

CRISPR-Cas systems have been engineered as powerful tools to control gene expression in bacteria. The most common strategy relies on the use of Cas effectors modified to bind target DNA without introducing DNA breaks. These effectors can either block the RNA polymerase or recruit it through activation domains. Here, we discuss the mechanistic details of how Cas effectors can modulate gene expression by blocking transcription initiation or acting as transcription roadblocks. CRISPR-Cas tools can be further engineered to obtain fine-tuned control of gene expression or target multiple genes simultaneously.
KEYWORDS: CRISPR, gene silencing, transcriptional regulation
SUMMARY
CRISPR-Cas systems have been engineered as powerful tools to control gene expression in bacteria. The most common strategy relies on the use of Cas effectors modified to bind target DNA without introducing DNA breaks. These effectors can either block the RNA polymerase or recruit it through activation domains. Here, we discuss the mechanistic details of how Cas effectors can modulate gene expression by blocking transcription initiation or acting as transcription roadblocks. CRISPR-Cas tools can be further engineered to obtain fine-tuned control of gene expression or target multiple genes simultaneously. Several caveats in using these tools have also been revealed, including off-target effects and toxicity, making it important to understand the design rules of engineered CRISPR-Cas effectors in bacteria. Alternatively, some types of CRISPR-Cas systems target RNA and could be used to block gene expression at the posttranscriptional level. Finally, we review applications of these tools in high-throughput screens and the progress and challenges in introducing CRISPR knockdown to other species, including nonmodel bacteria with industrial or clinical relevance. A deep understanding of how CRISPR-Cas systems can be harnessed to control gene expression in bacteria and build powerful tools will certainly open novel research directions.
INTRODUCTION
The ability to precisely control gene expression levels in bacteria is attractive for many reasons. Modifying the concentration of a protein or enzyme is a classical way of understanding gene function, as it allows comparison of the phenotypes of cells at different levels of a gene of interest. This is especially relevant in the case of essential genes, where a simple knockout is impossible. Controlling gene expression is also a way to discover regulatory circuits, by applying a perturbation and measuring how the expression of other genes adapts as a response. Furthermore, recent research in systems biology has aimed to understand the biological significance of gene expression levels in a broader context. Experimenting with gene expression levels has helped our understanding of the evolutionary dynamics of gene regulation (1, 2) and the importance of stochastic processes in protein expression (3, 4). Finally, the fine-tuning of gene expression levels has great potential for the engineering of organisms, with the aim of making them more efficient for industrial metabolite production (5, 6) or to create artificial regulatory circuits (7) to be used in biosensors and diagnostic tools (8, 9).
All these applications can benefit from the ability to change gene expression in a programmable way, i.e., the ability to set the expression of any protein of interest to arbitrary levels, according to some user-defined input in the form of a DNA sequence. However, the development of programmable biological devices has remained limited by a long-standing barrier: the complexity of macromolecule folding and their interactions make it very difficult to design sequences de novo such that they have the desired function in vivo. For this reason, biotechnological design has typically been limited to the recycling of components from nature or the artificial generation of random sequences followed by selection. In particular, inducible promoters are established as the standard tool for the control of gene expression in bacteria, allowing the expression level of a gene to be linked with the concentration of a chemical in the medium. While this method is extremely useful for a range of applications, the recent emergence of programmable methods has brought up new possibilities, especially in conjunction with high-throughput sequencing and DNA synthesis technologies. Such technologies offer many advantages in principle: as they are programmable, it is easy to create large-scale libraries or to make multiple orthogonal systems to be used all at once in the same cell. The sequence controlling gene expression can itself act as a barcode, allowing identification by sequencing in large libraries. The field of eukaryotic biology was the first to be revolutionized by the programmable control of gene expression, with the development of RNA interference (RNAi) during the 1990s (10). It took a few more years before similar methods were discovered for bacteria: the first tools based on antisense RNA (asRNA) were developed in the 2000s (11–13), followed by prokaryotic small RNA (sRNA) (14)- and, finally, CRISPR (clustered regularly interspaced short palindromic repeat)-based tools in the 2010s (15, 16).
Antisense RNAs act by forming a duplex with the ribosome-binding site (RBS) of cognate mRNA and preventing ribosomes from initiating translation (13, 17). They can be used on multiple targets at once and can be made more stable by using paired termini (13, 18). Small RNAs, on the other hand, use a scaffold to recruit the Hfq chaperone and lead to mRNA degradation (14). They too can be multiplexed, and the strength of knockdown is tunable by changing the energy of binding between the sRNA and the target (6). Unfortunately, the coupling between transcription and translation in prokaryotes makes it difficult to interfere with translation, since there is little time for the interfering RNA to find its target, resulting in moderate repression strength (14, 17). They also have limited portability from one species to another (14).
A few years ago, the discovery of the CRISPR bacterial immune systems brought an alternative repression method under the spotlight.
FROM THE CRISPR IMMUNE SYSTEM TO ARTIFICIAL TRANSCRIPTION FACTORS
Analogous with RNAi, gene knockdown using CRISPR effectors has been dubbed CRISPRi. However, unlike RNAi, which acts at the posttranscriptional level and prevents translation, CRISPRi typically works by stopping transcription. CRISPR-Cas systems are an extremely diverse ensemble of adaptive immune systems found in bacteria and archaea, which can be grouped in two classes, each comprised of many types and subtypes (reviewed in reference 19) with various mechanisms. To grant protection against foreign DNA, CRISPR-Cas systems work in three phases. The first one, called adaptation, is responsible for sampling short DNA sequences from invading genetic elements and storing them in the form of a series of fixed-length sequences intercalated with a constant motif. This collection of sequences is called the CRISPR array. In a second phase, the CRISPR array is expressed into a primary transcript, which is subsequently processed into small CRISPR RNAs (crRNAs), which form ribonucleoprotein complexes with Cas proteins. In the last phase, called interference, crRNAs guide Cas proteins to recognize and destroy invading DNA (or RNA).
The machinery involved in the interference step can be repurposed to artificially control gene expression by affecting either transcription or translation. In nature, the CRISPR array is first transcribed and processed into individual guide RNAs, which are then associated with one or several enzymes (the CRISPR effectors) to form a ribonucleoprotein complex with nuclease activity. Upon phage infection, this RNA-guided nuclease will recognize the invading genome through base pairing and introduce a double-strand break to stop the infection. The nature of CRISPR effectors can be very different depending on the class and type of CRISPR system. In class 2 systems, the effector is one single enzyme, making them very popular for biotechnological applications. Class 2 includes the type II systems, which use the famous Cas9 protein; type V systems with their effector Cas12 (formerly known as Cpf1); and type VI systems with their RNA-targeting effector Cas13 (formerly known as C2c2). In class 1 systems, the effector consists of a large multiprotein complex like the DNA-targeting Cascade complex of type I systems or the RNA-targeting Csm or Cmr complexes of type III systems. By engineering type I and II systems to eliminate their nuclease activity, it is possible to create artificial DNA-binding ribonucleoprotein complexes that can be directed to bind DNA sequences of interest by putting arbitrary addresses in the CRISPR array (15, 16, 20) (Fig. 1). In a similar fashion, effectors of types III and VI can be turned into programmable RNA-binding complexes (21, 22).
FIG 1.

General principle for the control of bacterial gene expression using reprogrammed CRISPR effectors. (A) Guide RNAs form complexes with natural or engineered CRISPR effectors. This results in a programmable ribonucleoprotein complex that will bind to either homologous DNA or RNA depending on the effector type. (B) Gene repression is possible by targeting either the promoter, the coding sequence, or mRNA. Gene activation is possible by fusing dCas9 to a transcriptional activator (such as ω, SoxS, or AsiA) and addressing it to a precise location upstream of the promoter.
To reprogram a CRISPR system, one can simply replace the native sequences in the CRISPR array with the desired target sequences and rely on natural processing and assembly of the ribonucleoprotein complex. Type II systems as well as some type V systems rely on a ternary complex comprising the effector (Cas9 or Cas12), the processed guide RNA, and a trans-activating CRISPR RNA (tracrRNA), which forms an RNA duplex with repeats in the primary transcript (23, 24). This duplex RNA is recognized by Cas9/12, leading to processing into short crRNAs. As an alternative, it is possible to build a chimeric RNA between the crRNA and the tracrRNA, referred to as the single guide RNA (sgRNA) (25). In some type V systems, the Cas12 effector carries RNase activity, enabling it to process the CRISPR primary transcript by itself, without the need for a tracrRNA or a host RNase (26). While the use of an sgRNA reduces the number of components required and can be advantageous, the use of CRISPR arrays makes it possible to easily perform multiplex targeting (27).
Not all positions can be targeted by CRISPR effectors. The target sequence of type I, II, and V complexes must be flanked by a DNA motif called the protospacer-adjacent motif (PAM). (“Protospacer” designates the part of phage DNA that will eventually be incorporated into the CRISPR array as a spacer during CRISPR adaptation. The PAM is required both for the selection of novel spacers to be acquired and for the identification of target sequences during the interference step [28–31].) The PAM sequence is different depending on which ortholog of CRISPR is being used but is typically between 2 and 8 nucleotides long, meaning that there are usually many discrete possible targeting sites throughout a given sequence. Some CRISPR effectors have strict PAM requirements, while others recognize a more variable range of motifs. For example, the canonical PAM used in the popular Cas9 from Streptococcus pyogenes (SpCas9) is an NGG motif on the 3′ side of the target (32), while Francisella novicida Cas12a prefers NTTN on the 5′ end of the target (33, 34). The length of the PAM determines the frequency of possible targets: on average, SpCas9 has one target every 8 bp in Escherichia coli’s genome if both DNA strands are considered. This, of course, depends on the characteristics of the target genome: as SpCas9’s canonical PAM is NGG, it is not as common in genomes with a low GC content. After binding to the DNA, the CRISPR effector starts to unwrap the DNA starting from the side of the PAM (35). As the DNA unwinds, the guide RNA progressively forms a hybrid with the target DNA (36, 37). This structure is called the R loop. For type II and V effectors, when the entire guide is annealed, a conformational shift occurs, and a double-strand break is introduced in the DNA. Cas9 uses two different catalytic domains to nick the two DNA strands, while Cas12a uses a single domain to cleave each strand sequentially (38, 39). For type I effectors, the Cas3 nuclease is recruited at the bound Cascade complex and unwinds and chops the target strand of DNA (40, 41).
To simply block gene expression without cutting the target DNA, the nuclease activity of the CRISPR effectors must be eliminated. For type II and V systems, this is done by mutating the catalytic residues (15, 16, 34). The resulting enzymes are marked with the letter “d” (for “dead”); for example, the inactive variant of Cas9 is called dCas9. For type I, the Cas3 nuclease can simply be deleted (20). Finally, gene knockdown can also be achieved by addressing RNA-binding effectors (of types III or VI) to mRNA (21, 22, 42).
Thanks to the remarkable effectiveness of CRISPR-Cas systems, many applications were quickly developed, even with limited knowledge of their exact mechanism. As a result, many rules of thumb were discovered during the testing and optimization of particular applications, and research is still ongoing to develop a unified, predictive biophysical model of how dead Cas effectors block RNA transcription.
THE MECHANICS OF NUCLEASE-DEFICIENT CRISPR-Cas SYSTEMS
A Roadblock in the Way of the RNA Polymerase
To repress a gene, multiple target choices are possible. If the effector (e.g., the dCas9/sgRNA complex) binds to the promoter region of the target, it lowers the number of mRNA transcripts by blocking the initiation of transcription. If the target is located within the coding sequence of the target gene, it interrupts elongation by standing in the way of the RNA polymerase (RNAP) (15, 43). After halting, it is unclear how fast the RNAP dissociates from the template. In vitro experiments indicated that the stalled RNAP remains bound to the template after encountering dCas9 (44). It is likely that something similar occurs in vivo. In fact, sequencing of the incomplete transcripts suggested that after encountering dCas9, the RNAP remains in place for some time, giving birth to trains of stalled polymerases (43). There is also evidence that, depending on the strength of the interaction between the dCas9/sgRNA complex and the target, the RNAP can actively displace dCas9 and continue elongation to produce a full mRNA transcript (45).
When the target is in the promoter sequence, any DNA strand can be targeted, and the repression will be equally strong (Fig. 2, left). This has been observed for all types of CRISPRs used so far: type I (20), type II (16, 46), and type V (33, 47). On the contrary, when the target is downstream of the promoter, within the coding sequence, target orientation has crucial importance, and the best orientation varies depending on the type of CRISPR system. For dCas9 (type II), repression is effective only if the guide RNA pairs with the nontemplate strand (i.e., when the guide RNA is homologous to the template strand) (Fig. 2, top right). If dCas9 is in the wrong orientation, the repressive effect is much weaker (1.5- to 3-fold repression, instead of 10- to 100-fold in the correct orientation) (46). Interestingly, an opposite behavior has been observed for dCas12a (type V), which produces stronger repression when it binds to the template strand (33, 47, 48) (Fig. 2, middle right). For the type I CRISPR of E. coli, the guide RNA must bind to the nontemplate strand to produce strong repression (20) (Fig. 2, bottom right). Together, these data indicate that no simple rule determines which orientation is most effective for a given type of CRISPR system, be it the PAM position, the direction of the R loop extension, or the nature of the targeted strand. The exact mechanism of the collision thus remains to be elucidated on a more structural level, especially for effectors other than S. pyogenes dCas9. Importantly, for dCas9, the ability to stop the RNAP strongly depends on temperature, with higher temperatures leading to weaker repression (45, 49). Repression at high temperatures is still possible, using a variant of dCas9 isolated from a thermophilic organism (50). It is not known if other effectors (such as dCas12a) are affected in the same way.
FIG 2.

Targeting the correct strand for efficient repression. When the target is within the promoter sequence, any strand can be targeted for strong repression. When the target is within the coding sequence, one orientation is typically much more effective than the other. The configuration depicted here is the one that leads to the strongest repression for each of the CRISPR types that have been tested.
Consequences of the Collision for the RNA Transcript
Early experiments on the effect of target position within a gene suggested that targets farther away from the initiation codon tend to be less efficient for target repression (15). More recent studies have since contradicted this phenomenon (46, 51). Targeting within the promoter or in a small sequence window downstream shows strong repression and likely inhibits transcription initiation by either blocking access to the promoter or interfering with the transition of the RNAP from its initiation state into a processive elongation complex. However, when the target is far enough from the promoter to avoid interactions with the initiation step, the position of the target does not systematically affect the repression strength.
When elongation by RNAP is interrupted, an incomplete mRNA is generated (52). If the target is inside the coding sequence of a protein, the incomplete transcript may have a ribosome-binding site and a start codon but no stop codon. In that case, it will likely be subject to degradation to avoid ribosome stalling (53), usually relying on a mechanism called trans-translation. As a result, both the non-stop mRNA and the nascent peptide are targeted for degradation (54, 55).
Another crucial aspect of transcription interruption in bacteria is that genes are often cotranscribed in operons. As a result, interfering with one gene will likely have effects on the other genes from the same mRNA molecule, referred to as polar effects. (It should also be noted that antisense RNAs, in spite of acting at the translation step, are also subject to polar effects since they trigger RNA degradation [13, 56], although this effect is only partial.) Repression by CRISPR with a target inside an operon has a clear effect on all downstream genes, which are also repressed (57), a property that has been exploited to reveal the presence of cryptic promoters in the middle of operons (57).
There is also some evidence that genes upstream of the target in an operon can be affected, to various degrees depending on the organism. This “reverse polar effect” appears to be strong in Bacillus subtilis (43) but much milder in Staphylococcus aureus, where the level of the upstream transcript is reduced by only ∼50% (58). Similarly, in E. coli, reverse polar effects are apparent only when the target is within 100 bp from the gene end (46, 51). This is probably due to changes in mRNA stability; however, the exact process is not fully understood and might be specific to each operon due to different mRNA regulation motifs (59, 60).
In some cases, polar effects may be troublesome for singling out the effect of one gene. They may also be an advantage, as operons usually consist of genes of related functions, from the same metabolic pathway or machinery. As a result, polar effects allow the reduction of the number of targets needed in screening libraries.
Other factors may interfere with dCas9 binding to its target. One potentially important contextual factor is transcription on the opposite DNA strand in the case of two convergent promoters (61). If dCas9 is set to block RNAP in one direction, other RNAPs coming in the opposite direction will eject it from the DNA with a very high probability. This could possibly interfere with the desired repression. Positive DNA supercoiling has also been shown to affect dCas9’s binding, as it increases the force required to unwind the DNA for target recognition (62). This may be important when using multiple binding sites on the same target in the hopes of increasing the repression strength: if the targets are too close to each other, they might exhibit anticooperative behavior (62).
Taming CRISPR Guides with Imperfect Complementarity
Large data sets have been produced to understand the effect of mismatches between the guide and the target (63–67). A particularly creative strategy was to tether many targets to a sequencing flow cell, locate the different targets by next-generation sequencing, and then measure the binding of fluorescence-labeled CRISPR effectors directly on the same flow cell (68, 69). The in vitro transcription/translation system (tx/tl) was also used to measure DNA cleavage and repression for many targets (70). Finally, recent studies employed libraries of CRISPR guides combined with fluorescence-activated cell sorting (FACS) to measure how the target sequence and the presence of mismatches affect the repression strength (65, 67).
In most cases, the seed sequence (the first 5 to 12 bp next to the PAM) is of essential importance for stable binding to the target. This, however, is not a universal rule, as mismatches in the seed do not necessarily abolish repression when the target is in a promoter (71).
The other end of the guide, the PAM-distal side, is typically important for DNA cleavage but not necessary for strong binding, and guides with up to 11 mismatches on that side can still have a significant repressive effect (16, 46). For both dCas9 and dCas12a, the complementarity of the PAM-distal region influences the dissociation rate, to the point that it was called the reversibility-defining region (72). This must be taken into account during guide design, in order to avoid off-targets in essential genes that could have a major impact on growth or that could be related to the phenotype of interest.
Kinetic models have been used to understand the behavior of CRISPR effectors in the presence of a mismatched target. In these models, the system is broken up into a finite number of states, with kinetic parameters associated with the different transitions (73, 74). As the R loop extends from the PAM-proximal to the PAM-distal end of the guide, mismatches between the guide and the target can be seen as high potential barriers that can be overcome, in agreement with the observation that when the R loop has extended past a mismatch, it can continue to extend normally (75). Such models make interesting predictions; for example, they have been used to explain the somewhat paradoxical result that Cas9 variants with more relaxed PAM recognition tend to have fewer off-target cutting sites despite having a higher number of potential targets (73, 76). Briefly, the total frequency of off-target cutting will depend on the number of potential binding sites and the probability, for each of these sites, that the R loop will extend until the end. Cas9 mutants selected for relaxed PAM recognition have more potential binding sites, but the interaction between Cas9 and the DNA is weaker, so only guides with high complementarity have a chance of causing cleavage. Interestingly, due to the long-lasting binding of Cas9 to the target, the dissociation rate is usually much lower than the catalytic rate (73). The consequence is that, for DNA cleavage, kinetic equilibrium (where equal numbers of molecules bind and unbind at any given moment) will never be reached in most cases. This is a major difference with repression by deactivated nucleases, where the system may eventually reach equilibrium, and is an important reason why repression efficiency cannot be extrapolated from cutting efficiency.
Interfering with Translation
In most applications so far, CRISPR knockdown systems were acting on transcription. However, some CRISPR immune systems act by cleaving RNA, making it conceivable to prevent translation rather than transcription. Such an activity is naturally present in type III and VI systems. In type III systems, the Cas ribonucleoprotein complex binds to target RNA, which activates the DNA nuclease activity of Cas10 as well as the production of cyclic oligoadenylate messengers, which in turn activate other effectors such as the Csm6 RNase (77–79). Since the crRNAs of type III systems bind RNA targets rather than DNA, there is no risk to target the CRISPR array itself, and type III systems do not require a PAM to prevent autoimmunity. Mechanisms have nonetheless been described that enable type III systems to discriminate between target sequences and transcripts generated from the CRISPR array in the antisense direction. This discrimination seems to be achieved both by base pair interactions between the 5′ end of the guide RNA and the target and by non-base pair interactions, with possible differences between type III subtypes (79–81). Typically, when the 5′ end of the crRNA is fully complementary to the target, the DNase activity is not triggered. This property was harnessed in the archaeon Sulfolobus solfataricus to obtain 2-fold repression of a reporter using the organism’s own type III CRISPR-Cas systems (21). Alternatively, it might be possible to mutate the Cas10 protein and other effectors to maintain a specific RNase activity while blocking DNA cleavage. Controlling gene expression with type VI systems is an attractive option as these systems target RNA exclusively. However, these effectors indiscriminately degrade the surrounding RNA molecules once they are activated by their target. When type VI Cas13a was used to repress a reporter in E. coli, the growth rate of the bacterium was severely diminished due to the collateral cleavage of other mRNAs in the cell (22). While this problem has not been observed in human cells (82), engineering of Cas13 effectors might therefore be necessary to make this a viable strategy in bacteria.
In addition to effectors whose natural target is RNA, certain variants of the DNA-targeting Cas9, such as the ones from S. aureus, Campylobacter jejuni, and Neisseria meningitidis, can also target RNA, with the peculiar property that a PAM is not required. As a result, one can direct Cas9 specifically to RNA by choosing a target that is not flanked by a PAM (42, 83). This also results in gene repression, although it is not clear whether the repression comes from the cleavage and degradation of the mRNA or just from interference with the ribosomes. In all cases, the effectiveness of the repression is strongly influenced by the accessibility of the target, which depends on the secondary structure of the mRNA (22, 42).
Switching CRISPR On and Off: a Matter of Timing
Many points of the mechanism of effector assembly and association with the guide RNA remain to be elucidated. It is likely that the assembly of Cas9 and the guide RNA (crRNA or sgRNA) is quite stable, as free Cas9 has a quite flexible structure and is stabilized by the crRNA (84–86).
The process by which the CRISPR effectors find their target has also received much attention. Most known transcriptional regulators, such as those involved in inducible promoters, recognize their binding motif from the side, without unwrapping the two strands of the DNA. They can thus slide along the DNA groove until the operator is found. For CRISPR-Cas systems, however, this is not possible, as target recognition involves complementary Watson-Crick base pairing, and double-stranded DNA has to be open. Experiments with DNA curtains and fluorescent effectors allow monitoring of the target search process in vitro. Such data exist for type II (Cas9) and type V (Cas12a) (35, 37, 87, 88). For both types, there is a first step of three-dimensional (3D) diffusion until a DNA molecule is encountered (35). This is followed by one-dimensional (1D) diffusion along DNA (87), although the contact with DNA is only intermittent. For Cas9, 1D diffusion is limited to very short distances (around 20 bp) but allows it to jump from one PAM to another. For Cas12a, however, the 1D diffusion step is dominant (88), and a molecule of Cas12a can appear to diffuse along the DNA molecule for extended periods of time. Another core difference between Cas9 and Cas12a regarding target searching is that binding to the PAM seems to have a limited contribution to DNA unwinding during R-loop formation for Cas12a (59). Cas12a probably dwells on PAM sequences for only very short periods of time if the guide does not match the target, while the transient association of Cas9 with PAM sites can easily be observed (35). Cas9 transiently associates with nonmatching sites with a residence time that depends on complementarity. As a result, it spends a few milliseconds on each potential binding site, and a single dCas9 molecule may take several hours to find its target (89). This search time has been evaluated to be about 6 h for E. coli’s 4-Mbp genome. Thus, a large number of Cas9 complexes might be required for a quick response, which is of particular importance for antiphage defense. The importance of this for gene knockdown is still unclear.
The dissociation of dCas9 from the target is also very slow (35), in the order of a few hours in vitro. Because of this, the amount of effector that is required for gene silencing is very small. In fact, one single complex per target locus might be sufficient to shut down expression until it gets kicked out by the RNAP or the replication fork. After stopping the expression of dCas9, it takes up to 5 h to reach the original expression level of the target gene (15, 90), which presumably corresponds to the time required for dCas9 to be lost through dilution and cell division.
HACKING CRISPR FOR BETTER CONTROL OF EXPRESSION
While the repurposing of natural CRISPR-Cas systems quickly led to impressive results, much effort has been undertaken to extend their capabilities. Here, we discuss recent advances in improving the repression strength, controlling multiple targets at the same time, and extending the application of CRISPR knockdown to a broader range of species, including some organisms that are not standard laboratory models. We also review the efforts that have been made to detect the potential undesirable effects of CRISPR-Cas systems and strategies to mitigate them.
Tunable Gene Repression and Activation
To obtain partial repression of the gene of interest, multiple approaches have been undertaken. The most straightforward one is to express the CRISPR system from an inducible promoter such as Pbad in E. coli (91, 92) or Pxyl in B. subtilis (43). In this case, the repression strength is controlled by using variable amounts of a chemical in the medium. Changing the amount of sgRNA, rather than of dCas9, may provide better control. Alternatively, by expressing a mismatched guide RNA and a constant amount of effector, it is possible to repress genes by a well-defined fraction (45). While the latter strategy requires one guide RNA design for each repression level, it is easier to multiplex and is less noisy.
Inducible promoters, which are often used to turn CRISPR knockdown on or off, usually produce a basal rate of leaky expression even when they are not induced. As discussed above, due to the long residence time of CRISPR effectors on the DNA, a small number of repressor complexes is enough to silence a target. Thus, even when under the control of a tight promoter, dCas9 can produce significant repression of the target in the absence of an inducer (43, 47, 91). One way to improve the dynamic range is to use a genetically recoded organism, with a nonnatural amino acid in dCas9. In this case, leaky repression is strongly alleviated unless the nonnatural amino acid is also present (93). While effective, this method is currently limited to one heavily modified strain of E. coli and cannot be easily adapted to other organisms. Using antisense RNA targeted at the CRISPR guide itself, it is possible to antagonize repression, possibly by impairing the formation of the complex, actively triggering the degradation of the guide RNA, or blocking the binding of dCas9 to the target (94). This provides another layer of regulation to construct genetic circuits and a way to quickly recover gene expression once repression is established.
Finally, CRISPR-based programmable transcription factors are not restricted to gene repression. By linking dCas9 to a transcription activator, it is also possible to increase the expression level of a target. Initial studies used the ω subunit from the RNA polymerase to induce expression in an rpoZ deletion strain (16); however, subsequent screening found more potent activators, in particular SoxS (95), an activator normally involved in the oxidative stress response which does not require the rpoZ deletion. When SoxS is tethered to dCas9 and the complex is targeted upstream of a promoter, the expression of the reporter can be increased by more than 10-fold, making it possible to look at overexpression phenotypes for many genes in an easy way.
An interesting application of CRISPR-based activation was to create a positive link between dCas9 binding and the expression of a selection marker, allowing the continuous evolution of improved ω-dCas9 fusions and novel dCas9 variants with modified PAM recognition specificity (76). Gene activation is also possible in B. subtilis using a similar approach (96). A potential drawback of this method is that it requires a PAM at the correct distance from the promoter, as the range of target positions that are effective for activation is very narrow, highlighting the importance of developing CRISPR effector variants with different PAM specificities. More recently, the AsiA anti-σ70 protein from bacteriophage T4 has been identified as a potent activator with a more relaxed binding window to activate target promoters (97). Protein fusions between dCas9 and AsiA could further be evolved to improve their activity, leading to a promising tool that is able to activate endogenous genes and is functional in several bacterial species.
A strategy to activate σ54 promoters in bacteria using dCas9 was also demonstrated (98). The σ54 factor binds to its promoters in a closed inactive form. In order to be activated, σ54 promoters require an activator protein to bind an upstream activating sequence (UAS) and remodel the RNAP-σ54-DNA complex in a process involving ATP hydrolysis. The authors of that study were able to recruit σ54 activators to the guide RNA scaffold using aptamers and activate target promoters by directing dCas9 to bind to the UAS (98). Their use of a Cas9 variant with an extended PAM (xdCas9) also enabled them to increase the range of promoters that can be activated in this manner.
Simultaneous Control of Multiple Targets
Natural CRISPR arrays often contain several dozens, sometimes hundreds, of guides (99). Accordingly, it is possible to express multiple guides with a deactivated effector to repress multiple targets at the same time. In this way, more than 20 CRISPR guides have been used simultaneously (100). Multiplex gene repression has been useful to find pairs of synthetic lethal genes and measure epistatic interactions (43, 101) as well as for metabolic engineering. Moreover, activation and repression can be used in the same cell by using an sgRNA-linked aptamer to recruit the activation tag only on certain targets (95). Another approach is to use composite CRISPR arrays: guides with different scaffolds can be coexpressed within the same array so that after processing, they associate with different effectors (27). Coexpressing sgRNAs can be quite cumbersome, as each of them requires its own promoter and transcription terminator. CRISPR arrays, similar to what is found in nature, might be more practical for the expression of many guides (27). dCas12a is particularly attractive for this purpose, owing to its ability to process the primary transcript of the CRISPR array without relying on host factors (27, 48). Due to their very repetitive nature, it is challenging to produce long CRISPR arrays with multiple spacers using standard DNA synthesis. However, by splitting the array into multiple parts to separate the repeats, it is possible to assemble a series of oligonucleotides into CRISPR arrays. Single-pot methods have been developed to create large CRISPR arrays in one step (27). Assembly can also be done iteratively, adding the guides one by one (20, 102). Finally, by using a set of diverse sgRNA handles, it is possible to avoid repetition and construct long arrays of sgRNA by DNA synthesis (100). As the pool of effectors is shared between all CRISPR guides, having more guides in one array makes each individual complex less abundant, requiring the expression of the effector at a higher level (20, 100). The number of complexes carrying each guide may also be different depending on the guide’s abundance, which is influenced by its position within the array and secondary structure (27). However, by choosing the target in the coding region and having a sufficient dCas9 concentration, it is possible to reach a saturation regime, where the strength of repression is not affected by crRNA abundance (45).
From One Bacterium to All Others
One reason for the popularity of CRISPR-Cas systems is the fact that they are remarkably portable. Within just a few years after their discovery, CRISPR-based technologies were successfully used in all kingdoms of life (32, 103–107). Systems for gene knockdown are readily available for many bacteria, including species of clinical or industrial interest for which available genetic tools are limited (108–110). By using easily transferable, modular plasmid systems relying on bacterial conjugation, it is possible to perform genetic screens on nonmodel bacteria, including many human pathogens (111). Furthermore, a thermostable variant of dCas9 was characterized for use in thermophilic organisms (50). CRISPR activation was also shown to work on nonmodel bacterial species (98).
Since about 45% of bacterial species possess a native CRISPR immune system (99), it may also be possible to use it for repression, rather than expressing a heterologous system. This has been done with the E. coli endogenous type I CRISPR system, where a simple deletion of the Cas3 protein, which carries the nuclease activity, allowed the creation of a programmable repressor (20).
Understanding and Addressing Side Effects
When expressed at an excessive level, CRISPR effectors themselves can have some toxic effects on bacteria. The extent of toxicity depends on the host organism and on the nature of the CRISPR effector. In E. coli, growth defects (112) as well as morphological defects (113) have been reported upon the overexpression of S. pyogenes dCas9. SpdCas9 is also toxic to mycobacteria, but this toxicity can be alleviated using a different Cas9 ortholog from Streptococcus thermophilus (114). Similarly, SpCas9 is toxic to cyanobacteria (115), but Cas12a from Francisella novicida is not (116).
Aside from direct toxicity, an important source of undesired side effects when using CRISPR knockdown is off-target binding. Even if good complementarity between the guide and the target is necessary for strong repression, only a few matching bases in the PAM-proximal region can be enough to produce a small repressive effect (46). If this effect happens on an essential gene, a severe growth defect may ensue. For some applications, off-target binding can be greatly reduced by replacing the PAM-binding domain of dCas9 with a binding domain from the PhlF repressor (117). Consequently, dCas9 can be targeted only at regions containing the PhlF operator sequence, which restricts the application of this system to synthetic biology.
Another unresolved problem is the “bad-seed effect” (BSE) (46). Among all the possible seed sequences (the last 5 nucleotides at the 3′ end of a guide), a few of them systematically cause a strong fitness defect when used with S. pyogenes dCas9 in E. coli. For instance, when a guide finishing in ACCCA is expressed in E. coli K-12, it causes a nearly complete growth arrest of the population. About 130 seed sequences (out of 1,024 possible) cause this kind of sickness. A list of the 10 most toxic ones is provided in Table 1. The origin of this effect is still unknown. It does not appear to be due to off-target binding to a particularly critical locus but may involve multiple simultaneous binding events or binding to substrates other than genomic DNA (46). This effect is very pervasive and can go unnoticed; for example, we found evidence for the BSE in data previously reported by others (51) (Fig. 3), showing that it could be an unsuspected source of noise in the data. To date, the best solution to avoid the BSE is simply to avoid bad seeds when designing a CRISPR guide. However, it was also shown that the BSE depends on dCas9’s concentration and that it can be alleviated by using a carefully chosen expression cassette (46) while still retaining a strong repression capacity. We hope that future work will elucidate the origin of the BSE.
TABLE 1.
The 10 seed sequences that have the greatest toxic effect on E. colia
| Seed sequence | Mean fitness effect (avg log2-transformed fold change)b
|
|
|---|---|---|
| High dCas9 expression | Low dCas9 expression | |
| AGGAA | −6.50 | −2.98 |
| TGACT | −5.90 | −1.34 |
| ACCCA | −5.87 | −2.46 |
| AAAGG | −5.63 | −1.68 |
| GAGGC | −5.41 | −1.91 |
| CGGAA | −5.40 | −1.51 |
| ATATG | −5.37 | −1.08 |
| AACTA | −5.10 | −0.88 |
| TGGAA | −5.07 | −1.26 |
| CACTC | −5.01 | −1.76 |
See reference 46. The use of an optimized strain with a low dCas9 expression level greatly reduces the bad-seed effect (right column) but does not completely abolish it. CRISPR guides with these seed sequences should be avoided if possible.
Shown are average log2-transformed fold changes in guide abundances through 17 generations, measured by high-throughput sequencing.
FIG 3.
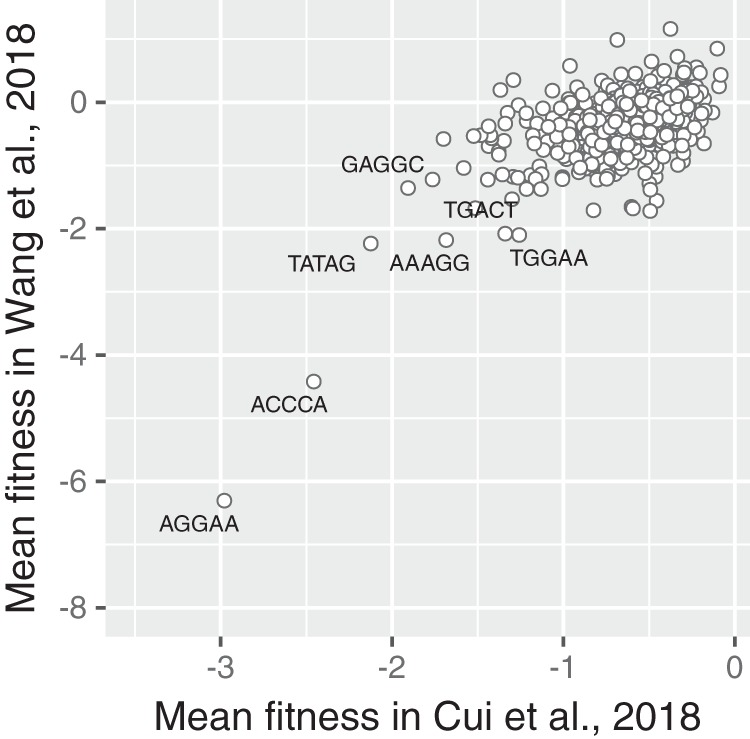
Unnoticed “bad-seed” effect in the CRISPR screen of Wang et al. (51). Using CRISPR guides that target only nonessential genes, we calculated the average fitness scores (log2 fold change) of guides depending on their seed sequence. The sequences AGGAA and ACCCA are the two most toxic bad seeds discovered by Cui et al. (46). In the CRISPR screen of Wang et al. (51), guides carrying a bad seed also have a strong average fitness defect, regardless of the genes that they target.
CURRENT SUCCESSES IN CONTROLLING TRANSCRIPTION
Genome-Wide CRISPR Screens in Bacteria
For a few model organisms, large collections of knockout strains have been developed and used successfully. These include the Keio collection for E. coli (118) and two barcoded deletion libraries for B. subtilis (119). While these libraries are extremely valuable for people working on these two model bacteria, creating such libraries is very work-intensive, and most importantly, they are limited to nonessential genes.
Early screens for essential genes were conducted using antisense RNA (11, 12, 56) and transposon insertion libraries (120). Recent improvements in sequencing capabilities and molecular biology techniques have enabled the screening of very dense transposon insertion libraries more easily, yielding useful data for many bacterial species and conditions (121–125). However, transposon insertion is random and not necessarily homogeneous across the entire chromosome. Long genes are more likely to be disrupted, while short sequences like noncoding RNAs are unlikely to be targeted, requiring very large and dense insertion libraries as well as a large amount of sequencing power to reach the desired precision in fitness measurement.
As an alternative, the ability to control the expression of genes in trans using CRISPR knockdown and activation makes it a promising tool for genome-wide screens. To create a CRISPR guide, one just needs to insert a short sequence (20 bp in the case of SpdCas9) in a well-defined locus. Thus, the construction procedure can be standardized and streamlined so that a large number of guides can be assembled in parallel, allowing the creation of customized screening libraries using on-chip oligonucleotide synthesis. Several cloning methods using homologous assembly (102), Golden Gate assembly (46, 126), or direct oligonucleotide integration coupled with negative selection (91) have made it possible to assemble CRISPR guides in a single step. Moreover, strains repressed by a CRISPR effector can easily be genotyped by simply sequencing the CRISPR guide, eliminating the need for a barcode or for a complex amplification protocol to locate an inserted sequence in the genome.
Multiple CRISPR screens have already been used to find essential genes under various conditions (51, 57, 94, 127). The applications of these data include the discovery of drug targets, searches for synthetic lethal pairs, and genome minimization. Partial repression can also be useful to find phenotypes for essential genes (43, 67). CRISPR screens are also more versatile than transposon sequencing (TnSeq), as one can target only a subset of genes of interest. Guide RNAs can also easily be designed to simultaneously target all copies of a duplicated gene or closely related paralogs that are likely to be functionally identical (51). If CRISPR guides are expressed from a plasmid, the same library can easily be reused on many strains and under many conditions, making the method more cost-effective in the long run.
Measuring fitness through growth-based enrichment or depletion of specific guides is perhaps the most evident output for genome-wide CRISPR screening, but it is not the only one. For example, by performing a complete phage replication cycle in a population repressed by such a screening library, it was possible to identify host factors necessary for the production of infectious phage particles (57). A CRISPR library has been combined with high-content microscopy to find the effect on growth and morphology and identify the function of unknown genes in the pathogen Streptococcus pneumoniae (128). In another study, comparison of the growth characteristics of a CRISPR library in the presence of a variety of chemicals allowed the reconstruction of genetic networks and the identification of the target of antibiotics (43). The last two approaches required isolated cultures of the different library members, greatly limiting the throughput of the screen. Recently, a method was developed to identify the genotype of CRISPR-repressed strains in situ during a pooled assay by using fluorescence measurements (129, 130). Currently limited to a few hundred strains, this principle could be scaled up to analyze large-scale libraries over many generations by high-content microscopy.
Synthetic Biology and Metabolic Engineering
The programmability of CRISPR has made it popular among synthetic biologists. By expressing multiple guides at the same time and targeting them at each other, it is possible to construct predictable genetic circuits (Fig. 4). As these circuits can interface with native chromosomal genes, this makes it easy to build artificial regulatory networks that control the bacterium’s natural functions (112). Such genetic circuits have been used to improve the yield of protein expression by creating feedback between the metabolic burden and transcription (131) so that protein expression is regulated to an optimal level.
FIG 4.

Overview of regulatory circuits that can be built using CRISPR repressors. As inputs, chemical inducers were used by Nielsen and Voigt (112). Metabolic burden was used by Ceroni et al. (131). The logic gates are from the study by Nielsen and Voigt (112). Since sgRNAs act as a NOT gate, and two identical sgRNAs act as NOR gates, all other basic logic gates like AND and OR can be constructed. As outputs, metabolic production is discussed by Ceroni et al. (131), replication is discussed by Wiktor et al. (49), filamentation is discussed by Mückl et al. (132), biofilm formation is discussed by Nielsen and Voigt (112), and cell shape is discussed by Elhadi et al. (136).
Aside from gene repression or activation, dCas9 has been programmed to interfere with various processes in the cell. These include modifying the spatial structure of E. coli’s chromosome to create artificial DNA loops (61), blocking the initiation of replication to take control of the cell cycle (49), and triggering cell filamentation in a reversible manner (132).
Another successful domain of application of CRISPR knockdown is metabolic engineering. CRISPR-based methods allow the quick identification of competing pathways and the optimization of metabolic fluxes (133) for the production of a compound of interest. CRISPRi has been set up in multiple industrially relevant organisms, such as Lactococcus lactis (108), Clostridium beijerinckii (134), and Corynebacterium glutamicum (135). Cho et al. previously reviewed other industrial strains where CRISPRi has been used (5), along with strategies used to improve production yield. Guides targeted at genes involved in rod shape maintenance were used to diversify the morphology of cells and optimize the production of biodegradable plastic (136). Finally, by inhibition of cell growth, one can optimize the balance between the production of biomass and the synthesis of metabolites (90).
CHALLENGES AND OPPORTUNITIES
CRISPR-Cas systems have not yet reached their full potential. There is a wide disparity in how much we know about different types of CRISPR-Cas systems. Compared to the vast literature surrounding type II or V, some other types (like type IV) are still largely unknown. More generally, the field of bacterial immunity against phages is in its early stages and will probably uncover more valuable biotechnological tools (137). Additionally, most applications so far have used wild-type proteins or simple deactivated mutants, but the artificial evolution of engineered variants offers promising perspectives, as has already been seen for expanded PAM recognition (76). Regarding the control of gene expression, it remains difficult to predict the effect of a given guide in silico, at the design stage. Some models are already available for E. coli and B. subtilis (67), but more research is needed to develop tools that are reliable and easy to use. In particular, a major challenge will be to devise predictable repression tools for nonmodel species, which would require a better understanding of polar effects and the role of host factors. Finally, the use of engineered CRISPR-Cas systems for gene activation in bacteria has so far remained limited to a few proofs of concept. We can expect that these tools will be adapted to high-throughput screens and used to unravel interesting biology in the coming years.
CONCLUSION
Compared to the usual speed of biotechnological development, CRISPR-Cas systems were repurposed as tools in a particularly short time after their discovery. Due to the multiplicity of their components, many degrees of freedom, like the sequence of the guide RNA scaffold or the stoichiometry of different parts, were not fully explored. After a few years, we now know better the importance of each part and begin to have a clear overview of how CRISPR interference happens, including the assembly of the dCas9 complex, the search for the target, the extension of the R loop, and the outcome of collisions with RNA polymerase.
Modern biology is in need of precise and quantitative results, as opposed to qualitative differences that simply pass the test of significance. Hence, it is crucial to identify all potential artifacts and false positives that are likely to arise when using CRISPR-Cas systems. Off-target binding, time and concentration dependence, and the bad-seed effect are all important challenges that need to be understood better to take full advantage of CRISPR’s capabilities. While many questions remain open, the control of transcription by CRISPR is becoming a mature technology that can now be used for more ambitious large-scale projects.
REFERENCES
- 1.Keren L, Hausser J, Lotan-Pompan M, Vainberg Slutskin I, Alisar H, Kaminski S, Weinberger A, Alon U, Milo R, Segal E. 2016. Massively parallel interrogation of the effects of gene expression levels on fitness. Cell 166:1282–1294.e18. doi: 10.1016/j.cell.2016.07.024. [DOI] [PubMed] [Google Scholar]
- 2.Schaerli Y, Jiménez A, Duarte JM, Mihajlovic L, Renggli J, Isalan M, Sharpe J, Wagner A. 2018. Synthetic circuits reveal how mechanisms of gene regulatory networks constrain evolution. Mol Syst Biol 14:e8102. doi: 10.15252/msb.20178102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones DL, Brewster RC, Phillips R. 2014. Promoter architecture dictates cell-to-cell variability in gene expression. Science 346:1533–1536. doi: 10.1126/science.1255301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolf L, Silander OK, van Nimwegen E. 2015. Expression noise facilitates the evolution of gene regulation. Elife 4:e05856. doi: 10.7554/eLife.05856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho S, Shin J, Cho B-K. 2018. Applications of CRISPR/Cas system to bacterial metabolic engineering. Int J Mol Sci 19:1089. doi: 10.3390/ijms19041089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Na D, Yoo SM, Chung H, Park H, Park JH, Lee SY. 2013. Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nat Biotechnol 31:170–174. doi: 10.1038/nbt.2461. [DOI] [PubMed] [Google Scholar]
- 7.Ghodasara A, Voigt CA. 2017. Balancing gene expression without library construction via a reusable sRNA pool. Nucleic Acids Res 45:8116–8127. doi: 10.1093/nar/gkx530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mimee M, Tucker AC, Voigt CA, Lu TK. 2015. Programming a human commensal bacterium, Bacteroides thetaiotaomicron, to sense and respond to stimuli in the murine gut microbiota. Cell Syst 1:62–71. doi: 10.1016/j.cels.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mimee M, Nadeau P, Hayward A, Carim S, Flanagan S, Jerger L, Collins J, McDonnell S, Swartwout R, Citorik RJ, Bulović V, Langer R, Traverso G, Chandrakasan AP, Lu TK. 2018. An ingestible bacterial-electronic system to monitor gastrointestinal health. Science 360:915–918. doi: 10.1126/science.aas9315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sen GL, Blau HM. 2006. A brief history of RNAi: the silence of the genes. FASEB J 20:1293–1299. doi: 10.1096/fj.06-6014rev. [DOI] [PubMed] [Google Scholar]
- 11.Ji Y, Zhang B, Van Horn SF, Null H, Warren P, Woodnutt G, Burnham MK, Rosenberg M. 2001. Identification of critical staphylococcal genes using conditional phenotypes generated by antisense RNA. Science 293:2266–2269. doi: 10.1126/science.1063566. [DOI] [PubMed] [Google Scholar]
- 12.Forsyth RA, Haselbeck RJ, Ohlsen KL, Yamamoto RT, Xu H, Trawick JD, Wall D, Wang L, Brown-Driver V, Froelich JM, Kedar GC, King P, McCarthy M, Malone C, Misiner B, Robbins D, Tan Z, Zhu Z, Carr G, Mosca DA, Zamudio C, Foulkes JG, Zyskind JW. 2002. A genome-wide strategy for the identification of essential genes in Staphylococcus aureus. Mol Microbiol 43:1387–1400. doi: 10.1046/j.1365-2958.2002.02832.x. [DOI] [PubMed] [Google Scholar]
- 13.Nakashima N, Tamura T, Good L. 2006. Paired termini stabilize antisense RNAs and enhance conditional gene silencing in Escherichia coli. Nucleic Acids Res 34:e138. doi: 10.1093/nar/gkl697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Man S, Cheng R, Miao C, Gong Q, Gu Y, Lu X, Han F, Yu W. 2011. Artificial trans-encoded small non-coding RNAs specifically silence the selected gene expression in bacteria. Nucleic Acids Res 39:e50. doi: 10.1093/nar/gkr034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. 2013. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bikard D, Jiang W, Samai P, Hochschild A, Zhang F, Marraffini LA. 2013. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res 41:7429–7437. doi: 10.1093/nar/gkt520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen H, Ferbeyre G, Cedergren R. 1997. Efficient hammerhead ribozyme and antisense RNA targeting in a slow ribosome Escherichia coli mutant. Nat Biotechnol 15:432–435. doi: 10.1038/nbt0597-432. [DOI] [PubMed] [Google Scholar]
- 18.Nakashima N, Tamura T. 2009. Conditional gene silencing of multiple genes with antisense RNAs and generation of a mutator strain of Escherichia coli. Nucleic Acids Res 37:e103. doi: 10.1093/nar/gkp498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koonin EV, Makarova KS, Zhang F. 2017. Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol 37:67–78. doi: 10.1016/j.mib.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo ML, Mullis AS, Leenay RT, Beisel CL. 2015. Repurposing endogenous type I CRISPR-Cas systems for programmable gene repression. Nucleic Acids Res 43:674–681. doi: 10.1093/nar/gku971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zebec Z, Manica A, Zhang J, White MF, Schleper C. 2014. CRISPR-mediated targeted mRNA degradation in the archaeon Sulfolobus solfataricus. Nucleic Acids Res 42:5280–5288. doi: 10.1093/nar/gku161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abudayyeh OO, Gootenberg JS, Konermann S, Joung J, Slaymaker IM, Cox DBT, Shmakov S, Makarova KS, Semenova E, Minakhin L, Severinov K, Regev A, Lander ES, Koonin EV, Zhang F. 2016. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353:aaf5573. doi: 10.1126/science.aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E. 2011. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan WX, Hunnewell P, Alfonse LE, Carte JM, Keston-Smith E, Sothiselvam S, Garrity AJ, Chong S, Makarova KS, Koonin EV, Cheng DR, Scott DA. 2019. Functionally diverse type V CRISPR-Cas systems. Science 363:88–91. doi: 10.1126/science.aav7271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, Virgin HW, Listgarten J, Root DE. 2016. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fonfara I, Richter H, Bratovič M, Le Rhun A, Charpentier E. 2016. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 532:517–521. doi: 10.1038/nature17945. [DOI] [PubMed] [Google Scholar]
- 27.Liao C, Ttofali F, Slotkowski RA, Denny SR, Cecil TD, Leenay RT, Keung AJ, Beisel CL. 2019. Modular one-pot assembly of CRISPR arrays enables library generation and reveals factors influencing crRNA biogenesis. Nat Commun 10:2948. doi: 10.1038/s41467-019-10747-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mojica FJM, Díez-Villaseñor C, García-Martínez J, Almendros C. 2009. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155:733–740. doi: 10.1099/mic.0.023960-0. [DOI] [PubMed] [Google Scholar]
- 29.Deveau H, Barrangou R, Garneau JE, Labonté J, Fremaux C, Boyaval P, Romero DA, Horvath P, Moineau S. 2008. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol 190:1390–1400. doi: 10.1128/JB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, Koonin EV, Zhang F. 2015. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leenay RT, Beisel CL. 2017. Deciphering, eommunicating, and engineering the CRISPR PAM. J Mol Biol 429:177–191. doi: 10.1016/j.jmb.2016.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. 2013. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim SK, Kim H, Ahn W-C, Park K-H, Woo E-J, Lee D-H, Lee S-G. 2017. Efficient transcriptional gene repression by type V-A CRISPR-Cpf1 from Eubacterium eligens. ACS Synth Biol 6:1273–1282. doi: 10.1021/acssynbio.6b00368. [DOI] [PubMed] [Google Scholar]
- 34.Leenay RT, Maksimchuk KR, Slotkowski RA, Agrawal RN, Gomaa AA, Briner AE, Barrangou R, Beisel CL. 2016. Identifying and visualizing functional PAM diversity across CRISPR-Cas systems. Mol Cell 62:137–147. doi: 10.1016/j.molcel.2016.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. 2014. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507:62–67. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiao Y, Luo M, Hayes RP, Kim J, Ng S, Ding F, Liao M, Ke A. 2017. Structure basis for directional R-loop formation and substrate handover mechanisms in type I CRISPR-Cas system. Cell 170:48–60.e11. doi: 10.1016/j.cell.2017.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szczelkun MD, Tikhomirova MS, Sinkunas T, Gasiunas G, Karvelis T, Pschera P, Siksnys V, Seidel R. 2014. Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc Natl Acad Sci U S A 111:9798–9803. doi: 10.1073/pnas.1402597111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sternberg SH, LaFrance B, Kaplan M, Doudna JA. 2015. Conformational control of DNA target cleavage by CRISPR-Cas9. Nature 527:110–113. doi: 10.1038/nature15544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dagdas YS, Chen JS, Sternberg SH, Doudna JA, Yildiz A. 2017. A conformational checkpoint between DNA binding and cleavage by CRISPR-Cas9. Sci Adv 3:eaao0027. doi: 10.1126/sciadv.aao0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loeff L, Brouns SJJ, Joo C. 2018. Repetitive DNA reeling by the Cascade-Cas3 complex in nucleotide unwinding steps. Mol Cell 70:385–394.e3. doi: 10.1016/j.molcel.2018.03.031. [DOI] [PubMed] [Google Scholar]
- 41.Westra ER, van Erp PBG, Künne T, Wong SP, Staals RHJ, Seegers CLC, Bollen S, Jore MM, Semenova E, Severinov K, de Vos WM, Dame RT, de Vries R, Brouns SJJ, van der Oost J. 2012. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Mol Cell 46:595–605. doi: 10.1016/j.molcel.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Strutt SC, Torrez RM, Kaya E, Negrete OA, Doudna JA. 2018. RNA-dependent RNA targeting by CRISPR-Cas9. Elife 7:e32724. doi: 10.7554/eLife.32724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peters JM, Colavin A, Shi H, Czarny TL, Larson MH, Wong S, Hawkins JS, Lu CHS, Koo B-M, Marta E, Shiver AL, Whitehead EH, Weissman JS, Brown ED, Qi LS, Huang KC, Gross CA. 2016. A comprehensive, CRISPR-based functional analysis of essential genes in bacteria. Cell 165:1493–1506. doi: 10.1016/j.cell.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Widom JR, Rai V, Rohlman CE, Walter NG. 2019. Versatile transcription control based on reversible dCas9 binding. RNA 25:1457–1469. doi: 10.1261/rna.071613.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vigouroux A, Oldewurtel E, Cui L, Bikard D, van Teeffelen S. 2018. Tuning dCas9’s ability to block transcription enables robust, noiseless knockdown of bacterial genes. Mol Syst Biol 14:e7899. doi: 10.15252/msb.20177899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cui L, Vigouroux A, Rousset F, Varet H, Khanna V, Bikard D. 2018. A CRISPRi screen in E. coli reveals sequence-specific toxicity of dCas9. Nat Commun 9:1912. doi: 10.1038/s41467-018-04209-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miao C, Zhao H, Qian L, Lou C. 2019. Systematically investigating the key features of the DNase deactivated Cpf1 for tunable transcription regulation in prokaryotic cells. Synth Syst Biotechnol 4:1–9. doi: 10.1016/j.synbio.2018.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang X, Wang J, Cheng Q, Zheng X, Zhao G, Wang J. 2017. Multiplex gene regulation by CRISPR-ddCpf1. Cell Discov 3:17018. doi: 10.1038/celldisc.2017.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wiktor J, Lesterlin C, Sherratt DJ, Dekker C. 2016. CRISPR-mediated control of the bacterial initiation of replication. Nucleic Acids Res 44:3801–3810. doi: 10.1093/nar/gkw214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mougiakos I, Mohanraju P, Bosma EF, Vrouwe V, Bou MF, Naduthodi MIS, Gussak A, Brinkman RBL, van Kranenburg R, van der Oost J. 2017. Characterizing a thermostable Cas9 for bacterial genome editing and silencing. Nat Commun 8:1647. doi: 10.1038/s41467-017-01591-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang T, Guan C, Guo J, Liu B, Wu Y, Xie Z, Zhang C, Xing X-H. 2018. Pooled CRISPR interference screening enables genome-scale functional genomics study in bacteria with superior performance. Nat Commun 9:2475. doi: 10.1038/s41467-018-04899-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mosrin-Huaman C, Turnbough CL, Rahmouni AR. 2004. Translocation of Escherichia coli RNA polymerase against a protein roadblock in vivo highlights a passive sliding mechanism for transcript elongation. Mol Microbiol 51:1471–1481. doi: 10.1111/j.1365-2958.2003.03926.x. [DOI] [PubMed] [Google Scholar]
- 53.Keiler KC. 2015. Mechanisms of ribosome rescue in bacteria. Nat Rev Microbiol 13:285–297. doi: 10.1038/nrmicro3438. [DOI] [PubMed] [Google Scholar]
- 54.Keiler KC, Waller PR, Sauer RT. 1996. Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA. Science 271:990–993. doi: 10.1126/science.271.5251.990. [DOI] [PubMed] [Google Scholar]
- 55.Yamamoto Y, Sunohara T, Jojima K, Inada T, Aiba H. 2003. SsrA-mediated trans-translation plays a role in mRNA quality control by facilitating degradation of truncated mRNAs. RNA 9:408–418. doi: 10.1261/rna.2174803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meng J, Kanzaki G, Meas D, Lam CK, Crummer H, Tain J, Xu HH. 2012. A genome-wide inducible phenotypic screen identifies antisense RNA constructs silencing Escherichia coli essential genes. FEMS Microbiol Lett 329:45–53. doi: 10.1111/j.1574-6968.2012.02503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rousset F, Cui L, Siouve E, Becavin C, Depardieu F, Bikard D. 2018. Genome-wide CRISPR-dCas9 screens in E. coli identify essential genes and phage host factors. PLoS Genet 14:e1007749. doi: 10.1371/journal.pgen.1007749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao C, Shu X, Sun B. 2017. Construction of a gene knockdown system based on catalytically inactive (“dead”) Cas9 (dCas9) in Staphylococcus aureus. Appl Environ Microbiol 83:e00291-17. doi: 10.1128/AEM.00291-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dar D, Sorek R. 2018. Extensive reshaping of bacterial operons by programmed mRNA decay. PLoS Genet 14:e1007354. doi: 10.1371/journal.pgen.1007354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Evguenieva-Hackenberg E, Klug G. 2011. New aspects of RNA processing in prokaryotes. Curr Opin Microbiol 14:587–592. doi: 10.1016/j.mib.2011.07.025. [DOI] [PubMed] [Google Scholar]
- 61.Hao N, Shearwin KE, Dodd IB. 2017. Programmable DNA looping using engineered bivalent dCas9 complexes. Nat Commun 8:1628. doi: 10.1038/s41467-017-01873-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Farasat I, Salis HM. 2016. A biophysical model of CRISPR/Cas9 activity for rational design of genome editing and gene regulation. PLoS Comput Biol 12:e1004724. doi: 10.1371/journal.pcbi.1004724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anderson EM, Haupt A, Schiel JA, Chou E, Machado HB, Strezoska Ž, Lenger S, McClelland S, Birmingham A, Vermeulen A, van Brabant Smith A. 2015. Systematic analysis of CRISPR-Cas9 mismatch tolerance reveals low levels of off-target activity. J Biotechnol 211:56–65. doi: 10.1016/j.jbiotec.2015.06.427. [DOI] [PubMed] [Google Scholar]
- 64.Fu BXH, Hansen LL, Artiles KL, Nonet ML, Fire AZ. 2014. Landscape of target:guide homology effects on Cas9-mediated cleavage. Nucleic Acids Res 42:13778–13787. doi: 10.1093/nar/gku1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jost M, Santos DA, Saunders RA, Horlbeck MA, Hawkins JS, Scaria SM, Norman TM, Hussmann JA, Liem CR, Gross CA, Weissman JS. 13 January 2020. Titrating gene expression using libraries of systematically attenuated CRISPR guide RNAs. Nat Biotechnol doi: 10.1038/s41587-019-0387-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F. 2013. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hawkins JS, Silvis MR, Koo B-M, Peters JM, Jost M, Hearne CC, Weissman JS, Todor H, Gross CA. 2019. Modulated efficacy CRISPRi reveals evolutionary conservation of essential gene expression-fitness relationships in bacteria. bioRxiv doi: 10.1101/805333. [DOI] [PMC free article] [PubMed]
- 68.Jung C, Hawkins JA, Jones SK, Xiao Y, Rybarski JR, Dillard KE, Hussmann J, Saifuddin FA, Savran CA, Ellington AD, Ke A, Press WH, Finkelstein IJ. 2017. Massively parallel biophysical analysis of CRISPR-Cas complexes on next generation sequencing chips. Cell 170:35–47.e13. doi: 10.1016/j.cell.2017.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jones SK, Hawkins JA, Johnson NV, Jung C, Hu K, Rybarski JR, Chen JS, Doudna JA, Press WH, Finkelstein IJ. 2019. Massively parallel kinetic profiling of natural and engineered CRISPR nucleases. bioRxiv doi: 10.1101/696393. [DOI] [PMC free article] [PubMed]
- 70.Marshall R, Maxwell CS, Collins SP, Jacobsen T, Luo ML, Begemann MB, Gray BN, January E, Singer A, He Y, Beisel CL, Noireaux V. 2018. Rapid and scalable characterization of CRISPR technologies using an E. coli cell-free transcription-translation system. Mol Cell 69:146–157.e3. doi: 10.1016/j.molcel.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cress BF, Jones JA, Kim DC, Leitz QD, Englaender JA, Collins SM, Linhardt RJ, Koffas MAG. 2016. Rapid generation of CRISPR/dCas9-regulated, orthogonally repressible hybrid T7-lac promoters for modular, tuneable control of metabolic pathway fluxes in Escherichia coli. Nucleic Acids Res 44:4472–4485. doi: 10.1093/nar/gkw231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Boyle EA, Andreasson JOL, Chircus LM, Sternberg SH, Wu MJ, Guegler CK, Doudna JA, Greenleaf WJ. 2017. High-throughput biochemical profiling reveals sequence determinants of dCas9 off-target binding and unbinding. Proc Natl Acad Sci U S A 114:5461–5466. doi: 10.1073/pnas.1700557114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bisaria N, Jarmoskaite I, Herschlag D. 2017. Lessons from enzyme kinetics reveal specificity principles for RNA-guided nucleases in RNA interference and CRISPR-based genome editing. Cell Syst 4:21–29. doi: 10.1016/j.cels.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Klein M, Eslami-Mossallam B, Arroyo DG, Depken M. 2018. Hybridization kinetics explains CRISPR-Cas off-targeting rules. Cell Rep 22:1413–1423. doi: 10.1016/j.celrep.2018.01.045. [DOI] [PubMed] [Google Scholar]
- 75.Strohkendl I, Saifuddin FA, Rybarski JR, Finkelstein IJ, Russell R. 2018. Kinetic basis for DNA target specificity of CRISPR-Cas12a. Mol Cell 71:816–824.e3. doi: 10.1016/j.molcel.2018.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hu JH, Miller SM, Geurts MH, Tang W, Chen L, Sun N, Zeina CM, Gao X, Rees HA, Lin Z, Liu DR. 2018. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556:57–63. doi: 10.1038/nature26155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kazlauskiene M, Kostiuk G, Venclovas Č, Tamulaitis G, Siksnys V. 2017. A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science 357:605–609. doi: 10.1126/science.aao0100. [DOI] [PubMed] [Google Scholar]
- 78.Niewoehner O, Garcia-Doval C, Rostøl JT, Berk C, Schwede F, Bigler L, Hall J, Marraffini LA, Jinek M. 2017. Type III CRISPR-Cas systems produce cyclic oligoadenylate second messengers. Nature 548:543–548. doi: 10.1038/nature23467. [DOI] [PubMed] [Google Scholar]
- 79.Johnson K, Learn BA, Estrella MA, Bailey S. 2019. Target sequence requirements of a type III-B CRISPR-Cas immune system. J Biol Chem 294:10290–10299. doi: 10.1074/jbc.RA119.008728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Marraffini LA, Sontheimer EJ. 2010. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature 463:568–571. doi: 10.1038/nature08703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Elmore JR, Sheppard NF, Ramia N, Deighan T, Li H, Terns RM, Terns MP. 2016. Bipartite recognition of target RNAs activates DNA cleavage by the type III-B CRISPR-Cas system. Genes Dev 30:447–459. doi: 10.1101/gad.272153.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Belanto JJ, Verdine V, Cox DBT, Kellner MJ, Regev A, Lander ES, Voytas DF, Ting AY, Zhang F. 2017. RNA targeting with CRISPR-Cas13. Nature 550:280–284. doi: 10.1038/nature24049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rousseau BA, Hou Z, Gramelspacher MJ, Zhang Y. 2018. Programmable RNA cleavage and recognition by a natural CRISPR-Cas9 system from Neisseria meningitidis. Mol Cell 69:906–914.e4. doi: 10.1016/j.molcel.2018.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shibata M, Nishimasu H, Kodera N, Hirano S, Ando T, Uchihashi T, Nureki O. 2017. Real-space and real-time dynamics of CRISPR-Cas9 visualized by high-speed atomic force microscopy. Nat Commun 8:1430. doi: 10.1038/s41467-017-01466-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jiang F, Zhou K, Ma L, Gressel S, Doudna JA. 2015. Structural biology. A Cas9-guide RNA complex preorganized for target DNA recognition. Science 348:1477–1481. doi: 10.1126/science.aab1452. [DOI] [PubMed] [Google Scholar]
- 86.Jiang F, Doudna JA. 2017. CRISPR-Cas9 structures and mechanisms. Annu Rev Biophys 46:505–529. doi: 10.1146/annurev-biophys-062215-010822. [DOI] [PubMed] [Google Scholar]
- 87.Globyte V, Lee SH, Bae T, Kim J-S, Joo C. 2019. CRISPR/Cas9 searches for a protospacer adjacent motif by lateral diffusion. EMBO J 38:e99466. doi: 10.15252/embj.201899466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jeon Y, Choi YH, Jang Y, Yu J, Goo J, Lee G, Jeong YK, Lee SH, Kim I-S, Kim J-S, Jeong C, Lee S, Bae S. 2018. Direct observation of DNA target searching and cleavage by CRISPR-Cas12a. Nat Commun 9:2777. doi: 10.1038/s41467-018-05245-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jones DL, Leroy P, Unoson C, Fange D, Ćurić V, Lawson MJ, Elf J. 2017. Kinetics of dCas9 target search in Escherichia coli. Science 357:1420–1424. doi: 10.1126/science.aah7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li S, Jendresen CB, Grünberger A, Ronda C, Jensen SI, Noack S, Nielsen AT. 2016. Enhanced protein and biochemical production using CRISPRi-based growth switches. Metab Eng 38:274–284. doi: 10.1016/j.ymben.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 91.Li X-T, Jun Y, Erickstad MJ, Brown SD, Parks A, Court DL, Jun S. 2016. tCRISPRi: tunable and reversible, one-step control of gene expression. Sci Rep 6:39076. doi: 10.1038/srep39076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Si F, Li D, Cox SE, Sauls JT, Azizi O, Sou C, Schwartz AB, Erickstad MJ, Jun Y, Li X, Jun S. 2017. Invariance of initiation mass and predictability of cell size in Escherichia coli. Curr Biol 27:1278–1287. doi: 10.1016/j.cub.2017.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Koopal B, Kruis AJ, Claassens NJ, Nobrega FL, van der Oost J. 2019. Incorporation of a synthetic amino acid into dCas9 improves control of gene silencing. ACS Synth Biol 8:216–222. doi: 10.1021/acssynbio.8b00347. [DOI] [PubMed] [Google Scholar]
- 94.Lee YJ, Hoynes-O’Connor A, Leong MC, Moon TS. 2016. Programmable control of bacterial gene expression with the combined CRISPR and antisense RNA system. Nucleic Acids Res 44:2462–2473. doi: 10.1093/nar/gkw056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dong C, Fontana J, Patel A, Carothers JM, Zalatan JG. 2018. Synthetic CRISPR-Cas gene activators for transcriptional reprogramming in bacteria. Nat Commun 9:4318. doi: 10.1038/s41467-018-06909-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lu Z, Yang S, Yuan X, Shi Y, Ouyang L, Jiang S, Yi L, Zhang G. 2019. CRISPR-assisted multi-dimensional regulation for fine-tuning gene expression in Bacillus subtilis. Nucleic Acids Res 47:e40. doi: 10.1093/nar/gkz072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ho H-I, Fang J, Cheung J, Wang HH. 2020. Programmable and portable CRISPR-Cas transcriptional activation in bacteria. bioRxiv doi: 10.1101/2020.01.03.882431. [DOI] [PMC free article] [PubMed]
- 98.Liu Y, Wan X, Wang B. 2019. Engineered CRISPRa enables programmable eukaryote-like gene activation in bacteria. Nat Commun 10:3693. doi: 10.1038/s41467-019-11479-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Barrangou R, Marraffini LA. 2014. CRISPR-Cas systems: prokaryotes upgrade to adaptive immunity. Mol Cell 54:234–244. doi: 10.1016/j.molcel.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Reis AC, Halper SM, Vezeau GE, Cetnar DP, Hossain A, Clauer PR, Salis HM. 2019. Simultaneous repression of multiple bacterial genes using nonrepetitive extra-long sgRNA arrays. Nat Biotechnol 37:1294–1301. doi: 10.1038/s41587-019-0286-9. [DOI] [PubMed] [Google Scholar]
- 101.Zhao Y, Li L, Zheng G, Jiang W, Deng Z, Wang Z, Lu Y. 2018. CRISPR/dCas9-mediated multiplex gene repression in Streptomyces. Biotechnol J 13:e1800121. doi: 10.1002/biot.201800121. [DOI] [PubMed] [Google Scholar]
- 102.Cress BF, Toparlak ÖD, Guleria S, Lebovich M, Stieglitz JT, Englaender JA, Jones JA, Linhardt RJ, Koffas MAG. 2015. CRISPathBrick: modular combinatorial assembly of type II-A CRISPR arrays for dCas9-mediated multiplex transcriptional repression in E. coli. ACS Synth Biol 4:987–1000. doi: 10.1021/acssynbio.5b00012. [DOI] [PubMed] [Google Scholar]
- 103.Nayak DD, Metcalf WW. 2017. Cas9-mediated genome editing in the methanogenic archaeon Methanosarcina acetivorans. Proc Natl Acad Sci U S A 114:2976–2981. doi: 10.1073/pnas.1618596114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shan Q, Wang Y, Li J, Zhang Y, Chen K, Liang Z, Zhang K, Liu J, Xi JJ, Qiu J-L, Gao C. 2013. Targeted genome modification of crop plants using a CRISPR-Cas system. Nat Biotechnol 31:686–688. doi: 10.1038/nbt.2650. [DOI] [PubMed] [Google Scholar]
- 105.Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. 2013. RNA-programmed genome editing in human cells. Elife 2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jacobs JZ, Ciccaglione KM, Tournier V, Zaratiegui M. 2014. Implementation of the CRISPR-Cas9 system in fission yeast. Nat Commun 5:5344. doi: 10.1038/ncomms6344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ghorbal M, Gorman M, Macpherson CR, Martins RM, Scherf A, Lopez-Rubio J-J. 2014. Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat Biotechnol 32:819–821. doi: 10.1038/nbt.2925. [DOI] [PubMed] [Google Scholar]
- 108.Berlec A, Škrlec K, Kocjan J, Olenic M, Štrukelj B. 2018. Single plasmid systems for inducible dual protein expression and for CRISPR-Cas9/CRISPRi gene regulation in lactic acid bacterium Lactococcus lactis. Sci Rep 8:1009. doi: 10.1038/s41598-018-19402-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tak YE, Kleinstiver BP, Nuñez JK, Hsu JY, Horng JE, Gong J, Weissman JS, Joung JK. 2017. Inducible and multiplex gene regulation using CRISPR-Cpf1-based transcription factors. Nat Methods 14:1163–1166. doi: 10.1038/nmeth.4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sato’o Y, Hisatsune J, Yu L, Sakuma T, Yamamoto T, Sugai M. 2018. Tailor-made gene silencing of Staphylococcus aureus clinical isolates by CRISPR interference. PLoS One 13:e0185987. doi: 10.1371/journal.pone.0185987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Peters JM, Koo B-M, Patino R, Heussler GE, Hearne CC, Qu J, Inclan YF, Hawkins JS, Lu CHS, Silvis MR, Harden MM, Osadnik H, Peters JE, Engel JN, Dutton RJ, Grossman AD, Gross CA, Rosenberg OS. 2019. Enabling genetic analysis of diverse bacteria with mobile-CRISPRi. Nat Microbiol 4:244–250. doi: 10.1038/s41564-018-0327-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nielsen AA, Voigt CA. 2014. Multi‐input CRISPR/Cas genetic circuits that interface host regulatory networks. Mol Syst Biol 10:763. doi: 10.15252/msb.20145735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cho S, Choe D, Lee E, Kim SC, Palsson B, Cho B-K. 2018. High-level dCas9 expression induces abnormal cell morphology in Escherichia coli. ACS Synth Biol 7:1085–1094. doi: 10.1021/acssynbio.7b00462. [DOI] [PubMed] [Google Scholar]
- 114.Rock JM, Hopkins FF, Chavez A, Diallo M, Chase MR, Gerrick ER, Pritchard JR, Church GM, Rubin EJ, Sassetti CM, Schnappinger D, Fortune SM. 2017. Programmable transcriptional repression in mycobacteria using an orthogonal CRISPR interference platform. Nat Microbiol 2:16274. doi: 10.1038/nmicrobiol.2016.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wendt KE, Ungerer J, Cobb RE, Zhao H, Pakrasi HB. 2016. CRISPR/Cas9 mediated targeted mutagenesis of the fast growing cyanobacterium Synechococcus elongatus UTEX 2973. Microb Cell Fact 15:115. doi: 10.1186/s12934-016-0514-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ungerer J, Pakrasi HB. 2016. Cpf1 is a versatile tool for CRISPR genome editing across diverse species of cyanobacteria. Sci Rep 6:39681. doi: 10.1038/srep39681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang S, Voigt CA. 2018. Engineered dCas9 with reduced toxicity in bacteria: implications for genetic circuit design. Nucleic Acids Res 46:11115–11125. doi: 10.1093/nar/gky884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Koo B-M, Kritikos G, Farelli JD, Todor H, Tong K, Kimsey H, Wapinski I, Galardini M, Cabal A, Peters JM, Hachmann A-B, Rudner DZ, Allen KN, Typas A, Gross CA. 2017. Construction and analysis of two genome-scale deletion libraries for Bacillus subtilis. Cell Syst 4:291–305.e7. doi: 10.1016/j.cels.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gerdes SY, Scholle MD, Campbell JW, Balázsi G, Ravasz E, Daugherty MD, Somera AL, Kyrpides NC, Anderson I, Gelfand MS, Bhattacharya A, Kapatral V, D’Souza M, Baev MV, Grechkin Y, Mseeh F, Fonstein MY, Overbeek R, Barabási A-L, Oltvai ZN, Osterman AL. 2003. Experimental determination and system level analysis of essential genes in Escherichia coli MG1655. J Bacteriol 185:5673–5684. doi: 10.1128/jb.185.19.5673-5684.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wetmore KM, Price MN, Waters RJ, Lamson JS, He J, Hoover CA, Blow MJ, Bristow J, Butland G, Arkin AP, Deutschbauer A. 2015. Rapid quantification of mutant fitness in diverse bacteria by sequencing randomly bar-coded transposons. mBio 6:e00306-15. doi: 10.1128/mBio.00306-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Price MN, Wetmore KM, Waters RJ, Callaghan M, Ray J, Liu H, Kuehl JV, Melnyk RA, Lamson JS, Suh Y, Carlson HK, Esquivel Z, Sadeeshkumar H, Chakraborty R, Zane GM, Rubin BE, Wall JD, Visel A, Bristow J, Blow MJ, Arkin AP, Deutschbauer AM. 2018. Mutant phenotypes for thousands of bacterial genes of unknown function. Nature 557:503–509. doi: 10.1038/s41586-018-0124-0. [DOI] [PubMed] [Google Scholar]
- 123.Goodall ECA, Robinson A, Johnston IG, Jabbari S, Turner KA, Cunningham AF, Lund PA, Cole JA, Henderson IR. 2018. The essential genome of Escherichia coli K-12. mBio 9:e02096-17. doi: 10.1128/mBio.02096-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Langridge GC, Phan M-D, Turner DJ, Perkins TT, Parts L, Haase J, Charles I, Maskell DJ, Peters SE, Dougan G, Wain J, Parkhill J, Turner AK. 2009. Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Res 19:2308–2316. doi: 10.1101/gr.097097.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.van Opijnen T, Bodi KL, Camilli A. 2009. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat Methods 6:767–772. doi: 10.1038/nmeth.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Dion MF, Kapoor M, Sun Y, Wilson S, Ryan J, Vigouroux A, van Teeffelen S, Oldenbourg R, Garner EC. 2019. Bacillus subtilis cell diameter is determined by the opposing actions of two distinct cell wall synthetic systems. Nat Microbiol 4:1294–1305. doi: 10.1038/s41564-019-0439-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liu X, Gallay C, Kjos M, Domenech A, Slager J, van Kessel SP, Knoops K, Sorg RA, Zhang J-R, Veening J-W. 2017. High-throughput CRISPRi phenotyping identifies new essential genes in Streptococcus pneumoniae. Mol Syst Biol 13:931. doi: 10.15252/msb.20167449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Liu Y, Han J, Chen Z, Wu H, Dong H, Nie G. 2017. Engineering cell signaling using tunable CRISPR-Cpf1-based transcription factors. Nat Commun 8:2095. doi: 10.1038/s41467-017-02265-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lawson MJ, Camsund D, Larsson J, Baltekin Ö, Fange D, Elf J. 2017. In situ genotyping of a pooled strain library after characterizing complex phenotypes. Mol Syst Biol 13:947. doi: 10.15252/msb.20177951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Camsund D, Lawson MJ, Larsson J, Jones D, Zikrin S, Fange D, Elf J. 2020. Time-resolved imaging-based CRISPRi screening. Nat Methods 17:86–92. doi: 10.1038/s41592-019-0629-y. [DOI] [PubMed] [Google Scholar]
- 131.Ceroni F, Boo A, Furini S, Gorochowski TE, Borkowski O, Ladak YN, Awan AR, Gilbert C, Stan G-B, Ellis T. 2018. Burden-driven feedback control of gene expression. Nat Methods 15:387–393. doi: 10.1038/nmeth.4635. [DOI] [PubMed] [Google Scholar]
- 132.Mückl A, Schwarz-Schilling M, Fischer K, Simmel FC. 2018. Filamentation and restoration of normal growth in Escherichia coli using a combined CRISPRi sgRNA/antisense RNA approach. PLoS One 13:e0198058. doi: 10.1371/journal.pone.0198058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tian T, Kang JW, Kang A, Lee TS. 2019. Redirecting metabolic flux via combinatorial multiplex CRISPRi-mediated repression for isopentenol production in Escherichia coli. ACS Synth Biol 8:391–402. doi: 10.1021/acssynbio.8b00429. [DOI] [PubMed] [Google Scholar]
- 134.Wang Y, Zhang Z-T, Seo S-O, Lynn P, Lu T, Jin Y-S, Blaschek HP. 2016. Gene transcription repression in Clostridium beijerinckii using CRISPR-dCas9. Biotechnol Bioeng 113:2739–2743. doi: 10.1002/bit.26020. [DOI] [PubMed] [Google Scholar]
- 135.Park J, Shin H, Lee S-M, Um Y, Woo HM. 2018. RNA-guided single/double gene repressions in Corynebacterium glutamicum using an efficient CRISPR interference and its application to industrial strain. Microb Cell Fact 17:4. doi: 10.1186/s12934-017-0843-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Elhadi D, Lv L, Jiang X-R, Wu H, Chen G-Q. 2016. CRISPRi engineering E. coli for morphology diversification. Metab Eng 38:358–369. doi: 10.1016/j.ymben.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 137.Doron S, Melamed S, Ofir G, Leavitt A, Lopatina A, Keren M, Amitai G, Sorek R. 2018. Systematic discovery of antiphage defense systems in the microbial pangenome. Science 359:eaar4120. doi: 10.1126/science.aar4120. [DOI] [PMC free article] [PubMed] [Google Scholar]