

Abstract
DNA Double-Strand Break (DSB) repair is essential to safeguard genome integrity. Upon DSBs, the ATM PI3K kinase rapidly triggers the establishment of megabase-sized, γH2AX-decorated chromatin domains which further act as seeds for the formation of DNA Damage Response (DDR) foci1. How these foci are rapidly assembled in order to establish a “repair-prone” environment within the nucleus is yet unclear. Topologically Associating Domains (TADs) are a key feature of 3D genome organization that compartmentalize transcription and replication, but little is known about their contribution to DNA repair processes2,3. Here we found that TADs are functional units of the DDR, instrumental for the correct establishment of γH2AX/53BP1 chromatin domains in a manner that involves one-sided cohesin-mediated loop extrusion on both sides of the DSB. We propose a model whereby H2AX-containing nucleosomes are rapidly phosphorylated as they actively pass by DSB-anchored cohesin. Our work highlights the critical impact of chromosome conformation in the maintenance of genome integrity and provides the first example of a chromatin modification established by loop extrusion.
Keywords: Cohesin, Loop extrusion, DNA Double-Strand Breaks, γH2AX, DNA Damage Response, Topologically Associated Domains.
DNA double-strand breaks induce the formation of DDR foci, which are microscopically visible and characterized by specific chromatin modifications (γH2AX, ubiquitin accumulation, histone H1 depletion) and the accumulation of DDR factors (53BP1, MDCI)4–6. Previous evidence indicated that chromosome architecture may control γH2AX spreading. Indeed, γH2AX domain boundaries were found in some instances to coincide with TAD boundaries7. Moreover, super-resolution light microscopy revealed that CTCF, which binds at TAD boundaries and thereby constrains the loop-extruding activity of the cohesin complex that shapes these domains in undamaged cells, is juxtaposed to γH2AX foci8. In addition, 53BPI can form nanodomains which frequently overlap with a TAD, as detected by DNA-FISH9. High-resolution ChIP-seq mapping following the induction of multiple DSB at annotated positions (using human DIvA cells)10 revealed that the spreading of these DDR foci components on nearby chromatin follows a highly stereotyped pattern (one example shown in Fig. 1a)5. We hypothesized that such patterns could be governed by pre-existing high-order chromatin structure established prior to DSB induction.
Figure 1. TADs are functional units governing DDR chromatin domains establishment.

(a) 4C-seq track in undamaged cells (-DSB) and ChIP-seq tracks of histone H1 (H1.2) and Ubiquitin (FK2) (log2 (+DSB/-DSB)) as well as γH2AX, MDC1 and 53BP1 (+DSB) as indicated. ChIP-seq data were smoothed using 50kb span, 4C-seq using a 10kb span. The AsiSI site is indicated by an arrow. (b) 4C-seq before DSB induction (-DSB) and γH2AX ChIP-seq after DSB induction (+DSB) tracks (smoothed using a 50kb span) for different viewpoints localized at three AsiSI sites or a Control region. One representative experiment is shown (out of n=3). Arrows: AsiSI sites. (c) γH2AX ChIP-seq (+DSB) and 4C-seq (-DSB) tracks (10 kb smoothed) for two viewpoints localized at the AsiSI site or 470 kb upstream of the AsiSI site. Viewpoints: red arrows, DSB: black arrows. (d) Hi-C contact matrix of a region of the chromosome 1 in DIvA cells before DSB induction (top panel). γH2AX ChIP-seq after DSB induction, 4C-seq signal, TAD borders computed from Hi-C data and CTCF ChIP-seq peaks position before DSB induction are shown. Peaks in blue contain a CTCF motif in the forward orientation and peaks in red a CTCF motif in the reverse orientation. (e) Average profile of γH2AX ChIP-seq after DSB induction centered on the closest TAD border to the 174 best-induced DSBs (damaged TAD on the right). (f) 4C-seq track (10 kb smoothed) before DSB induction (-DSB) (in blue) using view points as indicated (red arrows). γH2AX ChIP-chip tracks (log2 sample/input, smoothed using 500 probes span) after DSB induction with CRISPR/Cas9 (black arrows) are shown in red. (g) γH2AX and pATM (S1981) ChIP-seq tracks after DSB induction (+DSB) on an 8 Mb window (top panel) and a 15 kb window (bottom panel) around an AsiSI site (black arrow).
γH2AX spreads within TADs
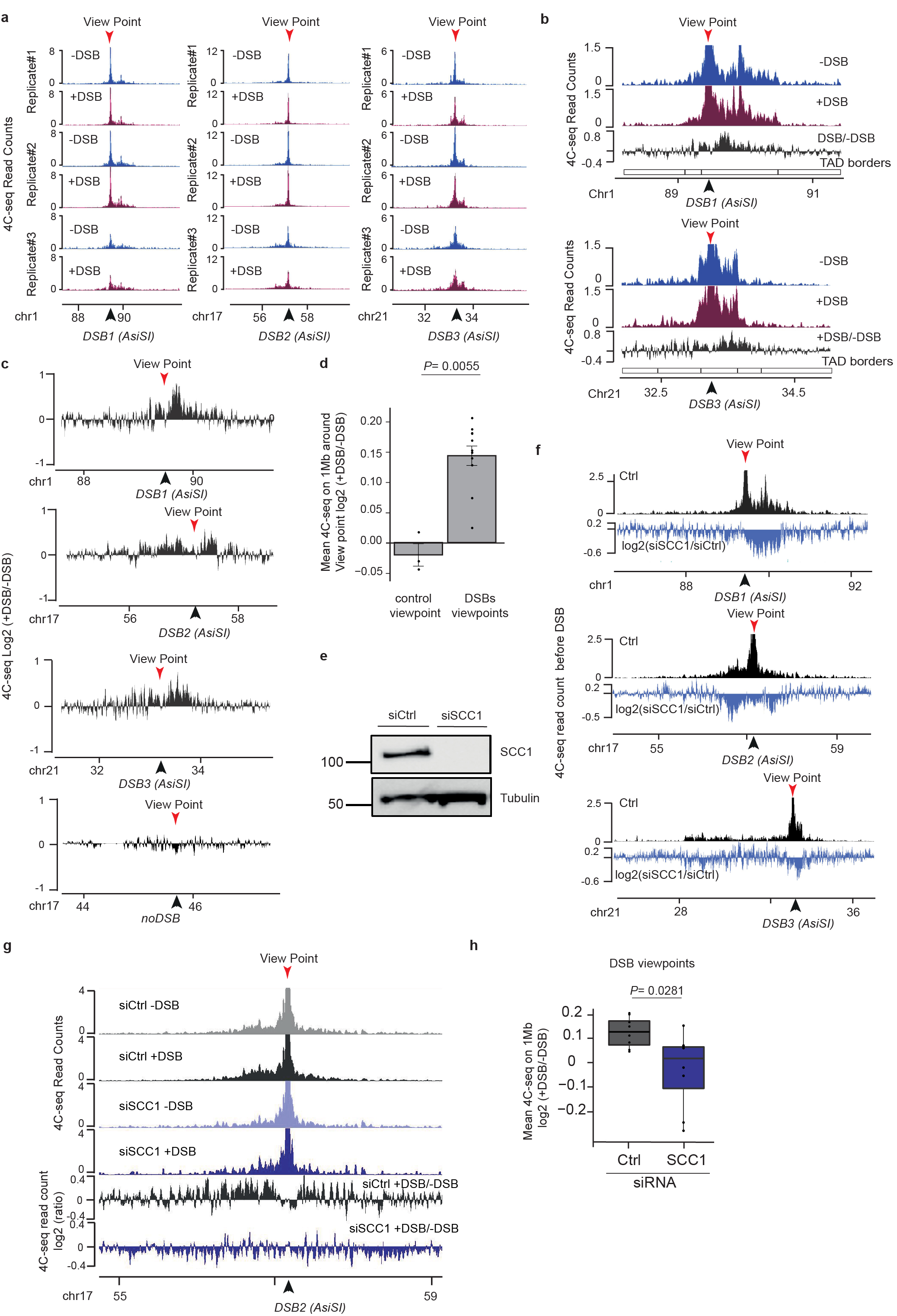
To relate the spreading of DDR foci components with chromosome conformation, we performed high-resolution 4C-seq experiments in undamaged human DIvA cells. As viewpoints we selected three genomic locations that are damaged in DIvA cells following activation of the AsiSI restriction enzyme as well as one undamaged control region. The chromatin conformation around these three viewpoints in undamaged condition was remarkably similar to the distribution of γH2AX determined post DSB induction (Fig. 1a-b, Extended Data Fig. 1a), suggesting that initial chromosome architecture dictates γH2AX spreading and downstream events such as accumulation of MDC1, ubiquitin and 53BP1 following DSB. To prove that DDR domains do not spread into neighboring self-interacting domains, we focused on a DSB located on chr1, for which spreading of DDR foci components is profoundly asymmetrical (Fig. 1c, red track). 4C-seq performed at two viewpoints separated by 470 kb revealed the existence of two adjacent self-interacting domains with a boundary corresponding to the abrupt drop in γH2AX (Fig. 1c, blue track, TAD boundary is indicated by the dotted line). This strongly suggests that pre-existing chromatin domains, established before any damage occurs, constrain the spread of DDR foci. To generalize this finding, we performed Hi-C and CTCF ChIP-seq in undamaged DIvA cells (Extended Data Fig. 1b-d). Strikingly, computed Topologically Associating Domains (TADs) borders and CTCF-bound genomic loci coincided with a sharp decrease in γH2AX signals (Fig. 1d-e, Extended Data Fig. 1e). In agreement, γH2AX, MDC1 and 53BP1 were significantly more enriched within the damaged TADs compared to neighboring TADs (Extended Data Fig. 1f), although spreading through boundaries was observed to some extent, in agreement with the rather moderate insulation properties of TAD boundaries11.
To further investigate if TADs dictate γH2AX spreading, we used the CRISPR/Cas9 system to induce a single DSB at designed positions within the same TAD, and investigated both chromosome conformation and γH2AX distribution. Cas9-induced DSB recapitulated the γH2AX spreading observed when inducing a DSB at the same genomic location by AsiSI (Extended Data Fig. 1g), thus confirming that γH2AX spreading is independent of the DSB induction method. Moving the DSB to a further downstream position in the TAD triggered a change in the γH2AX profile that was notably similar to the 3D interaction pattern of this genomic region, yet it remained constrained within the same TAD (Fig. 1f). Altogether these data indicate that mechanisms governing the spatial organization of chromosomes into self-interacting domains facilitates and demarcates the formation of γH2AX domains. Given that γH2AX is seeding further signaling events leading to the stable assembly of DDR foci, this suggests that genome organization within TADs is critical for the response to DNA damage.
In human cells, ATM is the main DDR kinase that catalyzes H2AX phosphorylation upon DSB detection, as indicated by a strong decrease of γH2AX upon ATM inhibition12 (Extended Data Fig. 1h-j) but not upon DNAPK12 or ATR inhibition (Extended Data Fig. 1i-j). In order to gain more insights into the mechanism that mediates γH2AX establishment on entire self-interacting domains, we further profiled ATM. Activated ATM (autophosphorylated on S1981) binding was restricted to the immediate vicinity of the DSB (< 5kb) in sharp contrast with the pattern observed for γH2AX (Fig. 1g, Extended Data Fig. 1k). This indicates that H2AX phosphorylation is not mediated by the linear spreading of the kinase on entire TADs.
Cohesin-mediated loop extrusion at DSBs
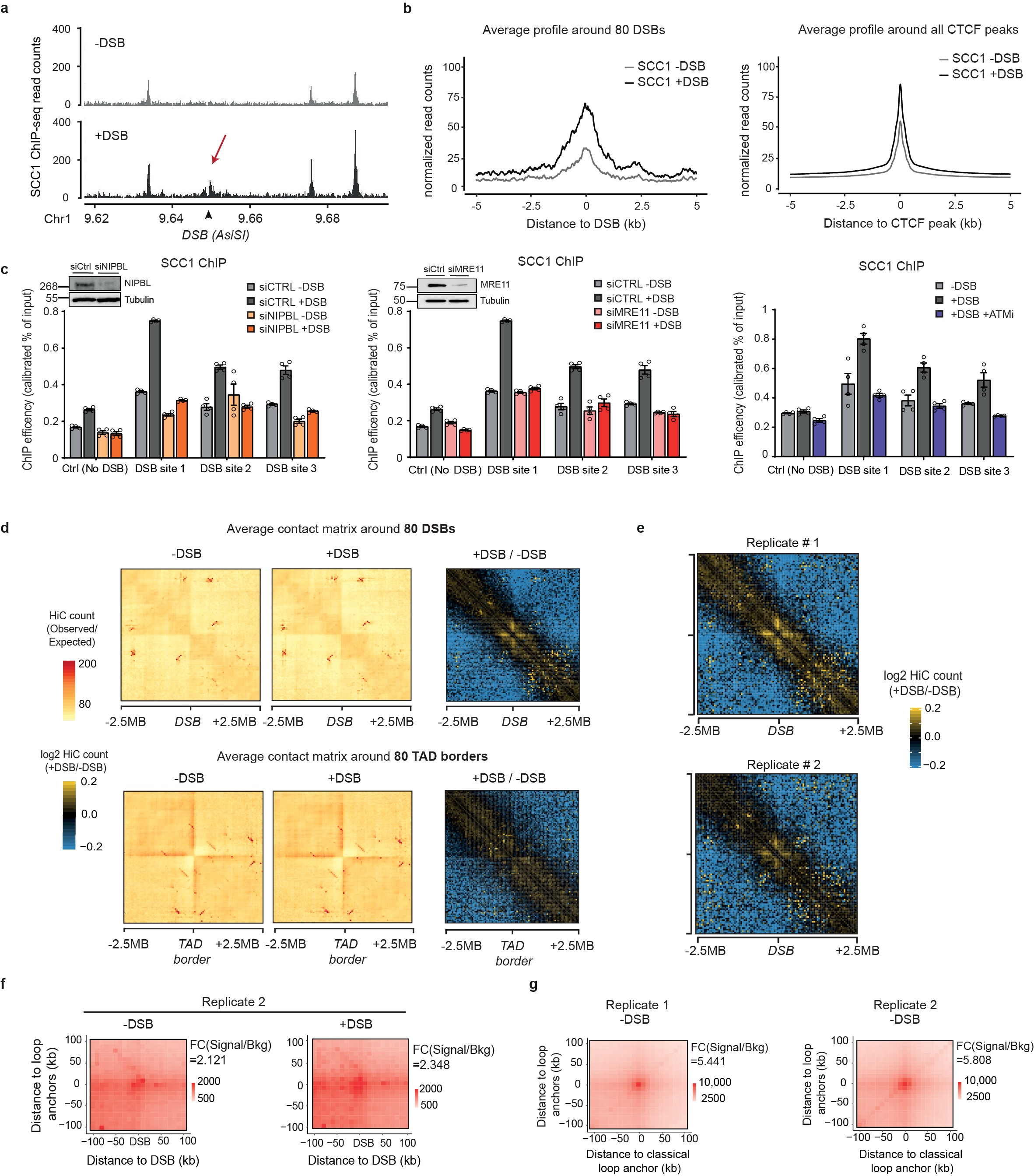
The organisation of the genome into TADs is driven by activity of cohesin13,14, a ring-shaped protein complex, which was initially identified for its essential role in sister chromatid cohesion. Of importance, strong evidence support a role of cohesin in the maintenance of genome integrity15,16 and cohesin accumulates at sites of damage, which may be in line with a role in sister chromatid cohesion during Homologous Recombination (HR) in S/G217–20. Yet, cohesin enrichment has been identified at DSBs throughout the cell cycle, which argues against an exclusive role for cohesin in HR7,16. To get insights in cohesin binding at DSBs at high resolution, we performed calibrated ChIP-seq profiling of the SCC1 cohesin subunit, in both undamaged and damaged conditions. Notably, cohesin was enriched at sites of damage spanning 2-5kb around the DSB (Fig. 2a) leading to the formation of peaks at DSB sites nearly as high as pre-existing cohesin peaks at CTCF binding sites (Extended Data Fig. 2a-b), and in a manner that depends on the cohesin loader NIPBL, on ATM activity and on the MRN complex subunit MRE11 (Extended Data Fig. 2c).
Figure 2. DSB-anchored cohesin mediates loop extrusion.

(a) Genomic tracks of SCC1 and XRCC4 ChIP-seq at two DSBs, indicated by black arrows. (b) Averaged Hi-C contact matrix of the log2 (+DSB/-DSB) (n=2 biological replicates) centered on the 80 best-induced DSBs (50 kb resolution, 5 Mb window). Stripes are indicated by white arrows. (c) Mean aggregate Peak Analysis (APA) plot on a 200kb window (10 kb resolution) before and after DSB induction, calculated between the DSBs and nearby loops anchors (n=525 pairs). The fold change between the signal (central pixel) and the background (upper left corner 5×5 pixels) is indicated. (d) Averaged differential Hi-C contact matrix (+DSB/-DSB) (n=2 biological replicates) around 30 HR-repaired DSBs, 30 NHEJ-repaired DSBs and 30 random undamaged sites. (e) Box plot of the SCC1 ChIP-seq enrichment before and after DSB on 4 kb around DSBs repaired by HR (yellow), NHEJ (green) and random undamaged sites (grey) (n=30). P values are indicated (paired two-sided Wilcoxon test). Center line: median; Box limits: 1st and 3rd quartiles; Whiskers: Maximum and minimum without outliers; Points: outliers. (f) Differential 4C-seq track in control (black) or in SCC1 siRNA condition (blue). (g) Averaged log2 (+DSB/-DSB) Hi-C matrix upon CTRL or SCC1 siRNA, around eighty best-induced DSBs (100 kb resolution).
Cohesins structure TADs by an active, ATP-dependent, loop extrusion mechanism21–24. Once loaded onto chromatin, cohesin leads to the formation and enlargement of DNA loops, eventually arrested at boundary elements, of which a large fraction is bound by the CTCF insulator protein. Increased cohesin around DSBs could thus indicate locally increased loop extrusion at site of damage. We hence set to analyze 3D genome organization by Hi-C before and after DSB induction in DIvA cells, focusing on cis interaction frequencies around DSBs. Differential (+DSB/-DSB) aggregate Hi-C maps were further computed around DSBs and around TAD borders as a control (Extended data Fig. 2d). Interestingly, a unique pattern of “stripes” appears at both sides of the DSBs following DSB induction (Fig. 2b, white arrows, Extended Data Fig. 2d-e). “Stripes” or “lines” were previously reported to arise from arrested loop-extrusion at CTCF-bound loci22,24–27. Indeed, our averaged Hi-C contact matrixes around TAD borders revealed, as expected, similar stripes, but those were independent of DSB induction (Extended Data Fig. 2d). We further performed Aggregate Plot Analysis (APA) to assess looping between the DSB position and neighboring anchors. Notably, the APA score increased following production of DSBs (Fig. 2c, Extended Data Fig. 2f) indicating that the DSBs themselves display the potential to arrest loop-extrusion, although to a lesser extent than classical loop anchors (CTCF bound loci) (Extended Data Fig. 2g).
We previously determined which repair pathway (i.e. Homologous Recombination (HR) or Non Homologous End Joining (NHEJ)) is preferentially utilized at different DSBs induced by AsiSI in DIvA cells28. Importantly, an equivalent stripe pattern was observed at both DSBs repaired by HR or those repaired by NHEJ (Fig. 2d). In agreement, SCC1 accumulates on a 4 kb window around DSBs irrespective of the pathway used for repair (Fig. 2e). Altogether these data suggest that cohesin accumulates at either side of a DSB, irrespective of the pathway used for repair, to induce a divergent one-sided loop extrusion towards (and thus increased contacts with) the surrounding regions on both sides of the break.
In order to further investigate DSB-anchored loop extrusion, we performed 4C-seq before and after DSBs induction, using viewpoints located at the exact positions of three DSBs induced in DIvA cells (same viewpoints as in Figure 1). Importantly, the overall TADs structure and boundaries were well maintained post DSB induction (Extended Data Fig. 3a) indicating that chromosome conformation within TAD is not completely reshuffled upon damage induction. Yet, as expected from Hi-C data, we could detect increased interactions between viewpoints and surrounding loci post-DSB (Extended Data Fig. 3b-d), which was not the case when using a control undamaged sequence as a viewpoint (Extended Data Fig. 3c-d). If DSB-anchored, cohesin-mediated loop extrusion is responsible for enhanced interaction frequency of the DSB with neighboring sequences post DSB induction, such behavior should be abolished following cohesin depletion. Indeed, 4C-seq experiments revealed that depletion of SCC1 (Extended Data Fig. 3e-f) strongly impaired the overall increase in contacts between the DSBs and its neighboring sequences in damaged TADs (Fig. 2f, Extended Data Fig. 3g-h). We further performed Hi-C in damaged and undamaged conditions following depletion of SCC1. As expected from previous studies14,29, SCC1 depletion led to the dissolution of TADs and to a stronger compartmentalization (plaid pattern) on Hi-C maps (Extended Data Fig. 4a). Notably, SCC1 depletion abolished the stripe pattern induced at DSBs following damage (Fig. 2g). Given that ATM is involved in SCC1 recruitment at DSBs (Extended Data Fig. 2c) we further assessed the consequences of pharmaceutical inhibition of the ATM kinase activity on the interaction frequency post-DSB by 4C-seq. Notably, upon ATM inhibition the ability of the DSB to engage contacts with proximal sequences within damaged TADs was strongly reduced (Extended Data Fig. 4b-c) in agreement with defective SCC1 recruitment at DSBs in these conditions (Extended Data Fig. 2c).
Altogether these data indicate that the ability of the DSB to contact neighboring loci within the damaged TAD is a proper DNA Damage Response and cannot solely be explained by the physical disruption of the DNA. Importantly, it depends on ATM activity and on the cohesin complex, in agreement with a DSB-anchored loop extrusion mechanism.
Loop extrusion in γH2AX domain formation
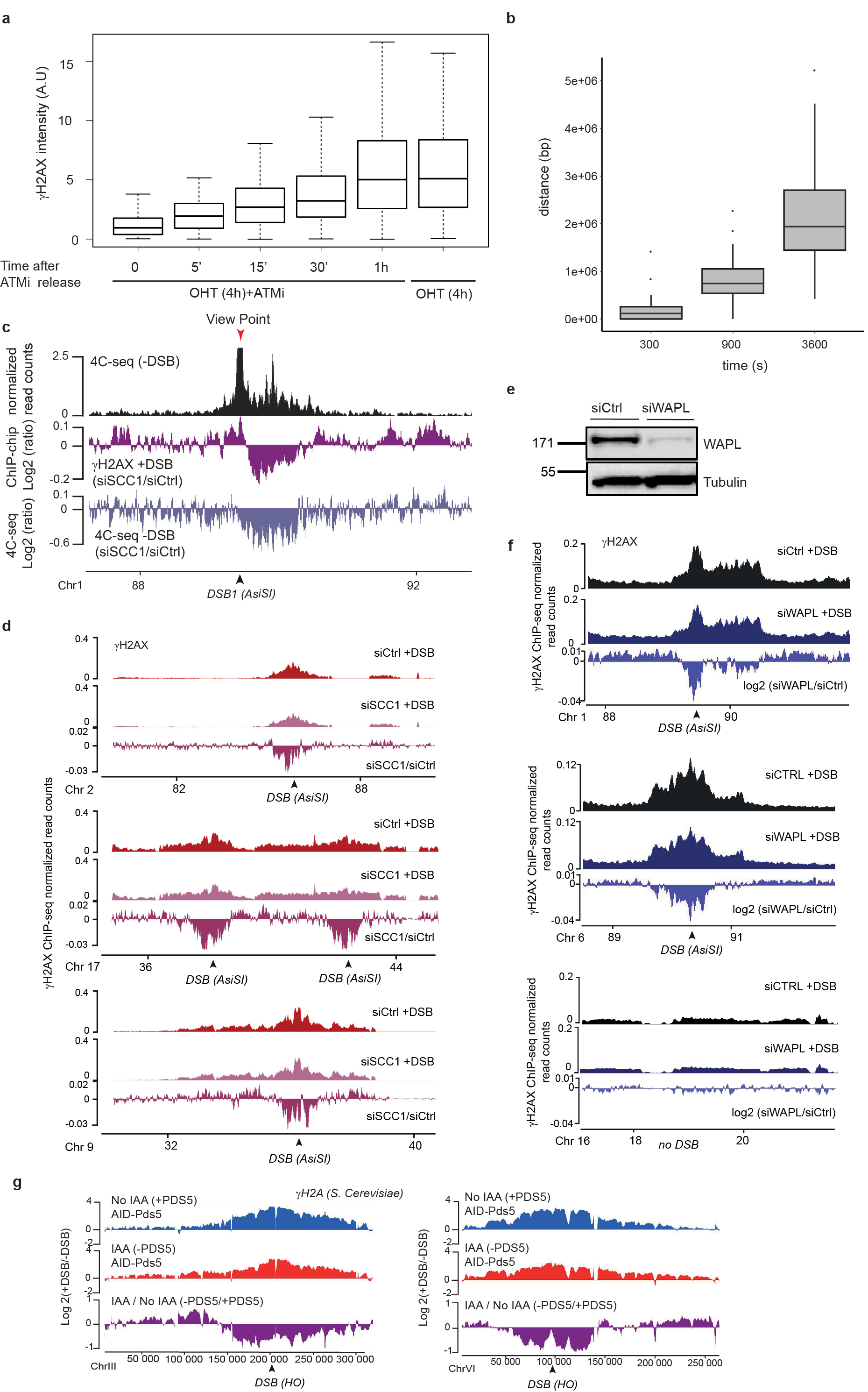
We further investigated whether cohesin-mediated loop extrusion that takes place at DSBs is instrumental for γH2AX deposition. In this scenario γH2AX should spread linearly from the DSB site across time. To achieve high synchronization of γH2AX deposition within the cell population, we induced DSBs (by OHT treatment) but concomitantly inhibited ATM activity (using an ATM inhibitor) allowing to “poise” γH2AX establishment. Relieving ATM inhibition allowed for a fast and synchronous accumulation of γH2AX (Extended Data Fig. 5a). With this experimental set up we observed a linear and bidirectional spreading of γH2AX from the DSBs by ChIP-seq, that proceeds at a speed of approximately 0.6kb/s, in agreement with a loop-extrusion dependent mechanism (Fig. 3a, Extended Data Fig. 5b)21,23.
Figure 3. DSB-anchored loop extrusion mediates γH2AX spreading.

(a) γH2AX ChIP-seq tracks at three DSB sites upon DSB induction at different time points after ATMi release (expressed as log2 (+DSB+ATMi+time after washes/ +DSB+ATMi+0min after washes)) (20 kb smoothed). (b) Genomic track showing differential (log2 ratio siSCC1/ siCtrl) γH2AX enrichment obtained after DSB induction (purple) (20kb smoothed). Light blue track shows the differential 4C-seq signal obtained in SCC1-depleted versus control cells before DSB induction (log2 siSCC1/siCtrl). (c) Genomic tracks showing the CTCF signal before DSB induction, the γH2AX ChIP-seq signal after DSB induction in Ctrl or WAPL-depleted cells and the differential γH2AX signal obtained after DSB induction (expressed as the log2 ratio siWAPL/siCtrl, 20kb smoothed) at two DSB sites. (d) Genomic tracks showing the differential γH2A ChIP-seq signal (log2 +DSB/-DSB) before (no IAA) or after PDS5 degradation (IAA) at one DSB site (HO site) (top panel) and in a control region (without DSB) (bottom panel) in S.Cerevisiae PDS5-AID. The differential signal between after and before PDS5 degradation (IAA / No IAA) is also shown (purple). Data are smoothed with a 2kb span.
To investigate whether cohesin-mediated loop extrusion contributes to DDR foci formation, we analyzed γH2AX profiles in SCC1-deficient cells. Both γH2AX ChIP-chip7 and ChIP-seq showed altered γH2AX spreading in SCC1-deficient cells compared to SCC1-proficient cells (Fig. 3b, Extended Data Fig. 5c-d) in a manner that coincided with a loss of cis contacts upon cohesin depletion (Fig. 3b, Extended Data Fig. 5c). Of note, the decreased γH2AX level in cohesin depleted cells was weak (~5-10%) compared to the decreased in 4C-seq signal (30%), which may indicate that other factors (e.g. SMC5/6) could contribute in loop extrusion mediated γH2AX establishment and/or that intra-TAD chromatin dynamic contributes to γH2AX deposition.
Cohesin are released from chromatin by the accessory WAPL and PDS5 factors. Consequently their depletion trigger longer chromatin loops proposed to arise from a more processive cohesin-mediated, loop extrusion29,30. Notably, we observed extended spreading of γH2AX in WAPL-depleted cells (Fig. 3c, Extended Data Fig 5e) in agreement with a contribution of loop extrusion in γH2AX deposition. Of note, this was also accompanied by a decreased level of γH2AX within TAD (Extended Data Fig. 5f). Given that WAPL depletion, while enlarging loops, also decreases intra-TAD chromatin interaction30, this suggests that intra-TAD chromosome dynamics also contributes to γH2AX full deposition.
To investigate if such a cohesin-dependent mechanism could account for the establishment of DDR foci in budding yeast, we depleted PDS5 thanks to an auxin-inducible system in a S. cerevisiae strain31 that carries three HO endonuclease cleavage sites32. Consistent with our observations in human WAPL-depleted cells, extended spreading of γH2A in yeast occurred following depletion of PDS5 (Fig. 3d). Notably, PDS5 deficiency triggered decreased γH2A levels adjacent to the DSBs (Extended Data Fig. 5g), similarly to WAPL depletion in human cells.
Taken together these data suggest that cohesin accumulation at DSBs initiate a one-sided loop extrusion process at either side of the break that contributes in establishing the phosphorylation of H2AX and spreads until it reaches a strong boundary element (i.e. a TAD border). Interestingly, this cohesin-dependent mechanism is conserved from yeast to human.
Cohesin changes in damaged TADs
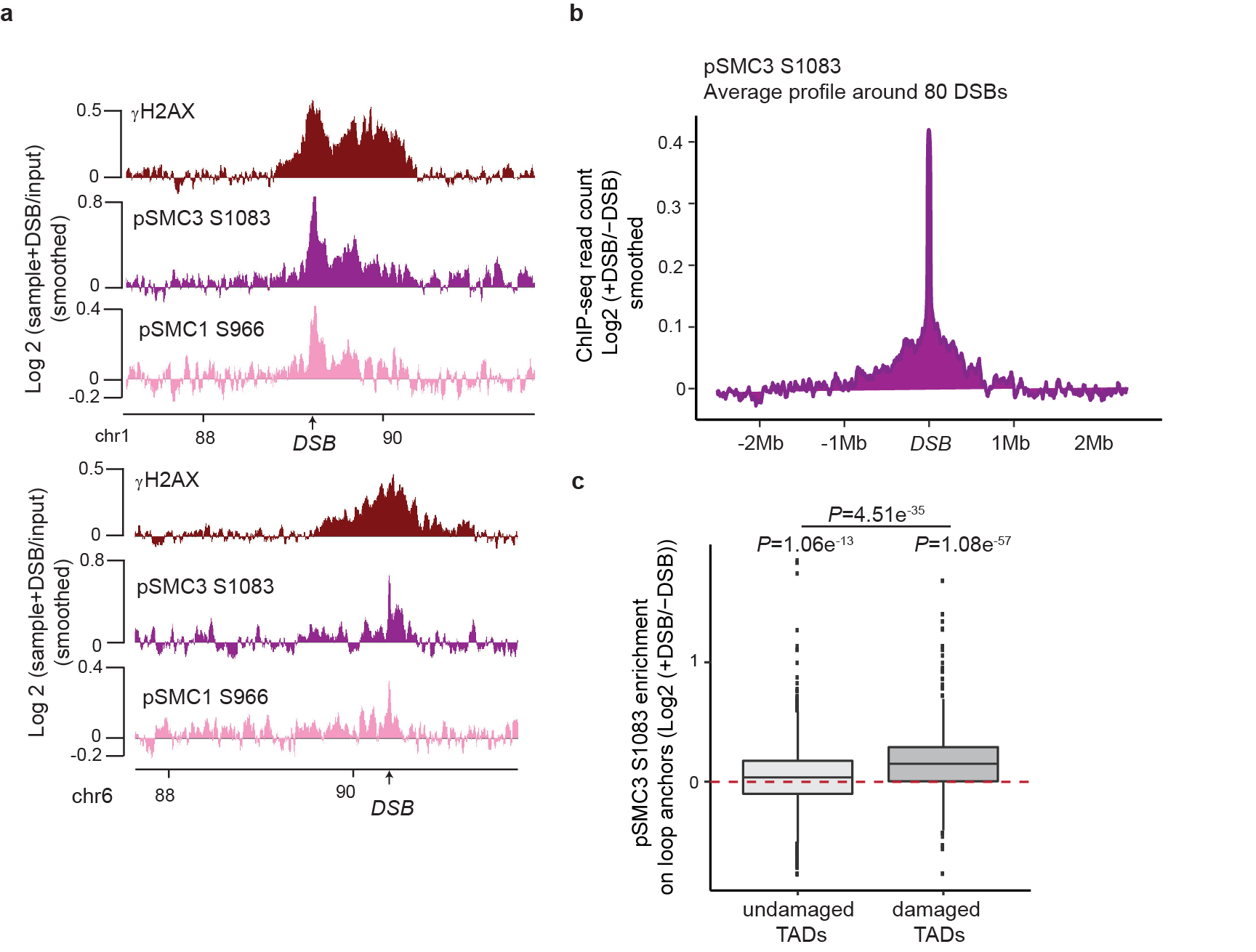
Previous work indicated a genome-wide increase of cohesin and reinforcement of TADs following exposure to irradiation33,34. In agreement, we found that SCC1 enrichment was increased at cohesin-binding sites post-break induction, coinciding with an increased loop strength (Extended Data Fig. 6a-b). Of interest, DSB-induced increased loop strength and SCC1 accrual were more pronounced within damaged TADs than undamaged TADs and decreased with the distance to DSBs (Fig. 4a, Extended Data Fig. 6c-g). Thus, our data indicate a generalized increase in SCC1 occupancy and loop strength throughout the genome after DSB production in a manner that is weakly exacerbated within the TADs subjected to DSB. The SMC1 and SMC3 cohesin subunits have been previously reported to be phosphorylated by ATM following DSBs induction35 and these modifications are essential for cohesin reinforcement on the genome post irradiation34. ChIP-chip analyses indicated that phosphorylated SMC1 (pSMC1 S966) and SMC3 (pSMC3 S1083) accumulate on entire TADs around DSBs (Extended Data Fig. 7a). ChIP-seq against pSMC3 S1083 further confirmed that phosphorylated SMC3 increased at cohesin-bound sites and loop anchors within damaged TADs (Fig. 4b, Extended Data Fig. 7b-c). These DSB-induced, ATM-mediated cohesin modifications accumulating around DSBs, may regulate cohesin properties, such as loop extrusion velocity or chromatin unloading, which could translate into increased cohesin residence time at boundaries elements and further contribute to isolate DDR domains from adjacent chromatin.
Figure 4. DSBs trigger modifications of cohesin biology at a genome-wide scale and accentuated in damaged TADs.

(a) Boxplot showing the quantification of SCC1 recruitment on loop anchors before (grey) and after (red) DSB induction, within damaged (n=1456) or undamaged TADs (n=7804). Center line: median; Box limits: 1st and 3rd quartiles; Whiskers: Maximum and minimum without outliers; Points: outliers. P values are indicated (two-sided Wilcoxon test). The increased SCC1 enrichment on loop anchors following DSB is significantly higher in damaged TADs as compared to undamaged TADs (two-sided Wilcoxon test). (b) Genomics tracks showing the γH2AX ChIP-seq signal (50kb smoothed), SCC1 and phosphorylated SMC3 (pSMC3 S1083) ChIP-seq signal expressed as the log2 ratio (+DSB/-DSB) (20kb smoothed). (c) Model: Cohesin-mediated loop extrusion ensures γH2AX establishment on the entire damaged TAD. (i) Loop extrusion constantly occurs on the genome. (ii) The occurrence of a DSB creates a roadblock for cohesin-mediated loop extrusion leading to accumulation of cohesin at the site of damage. (iii) Cohesin blocked at DSB continues to mediate loop extrusion in a unidirectional manner (i.e. one-sided loop extrusion, arrows). ATM, recruited at the immediate vicinity of the break, phosphorylates H2AX-containing nucleosomes as they are extruded. Meanwhile, cohesin are also phosphorylated by ATM. (iv) The same process takes place on both sides of the DSB, leading to divergent one-sided loop extrusion at either side of the break ensuring a bidirectional spreading of γH2AX. (v) Loop extrusion triggers enlargement of γH2AX modified chromatin and halt at boundary elements such as CTCF-bound loci, that demarcate TAD borders. The speed of loop extrusion (measured in vitro as 0.5-2kb/s) ensures that the entire damaged TAD is phosphorylated in 10-30 minutes, giving rise to a DDR focus. NB: here the cohesin is shown as a ring encircling DNA for graphical reasons, but it is not known yet, whether and how cohesin ring entraps DNA during the loop extrusion process.
A model for γH2AX domain formation
In summary, our work unambiguously shows that TADs are the template for the spreading of many DSB repair signaling events such as the phosphorylation of H2AX, in agreement with a recent report36, the eviction of histone H1 and the accrual of 53BP1, MDC1 and ubiquitin, allowing a DSB signaling at the megabase scale. Our data suggest a DSB-anchored cohesin-mediated loop extrusion model that would mediate H2AX phosphorylation (Fig. 4c). In this model, cohesin rapidly accumulates on both sides of a DSB in a manner that is fostered by ATM, NIPBL and the MRN complex. Whether this is due to prior ongoing loop extrusion arresting at DSB or to de novo loading of the cohesin complex still need to be determined. Divergent one-sided loop extrusion further takes place at the DSB, which in turn allows the locally-recruited ATM to phosphorylate H2AX containing nucleosomes as the chromatin fiber is pulled by the cohesin ring. Given that current estimates of cohesin-mediated loop extrusion suggest a rate of 0.5-2 kb per second in vitro 21,23, such a mechanism would allow DDR foci assembly in a very rapid manner, with the entire megabase-sized chromatin domain being modified in about 10-30 min, which fits with the observed rate of γH2AX foci assembly9. This model would be in agreement with i) the finding that in yeast, the ATM ortholog Tel1 mediates H2A phosphorylation in a manner that agrees with a 1D sliding model rather than a 3D diffusion model37 and ii) the recent observation, using light-induced activation of Cas9, that γH2AX is established at a speed of ~150kb/min and can in some instance reach up to 30Mb38. Moreover, our data also indicate that upon DSB induction, the loop strength is reinforced, cohesin accumulates at loop anchors and the cohesin complex itself is modified by ATM within damaged TADs. We propose that ATM-mediated phosphorylation of the cohesin complex may alter the properties of cohesin such as loop extrusion velocity or its capability to load onto/unload from chromatin. This may contribute to further reinforce H2AX phosphorylation thanks to intra-TAD chromatin dynamics following an initial loop-extrusion dependent γH2AX establishment.
Recent work supports the key role of TADs borders and loop extrusion in the maintenance of genome architecture and stability, including immunoglobulin loci rearrangements39,40 and in DSB occurrence through topoisomerase reactions41,42. Our study shows that genome architecture is also instrumental for the correct establishment of γH2AX and DDR foci formation, expanding the function of genome organization within TADs to the response to DNA damage. We propose that arresting loop extrusion provides an efficient and rapid way to signal a DSB and assemble a DDR focus, while boundary elements help constraining DDR signaling to DSB-surrounding, self-interacting chromatin domains. This creates a specific repair-prone chromatin compartment, that displays modified dynamics properties, which may, for example reduce search time for DNA end rejoining and homology search, and/or concentrate repair factors.
Methods
Cell culture and treatments
DIvA (AsiSI-ER-U20S)10 cells were grown in Dubelcco’s modified Eagle’s medium (DMEM) supplemented with 10% SVF (InVitrogen), antibiotics and 1 μg/mL puromycin (DIvA cells) at 37 °C under a humidified atmosphere with 5% CO2. Cells were regularly tested negative for mycoplasma contamination. For DSB induction, cells were treated with 300nM 4OHT (Sigma, H7904) for 4 h. For ATM inhibition, cells were pretreated for 1 h with 20μM KU-55933 (Sigma, SML1109) and during subsequent 4OHT treatment. For ATR inhibition, cells were pretreated for 1 h with 2μM ETP-46464 (Sigma, SML1321) and during subsequent 4OHT or HU treatment (1h at 1mM (Sigma, H8627)). For kinetics experiment (Fig. 3a), cells were pretreated for 1h with 20μM KU-55933 (Sigma, SML1109) and during subsequent 4OHT treatment before to be washed three times with PBS 1X and released during 0min, 5min, 15min or 1h. siRNA transfections were performed with a siRNA Ctrl: CAUGUCAUGUGUCACAUCU and a siRNA targeting SCC1: GGUGAAAAUGGCAUUACGG or WAPL: CGGACUACCCUUAGCACAA or NIPBL: GCUCGGAACAAAGCAAUUA or MRE11: GCUAAUGACUCUGAUGAUA, using the 4D-Nucleofector™ and the SE cell line 4D-Nucleofector™ X kit L (Lonza) according to the manufacturer’s instructions and subsequent treatment(s) were performed 48 h later. For CRISPR/Cas9-mediated DSB induction, sgRNA (AsiSI site position: “CGCCGCGATCGCGGAATGGA” or Position further within the TAD: “GGGCCAGTCGCGGCACTCGC”) were delivered in U2OS cells using the “nanoblades” technology which relies on direct cell transduction with a viral-derived particle containing the Cas9/sgRNA ribonucleoprotein43,44. Cells were analyzed 24 h after transduction.
To make the S. cerevisiae strain yFZ014, a linearized TIR1 gene was obtained through a restriction enzyme digest of plasmid pJH2955 with PmeI1 and inserted into the leu2 locus of strain YSCL00432. Insertion of TIR1 45 was verified by PCR with primers internal to TIR1 and leu2. yFZ016 was made by PCR amplification of plasmid pJH2898 to produce a 9myc-AID::KAN PCR product with homologies at each end to the C-terminus of Pds5; this PCR product was inserted using standard yeast transformation protocols to produce a Pds5::9myc-AID fusion protein. A Western Blot was used to verify the degradation of Pds5::9myc-AID in yFZ014 and yFZ016 after auxin addition. DSB were induced as in33.
Immunofluorescence
DIvA cells were plated on glass coverslips and fixed with 4% paraformaldehyde during 15 min at room temperature, permeabilized with 0.5% Triton X-100 in PBS for 10 min then blocked with 3% BSA in PBS for 30min. Cells were then incubated with the primary antibody (referenced in Extended Data Table 1) diluted in PBS-BSA overnight at 4°C, washed with 1X PBS and incubated with the appropriate anti-mouse or anti-rabbit secondary antibodies (conjugated to Alexa 594 or Alexa 488, Invitrogen), diluted 1:1000 in PBS-BSA, for 1h at room temperature, followed by DAPI staining. Coverslips were mounted in Citifluor (Citifluor, AF-1). Image acquisition was performed with MetaMorph on a wide-field microscope (Leica, DM6000) equipped with a camera (DR-328G-C01-SIL-505, ANDOR Technology) using 40x or 100x objectives. For the quantification, cells were acquired with a 40x objective and analyzed using Columbus software (Perkin Elmer). γH2AX foci were detected using the method D.
Western Blot
For SCC1, WAPL, NIPBL and MRE11 detection, cells were incubated in RIPA buffer (50 mM Tris at pH 8, 150 mM NaCl, 0.5% deoxycholate, 1% NP-40, 0.1% SDS) for 20 min on ice and centrifuged at 13,000 rpm for 10 min to remove insoluble material. SDS loading buffer and reducing agent were then added to the supernatant. For pCHK1 detection, cells were resuspended in 100μL of histone extraction buffer (1% SDS, 1% Triton, 10mM Tris pH7.5, 0.5M NaCl, phosphatase 0.01X (Sigma, P5726) and Complete protease inhibitors 1X (Sigma, 11873580001)) and sonicated 2 times 10sec with an amplitude of 30% before addition of SDS loading buffer and reducing agent. All protein extracts were resolved on 3%–8% NuPAGE Tris-acetate gels (Invitrogen) and transferred onto PVDF membranes (Invitrogen) according to the manufacturer’s instructions. Membranes were blocked in TBS containing 0.1% Tween 20 (Sigma, P1379) and 3% nonfat dry milk for 1h followed by an overnight incubation at 4°C with primary antibodies (referenced in Extended Data Table 1). The appropriate horseradish peroxidase-coupled secondary antibodies were used to reveal the proteins (anti-mouse at 1:10,000 (Sigma, A2554) and anti-rabbit at 1: 10,000 (Sigma, A0545)) using a luminol-based enhanced chemiluminescence HRP substrate (Super Signal West Dura Extended Duration Substrate, Thermo Scientific). Pictures of the membranes were acquired with the ChemiDoc™ Touch Imaging System and were visualized using Image Lab Touch software. Uncropped blots are presented in Supplementary Information Figure 1.
Hi-C
Hi-C experiments were performed in DIvA cells using the Arima Hi-C kit (Arima Genomics) according to the manufacturer’s instructions. 1×106 cells were used by condition and experiments were performed in duplicates. Briefly, cells were cross-linked with 2% formaldehyde for 10 min at RT, lysed and chromatin was digested with two different restriction enzymes included in the kit. Ends were filled-in in the presence of biotinylated nucleotides, followed by subsequent ligation. Ligated DNA was sonicated using the Covaris S220 to an average fragment size of 350 bp with the following parameters (Peak incident power: 140; Duty factor: 10%; Cycles per burst: 200; Treatment time: 70s). DNA was then subjected to a double-size selection to retain DNA fragments between 200 and 600 bp using Ampure XP beads (Beckman Coulter). Biotin-ligated DNA was precipitated with streptavidin-coupled magnetic beads (included in the kit). Hi-C library was prepared on beads using the NEBNext® Ultra™ II DNA Library Prep Kit for Illumina and NEBNext® Multiplex Oligos for Illumina (New England Biolabs) following instructions from the Arima Hi-C kit. The final libraries were subjected to 75 bp paired-end sequencing on a Nextseq500 platform at the EMBL Genomics core facility (Heidelberg, Germany). Hi-C reads were mapped to hg19 and processed with Juicer using default settings (https://github.com/aidenlab/juicer). Matrix-balanced Hi-C count matrices were generated at multiple resolutions: 250kb, 100kb, 50kb, 25kb, 10kb and 5kb and visualized on Juicebox and on Hi-Glass.
4C-seq
The 4C-seq experiments were realized as in46 with minor modifications. Briefly, 15×106 DIvA cells were cross-linked with 2% formaldehyde for 10 min at RT, lysed and digested with MboI (New England Biolabs). Two or three rounds of 4 h of digestion with MboI were necessary. Digested DNA was then ligated with a T4 DNA ligase (HC) (Promega), purified and digested with NlaIII overnight (New England Biolabs). After a second ligation step, DNA was purified before proceeding to the library preparation. For DNA purification steps, AMPure XP beads (Beckman Coulter) were used diluted at 1:10 in 20% PEG solution (PEG 8000 (Sigma) 20%, 2.5M NaCl, Tween 20 20%, Tris pH8 10mM, EDTA 1mM). For 4C-seq library preparation, 800ng-900ng of 4C-seq template was amplified using 16 individual PCR reactions with inverse primers (PAGE-purified) including the Illumina adapter sequences and a unique index for each condition (primers in Extended Data Table 2). Libraries were purified with the QIAquick PCR Purification Kit (Qiagen), pooled and subjected to 75bp single-end sequencing on a Nextseq500 platform at the I2BC Next Generation Sequencing Core Facility (Gif-sur-Yvette, France). Each sample was then demultiplexed using a specific python script from the FourCSeq R package47 thus assigning each read to a specific viewpoint based to its primer sequence into separate fastQ files. bwa mem was then used for mapping and samtools for sorting and indexing. A custom R script (https://github.com/bbcf/bbcfutils/blob/master/R/smoothData.R)48 was used to build the coverage file in bedGraph format, to normalize using the average coverage and to exclude the nearest region from each viewpoint (viewpoint-containing restriction fragment and the two adjacent restriction fragments). Then the bedGraph file was converted in a BigWig file using bedGraphToBigWig program from UCSC.
ChlP-qPCR, ChIP-seq and ChlP-chip
For Fig. 1a, Ubiquitin, H1, γH2AX and 53BP1 ChIP-seq data were retrieved from5. ChIP experiments of pATM and MDC1 were performed in DIvA cells as in10 with 200μg of chromatin per immunoprecipitation. Prior to library preparation, samples from multiple ChIP experiments of pATM and MDC1 were pooled and sonicated for 15 cycles (30 sec ON, 30 sec OFF, high setting) with a Bioruptor (Diagenode) then concentrated with a vacuum concentrator (Eppendorf). CTCF, pSMC3 (S1083), and γH2AX (Fig.3–4, Extended Data Fig. 7 and Extended Data Fig. 1j) ChIP experiments were realized as follow: Briefly, cross-linked cells were first lysed 10min at 4°C in 500 μL of the Lysis Buffer 1 (10mM Tris pH8, 10mM NaCl, 0.5% NP-40, Complete protease inhibitor (Sigma, 11873580001)) then 10min at 4°C in the Lysis buffer 2 (50mM Tris pH8, 10mM EDTA, 0.5% NP-40, Complete protease inhibitor (Sigma)) and subsequently sonicated in 15ml conical tubes with a Bioruptor Pico (Diagenode) in the presence of 800 mg of sonication beads (20 cycles of 30 sec ON/30 sec OFF) to an average fragment size of 250pb. 200μg of chromatin were then immunoprecipitated as in10. The antibodies used are detailed in Extended Data Table 1. Sequencing libraries were prepared by using 10 ng of purified DNA (averaged size 250-300 bp) with the NEBNext® Ultra™ II Library Prep Kit for Illumina (New England Biolabs) using the application note for “Low input ChIP-seq.”, and subjected to 75 bp single-end sequencing on a Nextseq500 platform at the EMBL Genomics core facility (Heidelberg, Germany).
For the SCC1 calibrated ChIP-seq, we used a spike-in method49. Briefly, cross-linked cells from DIvA cells or from ES cells were lysed and fragmented as for CTCF, pSMC3 (S1083) and γH2AX. Prior to immunoprecipitation with SCC1 antibody, 20% of chromatin from mouse ES cells (40μg) was added to chromatin prepared from treated or untreated human DIvA cells (200μg). Sequencing libraries were prepared from IP and input samples using the NEBNext® Ultra™ II Library Prep Kit for Illumina and subjected to 75 bp single-end sequencing on a Nextseq500 platform at the EMBL Genomics core facility (Heidelberg, Germany). First, SCC1 was aligned on the mouse genome (mm10) with bwa in order to only map the reads used as a reference for the normalization (spike-in). Remaining unmapped reads were re-converted into a fastQ file using bam2fastq and mapped to the human genome (hg19) using bwa. Samtools was used for sorting and indexing, and reads mapped to the mouse genome were used as a normalization factor, as in49 and using the following formula.
where Inputctrl is the total number of reads mapped in ES Input (Mouse) and Inputexp the total number of reads in DIvA Input. readsctrl and readsexp were respectively the number of reads from immunoprecipitated samples mapped on the mm10 genome and hg19 genome.
For calibrated SCC1 ChIP-qPCR, the immunoprecipitated samples from DIvA cells were normalized by the signal of the immunoprecipitated sample from ES cells on a mouse cohesin positive site (using primers provided in Extended Data Table 2). Data were analyzed using the Bio-Rad CFX manager software.
For the ChIP-chip experiments, the immunoprecipitated samples of γH2AX, pSMC1 S966, pSMC3 S1083 and input samples were amplified as in10, labeled, and hybridized on Affymetrix tiling arrays covering human chromosomes 1 and 6 (at the Genotoul GeT-biopuces facility, Toulouse, France)). Scanned array data were normalized using Tiling Affymetrix Software (TAS) (quantile normalization, scale set to 500) and analyzed as described in10,12 and converted to.wig files using R/Bioconductor software, when necessary, for visualization using the Integrated Genome Browser (bioviz.org).
For the ChIP experiment in yeast, individual colonies of yFZ014 and yFZ016 were grown in YEP + 3% lactic acid (YEP-Lac) until log phase growth with a final cell concentration between 5 × 106 cells/ml and 8 × 106 cells/ml. Degradation of Pds5::9myc-AID in yFZ016 was induced by addition of auxin (indole-3-acetic acid (IAA) Sigma Aldrich # I3750) at a final concentration of 1 mM and confirmed by Western blotting. For chromatin immunoprecipitation, 45 ml of culture was fixed and cross-linked with 1% formaldehyde for 10 min, after which 2.5 ml of 2.5 M glycine was added for 5 min to quench the reaction. Cells were pelleted and washed 3 times with 4°C TBS. Yeast cell walls were disrupted by beating the cells with 425-600 μm glass beads for 1 h in lysis buffer at 4°C. The lysate was sonicated for 2 min to obtain chromatin fragments of ~500 bp in length. Debris was then pelleted and discarded, and equal volume of lysate was immunoprecipitated using γ-H2A antibody for 1 h at 4°C, followed by addition of Protein-A agarose beads (Sigma-Aldrich #1719408001) for 1 h at 4°C. The immunoprecipitate was then washed twice in 140 mM NaCl lysis buffer, once with 0.5 M NaCl lysis buffer, once with 0.25 M LiCl wash buffer and once with TE. Crosslinking was reversed at 65°C overnight followed by proteinase K and glycogen addition for 2 h. Protein and nucleic acids were separated by phenol extraction. LiCl was added to a final concentration of 400 mM LiCl. DNA was precipitated using 99.5% EtOH. A second precipitation step was carried out using 75% EtOH and the DNA resuspended in TE. Sequencing libraries were prepared and sequenced as for ChIP-seq in human cells.
Hi-C, 4C-seq and ChIP-seq analyses
Hi-C heatmaps
Hi-C heatmaps screenshots were generated using Juicebox stand-alone program (https://github.com/aidenlab/Juicebox/wiki/Download). To build the average heatmaps, sub-matrix for cis interactions around DSBs were extracted using Juicer, both for observed matrix and observed over expected matrix. Log2(ratio) after vs before DSB were computed, using both Hi-C replicates, and averaged for each bin of the final matrix.
Insulation score and TAD calling
Insulation score was computed using Hi-C matrices at 50 kb resolution with matrix2insulation.pl (https://github.com/dekkerlab/crane-nature-2015). As parameters, we used is=800000 and ids=100000. TADs were called using Hi-C matrices at 50 kb resolution with TopDom R package and window size parameter of 10 (https://github.com/HenrikBengtsson/TopDom). In order to filter out very weak TAD borders (corresponding to sub-TAD borders), we filtered TAD borders with an insulation score below a threshold of -0.05. For Extended Data Fig. 2d, 80 TADs were also randomly selected from TopDom output, which didn’t contain any of the best 80 cleaved DSBs, to be used as controls.
Loops anchors and APA
Loops were called using Juicer Tools HiCCUPS program at 10 kb and 25 kb resolutions (https://github.com/aidenlab/juicer/wiki/HiCCUPS). Aggregate Peak Analysis (APA) was done using Juicer Tools APA program at 10 kb resolution (https://github.com/aidenlab/juicer/wiki/APA). 525 loops were retrieved between the 174 best cleaved DSBs and nearby loop anchors (<1Mb) for replicate 1 (Fig. 2c), and 552 for replicate 2 (Extended Data Fig. 2f). Fold change between Signal (central pixel) and background (upper left corner 5×5 pixels) was computed. For Extended Data Fig. 5f, APA were generated for loops filtered on their size (<200kb) and around the best 80 cleaved DSBs. 597 and 17206 loops in damaged (80 damaged TADs) and undamaged TADs were retrieved respectively in Replicate 1, while 645 and 19150 were retrieved for Replicate 2. Fold change between Signal (central pixel) and background (lower left corner 5×5 pixels) was computed. APA heatmaps were reprocessed using ggplot2 in order to display counts at the same color scale between - DSB and +DSB conditions. For Extended Data Fig. 5g, loop strength was extracted from APA files enhancement.txt corresponding to enrichment fold-change (Peak to Mean, P2M). Differential loop strength was the log-ratio of two conditions loop strengths (+DSB/-DSB).
ChIP-seq analyses
ChIP-seq data were processed using same procedure as in5, except for yeast ChIP-seq, which was aligned on Saccharomyces cerevisiae R64-1-1 assembly, and without PCR duplicate removal. SCC1 and CTCF Peaks were identified using MACS2 program with callpeak algorithm, with default setting, using Input as control and the SCC1 ChIP-seq data before break induction as sample. For SCC1, before breaks, 46184 peaks were identified, with a median and a mean size of 628 and 742 respectively. For CTCF before breaks, 96801 peaks were identified, with a median and a mean size of 339 and 500 respectively. Overlap between CTCF peaks and CTCF motifs was then performed, in order to associate a peak with the orientation of its motif. For representation of genomic tracks, the data were further smoothed using slidding windows as indicated. bamCompare from deeptools, with the parameters --binSize=50, --operation=log2 and with default normalization (readCount) was used to generate differential tracks. For kinetics analysis (Extended Data Fig. 5b), γH2AX domains boundaries around the best cleaved DSBs were manually retrieved thanks to visualization of the 50kb smoothed data on a genome browser (IGB) at different time points. The distribution of γH2AX spread is further showed as a boxplot (n=71).
4C-seq
For differential analyses of the 4C-seq data, the log2 ratio between two bam files were computed using bamCompare from deeptools, with the parameters --binSize=50 and -- operation=log2. Extended Data Fig. 3d shows the mean and s.e.m of the 4C-seq ratio on 1 MB around each viewpoint, obtained across four independent experiments (ctrl viewpoint: n=3 DSB viewpoints, n=11). Extended Data Fig. 3h and Extended Data Fig. 4c show the distribution (box plots) of the 4C-seq ratio on 1 MB around DSB viewpoints obtained across two (siSCC1) or three (ATMi) independent experiments (n=8).
Extended Data
Extended Data Figure 1. γH2AX spreads within prior TAD as revealed by 4C-seq.

(a) 4C-seq tracks before DSB induction obtained for three independent biological replicates and γH2AX ChIP-seq track after DSB induction for different viewpoints (red arrows) localized at three AsiSI sites (black arrows). ChIP-seq data are smoothed using 100 kb span, 4C-seq using a 50 kb span. (b) Example of the Hi-C pattern obtained on chromosome 1 at a 500kb resolution (left) together with a magnification at a 10kb resolution (right). (c) CTCF and calibrated-SCC1 ChIP-seq tracks. (d) Average profile of CTCF ChIP-seq around all loop anchors on the genome (determined using this Hi-C dataset, see Methods), validating both CTCF ChIP-seq and Hi-C datasets. (e) γH2AX ChIP-seq after DSB induction, 4C-seq and CTCF ChIP-seq peak position before DSB induction are shown (peaks in blue contain a CTCF motif in the forward orientation and peaks in red a CTCF motif in the reverse orientation). (f) Box plot showing γH2AX (top), 53BP1 (middle) and MDC1 (bottom) ChIP-seq quantification within the damaged TAD and neighboring TADs for the best cleaved DSBs in DIvA cells (see methods). Center line: median; Box limits: 1st and 3rd quartiles; Whiskers: Maximum and minimum without outliers; Points: outliers. (n=153). (g) γH2AX tracks around a DSB induced by CRISPR/Cas9 (upper panel, ChIP-chip, expressed as the log2 sample/input, smoothed using 100 probes windows) and by AsiSI at the same position (lower panel, ChIP-seq, 50kb smoothed). (h) Top: Immunofluorescence experiment showing γH2AX and DAPI staining before and after DSB induction with or without ATM inhibitor as indicated (scale bar, 10μm). Bottom: Quantification of γH2AX intensity (expressed in A.U: Arbitrary Unit) in the above conditions. One representative experiment is shown (out of n=3 biological replicates. Center line: median; Box limits: 1st and 3rd quartiles; Whiskers: Maximum and minimum without outliers; Points: outliers. (−DSB n=117 nuclei; +DSB n=97 nuclei; +DSB+ATMi n=95 nuclei) (i) Validation of the ATR inhibitor efficiency: Western Blot showing the effect of ATRi on the phosphorylation of CHK1 following a treatment with hydroxyurea (HU) (n=2). For gel source data, see Supplementary Figure 1. (j) γH2AX ChIP-seq tracks after DSB induction in untreated cells or in cells treated with an inhibitor of ATM or ATR at two DSB sites (20kb smoothed). The differential γH2AX signal obtained after DSB induction (expressed as the log2 ratio ATMi/untreated or ATRi/untreated, grey tracks) is also shown. (k) Average profile of pATM (S1981) (left panel) and γH2AX (right panel) ChIP-seq on a 2 Mb window around the eighty best-cleaved DSBs in DIvA cells.
Extended Data Figure 2. Cohesin recruitment and loop extrusion occurs at DSBs.

(a) Calibrated SCC1 ChIP-seq tracks before (grey) and after (black) DSB induction. SCC1 enrichment at DSB site is indicated by a red arrow. (b) Average profile of SCC1 ChIP-seq signal centered on 80 best-induced DSBs (left panel) or centered on all CTCF peaks of the genome (right panel) on a 10 kb window. (c) Calibrated ChIP-qPCR of SCC1 in the indicated conditions at three DSB sites or a control negative region. Western Blot validating the depletion of the proteins NIPBL (n=1) and MRE11 (n=2) by the corresponding siRNA are shown. For gel source data, see Supplementary Figure 1. S.e.m and mean for technical replicates (n=4) of a representative experiment is shown (out of n=2 biological replicates). (d) Averaged Hi-C matrix before (-DSB) and after DSB induction (+DSB) (observed/expected) and of the log2 ratio between damaged versus undamaged cells centered on the eighty best-induced DSBs (top panels) or centered on eighty random TAD borders (bottom panels) (50 kb resolution, 5 Mb window) (combined replicates). (e) Averaged Hi-C contact matrix of the log2 +DSB/-DSB centered on the eighty best-induced DSBs in the two independent biological replicates. (f) Aggregate Peak Analysis (APA) plot on a 200kb window (10 kb resolution) before (-DSB) and after DSB induction (+DSB) in the biological replicate #2 (replicate#1 shown as main Fig.2c). APA are calculated between the DSBs and loops anchors (n=552 pairs). The fold change between the signal (central pixel) and the background (upper left corner 5x5 pixels) is indicated. (g) For comparison with ED. Fig. 2f, APA plot on a 200kb window (10 kb resolution) before DSB induction were computed between classical loop anchors that are near DSB sites (<500kb) (n=674 pairs for replicate 1 and n=737 pairs for replicate 2). The fold change between the signal (central pixel) and the background (upper left corner 5x5 pixels) is indicated. The loop strength (quantified by the FC between signal and background on APA plot) is higher at loop anchors (ED Fig. 2g, Rep#1 FC=5.4 and Rep#2 FC=5.8) than the loop strength observed at DSB post break induction (Fig. 2c, Rep#1, FC=2 and ED Fig. 2f, Rep#2 FC=2.3).
Extended Data Figure 3. Loop extrusion at DSBs detected by 4C-seq.

(a) 4C-seq tracks (10 kb smoothed) before and after DSB induction as indicated, obtained for three biological replicates using viewpoints localized at three DSB sites. The DSBs are indicated by arrows. (b) 4C-seq tracks before (blue) and after (purple) DSB induction, at two DSB viewpoints. Differential 4C-seq (Log2 +DSB/-DSB) is also shown (black). (c) Differential 4C-seq (log2 +DSB/-DSB) is shown for three viewpoints located at DSB sites and on a Ctrl region as indicated. (d) Bar plot showing the differential 4C-seq signal (log2 +DSB/-DSB) computed on 1 Mb around four independent viewpoints located at DSBs (“DSBs viewpoints”, n=11) and one control region (“Control viewpoint”, n=3), across four independent biological experiments (see Methods). P value between control and DSBs viewpoints is indicated (two-sided Wilcoxon test). S.e.m and mean values are plotted. (e) Western Blot showing the depletion of SCC1 by siRNA (n=3). For gel source data, see Supplementary Figure 1. (f) Differential (log2) 4C-seq track in SCC1 siRNA treated cells versus control siRNA-treated cells (in undamaged conditions) for three viewpoints. (g) Genomics tracks showing the 4C-seq signals before and after DSB induction in siRNA Ctrl-or SCC1-treated cells and the differential 4C-seq signal in Ctrl or SCC1 siRNA treated cells (log2 +DSB/-DSB) (10kb smoothed). (h) Boxplot showing the average Log2 +DSB/-DSB 4C-seq, on 1 Mb around four DSBs viewpoints (two biological experiments) upon control or SCC1 depletion by siRNA (see Methods) (n=8). P value: two-sided Wilcoxon test. Center line: median; Box limits: 1st and 3rd quartiles; Whiskers: Maximum and minimum without outliers.
Extended Data Figure 4. ATM activity is required for loop extrusion at DSB.

(a) Hi-C maps before DSB induction of a region of the chromosome 17 in Ctrl- and SCC1-depleted cells. Left panel: 100kb resolution, right panel (magnification): 25kb resolution. (b) Genomic tracks of 4C-seq before and after DSB induction in untreated or ATM inhibitor treated cells and of differential 4C-seq signal (log2 +DSB/-DSB or +DSB+ATMi/-DSB) (10kb smoothed). (c) cis interactions computed as in ED. Fig. 3h for four DSBs viewpoints across three biological experiments, in control condition or upon ATM inhibition. P value: two-sided Wilcoxon test. Center line: median; Box limits: 1st and 3rd quartiles; Whiskers: Maximum and minimum without outliers; (n=8)
Extended Data Figure 5. Altered loop extrusion modifies γH2AX spreading.

(a) Quantification of γH2AX intensity (expressed in A.U: Arbitrary Unit) after DSB induction (OHT 4h) and upon ATM inhibition followed by different time points after ATMi release (0min n=172 nuclei; 5min n=183 nuclei, 15 min n=171 nuclei; 30min n=197 nuclei; 1h n=189 nuclei). Treatment with OHT for 4h without ATMi is also shown (n=182 nuclei). One representative experiment is shown (out of n=2 biological replicates). Center line: median; Box limits: 1st and 3rd quartiles; Whiskers: Maximum and minimum without outliers. (b) boxplot showing the spread of γH2AX (in bp) at the indicated time points after release from ATMi around the best cleaved DSBs (n=71). Center line: median; Box limits: 1st and 3rd quartiles; Whiskers: Maximum and minimum; points: outliers. (c) Black: 4C-seq track before DSB induction using a DSB viewpoint (DSB is indicated by a black arrow, Viewpoint by a red arrow). Purple (middle) track shows the differential γH2AX signal obtained after DSB induction by ChIP-chip in SCC1-depleted versus control cells (expressed as the γH2AX log2 ratio siSCC1/ siCtrl). Light blue track (bottom) shows the differential 4C-seq signal obtained in SCC1-depleted versus control cells before DSB induction (log2 siSCC1/ siCtrl). (d) Genomic tracks of γH2AX ChIP-seq signal after DSB induction in Ctrl (red) or SCC1 (pink) depleted cells and of the differential γH2AX signal obtained after DSB induction (expressed as the log2 ratio siSCC1/siCtrl, purple) at two DSB sites. (e) Western blot validating the effect of the siRNA targeting WAPL on the WAPL protein level (n=2). For gel source data, see Supplementary Figure 1. (f) Genomics tracks of γH2AX ChIP-seq after DSB induction in Ctrl or WAPL depleted cells and of the differential γH2AX signal obtained after DSB induction (expressed as the log2 ratio siWAPL/siCtrl) at two DSB sites and one control (bottom panel) genomic locus (no DSB) (20kb smoothed). (g) Genomics tracks of the differential γH2A ChIP-seq signal (log2 +DSB/-DSB) before (no IAA) or after PDS5 degradation (IAA) at two DSB sites (HO sites) in S. Cerevisiae (SacCer3, coordinates in bp).
Extended Data Figure 6. Increased genome-wide, DSB-induced, cohesin binding is enhanced within damaged TADs.

(a) Upper panel: Contact matrix (5kb resolution) showing the log2 (observed/expected) before or after DSB induction as indicated, on a region showing a loop on chromosome 20 and devoid of AsiSI site (no DSB). Loops anchors are circled and indicated by red and blue bars. Lower panel: Genome browser screenshot showing the SCC1 calibrated ChIP-seq on the same region before and after DSB induction as indicated. Cohesin enrichment at the loop anchors (blue and red bars) is increased after DSB (black arrows) compared to before DSB (grey arrows) in agreement with an increased loop strength (grey and black circles). (b) Violin plots showing the SCC1 enrichment at cohesin peaks (n=46194) before and after DSB induction as indicated. P values are indicated (paired one-sided Wilcoxon test). (c) Genomic tracks of γH2AX (red) and SCC1 ChIP-seq signal before (blue) and after (purple) DSB 862 induction. The ratio between before and after DSB induction (grey) is also shown (expressed 863 as the log2 +DSB/-DSB) (10kb smoothed). (d) Boxplot showing the quantification of SCC1 864 recruitment on loop anchors, at different distances from DSB sites as indicated (from left to 865 right, n=1610, 3161, 1930, 3232, 4786, 25263, 114461). Center line: median; Box limits: 1st and 3rd quartiles; Whiskers: Maximum and minimum; points: outliers. (e) γH2AX ChIP-seq signal and Hi-C signal at different distances from a damaged TAD of the chromosome 1 before (-DSB) and after DSB induction (+DSB). Green circles represent the chromatin loops. (f) Aggregate Peak Analysis (APA) plot on a 200 kb window (10 kb resolution) before (-DSB) and after DSB induction (+DSB) calculated for all loop anchors, in damaged and undamaged TAD as indicated. The fold change between the signal (central pixel) and the background (lower left corner 5x5 pixels) is indicated. (g) Boxplots showing the differential loops strength in undamaged or damaged TADs as indicated (see methods), computed from Hi-C data obtained before and after DSB, from replicate #1 (left panel) ad replicate #2 (right panel). P values between before and after DSB are indicated (Wilcoxon 876 test, μ=0). The increased loop strength following DSB is significantly higher in damaged TADs as compared to undamaged TADs (paired two-sided Wilcoxon test) in both Hi-C replicates experiments. Replicate #1: undamaged n=2936; damaged n=264. Replicate #2, undamaged n=3181, damaged n=302. Center line: median; Box limits: 1st and 3rd quartiles; Whiskers: Maximum and minimum without outliers; Points: outliers.
Extended Data Figure 7. DSB-induced phosphorylation of cohesin occurs in damaged TADs.

(a) Genomic tracks showing γH2AX, phosphorylated SMC3 (pSMC3 S1083) and phosphorylated SMC1 (pSMC1 S966) ChIP-chip signal expressed as the log2 ratio sample/input after DSB induction. Two damaged genomic locations are shown. (b) Average profile of pSMC3 S1083 (expressed as the log2 (+DSB/-DSB) ChIP-seq signal) around the eighty best-induced DSBs on a 4Mb window. (c) Boxplot showing the quantification of pSMC3 S1083 signal on loop anchors, within damaged or undamaged TADs as indicated. P values between before and after DSB are indicated (paired two-sided Wilcoxon test). The increased pSMC3 S1083 enrichment on loop anchors following DSB is significantly higher in damaged TADs as compared to undamaged TADs (two-sided Wilcoxon test). undamaged n=9040; damaged n=1626. Center line: median; Box limits: 1st and 3rd quartiles; Whiskers: Maximum and minimum without outliers; Points: outliers.
Extended Data Table 1. Antibody used in this study.
| Target | Application | Reference | Quantity |
| γH2AX(S139) | ChIP | Merck Millipore 07-164 | 2 μg |
| γH2AX (S139) | IF | Merck Millipore 05-636 (clone JBW301) | 1:1000 |
| P-ATM (S1981) | ChIP | Abeam ab81292 | 2 μg |
| MDC1 | ChIP | Abeam ab11171 | 3 μg |
| SCC1 | ChIP | Abeam ab992 | 4 μg |
| SCC1 | Western Blot | Abeam ab992 | 1:500 |
| WAPL | Western Blot | Santa Cruz sc-365189 | 1:500 |
| NIPBL | Western Blot | Bethyl Laboratories A301-779A | 1:1000 |
| MRE11 | Western Blot | GeneTex GTX70212 (clone 12D7) | 1:4000 |
| Tubulin | Western Blot | Sigma T6199 | 1:10000 |
| Myosin | Western Blot | Sigma M3567 | 1:2000 |
| Phospho-CHK1 (ser345) | Western Blot | Cell Signaling 2348S | 1:1000 |
| P-SMC1 (S966) | ChIP | Epitomics EP2858Y | 2 μL |
| P-SMC3 (S1083) | ChIP | Bethyl Laboratories A300-480A | 2 μg |
| CTCF | ChIP | Millipore 07-729 | 4 μL |
| γH2A (yeast) | ChIP | Abeam ab15083 | 2μg |
Extended Data Table 2. Primers and siRNA used in this study.
NNN is the position of the optional index
| Application | Name | Forward primer | Reverse primer |
| 4C-seq | ViewpointDSB1 | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTAACCTGGCAACTTATGAATCAGGA | CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTATGTCAAAAGCCAAGGGGACA |
| 4C-seq | ViewpointDSB2 | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTTCCTTACGATTATTTGTGAATTTTG | CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTAAGCTAATTCTGAGTTACATACATT |
| 4C-seq | ViewpointDSB3 | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGATTACGTAGAAGGGTGCC | CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTAAGGCAAATGATAACCCTGT |
| 4C-seq | Viewpoint Ctrl region | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTTCCTCAGGTTATCATCCCAA | CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTCACCTTCGCTGTACCTTTG |
| 4C-seq | Viewpoint CRISPR site | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTTTAAGCACCCTCCTCCTAG | CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTACCTTTACACCTCAAAACCT |
| 4C-seq | Viewpoint470 kb upstream (Fig. 1c) | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTACAAGGAAGAAGCAGGCATTCA | CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTTTGAAATGAGTACTCTGCCATCCA |
| ChlP-qPCR | Ctrl region | AGCACATGGGATTTTGCAGG | TTCCCTCCTTTGTGTCACCA |
| ChlP-qPCR | DSB site 1 | TCCCCTGTTTCTCAGCACTT | CTTCTGCTGTTCTGCGTCCT |
| ChlP-qPCR | DSB site 2 | CCGCCAGAAAGTTTCCTAGA | CTCACCCTTGCAGCACTTG |
| ChlP-qPCR | DSB site 3 | CCTAGCTGAGGTCGGTGCTA | GAAGAGTGAGGAGGGGGAGT |
| ChlP-qPCR | Cohesin positive site (mouse) | CAGAGATTTGCGGTGTTCGG | TTCACACCTAGAGGAGGGGT |
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank the genomics core facility of EMBL for high throughput sequencing as well as the facilities and expertise of the high throughput sequencing core facility of the I2BC (Centre de Recherche de Gif), F. Beckouet (CBI) for advice on the yeast work, J. Rispal for occasional experimental help, and C. Normand (CBI) for continuous helpful discussions. Work in J.E.H.’s lab was funded by grant R35 GM127029 from the U.S. National Institutes of Health. F.Z. was supported by the National Institute of General Medical Sciences Training Grant TM32GM007122. E.R. is supported by Labex Ecofect (ANR-11-LABX-0048) of the Université de Lyon, Fondation FINOVI and by the European Research Council (ERC-StG-LS6-805500) under the European Union’s Horizon 2020 research and innovation programs. Funding in G.L.’s laboratory was provided by grants from the European Research Council (ERC-2014-CoG 647344), the Agence Nationale pour la Recherche (ANR-14-CE10-0002-01 and ANR-18-CE12-0015), the Institut National Contre le Cancer (INCA), the Ligue Nationale Contre le Cancer (LNCC). C.A is a recipient of a FRM fellowship (FRM FDT201904007941).
Footnotes
Author contributions
C.A. performed 4C-seq, Hi-C, and ChIP-seq/ChIP-chip/ChIP-qPCR experiments. A-L.F. contributed to siRNA experiments and performed CTCF ChIP-seq. K.L. and F.Z. performed yeast strain construction and γH2A ChIP. P.C. performed ChIP-chip in SCC1 siRNA. V.R. and R.M. performed bioinformatic analyses of 4C-seq, Hi-C and ChIP-seq data sets. E.P.R. and P.E.M. provided nanoblades for CRISPR/cas9 experiments. D.N. helped to realize and analyze 4C-seq experiments. G.L. conceived experiments and G.L. and T.C. supervised experiments in human cells and helped with library preparation. J.E.H. conceived and supervised work in yeast. G.L. wrote the manuscript. All authors commented and edited the manuscript.
Competing interest statement
The authors declare no competing interests.
Additional information
Correspondence can be addressed to gaelle.legube@univ-tlse3.fr
Data Availability Statement
All high throughput sequencing data (Hi-C, ChIP-seq, 4C-seq) have been deposited to Array Express under accession number E-MTAB-8851. ChIP-chip data have been deposited to Array Express under accession number E-MTAB-8793. Uncropped blots are shown in Supplementary Information Figure 1. Other source data (ChIP-qPCR and raw microscopy data) are available upon request.
Code Availability Statement
Source codes are available on https://github.com/LegubeDNAREPAIR/LoopExtrusion
References
- 1.Clouaire T, Marnef A, Legube G. Taming Tricky DSBs: ATM on duty. DNA Repair (Amst) 2017;56:84–91. doi: 10.1016/j.dnarep.2017.06.010. [DOI] [PubMed] [Google Scholar]
- 2.McCord RP, Kaplan N, Giorgetti L. Chromosome conformation capture and beyond: toward an integrative view of chromosome structure and function. Mol Cell. 2020;77:688–708. doi: 10.1016/j.molcel.2019.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnould C, Legube G. The Secret Life of Chromosome Loops upon DNA Double-Strand Break. J Mol Biol. 2020;432:724–736. doi: 10.1016/j.jmb.2019.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clouaire T, et al. Comprehensive Mapping of Histone Modifications at DNA Double-Strand Breaks Deciphers Repair Pathway Chromatin Signatures. Mol Cell. 2018;72:250–262.e6. doi: 10.1016/j.molcel.2018.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stewart GS, Wang B, Bignell CR, Taylor AMR, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- 7.Caron P, et al. Cohesin protects genes against γH2AX Induced by DNA double-strand breaks. PLoS Genet. 2012;8:e1002460. doi: 10.1371/journal.pgen.1002460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Natale F, et al. Identification of the elementary structural units of the DNA damage response. Nat Commun. 2017;8 doi: 10.1038/ncomms15760. 15760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ochs F, et al. Stabilization of chromatin topology safeguards genome integrity. Nature. 2019;574:571–574. doi: 10.1038/s41586-019-1659-4. [DOI] [PubMed] [Google Scholar]
- 10.Iacovoni JS, et al. High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 2010;29:1446–1457. doi: 10.1038/emboj.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang L-H, Ghosh S, Noordermeer D. Tads and their borders: free movement or building a wall? J Mol Biol. 2020;432:643–652. doi: 10.1016/j.jmb.2019.11.025. [DOI] [PubMed] [Google Scholar]
- 12.Caron P, et al. Non-redundant Functions of ATM and DNA-PKcs in Response to DNA Double-Strand Breaks. Cell Rep. 2015;13:1598–1609. doi: 10.1016/j.celrep.2015.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwarzer W, et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature. 2017;551:51–56. doi: 10.1038/nature24281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rao SSP, et al. Cohesin loss eliminates all loop domains. Cell. 2017;171:305–320.e24. doi: 10.1016/j.cell.2017.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gelot C, et al. The Cohesin Complex Prevents the End Joining of Distant DNA Double-Strand Ends. Mol Cell. 2016;61:15–26. doi: 10.1016/j.molcel.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 16.Meisenberg C, et al. Repression of Transcription at DNA Breaks Requires Cohesin throughout Interphase and Prevents Genome Instability. Mol Cell. 2019;73:212–223.e7. doi: 10.1016/j.molcel.2018.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Potts PR, Porteus MH, Yu H. Human SMC5/6 complex promotes sister chromatid homologous recombination by recruiting the SMC1/3 cohesin complex to double-strand breaks. EMBO J. 2006;25:3377–3388. doi: 10.1038/sj.emboj.7601218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ström L, Lindroos HB, Shirahige K, Sjögren C. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol Cell. 2004;16:1003–1015. doi: 10.1016/j.molcel.2004.11.026. [DOI] [PubMed] [Google Scholar]
- 19.Unal E, et al. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol Cell. 2004;16:991–1002. doi: 10.1016/j.molcel.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 20.Covo S, Westmoreland JW, Gordenin DA, Resnick MA. Cohesin Is limiting for the suppression of DNA damage-induced recombination between homologous chromosomes. PLoS Genet. 2010;6:e1001006. doi: 10.1371/journal.pgen.1001006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davidson IF, et al. DNA loop extrusion by human cohesin. Science. 2019;366:1338–1345. doi: 10.1126/science.aaz3418. [DOI] [PubMed] [Google Scholar]
- 22.Fudenberg G, et al. Formation of chromosomal domains by loop extrusion. Cell Rep. 2016;15:2038–2049. doi: 10.1016/j.celrep.2016.04.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim Y, Shi Z, Zhang H, Finkelstein IJ, Yu H. Human cohesin compacts DNA by loop extrusion. Science. 2019;366:1345–1349. doi: 10.1126/science.aaz4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vian L, et al. The energetics and physiological impact of cohesin extrusion. Cell. 2018;173:1165–1178.e20. doi: 10.1016/j.cell.2018.03.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmitt AD, et al. A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Rep. 2016;17:2042–2059. doi: 10.1016/j.celrep.2016.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mirny LA, Imakaev M, Abdennur N. Two major mechanisms of chromosome organization. Curr Opin Cell Biol. 2019;58:142–152. doi: 10.1016/j.ceb.2019.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barrington C, et al. Enhancer accessibility and CTCF occupancy underlie asymmetric TAD architecture and cell type specific genome topology. Nat Commun. 2019;10 doi: 10.1038/s41467-019-10725-9. 2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aymard F, et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat Struct Mol Biol. 2014;21:366–374. doi: 10.1038/nsmb.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wutz G, et al. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 2017;36:3573–3599. doi: 10.15252/embj.201798004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haarhuis JHI, et al. The cohesin release factor WAPL restricts chromatin loop extension. Cell. 2017;169:693–707.e14. doi: 10.1016/j.cell.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dauban L, et al. Regulation of Cohesin-Mediated Chromosome Folding by Eco1 and Other Partners. Mol Cell. 2020;77:1279–1293.e4. doi: 10.1016/j.molcel.2020.01.019. [DOI] [PubMed] [Google Scholar]
- 32.Lee C-S, Lee K, Legube G, Haber JE. Dynamics of yeast histone H2A and H2B phosphorylation in response to a double-strand break. Nat Struct Mol Biol. 2014;21:103–109. doi: 10.1038/nsmb.2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanders JT, et al. Radiation-induced DNA damage and repair effects on 3D genome organization. Nat Commun. 2020;11 doi: 10.1038/s41467-020-20047-w. 6178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim B-J, et al. Genome-wide reinforcement of cohesin binding at pre-existing cohesin sites in response to ionizing radiation in human cells. J Biol Chem. 2010;285:22784–22792. doi: 10.1074/jbc.M110.134577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim S-T, Xu B, Kastan MB. Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev. 2002;16:560–570. doi: 10.1101/gad.970602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collins PL, et al. DNA double-strand breaks induce H2Ax phosphorylation domains in a contact-dependent manner. Nat Commun. 2020;11 doi: 10.1038/s41467-020-16926-x. 3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li K, Bronk G, Kondev J, Haber JE. Yeast ATM and ATR kinases use different mechanisms to spread histone H2A phosphorylation around a DNA double-strand break. Proc Natl Acad Sci USA. 2020 doi: 10.1073/pnas.2002126117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Y, et al. Very fast CRISPR on demand. Science. 2020;368:1265–1269. doi: 10.1126/science.aay8204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y, et al. The fundamental role of chromatin loop extrusion in physiological V(D)J recombination. Nature. 2019;573:600–604. doi: 10.1038/s41586-019-1547-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang X, et al. Fundamental roles of chromatin loop extrusion in antibody class switching. Nature. 2019;575:385–389. doi: 10.1038/s41586-019-1723-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gothe HJ, et al. Spatial chromosome folding and active transcription drive DNA fragility and formation of oncogenic MLL translocations. Mol Cell. 2019;75:267–283.e12. doi: 10.1016/j.molcel.2019.05.015. [DOI] [PubMed] [Google Scholar]
- 42.Canela A, et al. Topoisomerase II-Induced Chromosome Breakage and Translocation Is Determined by Chromosome Architecture and Transcriptional Activity. Mol Cell. 2019;75:252–266.e8. doi: 10.1016/j.molcel.2019.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mangeot PE, et al. Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat Commun. 2019;10 doi: 10.1038/s41467-018-07845-z. 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marnef A, et al. A cohesin/HUSH-and LINC-dependent pathway controls ribosomal DNA double-strand break repair. Genes Dev. 2019;33:1175–1190. doi: 10.1101/gad.324012.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morawska M, Ulrich HD. An expanded tool kit for the auxin-inducible degron system in budding yeast. Yeast. 2013;30:341–351. doi: 10.1002/yea.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matelot M, Noordermeer D. Determination of High-Resolution 3D Chromatin Organization Using Circular Chromosome Conformation Capture (4C-seq) Methods Mol Biol. 2016;1480:223–41. doi: 10.1007/978-1-4939-6380-5_20. [DOI] [PubMed] [Google Scholar]
- 47.Klein FA, et al. FourCSeq: analysis of 4C sequencing data. Bioinformatics. 2015;31:3085–3091. doi: 10.1093/bioinformatics/btv335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.David FPA, et al. HTSstation: a web application and open-access libraries for high-throughput sequencing data analysis. PLoS One. 2014;9:e85879. doi: 10.1371/journal.pone.0085879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kojic A, et al. Distinct roles of cohesin-SA1 and cohesin-SA2 in 3D chromosome organization. Nat Struct Mol Biol. 2018;25:496–504. doi: 10.1038/s41594-018-0070-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All high throughput sequencing data (Hi-C, ChIP-seq, 4C-seq) have been deposited to Array Express under accession number E-MTAB-8851. ChIP-chip data have been deposited to Array Express under accession number E-MTAB-8793. Uncropped blots are shown in Supplementary Information Figure 1. Other source data (ChIP-qPCR and raw microscopy data) are available upon request.
Source codes are available on https://github.com/LegubeDNAREPAIR/LoopExtrusion