkyl chains. Changes in cytotoxicity, however, were more consistent and indicated that the addition of longer alkyl substituents resulted in higher cytotoxicity. Although compounds 2a and 2d showed excellent inhibitory activity against influenza virus, their selectivity indices were unfavorable [21]. Noteworthy, some of these derivatives with amphiphilic, bulky substituents (2i, 2j) displayed anti-influenza virus activity without significant cytotoxicity, despite their high calculated logP values.
With the aim of achieving derivatives with more favorable biological properties, we decided to carry out a systematic structure-activity relationship analysis of teicoplanin pseudoaglycon derivatives resembling 1a, 1b, or e.g. 2d. A simple conjugation method for protein and peptide modification was reported [22], involving rapid and clean reaction of 3,4-dibromomaleimides and thiols, and giving bis-alkylthio maleimides in high yields through an addition-elimination mechanism. We envisioned the application of maleimide chemistry in combination with azide-alkyne click chemistry, to produce three sets of teicoplanin derivatives equipped with two lipophilic substituents similarly as in 1a and 1b. (In the Discussion, these derivatives are denoted as Series 1–3). Compounds 2a-j are referred to in the SAR analysis as Series 4. Two further sets of derivatives (Series 5 and 6) comparable to 2a-g, which contain only one lipophilic alkyl chain, were prepared by azide-alkyne 1,3-dipolar cycloaddition reaction and by N-sulfonylation. Here we describe the synthesis and antiviral investigation of these compounds.
2. Results and discussion
Chemistry. The first group of derivatives (12–18) was prepared by synthesizing various bis-alkylthio maleimides 4a-10a from 3,4-dibromomaleimide 3, followed by subsequent ethoxycarbonylation of the maleimide NH group [23] making compounds 4b-10b suitable for conjugation to the N-terminus of teicoplanin pseudoaglycon 11 (Scheme 1 ). Although we have described this path earlier [24] for the synthesis of compounds 4, 7, 8, 10, 12, 15, 16, 18, this time we also prepared the bis-n-butylthio and bis-n-hexylthio maleimide derivatives (5 and 6) for the sake of the systematic approach in this study. We also prepared the asymmetrically substituted maleimide variant 9 that has an n-propyl and n-dodecyl function.
Scheme 1.

Synthesis of maleimide derivatives 12–18 (Series 1) with double lipophilic tails by direct coupling of 4–10 to teicoplanin pseudoaglycon 11.
The second series of derivatives contains similar maleimide-derived side chains as the previous group, but these are attached through a triazole linker to the glycopeptide core, in order to increase the distance of the lipophilic chains from the pseudoaglycon. In this synthetic procedure we prepared N-propargyl maleimides 19–24 by reaction of the corresponding N-ethoxycarbonyl maleimides (4b-8b, 10b) with propargylamine. Finally, an azide-alkyne click reaction was carried out between azido teicoplanin pseudoaglycon 25 [9] and maleimides 19–24, yielding final products 26–31 (Scheme 2 ).
Scheme 2.

Conjugation of the bis-alkylthio maleimide derivatives 19–24 to azido teicoplanin pseudoaglycon 25 through a triazole moiety (Series 2).
The third group of derivatives carrying maleimide substituents differs from the previous one in the structure of the linker, which in this case contains a tetraethylene glycol segment besides the triazole ring as in compounds 1a and 1b. By introduction of the tetraethylene glycol fragment, the overall lipophilicity of the compounds is reduced, while the crucial lipophilic substituents are still incorporated in the structure. This linker modification proved to have a substantial effect on the biological profile of the molecules (see below).
The compounds suitable for this type of modification (33–39) were prepared by the simple reaction of N-ethoxycarbonyl maleimides 4b-8b, 10b, 32b and triethylene glycol 2-aminoethyl propargyl ether. Subsequent click reaction with azido teicoplanin pseudoaglycon 25 gave glycopeptides 40–46 (Scheme 3 ).
Scheme 3.

Conjugation of bis-alkylthio maleimide derivatives equipped with an N-TEG-propargyl moiety (33–39) to azido teicoplanin pseudoaglycon 25 gave Series 3 (40–46).
The fourth type of modification we describe here was based on our previous results mentioned above. Although compounds 2a and 2d possessing single, relatively long alkyl chains were highly active against influenza virus, they also displayed considerable cytotoxicity [21]. (For the sake of clarity, these compounds are listed as a separate series, i.e. Series 4 in Table 1 . The structures can be found in Fig. 1.)
Table 1.
Activity in influenza virus-infected MDCKa cells.
| Compound* |
Antiviral EC50b |
Cytotoxicityc |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A/H1N1 |
A/H1N1pdm |
A/H3N2 |
Influenza B |
|||||||
| CPE |
MTS |
CPE |
MTS |
CPE |
MTS |
CPE |
MTS |
CC50 |
MCC |
|
| (μM) | ||||||||||
| 1a | 0.80 | 1.2 | 1.4 | <0.80 | 1.8 | 1.2 | >100# | >100# | 11 | 20 |
| Series 1: Maleimide derivatives | ||||||||||
| 12 (2x C3) | >100 | >100 | 1.8 | 2.0 | >100 | >100 | >100 | 7.2 | 18 | 20 |
| 13 (2x C4) | >100 | >100 | 2.9 | 2.6 | >100 | >100 | >100 | >100 | 8.6 | 4 |
| 14 (2x C6) | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 9.8 | 4 |
| 15 (2x C8) | >100 | >100 | 2.1 | 1.9 | >100 | >100 | >100# | >100# | 11 | 20 |
| 16 (2x C12) | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | ≤1.1 | ≤4.0 |
| 17 (C3, C12) | >100 | 10 | 5.1 | 8.2 | 7.3 | 6.8 | 8.9 | 7.3 | 29 | ≥20 |
| 18 (2 x Galp) | >100 | 9.1 | 2.1 | 2.2 | 2.3 | 1.7 | >100 | 4.2 | 26 | 20 |
| Series 2: Maleimide derivatives with a triazole linker | ||||||||||
| 26 (2x C3) | >100 | >100 | >100 | >100 | >100 | >100 | >100# | >100# | 22 | 20 |
| 27 (2x C4) | 2.1 | 3.2 | 9.8 | 7.4 | >100 | 4.9 | 2.5 | 3.5 | 28 | 20 |
| 28 (2x C6) | 3.1 | >100 | 4.0 | 2.9 | 1.9 | 1.9 | 2.3 | 2.7 | 12 | ≥4 |
| 29 (2x C8) | 9.2 | 12 | 4.4 | 3.4 | 8.9 | 6.3 | 6.7 | 7.2 | 49 | 73 |
| 30 (2x C12) | >100 | >100 | >100 | >100 | >100 | >100 | >100# | >100# | 75 | 100 |
| 31 (2 x Galp) | >100 | >100 | >100 | >100 | >100 | >100 | >100# | >100# | 18 | 20 |
| Series 3: Maleimide derivatives with a triazolyl TEG linker | ||||||||||
| 40 (2x C3) | >100 | >100 | >100 | 7.8 | 6.2 | ≤7.8 | >100# | >100# | 38 | 20 |
| 41 (2x C4) | 4.8 | 5.8 | 9.5 | 8.6 | 5.3 | 6.0 | 5.5 | 4.9 | >100 | 100 |
| 42 (2x C6) | 2.0 | 3.5 | 2.7 | 6.0 | 2.0 | 2.9 | 1.8 | 3.6 | 65 | 20 |
| 43 (2x C8) | 1.3 | 2.0 | ≤1.4 | ≤0.80 | 0.92 | ≤0.80 | >20# | >20# | 19 | ≥4.0 |
| 44 (2x C12) | >100 | 13 | >100 | 2.8 | >100 | 2.4 | >100 | >100 | 79 | 20 |
| 45 (2x TEG) | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 3.0 | 4.0 |
| 46 (2 x Galp) | >100 | >100 | >100 | >100 | >100 | 23 | >100 | 42 | ≥32 | >100 |
| Series 4: Triazole derivatives [21] | ||||||||||
| 2a (C16) | 1.6 | 1.8 | 1.8 | 1.8 | ≤8.9 | ≤1.6 | 1.8# | 1.3# | 7.6 | ≥4.0 |
| 2b (C12) | >100 | >100 | >100 | >100 | >100 | >100 | >100# | >100# | 2.3 | 9.3 |
| 2c (C10) | >100 | >100 | >100 | >100 | >100 | >100 | >100# | >100# | 3.7 | 4 |
| 2d (C8) | >100 | 2.2 | 1.8 | 1.9 | 2.1 | 1.5 | 1.8# | 1.9# | 13 | ≥4.0 |
| 2e (C6) | >100 | >100 | >100 | >100 | 15 | 13 | 17# | 11# | 53 | ≥20 |
| 2f (Ph) | >100 | >100 | >100 | >100 | 11 | 11 | >100# | >100# | 41 | ≥20 |
| 2g (α-Np) | 11 | 11 | 11 | 4.4 | 15 | 8 | 15# | 6.6# | 47 | 20 |
| 2h (Galp) | 52 | 47 | 45 | 43 | 39 | 24 | 39# | 29# | >100 | ≥100 |
| 2i (lactose) | >100 | >100 | 8.9 | 7.3 | 8.9 | 20 | 6.6# | 4.0# | >100 | ≥20 |
| 2j (calixarene) | >100 | 43 | 8.9 | 6.7 | >100 | 5.2 | >100# | 5.7# | ≥58 | ≥20 |
| Series 5: Triazole derivatives with a TEG linker and single alkyl chains | ||||||||||
| 60 (C4) | >100 | 51 | >100 | 47 | >100 | 31 | >100 | 28 | >100 | 100 |
| 61 (C6) | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 51 | 100 |
| 62 (C8) | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 19 | 20 |
| 63 (C10) | 1.6 | 1.5 | 1.8 | 1.8 | 1.6 | 1.3 | >100 | <0.80 | ≥82 | ≥20 |
| Series 6: Sulfonamide derivatives | ||||||||||
| 72 (toluyl) | 9 | 11 | 30 | 32 | 41 | 19 | 11 | 12 | >100 | 100 |
| 73 (Ph) | 16 | 27 | 46 | 52 | 25 | 23 | 51 | 21 | >100 | 100 |
| 74 (PhNHAc) | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 47 | 60 | 100 |
| 75 (biphenyl) | 1.8 | 1.7 | 2.0 | 2.0 | 1.5 | 1.6 | 1.8 | 2.4 | 15 | 11 |
| 76 (dansyl) | 4.9 | 7.0 | 6.3 | 5.9 | 8.3 | 2.3 | 6.7 | 5.3 | 52 | 20 |
| 77 (C6) | 8.6 | 9.6 | 8.9 | 9.1 | 8.3 | 8.9 | 8.9 | 10.3 | 54 | 100 |
| 78 (C8) | 1.9 | 2.5 | >100 | >100 | 2.0 | 2.4 | 4.0 | >100 | 14 | 20 |
| 79 (C12) | 0.4 | 0.8 | >100 | 1.6 | <0.8 | <0.8 | <0.8 | <0.8 | 4.5 | 4.0 |
| Zanamivir | 0.041 | 0.19 | 1.9 | 30 | 20 | 3.2 | 0.0079 | 0.0062 | >100 | >100 |
| Ribavirin | 8.4 | 9.1 | 10 | 8.5 | 13 | 5.9 | 2.3 | 2.2 | >100 | >100 |
| Amantadine | >500 | >500 | >500 | >500 | 11 | 1.9 | >500 | >500 | >500 | >500 |
| Rimantadine | >500 | >500 | >500 | >500 | 0.20 | 0.17 | >500 | >500 | >500 | >500 |
*Lipophilic substituents are specified in brackets.
aMDCK, Madin-Darby canine kidney cells.
bAntiviral activity expressed as the EC50, i.e. the compound concentration producing 50% inhibition of virus replication, as estimated by microscopic scoring of the cytopathic effect (CPE) or by measuring cell viability in the formazan-based MTS assay. Influenza strains: A/PR/8/34 (A/H1N1); A/Virginia/ATCC3/2009 (A/H1N1pdm); A/HK/7/87 (A/H3N2); B/Ned/537/05 or B/HK/5/72 (data marked with #).
cCytotoxicity expressed as the minimum cytotoxic concentration (MCC; compound concentration producing minimal changes in cell morphology, as estimated by microscopy) or the 50% cytotoxic concentration (CC50; estimated by the MTS cell viability assay).
Data represent the means of two to five independent tests.
We speculated that this high cytotoxicity might be related to a combination of two factors, the first being high lipophilicity of the side chains in contrast to the hydrophilic pseudoaglycon part, which may cause a membrane-disrupting effect. This might be enhanced by the simple structure of the alkyl chains, which could facilitate insertion of these molecules into biological membranes. In our previous work [21] we attached two very bulky substituents (a perbenzylated disaccharide and a calix[4]arene carrying tert-butyl-substituents) to the same pseudoaglycon (2i and 2j). Calculated logP values for these compounds indicated higher lipophilicity than e.g. the n-hexadecyl or n-octyl derivatives 2a and 2d. These derivatives did not produce profound cytotoxic effect, supporting our hypothesis that cytotoxicity is determined by the combination of the lipophilicity and bulkiness of the substituents. It needs to be mentioned here, that in the A2 components of teicoplanin the ß-d-glucosamine on the D-ring is N-acylated by similarly simple acyl chains, yet these compounds seem to be harmless to mammalian cells. Therefore, the overall lipophilicity of the molecules may also influence cytotoxicity.
Since the introduction of alkyl chains proved to be promising in terms of antiviral activity, we decided to insert the tetraethylene glycol linker between the core and these substituents, in order to decrease lipophilicity as in the case of maleimide derivatives, which again caused a significant change in biological properties.
First, the mono-azido derivative of tetraethylene glycol 47 was utilized in a click reaction with alkyl propargyl ethers 48–51, yielding triazole derivatives 52–55. These were O-alkylated with propargyl bromide to provide compounds 56–59, which were used in the last step in a CuAAC reaction with azido teicoplanin pseudoaglycon 25 to obtain derivatives 60–63 (Scheme 4 ).
Scheme 4.

Another variant (Series 5) of teicoplanin pseudoaglycon modification (60–63).
Finally, by the reaction of teicoplanin pseudoaglycon with different sulfonyl chlorides 64–71 we prepared sulfonamide derivatives (72–79) (Scheme 5 ). This type of derivatization was attractive mainly because of its simplicity and high stability of the sulfonamide bond. Despite this, to our knowledge no one has thus far reported this type of derivatives of teicoplanin or related glycopeptides.
Scheme 5.

Synthesis of sulfonamide derivatives (Series 6) of teicoplanin pseudoaglycon (72–79).
Biological evaluation. The anti-influenza virus activity was determined in Madin-Darby canine kidney (MDCK) cells using a reported cytopathic effect (CPE) reduction method, in which CPE is assessed by microscopy plus formazan-based cell viability assay [25]. In parallel, cytotoxicity was determined in mock-infected cells. Table 1 summarizes the data for three human influenza A strains (including an A/H1N1 2009 pandemic strain) plus one influenza B strain.
Series 1, maleimide derivatives 12–18, showed variable activity against the four influenza virus strains tested, but neither of them proved as potent as compound 1a. Only compounds 17 and 18 displayed quite consistent activity against the four strains. Curiously enough, neither the double n-propyl nor the double n-dodecyl substituted maleimide moieties yielded active compounds, while the combination of n-propyl and n-dodecyl chains (compound 17) led to notable activity. The fair activity of compound 18 carrying a double galactose substituted maleimide was accompanied by modest selectivity with an SI of 5 [selectivity index (SI): ratio of MCC to average EC50]. This indicated the need for structural modification in order to reduce the cytotoxicity.
Series 2 (26–31) containing the triazole linker was, overall, more successful against influenza virus with somewhat lower cytotoxicity than the first series. Namely, antiviral activity was seen in the case of compounds 27, 28 and 29 which contain double n-butyl, n-hexyl and n-octyl chains, respectively. The presence of the linker highly altered the behavior of these compounds, since the analogous derivatives in the previous series (i.e. no triazole linker) with the same alkyl chain lengths were inactive. 29 displayed robust antiviral activity and a favorable SI of 10, whereas 27 and in particular 28 still had poor selectivity. 30 with the double n-dodecyl moiety remained inactive, but became less cytotoxic, which was a general finding in Series 2. Sadly, the double galactose substituted derivative lost its activity on introduction of the triazole linker.
The biological results for Series 3 (40–46) demonstrated that by introducing the tetraethylene glycol linker, cytotoxicity was generally reduced, while the anti-influenza virus activity was retained in the n-butyl (41) and n-hexyl (42) derivatives, and even increased in the n-octyl (43) analogue. Since the tetraethylene glycol element seemed beneficial, we also prepared a derivative with double tetraethylene glycol side chains (46), but this modification did not yield an active compound, probably because it misses the lipophilic moiety that proved to be essential for anti-influenza virus activity (see conclusions). Surprisingly, despite the lack of lipophilic moieties, compound 46 proved to be highly cytotoxic. Ultimately, 41 stood out as the most promising compound in this group, with very low cytotoxicity and robust activity against all tested strains, yielding a nice SI of 16.
The activities of triazole derivatives (2a-j) in Series 4 have already been published [21] elsewhere. These data are shown again, considering the systematic approach in our present report and the structural analogy to some of the compounds in this work. Surprisingly, among the alkyl substituted compounds, only the n-hexadecyl (2a) and n-octyl (2d) derivatives showed high activity, while the attachment of n-dodecyl (2b) and n-decyl (2c) side chains led to inactive compounds. However, all four derivatives had very high cytotoxicity. 2e with a n-hexyl side chain and compounds with aromatic substituents (2f, 2g) were only mildly active with moderate cytotoxicity. Interestingly, 2h and 2i (a perbenzylated lactose and a calix[4]arene derivative) displayed moderately high activity which was accompanied by only modest cytotoxicity, despite their very high calculated logP values indicating much higher lipophilicity when compared to 2a. As stated above, the explanation could be that, although these molecules are very lipophilic, their bulky structures may lead to an inability to disrupt cellular membranes as is the case for alkyl derivatives.
To further investigate how the tetraethylene glycol linker impacts the cytotoxicity, we synthesized Series 5, derivatives 60–63, which are analogous to the alkyl chain-containing triazole derivatives in Series 4. We speculated that introduction of the linker would boost the antiviral activity and reduce the cytotoxicity compared to the published compounds. Indeed, compound 63 (an analogue of 2c) showed excellent antiviral activity and reasonable cytotoxicity compared to 2c which is highly cytotoxic and devoid of anti-influenza virus activity. Interestingly, lowering the length of the alkyl chain (analogues 60–62) did not yield effective compounds.
Also for the sulfonamide derivatives (Series 6), the size and type of lipophilic substituents seemed to play an important role. The less hydrophobic tosyl (72), benzenesulfonyl (73), and p-acetamido-benzenesulfonyl (74) derivatives had rather low or no anti-influenza virus activity and were also not cytotoxic. However, the more lipophilic aryl substituted compounds, such as the biphenylsulfonyl (75) and dansyl (76) derivatives, displayed good activity. Unfortunately, they were also cytotoxic in the MDCK cells. The relationship between alkyl chain length, cytotoxicity and antiviral activity was very clear in case of alkylsulfonates 77–79. The hexanesulfonyl derivative (77) was the best compound showing moderate cytotoxicity and very consistent anti-influenza virus activity. With growing alkyl chain length (78, 79), the activity rapidly increased, but so did the cytotoxicity.
Inhibition of influenza HA- and NA-bearing lentiviral pseudoparticles. We previously published the anti-influenza virus mechanism of action of a lipophilic derivative of aglycoristocetin denoted SA-19 [16]. This compound proved to have nice activity in an assay with green fluorescent protein (GFP)-expressing lentiviral pseudoparticles bearing influenza virus HA and NA. This was consistent with other findings that SA-19 interferes with HA-mediated endocytosis. We now used the same procedure for two of the most potent teicoplanin pseudoaglycon derivatives (1a, 63). Strong and dose-dependent inhibition was seen with all four control compounds (Fig. 3), i.e. SA-19; chloroquine (an inhibitor of endosomal acidification); and two aniline-based influenza fusion inhibitors (80, 81, Fig. 2 ) which inhibit the conformational refolding of H1 HA at low endosomal pH [26]. With 63, the inhibition of pseudoparticle entry, as deduced from the reduction in GFP signal, was 92% at 10 μM and 56% at 2 μM. Likewise, 1a produced 54% reduction at 2 μM.
Fig. 3.

Inhibitory effect on cell entry of influenza virus HA- and NA-bearing lentiviral pseudoparticles. The GFP-expressing particles carrying H1-HA and N1-NA (from A/PR/8/34) were transduced into MDCK cells in the presence of compounds (X-axis: concentrations in μM), and GFP expression was quantified after three days incubation. The two test compounds, 1a and 63, were tested in parallel with four control compounds, i.e. SA-19, a lipophilic derivative of aglycoristocetin [16]; chloroquine; and two aniline-based influenza fusion inhibitors, 80 and 81 [26]. Data are the mean ± SEM of four independent experiments.
Fig. 2.
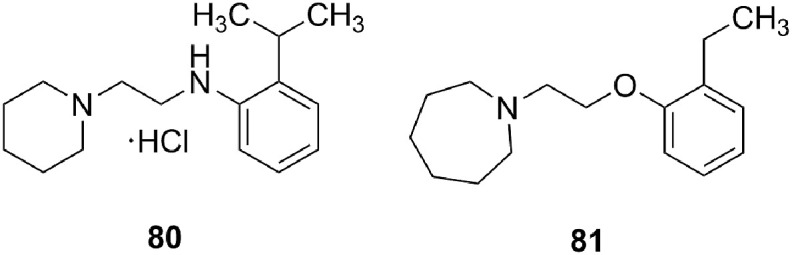
Structure of aniline-based influenza virus fusion inhibitors 80 and 81. (See ref. [26]. compounds 9d and 14a, respectively).
Activity against human coronavirus 229E. Given that teicoplanin and related glycopeptides were reported to have anti-coronavirus activity [10,11], we evaluated a subset of the newly synthesized compounds against human coronavirus 229E, using two complementary methods, i.e. CPE reduction in human embryonic lung (HEL) fibroblast cells and virus yield reduction in human lung carcinoma A549 cells. Among Series 1, 2 and 3, we tested the analogues carrying alkyl groups of intermediate length, i.e. n-butyl and n-hexyl; for Series 6, the entire series was tested. The antiviral activity values obtained (see Table S6 in Supporting Information) in HEL and A549 cells showed nice agreement. In A549 cells, several compounds produced 2-log10 (i.e. 100-fold) reduction in coronavirus yield at a concentration of ∼10 μM, with no or minimal cytotoxicity at 50 μM. The alkanesulfonamide derivatives 77 and 79 displayed potent anti-coronavirus activity in terms of EC50 (∼2 μM) and SI (ratio of MCC to EC50: 11 for 77 and 13 for 79). Within the same series, the p-toluenesulfonamide analogue 72 was also nicely active and selective (SI: 19).
3. Conclusion
In summary, we synthesized and evaluated several derivatives of teicoplanin pseudoaglycon in a systematic manner to obtain structure-activity relationships concerning the anti-influenza virus activity of the compounds. Many of the lipoglycopeptides exhibit remarkable activity against both influenza A and B viruses and showing, occasionally, favorable selectivity index.
Based on the biological data, it became evident that inhibition of influenza virus replication is not primarily dependent on the type or complexity of the chemical bond or functional group between the glycopeptide N-terminus and the newly introduced fragments. The structure of the side chains, but even more the overall lipophilicity of the compounds are the most influential on biological properties.
Hence, the presence of a lipophilic side chain is definitely a requirement for antiviral activity. The optimal length of alkyl substituents, which is somewhat dependent on the structure of the linker, is usually equivalent to 6–10 methylene groups. If the side chain carries two alkyl groups, reduced length might be enough, e. g. double butyl substitution. The incorporation of longer alkyl groups (>14 carbon atoms, Series 4) might also lead to active compounds, however most of these proved to be highly cytotoxic. The latter was even noted for some compounds with the optimum side chain lengths (e.g. 8–10). Reducing the cytotoxicity with preservation of antiviral activity was achieved by incorporation of the tetraethylene glycol linker. In some cases, this modification led to an improvement, while other derivatives became less active, probably due to a loss of required lipophilicity.
Analyzing the influence of the aryl substitution is more difficult, since this type of substituent is only present in two compound series (Series 4 and 6) in a smaller number. Nevertheless, these analogues seem to display a similar relationship between lipophilicity, anti-influenza virus activity and cytotoxicity, as the alkyl substituted compounds. Compounds containing one aromatic ring are usually not very active nor cytotoxic. Two aromatic rings seem to boost antiviral activity because of increased lipophilicity, but cytotoxicity also becomes prominent. Interestingly, the two compounds that carried numerous aromatic rings in the form of a highly bulky, lipo/hydrophilic side chain were effective against one or more influenza virus strains without causing serious cytotoxicity.
As mentioned earlier, it is likely that cytotoxicity of the lipoglycopeptides increases with the compounds' ability to solubilize cellular membranes and with net lipophilicity. With bulky and amphiphilic side chains, this effect might be weaker compared to more simple structures such as alkyl chains which could easily insert into lipid membranes. This could explain the apparent contradiction that some analogues displayed very high lipophilicity without severe cytotoxicity.
It is evident that the major challenge lies in designing the optimal lipoglycopeptide structures to provide sufficient lipophilicity for anti-influenza virus activity without causing serious cytotoxicity. As we have demonstrated, high activity goes hand in hand with high toxicity in many instances. Still, our study proved that once an active lead compound has been identified, it is possible to diminish its cytotoxicity by applying appropriate structural adjustments. Besides the modifications described here, there are unlimited variations that could lead to derivatives with excellent biological profiles. Moreover, as we have seen, the promising antiviral activity is not limited to influenza virus since some of our derivatives also displayed activity against human coronavirus. This makes this class of lipoglycopeptides a relevant class for further investigation [27].
4. Experimental section
4.1. General information
Propargylamine and alkyl thiols were purchased from Sigma Aldrich Chemical Co., dansyl chloride, benzenesulfonyl chloride, p-acetamidobenzenesulfonyl chloride, biphenylsulfonyl chloride were purchased from Tokyo Chemical Industry Co., Ltd., alkyl sulfonyl chlorides were prepared from the corresponding thiols using a literature method [28], triethylene glycol 2-aminoethyl propargyl ether, [29] 1-mercapto-11-hydroxy-3,6,9-trioxaundecane, [30] 1-azido-11-hydroxy-3,6,9-trioxaundecane, [31] 2,3-dibromomaleimide 3 [32], teicoplanin pseudoaglycon 11 [9] and azido teicoplanin pseudoaglycon 25 [9] were also prepared according to literature procedures. TLC analysis was performed on Kieselgel 60 F254 (Merck) silica gel plates with visualization by immersing in ammonium-molibdate solution followed by heating or Pauly-reagent in the case of teicoplanin-derivatives. Flash column chromatography was performed on silica gel 60 (Merck 0.04–0.063 mm). Organic solutions were dried over Na2SO4 and concentrated under vacuum. The 1H (360, 400 and 500 MHz) and 13C NMR (90.54, 100.28, 125.76 MHz) spectra were recorded with Bruker DRX-360, Bruker DRX-400 and Bruker Avance II 500 spectrometers. Chemical shifts are referenced to Me4Si or DSS (0.00 ppm for 1H) and to solvent signals (CDCl3: 77.16 ppm, DMSO‑d 6: 39.52 ppm for 13C). MS (MALDI-TOF) analysis was carried out in positive reflectron mode on a BIFLEX III mass spectrometer (Bruker, Germany) with delayed-ion extraction. The matrix solution was a saturated solution of 2,4,6-trihydroxyacetophenone (2,4,6-THAP) in DMF. Elemental analysis (C, H, N, S) was performed on an Elementar Vario MicroCube instrument.
4.2. General method A for the preparation of N-ethoxycarbonyl bis-alkylthio maleimides (5b, 6b, 9b, 32b)
To a stirred solution of bis-alkylthio maleimide (1.0 equiv.) in dry acetone (20 mL) K2CO3 (1.2 equiv.) and ethyl chloroformate (1.2 equiv.) were added under an argon atmosphere and stirred for 3 h at room temperature. The reaction mixture was diluted with CH2Cl2, filtered through a pad of Celite and concentrated. The crude product was used for further steps without purification.
4.3. General method B for preparing N-propargyl-bis-alkylthio maleimides or N-TEG-propargyl-bis-alkylthio maleimides
To a stirred solution of N-ethoxycarbonyl bis-alkylthio maleimide [24] (1.0 equiv.) in CH2Cl2 (30 mL) propargylamine or triethylene glycol 2-aminoethyl propargyl ether (1.25 equiv.) and Et3N (1.25 equiv.) were added under an argon atmosphere and stirred overnight at room temperature. The reaction mixture was diluted with CH2Cl2, washed with cc. NH4Cl and water twice, dried over Na2SO4, filtered and concentrated. The crude product was purified by flash chromatography to give the desired compound.
4.4. General method C for azide-alkyne click reaction
To a stirred solution of azide (1.0 equiv.) in dry DMF (5 mL) the alkyne compound (1.0–1.5 equiv.), Et3N (1.0 equiv.) and Cu(I)I (20–30 mol%) were added under an argon atmosphere and stirred for overnight at room temperature. The reaction mixture was concentrated, and the crude product was purified by flash chromatography. In the case of teicoplanin derivatives toluene/methanol (+1.0 v/v% acetic acid) or acetonitrile/water mixtures were used as eluent.
4.5. General method D for alkylation
To a stirred suspension of NaH (2.0 equiv.) in dry DMF (5 mL) the alcohol (1 equiv.) was added dropwise at 0 °C. After 30 min alkyl-bromide (1.5 equiv.) was added dropwise and stirred for 3 h. The reaction mixture was quenched with methanol, concentrated, diluted with DCM, washed twice with water, dried over Na2SO4, filtered and concentrated. The crude product was purified by flash chromatography to give the desired compound.
4.6. General method E for the preparation of sulfonamides
To a stirred solution of teicoplanin pseudoaglycon (1.0 equiv.) in dry pyridine (3–5 mL) the corresponding sulfonyl chloride (1.3–1.4 equiv.) was added at once and the reaction mixture was allowed to stir at room temperature for 4 h. Afterwards, methanol was added to quench the reaction, the solvent was evaporated, and the crude product was purified by flash chromatography using gradient elution starting from 100% acetonitrile to 90% acetonitrile 10% water.
Compounds 5a and 5b. To a stirred solution of 2,3-dibromomaleimide 3 [32] (255 mg, 1.0 mmol) in CH2Cl2 (20 mL) Et3N (279 μL, 2.0 mmol) and n-butyl-mercaptan (226 μl, 2.1 mmol) were added under an argon atmosphere and stirred for 3 h at room temperature. The reaction mixture was evaporated, and the crude product was purified by flash chromatography (hexanes:acetone = 9:1) to give the desired compound 5a (243 mg, 89%) as a yellow powder. 1H NMR (400 MHz, CDCl3): δ 8.07 (s, 1H, NH), 3.29 (t, J = 7.3 Hz, 4H, 2 x S-CH 2), 1.69–1.58 (m, 4H), 1.52–1.35 (m, 4H), 0.93 (t, J = 7.3 Hz, 6H, 2 x CH3). 13C NMR (101 MHz, CDCl3): δ 166.7 (2C, 2x C=O); 136.8 (C=C); 32.6, 31.6, 21.7 (6C, 6x CH2); 13.7 (2C, 2x CH3). MS (ESI): m/z calculated for C12H19NO2S2 + Na+ [M + Na+]: 296.075. Found: 296.073. Compound 5a was converted into the N-ethoxycarbonyl compound according to general method A. The crude compound 5b was used in further steps without purification.
Compounds 6a and 6b. To a stirred solution of 2,3-dibromomaleimide (510 mg, 2.0 mmol) in CH2Cl2 (20 mL) Et3N (558 μL, 4.0 mmol) and n-hexyl-mercaptan (600 μl, 4.2 mmol) were added under an argon atmosphere and stirred for 3 h at room temperature. The reaction mixture was evaporated, and the crude product was purified by flash chromatography (hexanes:ethyl acetate = 9:1) to give compound 6a (526 mg, 80%) as a yellow powder. 1H NMR (400 MHz, CDCl3): δ 7.88 (s, 1H), 3.28 (t, J = 7.4 Hz, 4H, 2 x -SCH 2), 1.70–1.59 (m, 4H, CH 2), 1.47–1.37 (m, 4H), 1.35–1.24 (m, 8H), 0.89 (t, J = 6.7 Hz, 6H, 2x CH 3). 13C NMR (101 MHz, CDCl3): δ 166.6 (2C); 136.8 (C=C); 31.9, 31.4, 30.6, 28.3 (8C, 8x CH2); 22.6 (2C, 2 x CH2S); 14.1 (2C, 2x CH3). MS (ESI): m/z calculated for C16H27NO2S2 + Na+ [M + Na+]: 352.138. Found: 352.201. Compound 6a was converted into N-ethoxycarbonyl compound according to general method A. The crude compound 6b was used in further steps without purification.
Compounds 9a and 9b. To a stirred solution of 2,3-dibromomaleimide (510 mg, 2.0 mmol) in CH2Cl2 (20 mL) Et3N (277 μl, 2.0 mmol) was added, then cooled to 0 °C and n-propyl-mercaptan (186 μl, 2.0 mmol) dissolved in CH2Cl2 (10 mL) was added dropwise under an argon atmosphere and stirred for 5 h. The reaction mixture was concentrated, and the crude product was purified by flash chromatography (hexanes:ethyl acetate = 9:1) to give the desired intermediate (261 mg, 52%) as a yellow powder. To a stirred solution of the intermediate (261 mg, 1.04 mmol) in CH2Cl2 (20 mL) Et3N (160 μl, 1.1 mmol) and n-dodecyl-mercaptan (264 μl, 1.1 mmol) were added under an argon atmosphere and stirred for 30 min. The reaction mixture was concentrated, and the crude product was purified by flash chromatography (hexanes:ethyl acetate = 95:5) to give compound 9a (273 mg, 71%) as a yellow powder. 1H NMR (400 MHz, CDCl3): δ 7.60 (s, 1H, NH), 3.34–3.22 (m, 4H, 2x S-CH 2), 1.74–1.59 (m, 4H), 1.45–1.37 (m, 2H), 1.30–1.21 (m, 16H), 1.03 (t, J = 7.4 Hz, 3H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 166.61, 166.40, (2C, 2 x C=O) 136.98, 136.71, (2C, 2 x C-S) 33.79, 32.04, 31.95, 30.59, 29.76, 29.69, 29.60, 29.48, 29.24, 28.62, 24.03, 22.82, (13C, 13 x CH2) 14.26, 13.23. (2C, 2 x CH3). MS (MALDI-TOF): m/z calculated for C19H33NO2S2 + Na+ [M + Na+]: 394.18. Found: 394.25. Compound 9a was converted into N-ethoxycarbonyl compound according to general method A. The crude compound 9b was used in further steps without purification.
Compound 13. To a stirred solution of teicoplanin pseudoaglycon [9] (224 mg, 0.16 mmol) in dry DMF (5 mL) N-ethoxycarbonyl bis-alkylthio maleimide 5b (43 mg, 0.16 mmol) and Et3N (22 μl, 0.16 mmol) were added under an argon atmosphere and stirred for overnight at room temperature. The reaction mixture was concentrated, and the crude product was purified by flash chromatography (toluene:methanol = 1:1) to give 13 (68 mg, 26%), as a yellow powder. NMR data can be found in Table S1(supporting information). MS (MALDI-TOF): m/z calculated for C78H74Cl2N8O25S2 + Na+ [M + Na+]: 1679.35. Found: 1679.5.
Compound 14. Compound 6b (53 mg, 0.16 mmol) was coupled to teicoplanin pseudoaglycon (224 mg, 0.16 mmol) according to the procedure described for compound 13. The crude product was purified by flash chromatography (toluene:methanol = 1:1) to give 14 (61 mg, 22%), as a yellow powder. NMR data can be found in Table S1(supporting information). MS (MALDI-TOF): m/z calculated for C82H82Cl2N8O25S2 + Na+ [M + Na+]: 1735.41. Found: 1734.99.
Compound 17. Compound 9b (27 mg, 71 μmol) was coupled to teicoplanin pseudoaglycon (100 mg, 71 μmol) according to the procedure described for compound 13. The crude product was purified by flash chromatography (toluene:methanol = 1:1) to give 17 (23 mg, 18%), as a yellow powder. NMR data can be found in Table S1 (supporting information). MS (MALDI-TOF): m/z calculated for C85H88Cl2N8O25S2 + Na+ [M + Na+]: 1777.46. Found: 1777.47.
Compound 19. The title compound was prepared by the reaction of 4b (63 mg, 0.2 mmol) and propargylamine according to general method B. The crude product was purified by flash chromatography in hexanes:acetone = 9:1, to give 19 (48 mg, 85%) as a yellow syrup. 1H NMR (360 MHz, CDCl3): δ 4.27 (d, J = 2.5 Hz, 2H, NCH 2), 3.38–3.16 (m, 4H, 2 x SCH 2), 2.23 (t, J = 2.5 Hz, CH2CCH), 1.80–1.60 (m, 4H, 2 x CH 2), 1.03 (t, J = 7.3 Hz, 6H, 2 x CH 3); 13C NMR (91 MHz, CDCl3): δ 165.3 (2C, 2 x C=O), 136.1 (2C, 2 x C=C), 71.6 (1C, CH), 33.8 (2C, 2 x SCH2) 27.6 (NCH2), 24.0 (1C, CH2), 13.2 (2C, 2x CH3). Elemental analysis calculated (%) for C13H17NO2S2: C 55.10, H 6.25, N 4.94, S 22.62. Found: C 54.96, H 6.42, N 4.79, S 22.45.
Compound 20. Compound 20 was prepared by the reaction of 5b (117 mg, 0.34 mmol) and propargylamine according to general method B. The crude product was purified by flash chromatography in hexanes:ethyl acetate = 9:1, to give the title compound (89 mg, 84%) as a yellow powder. 1H NMR (400 MHz, CDCl3): δ 4.27 (d, J = 3.3 Hz, 2H), 3.31 (t, J = 7.4 Hz, 4H), 2.23 (t, J = 2.5 Hz, 1H), 1.69–1.59 (m, 4H), 1.50–1.40 (m, 4H), 0.93 (t, J = 7.4 Hz, 6H, CH 3). 13C NMR (101 MHz, CDCl3): δ 165.4 (2C, 2x C=O); 136.1 (C=C); 71.6 (1C, C≡CH) 32.6, 31.7 (4C, 4x CH2); 27.6 (1C, N-CH2); 21.8 (2C, 2 x CH2S); 13.7 (2C, 2x CH3). MS (MALDI-TOF): m/z calculated for C15H21NO2S2 + Na+ [M + Na+]: 334.09. Found: 334.12.
Compound 21. Compound 6b (230 mg, 0.57 mmol) and propargylamine were reacted according to general method B. The crude product was purified by flash chromatography in hexanes:ethyl-acetate = 9:1, to give 21 (155 mg, 74%) as a yellow powder. 1H NMR (400 MHz, CDCl3): δ 4.27 (d, J = 2.3 Hz, 2H), 3.30 (t, J = 7.4 Hz, 4H), 2.22 (t, J = 2.3 Hz, 1H), 1.65 (m, 4H), 1.42 (m, 4H), 1.35–1.24 (m, 8H), 0.89 (t, J = 6.8 Hz, 6H). 13C (101 MHz, CDCl3): δ 165.4 (2C 2x C=O); 136.1 (C=C); 71.6 (1C, C≡CH) 32.0, 31.4, 30.5, 28.3, (8C, 8x CH2); 27.6 (1C, N-CH2); 22.6 (2C, 2 x CH2S); 14.1 (2C, 2x CH3). MS (MALDI-TOF): m/z calculated for C19H29NO2S2 + Na+ [M + Na+]: 390.15. Found: 390.3.
Compound 22. The desired compound was prepared by the reaction of 7b (341 mg, 0.74 mmol) and propargylamine according to general method B. The crude product was purified by silica gel chromatography in n-hexanes:ethyl acetate = 9:1, to give 22 (172 mg, 55%) as a yellow syrup. 1H NMR (400 MHz, CDCl3): δ 4.27 (4H, d, J = 2.4 Hz, 2 x CH 2), 3.34–3.26 (4H, m, 2 x SCH 2), 2.22 (1H, t, J = 2.5 Hz, CH), 1.69–1.60 (4H, m, 2 x CH 2), 1.45–1.36 (4H, m, 2 x CH 2), 1.27 (16H, br s, 8 x CH 2), 0.88 (6H, t, J = 6.8 Hz, 2 x CH 3); 13C NMR (101 MHz, CDCl3): δ 165.4 (2 x C=O), 136.1 (C=C), 71.6 (CH), 32.0 (N-CH2), 31.9, 30.6, 29.2, 29.2, 28.6, 27.5 (CH2), 22.7 (S-CH2), 14.2 (CH3). MS (MALDI-TOF): m/z calculated for C23H37NO2S2 + Na+ [M + Na+]: 446.22. Found: 446.19.
Compound 23. The reaction between compound 8b (114 mg, 0.2 mmol) and propargylamine was carried out according to general method B, followed by flash chromatography (hexanes:acetone = 8:2), which gave compound 23 (83 mg, 78%) as a yellow syrup. 1H NMR (360 MHz, CDCl3): δ 4.27 (d, J = 2.5 Hz, 2H), 3.36–3.22 (m, 4H), 2.21 (t, J = 2.5 Hz, 1H), 1.64 (dq, J = 13.6, 6.5, 5.9 Hz, 4H), 1.48–1.36 (m, 4H), 1.26 (s, 32H), 0.88 (t, J = 6.7 Hz, 6H). Elemental analysis calculated (%) for C31H53NO2S2: C 69.48, H 9.97, N 2.61, S 11.97. Found: C 69.32, H 10.18, N 2.55, S 11.86.
Compound 24. Compound 10b (107 mg, 0.15 mmol) was reacted with propargylamine and worked up according to general method B. The crude product was used in the next step without further purification (see compound 31 below).
Compound 26. Compound 19 (25 mg, 0.09 mmol) and azido teicoplanin pseudoaglycon 25 (107 mg, 0.075 mmol) were reacted according to general method C to give 26 (54 mg, 42%) as a yellow powder. NMR data can be found in Table S2 (supporting information). MS (MALDI-TOF): m/z calculated for C79H73Cl2N11O25S2 + Na+ [M + Na+]: 1732.35. Found: 1732.62.
Compound 27. The reaction of compound 20 (31 mg, 0.10 mmol) and azido teicoplanin pseudoaglycon 25 (107 mg, 0.075 mmol) according to general method C gave compound 27 (54 mg, 41%) as a yellow powder after purification. NMR data can be found in Table S2 (supporting information). MS (MALDI-TOF): m/z calculated for C81H77Cl2N11O25S2 + Na+ [M + Na+]: 1760.38. Found: 1760.60.
Compound 28. The glycopeptide 28 was prepared by the reaction of compound 21 (37 mg, 0.10 mmol) and azido teicoplanin pseudoaglycon 25 (107 mg, 0.075 mmol) according to general method C. After purification the title compound was obtained as a yellow powder (56 mg, 42%). NMR data can be found in Table S2 (supporting information). MS (MALDI-TOF): m/z calculated for C85H85Cl2N11O25S2 + Na+ [M + Na+]: 1816.44. Found: 1815.99.
Compound 29. Compound 22 (43 mg, 0.1 mmol) and azido teicoplanin pseudoaglycon 25 (107 mg, 0.075 mmol) were reacted according to general method C to give 29 (57 mg, 41%) as a yellow powder. NMR data can be found in Table S2 (supporting information). MS (MALDI-TOF): m/z calculated for C89H93Cl2N11O25S2 + Na+ [M + Na+]: 1872.51. Found: 1872.63.
Compound 30. The reaction of compound 23 (48 mg, 0.09 mmol) and azido teicoplanin pseudoaglycon 25 (107 mg, 0.075 mmol) according to general method C gave 30 (58 mg, 39%) as a yellow powder. NMR data can be found in Table S2. MS (MALDI-TOF): m/z calculated for C97H109Cl2N11O25S2 + Na+ [M + Na+]: 1984.63. Found: 1984.93.
Compound 31. Compound 24 (70 mg, ∼0.09 mmol) was reacted with azido teicoplanin pseudoaglycon 25 (107 mg, 0.075 mmol) according to general method C to give 31 (61 mg, 39%) as a yellow powder. NMR data can be found in Table S2 (supporting information). MS (MALDI-TOF): m/z calculated for C97H97Cl2N11O35S2 + Na+ [M + Na+]: 2132.49. Found: 2132.62.
Compounds 32a and b. To a stirred solution of 2,3-dibromomaleimide (255 mg, 1.0 mmol) in CH2Cl2 (20 mL) Et3N (2.0 mmol, 278 μL) and 1-mercapto-11-hydroxy-3,6,9-trioxaundecane (402 μl, 2.1 mmol) were added under an argon atmosphere and stirred for 3 h at room temperature. The reaction mixture was evaporated, and the crude product was converted into N-ethoxycarbonyl compound and worked up according to general method A yielding compound 32b which was used in further steps without purification.
Compound 33. The reaction of 4b (150 mg, 0.47 mmol) and triethylene glycol 2-aminoethyl propargyl ether was carried out according to general method B. After flash chromatography in hexanes:acetone = 9:1 the desired compound 33 was obtained (186 mg, 86%) as a yellow syrup. 1H NMR (400 MHz, CDCl3): δ 4.26–4.14 (m, 2H, CH 2), 3.76–3.56 (m, 16H, 8 x CH 2), 3.26 (t, J = 7.3 Hz, 4H, 2 x SCH 2), 2.48–2.41 (m, 1H, CH), 1.75–1.61 (m, 4H, 2 x CH 2), 1.03 (t, J = 7.3 Hz, 6H, 2 x CH 3). 13C NMR (101 MHz, CDCl3): δ 166.6 (2C, 2 x C=O), 135.9 (2C, 2 x C=C), 74.6 (CH≡C), 70.7, 70.7, 70.5, 70.1, 69.2, 67.9, 58.5 (8C, 8 x OCH2), 37.8 (NCH2), 33.8 (2C, 2 x SCH2), 24.0 (2C, 2 x CH2), 13.2 (2C, 2 x CH3). Elemental analysis calculated (%) for C21H33NO6S2: C 54.88, H 7.24, N 3.05, S 13.95. Found: C 54.80, H 7.35, N 2.99, S 13.78.
Compound 34. Compound 5b (117 mg, 0.34 mmol) was reacted with triethylene glycol 2-aminoethyl propargyl ether according to general method B. The crude product was purified by flash chromatography in hexanes:ethyl acetate = 7:3, to give 34 (154 mg, 93%) as a yellow syrup. 1H: (400 MHz, CDCl3): δ 4.21 (d, J = 2.4 Hz, 2H), 3.73–3.58 (m, 16H), 3.28 (t, J = 7.4 Hz, 4H), 2.44 (t, J = 2.4 Hz, 1H), 1.69–1.58 (m, 4H), 1.51–1.39 (m, 4H), 0.93 (t, J = 7.3 Hz, 6H). 13C NMR (101 MHz, CDCl3): δ 166.6 (2C, 2x C=O); 135.8 (2C, 2 x C=C); 74.6 (CH≡C), 70.7, 70.6, 70.5, 70.1, 69.2, 67.9, 58.5 (8C, 8 x OCH2), 37.8 (NCH2), 32.6, 31.6 (4C, 4x CH2); 21.7 (2C, 2 x CH2S); 13.7 (2C, 2x CH3) MS (ESI): m/z calculated for C23H37NO6S2 + Na+ [M + Na+]: 510.20. Found: 510.21.
Compound 35. Compound 35 was prepared by the reaction of 6b (230 mg, 0.57 mmol) and triethylene glycol 2-aminoethyl propargyl ether according to general method B. The crude product was purified by flash chromatography in hexanes:ethyl acetate = 85:15, yielding the title compound (236 mg, 76%) as a yellow syrup. 1H NMR (400 MHz, CDCl3): δ 4.20 (d, J = 2.4 Hz, 2H), 3.73–3.59 (m, 16H), 3.31–3.24 (m, 4H), 2.44 (t, J = 2.3 Hz, 1H), 1.64 (dt, J = 15.1, 7.4 Hz, 4H), 1.42 (dt, J = 14.3, 7.2 Hz, 4H), 1.36–1.23 (m, 8H), 0.89 (t, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3): δ 166.6 (2C, C=O), 135.7 (2C, 2 x C=C), 74.6 (CH≡C), 70.6, 70.6, 70.4, 70.0, 69.1, 67.9, 58.4 (8C, 8 x OCH2), 37.8 (NCH2), 31.9, 31.3, 30.4, 28.2, 22.5 (10C, 10 x CH2), 14.0 (2C, 2 x CH3). MS (MALDI-TOF): m/z calculated for C27H45NO6S2 + Na+ [M + Na+]: 566.26. Found: 566.25.
Compound 36. Compound 7b (150 mg, 0.33 mmol) and triethylene glycol 2-aminoethyl propargyl ether were reacted according to general method B. The crude product was purified by flash chromatography in hexanes:acetone = 8:2, to give 36 (112 mg, 57%) as a yellow syrup. 1H NMR (400 MHz, CDCl3): δ 4.21 (d, J = 2.4 Hz, 2H), 3.72–3.59 (m, 16H), 3.32–3.23 (m, 4H), 2.43 (t, J = 2.4 Hz, 1H), 1.64 (dt, J = 15.0, 7.4 Hz, 4H), 1.46–1.36 (m, 4H), 1.30 (dt, J = 8.7, 5.1 Hz, 16H), 0.88 (t, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3): δ 166.7 (2C, 2 x C=O), 135.9 (2C, 2 x C=C), 74.6 (CH≡C), 70.8, 70.7, 70.5, 70.1, 69.3, 68.0, 58.5 (8C, 8 x OCH2), 37.8 (NCH2), 32.0, 31.9 (2C, 2 x SCH2), 30.6, 29.3, 29.3, 28.7, 22.8 (10C, 10 x CH2), 14.2 (2C, 2 x CH3). Elemental analysis calculated (%) for C31H53NO6S2: C 62.07, H 8.91, N 2.33, S 10.69. Found: C 61.85, H 9.07, N 2.25, S 10.60.
Compound 37. Compound 8b (150 mg, 0.26 mmol) and triethylene glycol 2-aminoethyl propargyl ether were reacted according to general method B. The crude product was purified by silica gel chromatography in hexanes:acetone = 8:2, to give 37 (135 mg, 72%) as a yellow syrup. 1H NMR (400 MHz, CDCl3): δ 4.21 (d, J = 2.4 Hz, 2H), 3.73–3.57 (m, 16H), 3.27 (t, J = 7.4 Hz, 4H), 2.43 (t, J = 2.4 Hz, 1H), 1.69–1.59 (m, 4H), 1.40 (dd, J = 13.2, 7.5 Hz, 4H), 1.34–1.20 (m, 32H), 0.88 (t, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3): δ 166.7 (2C, 2 x C=O), 135.8 (2C, 2 x C=C), 74.6 (CH≡C), 70.7, 70.7, 70.5, 70.1, 69.2, 67.9, 58.5 (8C, 8 x OCH2), 37.6 (NCH2), 32.0 (2C, 2 x SCH2), 30.6, 29.8, 29.7, 29.6, 29.5, 29.2, 28.7, 22.8 (20C, 20 x CH2), 14.2 (2C, 2 x CH3). MS (MALDI-TOF): m/z calculated for C39H69NO6S2 + Na+ [M + Na+]: 734.45. Found: 735.01.
Compound 38. The reaction of 10b (381 mg, 0.53 mmol) and triethylene glycol 2-aminoethyl propargyl ether according to general method B gave a crude product that was purified by flash chromatography in hexanes:acetone = 7:3, yielding 38 (340 mg, 75%) as a yellow syrup. 1H NMR (400 MHz, CDCl3): δ 5.51 (d, J = 4.9 Hz, 2H, H-1), 4.62 (dd, J = 7.9, 2.1 Hz, 2H), 4.35–4.27 (m, 4H), 4.21 (d, J = 2.0 Hz, 2H), 3.98 (t, J = 6.8 Hz, 2H), 3.73–3.57 (m, 16H), 3.57–3.34 (m, 4H), 2.45 (t, J = 2.4 Hz, 1H, CH), 1.46 (d, J = 14.6 Hz, 12H), 1.32 (d, J = 3.9 Hz, 12H). 13C NMR (101 MHz, CDCl3): δ 166.3 (2C, 2 x C=O), 135.9 (2C, 2 x C=C), 109.5, 108.8 (4C, 4 x C q), 96.6 (2C, 2 x CH), 74.5 (1C, CH), 71.6, 71.0 (4C, 4 x CH), 70.7, 70.6 (3C, 3 x CH2) 70.5 (2C, 2 x CH), 70.5 (1C, CH2), 70.1, 69.2 (2C, 2 x CH2), 68.0 (2C, 2 x CH), 67.8 (1C, CH2), 58.5 (1C, CH2) 37.8 (1C, NCH2), 31.8 (2C, 2 x C-6,6’), 26.0 (4C, 4 x CH3), 25.0, 24.5 (4C, 4 x CH3). MS (MALDI-TOF): m/z calculated for C39H57NO16S2 + Na+ [M + Na+]: 882.30. Found: 882.30.
Compound 39. To a stirred solution of 32b (62 mg, 0.11 mmol) triethylene glycol 2-aminoethyl propargyl ether was added and the reaction was carried out according to general method B. The reaction was worked up and used in its crude form for the synthesis of the final product 46.
Compound 40 The reaction between compound 33 (46 mg, 0.1 mmol) and azido teicoplanin pseudoaglycon 25 (107 mg, 0.075 mmol) was carried out according to general method C to give 40 (59 mg, 42%) as a yellow powder. NMR data can be found in Table S3. MS (MALDI-TOF): m/z calculated for C87H89Cl2N11O29S2 + Na+ [M + Na+]: 1908.45. Found: 1908.60.
Compound 41. The reaction between compound 34 (49 mg, 0.1 mmol) and azido teicoplanin pseudoaglycon 25 (107 mg, 0.075 mmol) was carried out according to general method C to give 41 (34 mg, 24%) as a yellow powder. NMR data can be found in Table S3. MS (MALDI-TOF): m/z calculated for C89H93Cl2N11O29S2 + Na+ [M + Na+]: 1936.49. Found: 1936.70.
Compound 42. The reaction between compound 35 (54 mg, 0.1 mmol) and azido teicoplanin pseudoaglycon 25 (107 mg, 0.075 mmol) was carried out according to general method C to give 42 (27 mg, 18%) as a yellow powder. NMR data can be found in Table S3. MS (MALDI-TOF): m/z calculated for C93H101Cl2N11O29S2 + Na+ [M + Na+]: 1992.55. Found: 1991.9.
Compound 43. Glycopeptide derivative 43 was obtained by carrying out the reaction between compound 36 (60 mg, 0.1 mmol) and azido teicoplanin pseudoaglycon 25 (107 mg, 0.075 mmol) according to general method C, giving 43 (65 mg, 43%) as a yellow powder. NMR data can be found in Table S3. MS (MALDI-TOF): m/z calculated for C97H109Cl2N11O29S2 + Na+ [M + Na+]: 2048.61. Found: 2048.68.
Compound 44. Compound 37 (71 mg, 0.1 mmol) and azido teicoplanin pseudoaglycon 25 (107 mg, 0.075 mmol) were reacted according to general method C to give 44 (44 mg, 27%) as a yellow powder. NMR data can be found in Table S3. MS (MALDI-TOF): m/z calculated for C105H125Cl2N11O29S2 + Na+ [M + Na+]: 2160.74. Found: 2160.89.
Compound 45. To a stirred solution of azido teicoplanin pseudoaglycon 25 (107 mg, 0.075 mmol)in DMF, compound 38 (86 mg, 0.1 mmol) was added and the reaction was carried out according to general method C to give 45 (57 mg, 33%) as a yellow powder. NMR data can be found in Table S3. MS (MALDI-TOF): m/z calculated for C105H113Cl2N11O39S2 + Na+ [M + Na+]: 2308.59. Found: 2308.62.
Compound 46. Compound 39 (42 mg, 57 μmol) and azido teicoplanin pseudoaglycon 25 (82 mg, 57 μmol) were reacted according to general method C to give 46 (27 mg, 22%) as a yellow powder. NMR data can be found in Table S3. MS (MALDI-TOF): m/z calculated for C97H109Cl2N11O37S2 + Na+ [M + Na+]: 2176.57. Found: 2176.86.
Compound 52. 1-Azido-11-hydroxy-3,6,9-trioxaundecane 47 (413 mg, 1.89 mmol) was coupled with propargyl ether 48 (265 mg, 2.36 mmol) according to general method C. The crude product was purified by flash chromatography (hexanes:acetone = 6:4) resulting in 52 (405 mg, 52%) as a yellowish syrup. 1H NMR (360 MHz, CDCl3): δ 7.78 (s, 1H, C=CH), 4.62 (s, 2H, OCH 2C), 4.58–4.51 (m, 2H), 3.88 (t, J = 5.1 Hz, 2H), 3.76–3.70 (m, 2H) 3.67 (dd, J = 6.0, 2.7 Hz, 2H), 3.61 (q, J = 4.5 Hz, 8H), 3.53 (t, J = 6.6 Hz, 2H), 2.94 (s, 1H, OH), 1.58 (dt, J = 14.5, 6.7 Hz, 2H), 1.37 (dq, J = 14.5, 7.3 Hz, 2H), 0.91 (t, J = 7.3 Hz, 3H). 13C NMR (91 MHz, CDCl3): δ 145.3 (1C, C=CH), 123.7 (1C, C=CH), 72.6, 70.6, 70.5, 70.4, 70.3, 69.5, 64.3 (8C, 8 x OCH2), 61.6 (1C, CH2OH), 50.2 (1C, NCH2), 31.7 (1C, CH2), 19.3 (1C, CH2), 13.9 (1C, CH3). MS (ESI): m/z calculated for C15H29N3O5 + Na+ [M + Na+]: 354.200. Found: 354.201.
Compound 53. Compound 47 (444 mg, 2.03 mmol) and propargyl ether 49 (284 mg, 2.03 mmol) were reacted according to general method C. The crude product was purified by flash chromatography (hexanes:acetone = 6:4) resulting in 53 (542 mg, 74%) as a yellowish syrup. 1H NMR (360 MHz, CDCl3): δ 7.79 (s, 1H, C=CH), 4.62 (s, 2H, OCH 2), 4.55 (t, J = 5.0 Hz, 2H), 3.92–3.84 (m, 2H, OCH 2), 3.76–3.69 (m, 2H), 3.69–3.57 (m, 10H), 3.52 (t, J = 6.7 Hz, 2H), 3.09 (s, 1H, OH), 1.66–1.53 (m, 2H), 1.41–1.24 (m, 6H) 0.88 (t, J = 6.6 Hz, 3H, CH 3). 13C NMR (101 MHz, CDCl3): δ 145.2 (1C, C=CH), 123.8 (1C, C=CH), 72.6, 70.8, 70.5, 70.4, 70.2, 69.3, 64.2 (8C, 8 x OCH2), 61.5 (1C, CH2OH), 50.2 (1C, NCH2), 31.6, 29.6, 25.7, 22.6 (4C, 4 x CH2), 14.0 (1C, CH3). MS (ESI): m/z calculated for C17H33N3O5 + Na+ [M + Na+]: 382.231. Found: 382.226.
Compound 54. Compound 47 (400 mg, 1.83 mmol) and propargyl ether 50 (462 mg, 2.75 mmol) according to general method C. After purification by flash chromatography (hexanes:acetone = 7:3) 54 was obtained (480 mg, 68%) as a yellowish syrup. 1H NMR (360 MHz, CDCl3): δ 7.77 (s, 1H, C=CH), 4.62 (s, 2H, OCH 2), 4.55 (t, J = 5.0 Hz, 2H), 3.95–3.82 (m, 2H, OCH 2), 3.75–3.70 (m, 2H), 3.69–3.56 (m, 10H), 3.51 (t, J = 6.7 Hz, 2H), 2.94 (s, 1H, OH), 1.66–1.51 (m, 2H), 1.39–1.18 (m, 10H), 0.87 (t, J = 6.7 Hz, 3H, CH 3). 13C NMR (91 MHz, CDCl3): δ 145.3 (1C, C=CH), 123.7 (1C, C=CH), 72.6, 70.8, 70.6, 70.5, 70.3, 69.6, 64.3 (8C, 8 x OCH2), 61.7 (1C, CH2OH), 50.2 (1C, NCH2), 31.8, 29.7, 29.5, 29.3, 26.1, 22.7 (6C, 6 x CH2), 14.1 (1C, CH3). MS (ESI): m/z calculated for C19H37N3O5 + Na+ [M + Na+]: 410.263. Found: 410.258.
Compound 55. Compound 47 (260 mg, 1.19 mmol) was coupled with propargyl ether 51 (235 mg, 1.30 mmol) according to general method C. The crude product was purified by flash chromatography (hexanes:acetone = 7:3) resulting in 55 (463 mg, 86%) as a light yellow syrup. 1H NMR (360 MHz, CDCl3): δ 7.79 (s, 1H, C=CH), 4.62 (s, 2H, OCH 2), 4.55 (t, J = 5.0 Hz, 2H), 3.88 (t, J = 5.0 Hz, 2H), 3.75–3.70 (m, 2H, OCH 2), 3.68–3.64 (m, 2H), 3.65–3.57 (m, 8H), 3.52 (t, J = 6.7 Hz, 2H), 1.58 (dq, J = 10.1, 5.0, 3.4 Hz, 2H), 1.26 (br s, 14H, 7 x CH 2), 0.88 (t, J = 6.8 Hz, 3H, CH 3). 13C NMR (91 MHz, CDCl3): δ 72.6, 70.9, 70.5, 70.4, 70.3, 69.6, 64.3 (8C, 8 x OCH2), 61.7 (1C, CH2OH), 50.3 (1C, NCH2), 32.0, 29.8, 29.7, 29.6, 29.4, 29.2, 26.2, 22.7 (8C, 8 x CH2), 14.2 (1C, CH3). MS (MALDI-TOF): m/z calculated for C21H41N3O5 + Na+ [M + Na+]: 438.29. Found: 438.35.
Compound 56. Compound 52 (140 mg, 0.42 mmol) was O-propargylated according to general method D. The crude product was purified by flash chromatography (hexanes:acetone = 7:3) resulting in 56 (48 mg, 31%) as a light yellow syrup. 1H NMR (360 MHz, CDCl3): δ 7.73 (s, 1H, CH=C), 4.61 (s, 2H, OCH 2), 4.53 (t, J = 5.1 Hz, 2H), 4.18 (d, J = 2.3 Hz, 2H), 3.86 (t, J = 5.1 Hz, 2H), 3.71–3.57 (m, 12H, 6 x OCH 2), 3.51 (t, J = 6.7 Hz, 2H), 2.43 (t, J = 2.3 Hz, 1H), 1.57 (dt, J = 14.6, 6.7 Hz, 2H), 1.41–1.31 (m, 2H), 0.90 (t, J = 7.4 Hz, 3H, CH3). 13C NMR (91 MHz, CDCl3): δ 145.4 (1C, C=CH), 123.7 (1C, C=CH), 74.6 (1C, CH), 70.7, 70.6, 70.5, 69.6, 64.3 (9C, 9 x OCH2), 58.5 (1C, OCH2C≡CH), 50.3 (1C, NCH2), 31.8, 19.4 (2C, 2x CH2), 14.0 (1C, CH3). MS (ESI): m/z calculated for C18H31N3O5 + Na+ [M + Na+]: 392.216. Found: 392.216.
Compound 57. Compound 53 (140 mg, 0.39 mmol) was O-propargylated according to general method D. The crude product was purified by flash chromatography (hexanes:acetone = 7:3) resulting in 57 (60 mg, 39%) as a yellowish syrup. 1H NMR (360 MHz, CDCl3): δ 7.73 (s, 1H, C=CH), 4.61 (s, 2H, OCH 2), 4.54 (t, J = 5.1 Hz, 2H), 4.19 (d, J = 2.3 Hz, 2H), 3.88 (t, J = 5.1 Hz, 2H), 3.73–3.59 (m, 12H, 6 x OCH 2), 3.51 (t, J = 6.7 Hz, 2H), 2.44 (t, J = 2.4 Hz, 1H), 1.59 (p, J = 6.9 Hz, 2H), 1.40–1.21 (m, 6H, 3 x CH 2), 0.88 (t, J = 6.7 Hz, 3H, CH 3). 13C NMR (91 MHz, CDCl3): δ 145.3 (1C, C=CH), 123.5 (1C, C=CH), 74.6 (1C, CH), 70.8, 70.6, 70.5, 70.4, 69.5, 64.3 (9C, 9 x OCH2), 58.4 (1C, OCH2C≡CH), 50.3 (1C, NCH2), 31.7, 29.7, 25.8, 22.6 (4C, 4 x CH2), 14.1 (1C, CH3). MS (ESI): m/z calculated for C20H35N3O5 + Na+ [M + Na+]: 420.247. Found: 420.249.
Compound 58. Compound 54 (150 mg, 0.39 mmol) was O-propargylated according to general method D. The crude product was purified by flash chromatography (hexanes:acetone = 75:25) resulting in 58 (60 mg, 36%) as a yellow syrup. 1H NMR (360 MHz, CDCl3): δ 7.74 (s, 1H, C=CH), 4.61 (s, 2H, OCH 2), 4.54 (t, J = 5.1 Hz, 2H), 4.19 (d, J = 2.4 Hz, 2H), 3.92–3.84 (m, 2H), 3.73–3.57 (m, 12H, 6 x OCH 2), 3.51 (t, J = 6.7 Hz, 2H), 2.44 (t, J = 2.3 Hz, 1H, C≡CH), 1.59 (dt, J = 14.4, 6.8 Hz, 2H), 1.38–1.21 (m, 10H), 0.88 (t, J = 6.8 Hz, 3H, CH 3). 13C NMR (91 MHz, CDCl3): δ 123.6 (1C, C=CH), 74.6 (CH), 70.8, 70.6, 70.5, 70.4, 69.5, 69.2, 64.3 (9C, 9 x OCH2), 58.4 (1C, OCH2C≡CH), 50.3 (1C, NCH2), 31.8, 29.7, 29.5, 29.3, 26.2, 22.7 (6C, 6 x CH2), 14.1 (1C, CH3). MS (MALDI-TOF): m/z calculated for C22H39N3O5 + Na+ [M + Na+]: 448.28. Found: 448.30.
Compound 59. Compound 55 (300 mg, 0.72 mmol) was O-propargylated according to general method D. The crude product was purified by flash chromatography (hexanes:acetone = 7:3) resulting in 59 (284 mg, 87%) as a yellow syrup. 1H NMR (360 MHz, CDCl3): δ 7.73 (s, 1H, C=CH), 4.62 (s, 2H, OCH 2), 4.54 (t, J = 5.1 Hz, 2H), 4.20 (d, J = 2.4 Hz, 2H), 3.94–3.82 (m, 2H), 3.78–3.57 (m, 12H, 6 x OCH 2), 3.51 (t, J = 6.7 Hz, 2H), 2.44 (t, J = 2.4 Hz, 1H, C≡CH), 1.59 (p, J = 6.6 Hz, 2H), 1.26 (s, 14H), 0.88 (t, J = 6.7 Hz, 3H, CH 3). 13C NMR (91 MHz, CDCl3): δ 145.4 (1C, C=CH), 123.5 (1C, C=CH), 70.9, 70.6, 70.5, 69.6, 69.2, 64.4 (9C, 9 x OCH2), 58.5 (1C, OCH2C≡CH), 50.3 (1C, NCH2), 32.0, 29.7, 29.4, 26.2, 22.8 (8C, 8 x CH2), 14.2 (1C, CH3). MS (MALDI-TOF): m/z calculated for C24H43N3O5 + Na+ [M + Na+]: 476.31. Found: 476.30.
Compound 60. Azido teicoplanin pseudoaglycon 25 (86 mg, 0.06 mmol) and propargyl ether 56 (37 mg, 0.10 mmol) were allowed to react according to general method C. The crude product was purified by flash chromatography (acetonitrile:water = 9:1) resulting in 60 (28 mg, 26%) as a white powder. NMR data can be found in Table S4. MS (MALDI-TOF): m/z calculated for C84H87Cl2N13O28 + Na+ [M + Na+]: 1818.51. Found: 1818.4.
Compound 61. Azido teicoplanin pseudoaglycon 25 (86 mg, 0.06 mmol) and propargyl ether 57 (40 mg, 0.10 mmol) were allowed to react according to general method C. The crude product was purified by flash chromatography (acetonitrile:water = 9:1) resulting in 61 (39 mg, 36%) as a white powder. NMR data can be found in Table S4. MS (MALDI-TOF): m/z calculated for C86H91Cl2N13O28 + Na+ [M + Na+]: 1846.54. Found: 1846.6.
Compound 62. Azido teicoplanin pseudoaglycon 25 (86 mg, 0.06 mmol) and propargyl ether 58 (43 mg, 0.1 mmol) were allowed to react according to general method C. The crude product was purified by flash chromatography (acetonitrile:water = 9:1) resulting in 62 (22 mg, 20%) as a white powder. NMR data can be found in Table S4. MS (MALDI-TOF): m/z calculated for C88H95Cl2N13O28 + Na+ [M + Na+]: 1874.57. Found: 1874.6.
Compound 63. Compound 25 (100 mg, 0.07 mmol) azide was coupled with propargyl ether 59 (41 mg, 0.09 mmol) according to general method C. The crude product was purified by flash chromatography (toluene:MeOH = 1:1 + 1%v/v AcOH) resulting in 63 (35 mg, 27%). NMR data can be found in Table S4. MS (MALDI-TOF): m/z calculated for C90H99Cl2N13O28 + Na+ [M + Na+]: 1902.60. Found: 1902.60.
Compound 72. Teicoplanin pseudoaglycon 11 (100 mg, 0.071 mmol) and p-toluenesulfonyl chloride 64 (18 mg, 0.13 mmol) were allowed to react under the conditions described in general method E. The crude product was purified by flash chromatography to give 72 (59 mg, 54%) as a white powder. NMR data can be found in Table S5. MS (MALDI-TOF): m/z calculated for C73H64Cl2N8O25S + Na+ [M + Na+]: 1577.30. Found: 1577.4.
Compound 73. Teicoplanin pseudoaglycon (140 mg, 0.1 mmol) and benzenesulfonyl chloride 65 (18 μL, 0.14 mmol) were allowed to react under the conditions described in general method E. The crude product was purified by flash chromatography to give 73 (82 mg, 53%) as a white powder. NMR data can be found in Table S5. MS (MALDI-TOF): m/z calculated for C72H62Cl2N8O25S + Na+ [M + Na+]: 1563.28. Found: 1563.6.
Compound 74. Teicoplanin pseudoaglycon (140 mg, 0.1 mmol) and p-acetamidobenzenesulfonyl chloride 66 (33 mg, 0.14 mmol) were allowed to react under the conditions described in general method E. The crude product was purified by flash chromatography to give 74 (78 mg, 49%) as a white powder. NMR data can be found in Table S5. MS (MALDI-TOF): m/z calculated for C74H65Cl2N9O26S + Na+ [M + Na+]: 1620.30. Found: 1620.7.
Compound 75. Teicoplanin pseudoaglycon (140 mg, 0.1 mmol) and biphenylsulfonyl chloride 67 (35 mg, 0.14 mmol) were allowed to react under the conditions described in general method E. The crude product was purified by flash chromatography to give 75 (73 mg, 46%) as a white powder. NMR data can be found in Table S5. MS (MALDI-TOF): m/z calculated for C78H66Cl2N8O25S + Na+ [M + Na+]: 1639.31. Found: 1639.7.
Compound 76. Teicoplanin pseudoaglycon (140 mg, 0.1 mmol) and dansyl chloride 68 (38 mg, 0.14 mmol) were allowed to react under the conditions described in general method E. The crude product was purified by flash chromatography to give 76 (92 mg, 56%) as a yellow powder. NMR data can be found in Table S5. MS (MALDI-TOF): m/z calculated for C78H69Cl2N9O25S + Na+ [M + Na+]: 1656.34. Found: 1656.8.
Compound 77. Teicoplanin pseudoaglycon (100 mg, 0.071 mmol) and hexanesulfonyl chloride 69 (16 μL, 0.1 mmol) were allowed to react under the conditions described in general method E. The crude product was purified by flash chromatography to give 77 (28 mg, 25%) as a white powder. NMR data can be found in Table S5. MS (MALDI-TOF): m/z calculated for C72H70Cl2N8O25S + Na+ [M + Na+]: 1571.34. Found: 1571.7.
Compound 78. Teicoplanin pseudoaglycon (100 mg, 0.071 mmol) and octanesulfonyl chloride 70 (20 μL, 0.1 mmol) were allowed to react under the conditions described in general method E. The crude product was purified by flash chromatography to give 78 (32 mg, 29%) as a white powder. NMR data can be found in Table S5. MS (MALDI-TOF): m/z calculated for C74H74Cl2N8O25S + Na+ [M + Na+]: 1599.38. Found: 1599.4.
Compound 79. Teicoplanin pseudoaglycon (100 mg, 0.071 mmol) and dodecanesulfonyl chloride 71 (27 mg, 0.1 mmol) were allowed to react under the conditions described in general method E. The crude product was purified by flash chromatography to give 79 (36 mg, 31%) as a white powder. NMR data can be found in Table S5. MS (MALDI-TOF): m/z calculated for C78H82Cl2N8O25S + Na+ [M + Na+]: 1655.44. Found: 1655.9.
4.7. Influenza virus experiments
All details on the CPE reduction assay for influenza virus can be found in previous publications [16,25]. The virus strains were: A/PR/8/34 (A/H1N1); A/Virginia/ATCC3/2009 (A/H1N1pdm); A/HK/7/87 (A/H3N2); B/Ned/537/05; and B/HK/5/72. The infection medium consisted of Ultra-MDCK® medium (Lonza), supplemented with 0.0225% sodium bicarbonate, 2 mM l-glutamine and 2 μg/mL tosyl phenylalanyl chloromethyl ketone-treated trypsin. On day 0, semi-confluent cultures of Madin-Darby canine kidney (MDCK) cells in 96-well plates were infected with influenza virus at a multiplicity of infection (MOI) of 0.0004 plaque forming units (PFU) per cell. After three days incubation at 35 °C, virus-induced CPE and compound cytotoxicity were scored by microscopy, after which the data were confirmed by formazan-based MTS cell viability assay (CellTiter 96® AQueous One Solution Cell Proliferation Assay from Promega). The antiviral effect was expressed as the compound concentration producing 50% inhibition of the virus-induced CPE (EC50). Compound cytotoxicity was expressed as the compound concentration causing minimal changes in cell morphology (MCC), and 50% cytotoxic concentration based on MTS (CC50).
The plasmids for the lentiviral pseudoparticle assay [described in full detail elsewhere [16]] were: pCGGagPol (a gift from G. Maertens [33]); pQCXIP-AcGFP (kind gift from D. Daelemans); and two pCAGEN plasmids, i.e. pCAGEN backbone [kindly provided by C. Cepko (Boston, MA) via Addgene (plasmid No. 11160[34]] into which we cloned the A/PR/8/34 HA and NA cDNA sequences. The four plasmids were transfected into human HEK-293T cells to produce GFP-expressing lentiviral particles. Two days after transfection, the supernatant was collected and trypsin was added to activate the influenza HA0 protein [16] For lentiviral transduction, MDCK cells were seeded at 7500 cells per well in 96-well plates, and incubated one day later with lentiviral particles together with test compounds. After three days incubation, the cells were trypsinized, fixated with paraformaldehyde, and submitted to flow cytometry for GFP detection, using a BD FACSCanto II apparatus.
4.8. Coronavirus experiments
Human embryonic lung fibroblast (HEL) 299 cells; human lung carcinoma A549 cells; and human coronavirus (HCoV) 229E were obtained from ATCC. During virus infections, the medium contained 2% fetal calf serum. To conduct the CPE reduction assay, confluent HEL cell cultures in 96-well plates were exposed to HCoV-229E (MOI: 100 CCID50 per well) together with the test compounds. After 5 days incubation at 35 °C, microscopy was performed to determine the EC50 and MCC values (same definition as above).
To perform the qRT-PCR-based coronavirus yield assay, the materials were: HCoV-229E nucleoprotein-derived forward primer (5′-TTAGAGAGCGTGTTGAAGGTG-3′); reverse primer (5′-GTTCTGAATTCTTGCGCCTAAC-3′); probe (5′-6-FAM-TCTGGGTTG/ZEN/CTGTTGATGGTGCTA-IBFQ-3′; and a standardization plasmid with a 294-bp sequence of the HCoV-229E N-gene. As reference compound, the HCoV RNA synthesis inhibitor K22[35] (from ChemDiv) was used.
On day 0, confluent A549 cells in 96-well plates were exposed to the compounds together with HCoV-229E. One hour after infection, the virus inoculum was removed and the cells were further incubated at 35 °C in the presence of compounds. At day 3 p.i., the supernatants were frozen at −80 °C. Before qRT-PCR, virions were lysed by treating 2 μl sample with 10 μl resuspension buffer and 1 μl lysis enhancer (CellsDirect One-Step qRT-PCR kit; Invitrogen), and 10 min incubation at 75 °C. Next, quantification of CoV genome copies was performed by one-step qRT-PCR. The program was run on an Applied Biosystems 7500 Fast instrument and consisted of 15 min at 50 °C; 2 min at 95 °C; and 40 cycles of 15 s at 95 °C followed by 45 s at 60 °C. The EC99 and EC90 values were calculated by interpolation and defined as the compound concentration producing respectively a 2-log10 and 1-log10 reduction in viral RNA copy number, as compared to the virus control receiving no compound. To monitor compound cytotoxicity, microscopy was done on mock-infected cells incubated with the compounds for three days; the MCC value was defined as above.
Acknowledgments
The synthetic work was supported by National Research, Development and Innovation Office of Hungary (K119509, and TÉT_15_IN-1-2016-0071) and the EU and co-financed by the European Regional Development Fund under the project GINOP-2.3.2-15-2016-00008. The project was further supported by the János Bolyai Fellowship of the Hungarian Academy of Sciences (M. Csávás) and the Gedeon Richter's Talentum Foundation (1103 Budapest, Gyömrői út 19–21) for financial support. The research was also supported by the EU and co-financed by the European Regional Development Fund and European Social Fund under the project „Debrecen Venture Catapult Program” EFOP-3.6.1-16-2016-00022. L.N. acknowledges excellent technical assistance from Talitha Boogaerts and Benjamin Van Loy. We thank S. Vazquez (University of Barcelona) for the kind gift of the influenza virus fusion inhibitors 80 and 81, and G. Maertens and D. Daelemans for providing plasmid materials for the pseudoparticle assay.
Footnotes
Supplementary data related to this article can be found at https://doi.org/10.1016/j.ejmech.2018.08.058.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.Coleman B.L., Fadel S.A., Fitzpatrick T., Thomas S.M. Risk factors for serious outcomes associated with influenza illness in high- versus low- and middle-income countries: systematic literature review and meta-analysis. Influenza Other Respir. Viruses. 2018;12:22–29. doi: 10.1111/irv.12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vanderlinden E., Naesens L. Emerging antiviral strategies to interfere with influenza virus entry. Med. Res. Rev. 2014;34:301–339. doi: 10.1002/med.21289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Naesens L., Stevaert A., Vanderlinden E. Antiviral therapies on the horizon for influenza. Curr. Opin. Pharmacol. 2016;30:106–115. doi: 10.1016/j.coph.2016.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Dong G., Peng C., Luo J., Wang C., Han L., Wu B., Ji G., He H. Adamantane-resistant influenza A viruses in the world (1902-2013): frequency and distribution of M2 gene mutations. PLoS One. 2015;10 doi: 10.1371/journal.pone.0119115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Samson M., Pizzorno A., Abed Y., Boivin G. Influenza virus resistance to neuraminidase inhibitors. Antivir. Res. 2013;98:174–185. doi: 10.1016/j.antiviral.2013.03.014. [DOI] [PubMed] [Google Scholar]
- 6.Moscona A. Global transmission of oseltamivir-resistant influenza. N. Engl. J. Med. 2009;360:953–956. doi: 10.1056/NEJMp0900648. [DOI] [PubMed] [Google Scholar]
- 7.Zeng L.Y., Yang J., Liu S. Investigational hemagglutinin-targeted influenza virus inhibitors. Expet Opin. Invest. Drugs. 2017;26:63–73. doi: 10.1080/13543784.2017.1269170. [DOI] [PubMed] [Google Scholar]
- 8.Havers F.P., Campbell A.P., Uyeki T.M., Fry A.M. Commentary: a historical review of Centers for Disease Control and Prevention antiviral treatment and postexposure chemoprophylaxis guidance for human infections with novel influenza A viruses associated with severe human disease. J. Infect. Dis. 2017;216:S575–S580. doi: 10.1093/infdis/jix065. [DOI] [PubMed] [Google Scholar]
- 9.Pintér G., Batta G., Kéki S., Mándi A., Komáromi I., Takács-Novák K., Sztaricskai F., Rőth E., Ostorházi E., Rozgonyi F., Naesens L., Herczegh P. Diazo transfer−click reaction route to new, lipophilic teicoplanin and ristocetin aglycon derivatives with high antibacterial and anti-influenza virus activity: an aggregation and receptor binding study. J. Med. Chem. 2009;52:6053–6061. doi: 10.1021/jm900950d. [DOI] [PubMed] [Google Scholar]
- 10.Balzarini J., Keyaerts E., Vijgen L., Egberink H., De Clercq E., Van Ranst M., Printsevskaya S.S., Olsufyeva E.N., Solovieva S.E., Preobrazhenskaya M.N. Inhibition of feline (FIPV) and human (SARS) coronavirus by semisynthetic derivatives of glycopeptide antibiotics. Antivir. Res. 2006;72:20–33. doi: 10.1016/j.antiviral.2006.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou N., Pan T., Zhang J., Li Q., Zhang X., Bai C., Huang F., Peng T., Zhang J., Liu C., Tao L., Zhang H. Glycopeptide antibiotics potently inhibit cathepsin L in the late endosome/lysosome and block the entry of Ebola virus, Middle East respiratory syndrome coronavirus (MERS-CoV), and severe acute respiratory syndrome coronavirus (SARS-CoV) J. Biol. Chem. 2016;291:9218–9232. doi: 10.1074/jbc.M116.716100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y., Cui R., Li G., Gao Q., Yuan S., Altmeyer R. Teicoplanin inhibits Ebola pseudovirus infection in cell culture. Antivir. Res. 2016;125:1–7. doi: 10.1016/j.antiviral.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balzarini J., Pannecouque C., De Clercq E., Pavlov A.Y., Printsevskaya S.S., Miroshnikova O.V., Reznikova M.I., Preobrazhenskaya M.N. Antiretroviral activity of semisynthetic derivatives of glycopeptide antibiotics. J. Med. Chem. 2003;46:2755–2764. doi: 10.1021/jm0300882. [DOI] [PubMed] [Google Scholar]
- 14.Obeid S., Printsevskaya S.S., Olsufyeva E.N., Dallmeier K., Durantel D., Zoulim F., Preobrazhenskaya M.N., Neyts J., Paeshuyse J. Inhibition of hepatitis C virus replication by semi-synthetic derivatives of glycopeptide antibiotics. J. Antimicrob. Chemother. 2011;66:1287–1294. doi: 10.1093/jac/dkr104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Naesens L., Vanderlinden E., Rőth E., Jekő J., Andrei G., Snoeck R., Pannecouque C., Illyés E., Batta G., Herczegh P., Sztaricskai F. Anti-influenza virus activity and structure–activity relationship of aglycoristocetin derivatives with cyclobutenedione carrying hydrophobic chains. Antivir. Res. 2009;82:89–94. doi: 10.1016/j.antiviral.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vanderlinden E., Vanstreels E., Boons E., ter Veer W., Huckriede A., Daelemans D., Van Lommel A., Rőth E., Sztaricskai F., Herczegh P., Naesens L. Intracytoplasmic trapping of influenza virus by a lipophilic derivative of aglycoristocetin. J. Virol. 2012;86:9416–9431. doi: 10.1128/JVI.07032-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sipos A., Máté G., Rőth E., Borbás A., Batta G., Bereczki I., Kéki S., Jóna I., Ostorházi E., Rozgonyi F., Vanderlinden E., Naesens L., Herczegh P. Synthesis of fluorescent ristocetin aglycon derivatives with remarkable antibacterial and antiviral activities. Eur. J. Med. Chem. 2012;58:361–367. doi: 10.1016/j.ejmech.2012.10.030. [DOI] [PubMed] [Google Scholar]
- 18.Sipos A., Török Z., Rőth E., Kiss-Szikszai A., Batta G., Bereczki I., Fejes Z., Borbás A., Ostorházi E., Rozgonyi F., Naesens L., Herczegh P. Synthesis of isoindole and benzoisoindole derivatives of teicoplanin pseudoaglycon with remarkable antibacterial and antiviral activities. Bioorg. Med. Chem. Lett. 2012;22:7092–7096. doi: 10.1016/j.bmcl.2012.09.079. [DOI] [PubMed] [Google Scholar]
- 19.Bereczki I., Mándi A., Rőth E., Borbás A., Fizil Á., Komáromi I., Sipos A., Kurtán T., Batta G., Ostorházi E., Rozgonyi F., Vanderlinden E., Naesens L., Sztaricskai F., Herczegh P. A few atoms make the difference: synthetic, CD, NMR and computational studies on antiviral and antibacterial activities of glycopeptide antibiotic aglycon derivatives. Eur. J. Med. Chem. 2015;94:73–86. doi: 10.1016/j.ejmech.2015.02.028. [DOI] [PubMed] [Google Scholar]
- 20.Bereczki I., Kicsák M., Dobray L., Borbás A., Batta G., Kéki S., Nemes Nikodém É., Ostorházi E., Rozgonyi F., Vanderlinden E., Naesens L., Herczegh P. Semisynthetic teicoplanin derivatives as new influenza virus binding inhibitors: synthesis and antiviral studies. Bioorg. Med. Chem. Lett. 2014;24:3251–3254. doi: 10.1016/j.bmcl.2014.06.018. [DOI] [PubMed] [Google Scholar]
- 21.Szűcs Z., Csávás M., Rőth E., Borbás A., Batta G., Perret F., Ostorházi E., Szatmári R., Vanderlinden E., Naesens L., Herczegh P. Synthesis and biological evaluation of lipophilic teicoplanin pseudoaglycon derivatives containing a substituted triazole function. J. Antibiot. 2017;70:152–157. doi: 10.1038/ja.2016.80. [DOI] [PubMed] [Google Scholar]
- 22.Smith M.E.B., Schumacher F.F., Ryan C.P., Tedaldi L.M., Papaioannou D., Waksman G., Caddick S., Baker J.R. Protein modification, bioconjugation, and disulfide bridging using bromomaleimides. J. Am. Chem. Soc. 2010;132:1960–1965. doi: 10.1021/ja908610s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Castañeda L., Wright Z.V.F., Marculescu C., Tran T.M., Chudasama V., Maruani A., Hull E.A., Nunes J.P.M., Fitzmaurice R.J., Smith M.E.B., Jones L.H., Caddick S., Baker J.R. A mild synthesis of N-functionalised bromomaleimides, thiomaleimides and bromopyridazinediones. Tetrahedron Lett. 2013;54:3493–3495. doi: 10.1016/j.tetlet.2013.04.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Csávás M., Miskovics A., Szűcs Z., Rőth E., Nagy Z.L., Bereczki I., Herczeg M., Batta G., Nemes-Nikodém É., Ostorházi E., Rozgonyi F., Borbás A., Herczegh P. Synthesis and antibacterial evaluation of some teicoplanin pseudoaglycon derivatives containing alkyl- and arylthiosubstituted maleimides. J. Antibiot. 2015;68:579–585. doi: 10.1038/ja.2015.33. [DOI] [PubMed] [Google Scholar]
- 25.Vanderlinden E., Göktas F., Cesur Z., Froeyen M., Reed M.L., Russell C.J., Cesur N., Naesens L. Novel inhibitors of influenza virus fusion: structure-activity relationship and interaction with the viral hemagglutinin. J. Virol. 2010;84:4277–4288. doi: 10.1128/JVI.02325-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leiva R., Barniol-Xicota M., Codony S., Ginex T., Vanderlinden E., Montes M., Caffrey M., Luque F.J., Naesens L., Vázquez S. Aniline-based inhibitors of influenza H1N1 virus acting on hemagglutinin-mediated fusion. J. Med. Chem. 2018;61:98–118. doi: 10.1021/acs.jmedchem.7b00908. [DOI] [PubMed] [Google Scholar]
- 27.Colson P., Raoult D. Fighting viruses with antibiotics: an overlooked path. Int. J. Antimicrob. Agents. 2016;48:349–352. doi: 10.1016/j.ijantimicag.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bahrami K., Khodaei M.M., Soheilizad M. A novel, practical synthesis of sulfonyl chlorides from thiol and disulfide derivatives. Synlett. 2009;17:2773–2776. [Google Scholar]
- 29.Tran F., Odell A.V., Ward G.E., Westwood N.J. A modular approach to triazole-containing chemical inducers of dimerization for yeast three-hybrid screening. Molecules. 2013;18:11639–11657. doi: 10.3390/molecules180911639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lau J.L., Baksh M.M., Fiedler J.D., Brown S.D., Kussrow A., Bornhop D.J., Ordoukhanian P., Finn M.G. Evolution and protein packaging of small-molecule RNA aptamers. ACS Nano. 2011;5:7722–7729. doi: 10.1021/nn2006927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bollini M., Frey K.M., Cisneros J.A., Spasov K.A., Das K., Bauman J.D., Arnold E., Anderson K.S., Jorgensen W.L. Extension into the entrance channel of HIV-1 reverse transcriptase-Crystallography and enhanced solubility. Bioorg. Med. Chem. Lett. 2013;23:5209–5212. doi: 10.1016/j.bmcl.2013.06.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marminon C., Pierré A., Pfeiffer B., Pérez V., Léonce S., Renard P., Prudhomme M. Syntheses and antiproliferative activities of rebeccamycin analogues bearing two 7-azaindole moieties. Bioorg. Med. Chem. 2003;11:679–687. doi: 10.1016/s0968-0896(02)00532-1. [DOI] [PubMed] [Google Scholar]
- 33.Ulm J.W., Perron M., Sodroski J., Mulligan R.C. Complex determinants within the Moloney murine leukemia virus capsid modulate susceptibility of the virus to Fv1 and Ref1-mediated restriction. Virology. 2007;363:245–255. doi: 10.1016/j.virol.2006.09.048. [DOI] [PubMed] [Google Scholar]
- 34.Matsuda T., Cepko C. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc. Natl. Acad. Sci. Unit. States Am. 2004;101:16–22. doi: 10.1073/pnas.2235688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lundin A., Dijkman R., Bergström T., Kann N., Adamiak B., Hannoun C., Kindler E., Jónsdóttir H.R., Muth D., Kint J., Forlenza M., Müller M.A., Drosten C., Thiel V., Trybala E. Targeting membrane-bound viral RNA synthesis reveals potent inhibition of diverse coronaviruses including the middle East respiratory syndrome virus. PLoS Pathog. 2014;10 doi: 10.1371/journal.ppat.1004166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.