

Abstract
Coronavirus RNA synthesis is performed by a multienzymatic replicase complex together with cellular factors. This process requires the specific recognition of RNA cis-acting signals located at the ends of the viral genome. To identify cellular proteins involved in coronavirus RNA synthesis, transmissible gastroenteritis coronavirus (TGEV) genome ends, harboring essential cis-acting signals for replication, were used as baits for RNA affinity protein purification. Ten proteins were preferentially pulled down with either the 5′ or 3′ ends of the genome and identified by proteomic analysis. Nine of them, including members of the heterogeneous ribonucleoprotein family of proteins (hnRNPs), the poly(A)-binding protein (PABP), the p100 transcriptional co-activator protein and two aminoacyl-tRNA synthetases, showed a preferential binding to the 3′ end of the genome, whereas only the polypyrimidine tract-binding protein (PTB) was preferentially pulled down with the 5′ end of the genome. The potential function of the 3′ end-interacting proteins in virus replication was studied by analyzing the effect of their silencing using a TGEV-derived replicon and the infectious virus. Gene silencing of PABP, hnRNP Q, and glutamyl-prolyl-tRNA synthetase (EPRS) caused a significant 2 to 3-fold reduction of viral RNA synthesis. Interestingly, the silencing of glyceraldehyde 3-phosphate dehydrogenase (GAPDH), initially used as a control gene, caused a 2 to 3-fold increase in viral RNA synthesis in both systems. These data suggest that PABP, hnRNP Q, and EPRS play a positive role in virus infection that could be mediated through their interaction with the viral 3′ end, and that GAPDH has a negative effect on viral infection.
Keywords: Coronavirus, Proteomics, RNA-binding proteins, siRNAs, RNA synthesis
Introduction
Transmissible gastroenteritis virus (TGEV) is a member of the Coronaviridae family, included in the Nidovirales order (Enjuanes et al., 2000, Enjuanes et al., 2008). Coronaviruses (CoVs) have been classified in three different groups based on antigenic and genetic criteria. The most extensively studied group members are the TGEV and the human coronavirus (HCoV) 229E from group 1, the mouse hepatitis virus (MHV) and the severe acute respiratory syndrome (SARS) virus (SARS-CoV) from group 2, and the infectious bronchitis virus (IBV) from group 3. CoV infections cause a variety of diseases relevant in animal and human health, being of special relevance the SARS in humans (Drosten et al., 2003, Perlman et al., 2000). After the SARS outbreak in 2002 the interest to understand the molecular basis of CoV replication increased and, as a result, in the last years more than 30 new CoVs have been identified with a broad host distribution (Vijaykrishna et al., 2007).
CoVs contain the largest known single-stranded positive-sense infectious RNA genomes, varying between 27 and 31 kb in length (Enjuanes et al., 2008, Masters, 2006). The CoV genome resembles the structure of most cellular mRNAs, containing a cap structure at the 5′ end, a poly(A) tail at the 3′ end, and 5′ and 3′ untranslated regions (UTRs). The replicase gene, that occupies the 5′ two thirds of the genome, includes two overlapping open reading frames (ORF 1a and ORF 1b) that are translated into two large co-amino-terminal polyproteins, pp1a and pp1ab, the second one expressed by a ribosomal frameshift mechanism (Brierley et al., 1989). Both polyproteins are autoproteolytically cleaved into up to 16 mature products (nsp1 to nsp16), which are believed to be part of the replication–transcription complex (Ziebuhr et al., 2000). CoV replicase gene is extremely complex and, besides the RNA-dependent RNA-polymerase and helicase activities, it encodes other enzymes less frequent or exclusive among RNA viruses (Masters, 2006, Snijder et al., 2003, Ziebuhr, 2005), such as an endoribonuclease, a 3′–5′ exoribonuclease, a 2′-O-ribose methyltransferase, a ribose ADP 1″ phosphatase, and a second RNA-dependent RNA-polymerase (Imbert et al., 2006). In addition to the replicase components, the viral nucleoprotein has been shown to play a major role in CoV RNA synthesis (Almazán et al., 2004, Schelle et al., 2005). The resulting virus-encoded replication–transcription complex presumably associates with host-cell proteins to complete the synthesis of viral RNA (Enjuanes et al., 2006, Masters, 2006).
To date, several cellular proteins have been identified on the basis of their ability to interact in vitro with cis-acting replication signals of CoVs from group 2 (MHV and bovine CoV), including the polypyrimidine tract-binding protein (PTB) (Li et al., 1999), heterogeneous nuclear ribonucleoproteins (hnRNPs) A1 and Q (Choi et al., 2004, Li et al., 1997), mitochondrial aconitase (Nanda and Leibowitz, 2001), and poly(A)-binding protein (PABP) (Spagnolo and Hogue, 2000). Of these proteins, a functional effect on CoV RNA synthesis has been described for the hnRNP A1 (Shen and Masters, 2001, Shi et al., 2000), hnRNP Q (Choi et al., 2004) and PTB (Choi et al., 2002, Li et al., 1999). However, no interaction or functional study with cellular proteins has been reported for group 1 CoVs.
In this study, we identified a set of 10 host proteins preferentially interacting with either the 5′ or 3′ ends of the TGEV genome by RNA affinity chromatography and mass spectrometry analysis. Among these, the PTB was found to preferentially interact with the 5′ end of the TGEV genome while a subset of 9 proteins, including several hnRNPs (A1, A0, A2B1, Q, and U), the glutamyl-prolyl-tRNA synthetase (EPRS), the arginyl-tRNA synthetase, the PABP, and the p100 transcriptional co-activator, showed a preferential binding to the 3′ end. The relevance of the 3′ end-interacting proteins on TGEV infection was analyzed using RNA silencing approaches. A significant and highly reproducible reduction of 2 to 3-fold in RNA synthesis and virus titer was evidenced after silencing the expression of PABP, hnRNP Q and EPRS proteins, suggesting that these factors play a positive role in TGEV infection. Interestingly, silencing of the control gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) caused a 2 to 3-fold increase in TGEV RNA synthesis and virus titer, suggesting that this protein might have a role counteracting TGEV infection.
Results
Host proteins interacting with the 5′ and 3′ ends of TGEV genome
To identify host-cell proteins potentially involved in TGEV RNA synthesis, a screening based on the binding to 5′ and 3′ TGEV RNA genome ends was used. To this end, two synthetic RNAs containing the first 504 nt of the 5′ end of the TGEV genome and the last 493 nt of the 3′ end plus a poly(A) tail were biotin labeled during the in vitro transcription and used as baits for an RNA affinity protein isolation (Fig. 1 ). The length of the transcripts was chosen based on the sequences required for the amplification of TGEV-derived defective RNAs (Escors et al., 2003, Izeta et al., 1999; and unpublished results) and the biotin labeling was random and spaced out in order to minimize interference of the biotin groups with potential RNA structures.
Fig. 1.

Genetic structure of the TGEV genome and derived transcription templates. The bar on top represents the TGEV virus genome in which the different genes are illustrated as boxes. The bars below the genome represent the transcription templates for the generation of 5′ and 3′ RNA ends used in this study. The sequence relationship between the genome and the transcription templates is indicated by shadowed areas. The G nucleotides out of the boxes indicate additional non-viral sequences required for the in vitro transcription. The starting sites of transcription are indicated as + 1. Numbers below the bars indicate genome positions. TGEVg, TGEV genome; L, leader; ORF 1a and ORF 1b, replicase genes; S, spike gene; 3a and 3b, non-structural genes 3a and 3b; E, envelope protein gene; M, membrane protein gene; N, nucleoprotein gene; 7, non-structural gene 7; UTR, untranslated region; An, poly(A); SP6p and T7p, SP6 and T7 promoters, respectively.
The biotin labeled RNA baits were immobilized on a Streptavidin Sepharose resin and incubated with cell extracts from infected porcine ST and rodent BHK-pAPN (expressing TGEV receptor) cells that are highly susceptible to TGEV infection. In order to facilitate protein identification in the databases, extracts from the human hepatoma-derived cell line Huh-7, which is susceptible to the TGEV PUR46-C11 strain infection, were also used. RNA–protein complexes were isolated by centrifugation, resolved by electrophoresis in denaturing precasted gels and identified by mass spectrometry (Fig. 2A). From the pulled down proteins, a low proportion showed a preferential binding for one of the genome ends, that was reproduced for most of them in the three cell lines (Fig. 2B). Three independent RNA pull down assays were performed and all the proteins showing a preferential binding for one of the TGEV genome ends were excised from the gels and identified by mass spectrometry. A set of 10 proteins was reproducibly identified in at least two out of three experiments with significant MASCOT scores (p > 0.05). The percentage of protein identifications varied depending on the species of origin and it was 100% for human, 70% for rodent and 60% for porcine proteins (Table 1 ). That was probably a consequence of sequence variations with respect to the public database sequences for these species.
Fig. 2.

RNA affinity isolation of proteins preferentially interacting with the ends of the TGEV genome. (A) Scheme of the RNA affinity chromatography. (B) Proteins (600 μg) from infected Huh-7, BHK-pAPN, and ST cell extracts were pulled down, separated by SDS-PAGE and stained with Coomassie blue. All the bands showing a preferential interaction for one of the TGEV genome ends were excised (in both lanes and for the three cell lines) and analyzed by mass spectrometry. Differentially isolated proteins are listed and the band of origin indicated by arrows for the Huh-7 cells. The equivalent differentially isolated proteins are represented with arrowheads of the same pattern for BHK-pAPN and ST cells. The hnRNPs A1 and A2B1, which are not highlighted in the presented gel from infected ST cell extracts, were clearly identified in other experiments (Table 1). Molecular size markers are shown in kilodaltons.
Table 1.
Proteins differentially isolated with the ends of the TGEV genome.
| RNA bait | Accesion no. | Protein namea | Functionb | Mr | Masses matched/searched | Score/threshold/databasec | Speciesd | Ne |
|---|---|---|---|---|---|---|---|---|
| 3′TGEV | gi|4504445 | hnRNP A1 | RNA transport, processing, and splicing | 34289 | 11/32 | 107/70/m | H, R, P | 3 |
| 3′TGEV | gi|4504447 | hnRNP A2B1 | RNA transport and splicing | 36041 | 16/32 | 165/65/Hs | H, R, P | 3⁎ |
| 3′TGEV | gi|5803036 | hnRNP A0 | RNA splicing | 30993 | 5/18 | 76/65/Hs | H, R | 3 |
| 3′TGEV | gi|3202000 | hnRNP U | RNA processing and splicing | 91164 | 19/35 | 136/78/nr | H, R, P | 3⁎ |
| 3′TGEV | gi|44888326 | hnRNP Q (SYNCRIP) | RNA processing and splicing | 69818 | 27/66 | 321/76/nr | H, R, P | 3⁎ |
| 3′TGEV | gi|693937 | Polyadenylate binding protein (PABP) | RNA splicing and stabilization | 70671 | 9/13 | 84/69/m | H | 3⁎ |
| 3′TGEV | gi|5803036 | Glutamyl-prolyl tRNA synthetase (EPRS) | Translation | 172080 | 25/28 | 269/78/nr | H, R, P | 3⁎ |
| 3′TGEV | gi|30583627 | Arginyl-tRNA synthetase | Translation | 76129 | 17/84 | 111/76/nr | H, P | 2⁎ |
| 3′TGEV | gi|60415925 | staphylococcal nuclease domain containing 1; p100 co-activator | Transcription and RNA interference | 102709 | 29/40 | 363/76/nr | H, R | 3 |
| 5′TGEV | gi|131529 | Polypyrimidine tract-binding protein 1 (PTB) | RNA splicing | 56671 | 1/1 | 82/50/nr | H | 2 |
When alternative names are given, the one used in this study is shown in boldface.
Biological process according to Gene Ontology.
Mascot scores are given in boldface. Mascot threshold scores indicate the limit score from which the identification was significant (p < 0.05). Searches were performed against the NCBI database without restrictions (nr). In the indicated cases taxonomy was restricted to humans (Hs) or mammals (m).
Different species in which the protein was identified in parallel experiments, coming from human Huh-7 (H), rodent BHK-pAPN (R) or porcine ST (P) cell extracts.
Number of times that the protein was identified showing a clear binding preference for the indicated RNA bait. Asterisks indicate that the identification was confirmed by MS/MS.
Among the identified proteins, PTB was the only one showing a preferential binding to the 5′ end of the genome, whereas nine proteins were found to bind preferentially to the 3′ end (Fig. 2B and Table 1). These proteins include several hnRNPs (A1, A0, A2B1, Q, and U) involved in RNA processing and metabolism, the translation factors EPRS, arginyl-tRNA synthetase and PABP, and the p100 transcriptional co-activator.
Identification by Western-blot of host proteins differentially interacting with TGEV genome ends
To further confirm the preferential binding to one of the viral genome ends observed in the pull down experiments, a Western-blot analysis of the pulled down proteins from infected Huh-7 cell extracts was performed using specific antibodies. A strong binding preference for the 3′ end of the genome was confirmed for the hnRNPs U, A1, A2B1, and A0 proteins (Fig. 3A) while a moderate preference was observed for the EPRS, PABP, p100 and hnRNP Q proteins (Fig. 3B). In addition, a preferential interaction of the PTB with the 5′ end was also confirmed (Fig. 3C). In contrast, the viral nucleoprotein that was used as a loading control, was pulled down with a similar efficiency with both genome ends (Fig. 3D). Similar results were obtained with cell extracts from ST or BHK-pAPN infected cells (data not shown).
Fig. 3.
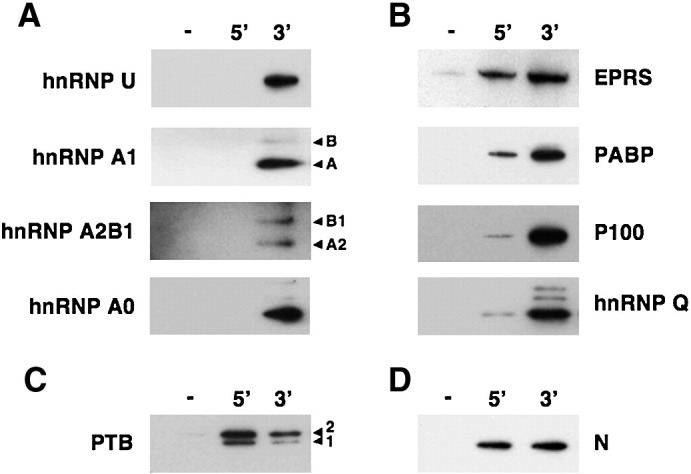
Western-blot analysis of the pulled down proteins. Proteins from infected Huh-7 cell extracts were pulled down with the TGEV 5′ and 3′ genome ends, separated in SDS-PAGE gels and analyzed by immunoblotting with specific antibodies. (A) Cellular proteins showing a strong preference for the 3′ end of the TGEV genome. (B) Cellular proteins showing a binding preference for the 3′ end of the genome. (C). Preferential binding of the PTB to the 5′ end of the genome. (D) Binding of the viral nucleoprotein to the 5′ and 3′ ends of the TGEV genome, as a loading control of the pull down output. In the case of hnRNPA1, hnRNPA2B1 and PTB, the different isoforms detected by the corresponding antibodies are indicated with arrowheads.
Effect of silencing the expression of 3′ end-interacting proteins on TGEV replicon activity
One of the initial steps of CoV RNA synthesis involves the specific recognition of the viral 3′ end to synthesize the negative-strand RNA. Therefore, proteins showing a preferential binding to the 3′ end were selected for further functional analysis. To evaluate the impact of these host proteins on TGEV RNA synthesis, their expression in 293T cells was knocked down with siRNAs and the effect on the TGEV replicon activity was analyzed by evaluating the production of subgenomic RNA 7 by real-time reverse transcription-PCR (Q-RT-PCR), in comparison with mRNA 7 levels in cells transfected with a commercially validated negative control siRNA (Ambion, Table 2 ).
Table 2.
siRNAs used for gene silencing analysis.
| siRNA IDa | Gene symbol | Full gene target name | Ref. no.b | siRNA sequence 5′ → 3′c |
|---|---|---|---|---|
| AM4623 | Cy3-GAPDH | Glyceraldehyde-3-phosphate dehydrogenase | Commercial positive control, sequence not available | |
| #4390843 | NEG Control #1 | Commercial negative control, sequence not available | ||
| #4390849 | GAPDH | Glyceraldehyde-3-phosphate dehydrogenase | Commercial positive control, sequence not available | |
| s25665 | PABP | Poly(A)-binding protein, cytoplasmic 1 | #1 | CCUAAAUGAUCGCAAAGUAtt |
| UACUUUGCGAUCAUUUAGGag | ||||
| s25664 | PABP | Poly(A)-binding protein, cytoplasmic 1 | #2 | AUCCAUUGAUAAUAAAGCAtt |
| UGCUUUAUUAUCAAUGGAUtt | ||||
| s20563 | hnRNP Q (SYNCRIP) | Synaptotagmin binding, cytoplasmic RNA interacting protein | #1 | GAACGAGUGAAGAAGUUAAtt |
| UUAACUUCUUCACUCGUUCca | ||||
| s20564 | hnRNP Q (SYNCRIP) | Synaptotagmin binding, cytoplasmic RNA interacting protein | #2 | CAAUAGAGGUUAUGCGUUUtt |
| AAACGCAUAACCUCUAUUGag | ||||
| s4767 | EPRS | Glutamyl-prolyl-tRNA synthetase | #1 | CGAUGUCAGCAUUUCCGUUtt |
| AACGGAAAUGCUGACAUCGtc | ||||
| s4768 | EPRS | Glutamyl-prolyl-tRNA synthetase | #2 | CGUUAGAGCUGAUUUACGAtt |
| UCGUAAAUCAGCUCUAACGcg |
ID provided by the manufacturer (Ambion).
Arbitrary reference numbers used in this study.
Sense and antisense sequences are given in upper and lower rows, respectively. All the siRNAs used are synthetic duplexes of 21 nt with 2 nt-protruding 3′ ends (in lower case).
To perform the silencing assays, human 293T cells were selected because they are efficiently transfected, support the replication of the TGEV replicon (Almazán et al., 2004), and because of the availability of pre-designed siRNA sequences and gene expression assays to monitor the silencing of human genes. Transfection efficiency of siRNAs and silencing were initially optimized using a Cy3-labeled validated-siRNA against the GAPDH gene, reaching transfection efficiencies close to 100% and a reduction of 80% at the mRNA level (data not shown). Synthetic siRNA duplexes against the GAPDH (silencing positive control) and the candidate genes were transfected twice into 293T cells to allow sustained mRNA silencing for at least 70 h in order to achieve detectable silencing at the protein level (Fig. 4A). Chemically modified siRNAs (Silencer Select siRNA, Ambion) were used in order to reduce the dose of transfected siRNAs and the potential off-target effects (Elmen et al., 2005). In addition, two different siRNAs per target were tested separately. Six hours after the second siRNA transfection, the TGEV replicon was transfected and total RNA and protein were isolated at 36 and 48 h posttransfection (90 and 102 h after the first siRNA transfection, respectively) to evaluate silencing of the target gene and the replicon activity (Fig. 4A). Efficiencies for the three consecutive transfections, cell viability, and silencing window during 3 days prior to the phenotypic assay were optimized by studying both mRNA and protein levels.
Fig. 4.

TGEV replicon activity in 293T cells after silencing the expression of TGEV 3′ end-interacting proteins. (A) Experimental design. The bar on top represents the TGEV-derived replicon (REP 2) that contains the 5′ and 3′ UTRs, the replicase (ORF 1a and ORF 1b), the nucleoprotein (N), and the 7 genes (Almazán et al., 2004). The time-line of the silencing and phenotypic assay is represented below. The time points refer to hours after the first transfection (hpt) of the siRNAs. 293T cells were transfected (T1) with either a validated negative control siRNA, an siRNA against the GAPDH gene (silencing positive control), or the siRNAs against the target genes (Table 2), seeded over poly-lysine-treated 96-well plates, and re-transfected with the same siRNAs (T2) before the transfection with the TGEV replicon (T3). The time points at which the RNA and proteins were extracted to evaluate the silencing and replicon activity are also indicated. (B) Silencing of cellular genes at the mRNA level. The percentage of mRNA remaining after the silencing of target genes PABP, hnRNP Q and EPRS or the control gene GAPDH was quantified by Q-RT-PCR, in comparison to reference levels from cells transfected with a validated negative control siRNA. Two different siRNA sequences (#1 and #2) were transfected separately to silence each target gene. Time points as in panel A. (C) Silencing of cellular genes at the protein level. Total cellular protein was extracted at 102 h after the first transfection of the siRNAs and the silencing was evaluated by immunoblotting. The β-actin was used as loading control. NEG, 293T cells transfected with a validated negative control siRNA (Table 2). (D) Replicon activity in silenced 293T cells. Total RNA was extracted at 36 and 48 h after the transfection of the TGEV replicon (90 and 102 h after the first transfection of the siRNAs, respectively) and the accumulation of viral mRNA 7 was quantified by Q-RT-PCR. Levels of mRNA 7 are represented as the fold-change in comparison to reference levels from cells transfected with a negative control siRNA. Only the time point (36 or 48 h after transfection of the replicon) in which the effect on the replicon activity was the highest is shown. The experiment was performed three times and the data represent the average of triplicates. Standard deviation is indicated as error bars.
For further studies PABP, hnRNP Q, and EPRS proteins were chosen because they showed good silencing efficiencies at both mRNA and protein levels at times posttransfection in which good cell viability (> 95%) was observed (data not shown). The silencing window of PABP, hnRNP Q, and EPRS at the mRNA level reached a maximum at 48 h after the first transfection (data not shown) that was prolonged at least until 102 h posttransfection (Fig. 4B). A reduction of 80% at the RNA level was detected for the positive control gene GAPDH. In the case of the target genes, the reduction in mRNA levels was dependent on siRNA sequence and it was within 70–95% for at least one siRNA of PABP and EPRS genes, and around 60% for both siRNAs of hnRNP Q gene (Fig. 4B). Silencing at the protein level was evaluated at 102 h after the first transfection of the siRNAs. A reduction of about 50% for hnRNP Q and between 65 and 85% for GAPDH, PABP and EPRS was observed (Fig. 4C).
Viral mRNA 7 expression was quantified as a measure of replicon activity at 36 and 48 h after the replicon transfection (90 and 102 h after the first siRNA transfection, respectively). A reproducible and significant reduction in replicon activity of 2 to 3-fold for PABP, hnRNP Q, and EPRS gene silencing was observed in comparison to reference levels from cells transfected with a validated negative control siRNA (Fig. 4D), suggesting that these factors have an important positive effect on replicon RNA synthesis. Reduction of replicon activity was transient, showing a good correlation with the lowest protein levels after silencing and a dose-depending effect in relation to the silencing potency of different siRNA sequences (Fig. 4D). Interestingly, a significant increment of about 3-fold in the replicon activity was observed after GAPDH silencing (Fig. 4D), indicating that GAPDH has a negative effect on replicon RNA synthesis. Although these effects were moderate, they were highly reproducible and statistically significant, and of similar magnitude than those reported in other viral systems (Hara et al., 2009, Pettit Kneller et al., 2009).
Effect of GAPDH, PABP, hnRNP Q and EPRS gene silencing on TGEV replication
To analyze the impact of silencing the expression of selected genes on TGEV RNA synthesis and infectious virus production, the human cell line Huh-7, which is susceptible to TGEV PUR46-C11 strain infection, was used. As described for the replicon system, the transfection efficiency of siRNAs and silencing were optimized in Huh-7 cells using a Cy3-labeled validated-siRNA against the GAPDH gene (data not shown). In addition, only the siRNAs providing higher silencing efficiencies for each target gene were chosen to perform the phenotypic assays (Table 2 and Fig. 4). Synthetic siRNAs were transfected into Huh-7 cells by reverse transfection and at 48 h posttransfection the cells were infected with the TGEV PUR46-C11 strain at a multiplicity of infection (MOI) of 5. A single siRNA transfection step was sufficient to achieve sustained mRNA and detectable protein silencing at the times of the phenotypic analysis. Total RNA was isolated at 36 and 72 h postinfection (84 and 120 h after transfection of the siRNAs, respectively) to evaluate silencing at the mRNA level and viral RNA synthesis by Q-RT-PCR with specific Taqman gene expression assays (Fig. 5 A). Silencing efficiency at mRNA level was within the 85–95% range for GAPDH, PABP, hnRNP Q, and EPRS genes (Fig. 5B). Silencing at the protein level was evaluated at 120 h after siRNAs transfection, and a reduction higher than 65% was observed for all tested proteins (Fig. 5C).
Fig. 5.

Effect of silencing the expression of 3′ end-interacting proteins on TGEV infection. (A) Time-line of the silencing and phenotypic assay in Huh-7 cells. The time points refer to hours after siRNAs transfection (hpt). The cells were transfected with siRNAs against the target genes (T) (Table 2) and infected with the TGEV PUR46-C11 strain at 48 h posttransfection. Total RNA and protein were recovered at the indicated times to analyze the silencing and the viral phenotype. (B) Analysis of candidate gene silencing at the mRNA level in Huh-7 cells. The percentage of mRNA remaining after the silencing of control gene GAPDH and the target genes PABP, hnRNP Q and EPRS was quantified by Q-RT-PCR, in comparison to reference levels from cells transfected with a validated negative control siRNA. (C) Silencing of candidate genes at the protein level. The silencing was evaluated at 120 h posttransfection by immunoblotting. The β-actin was used as loading control. NEG, Huh-7 cells transfected with a validated negative control siRNA (Table 2). (D) Virus RNA synthesis and virus production in silenced Huh-7 cells. Total RNA was extracted at 36 and 72 h postinfection (84 and 120 h posttransfection, respectively) and the accumulation of viral mRNA7 was quantified by Q-RT-PCR. Levels of mRNA 7 are represented as the fold-change in comparison to reference levels from cells transfected with a validated negative control siRNA. Virus production was quantified by titration of the supernatants on ST cells. The figure shows the effect observed on RNA synthesis and virus production at 72 h postinfection when the phenotype was more evident. The experiment was performed three times and the data represent the average of triplicates. Standard deviation is indicated as error bars.
Viral RNA synthesis was quantified at 36 and 72 h postinfection and a significant effect was observed at 72 h postinfection. This time point corresponds with maximum TGEV PUR46-C11 RNA synthesis in Huh-7 cells. Similar to the replicon system, a reproducible and significant reduction of 2-fold in the viral RNA synthesis was evidenced after silencing of PABP, hnRNP Q and EPRS genes, in comparison with the reference levels from cells transfected with a validated negative control siRNA (Fig. 5D). In accordance with the observation made with the TGEV replicon, a significant increment on viral RNA synthesis of about 2-fold was observed upon GAPDH silencing (Fig. 5D). Virus production was quantified at 72 h postinfection by titrating the supernatants from silenced Huh-7 cells on ST cells. In agreement with the effects observed in viral RNA synthesis, a reproducible reduction in virus titers to 36–50% was observed in cells silenced for PABP, hnRNP Q and EPRS genes, while a 340% increase was detected in cells silenced for the GAPDH gene (Fig. 5D). These results indicate that the silencing of PABP, hnRNP Q, EPRS, and GAPDH genes have a significant impact both on virus RNA synthesis and infectious virus production.
Discussion
RNA viruses have often evolved to subvert host-cell factors in favor of their own gene expression (Ahlquist et al., 2003, Lai, 1998). During the course of CoV infection, the interactions of specific RNA genome motifs with viral or cellular components probably regulate the initial replicase translation, the switch from translation to RNA synthesis and packaging of progeny genomes. In previous studies in group 2 CoVs, several cellular proteins interacting with different RNA domains at the 5′ or 3′ UTRs have been identified by UV-crosslinking and RNA affinity protein isolation approaches (Shi and Lai, 2005, Spagnolo and Hogue, 2000, Yu and Leibowitz, 1995). Nevertheless, limited studies have been performed with group 1 CoVs. In this study a set of 10 cellular proteins preferentially binding to the first 500 nt from either the 5′ or 3′ ends of the TGEV genome was identified and the relevance in virus infection of GAPDH and the 3′ end-interacting proteins PABP, hnRNP Q and EPRS, was determined. Considering that the 3′ end is the initiation site for replication that starts with the synthesis of the negative strand, the proteins interacting with the 3′-cis-acting replication signals could play a key role at the initial stage of RNA replication. In addition to the critical role of both 5′ and 3′ genome ends in CoV replication, the genome ends most likely also contain signals that regulate the translation, transcription and RNA stability (Masters, 2006, Ziebuhr, 2005). Thus, the proteins identified in this study could be involved in any of these processes.
A high proportion of the identified proteins belong to the cellular splicing machinery, including the PTB (hnRNP I), that was preferentially purified with the 5′ TGEV genome end, and five other hnRNPs (A1, A2B1, A0, U and Q) preferentially associated with the 3′ end. Although these proteins are predominantly nuclear factors, most of them can shuttle between the nucleus and the cytoplasm associated to mRNA (Caudy et al., 2003, Ghetti et al., 1992, Mizutani et al., 2000, Myer and Steitz, 1995, Pinol-Roma and Dreyfuss, 1993, Weighardt et al., 1995). On the other hand, nuclear proteins could be translocated to the cytoplasm during infection affecting viral replication, as described for other cytoplasmic RNA viruses (Lai, 1998). An example of this phenomenon was previously reported for the hnRNP A1 that relocates during infection from the nucleus to the sites of MHV RNA synthesis in the cytoplasm (Li et al., 1997). CoV transcription involves the fusion of non-continuous sequences by a discontinuous RNA synthesis mechanism different from conventional RNA splicing but with the same end result. The participation of cellular splicing factors could be critical for assisting this process during CoV transcription by bringing distant RNA sequences in close proximity. Among these splicing factors, PTB, hnRNP A1, and hnRNP Q have been previously shown to interact with MHV genome ends and to be involved in viral RNA synthesis (Choi et al., 2002, Choi et al., 2004, Li et al., 1999, Shen and Masters, 2001, Shi et al., 2000, Shi et al., 2003). In this study, we have found two additional splicing factors, hnRNP A2B1 and hnRNP A0, specifically associated to the 3′ end of TGEV genome. In addition, a preferential co-purification of the hnRNP Q with the 3′ end of the TGEV genome was observed, whereas the studies on MHV were limited to interaction with the 5′ end UTR (Choi et al., 2004). Furthermore, hnRNP Q silencing experiments showed a significant reduction in TGEV RNA synthesis and virus production in agreement with the effects observed after hnRNP Q silencing in MHV infected cells that produced a delay in viral RNA synthesis and syncytium formation (Choi et al., 2004).
In this report, we have described the specific co-purification of p100 with the 3′ end of the TGEV genome. An interaction of the p100 with the equine arteritis virus nsp 1, which is essential for mRNA synthesis, has been described (Tijms and Snijder, 2003), but no functional studies were reported on the role of p100 on arterivirus transcription.
PABP was specifically associated with the 3′ end of the TGEV genome and its silencing caused a significant reduction in viral RNA synthesis that correlated with a detectable reduction in virus titers. Binding of PABP to bovine CoV poly(A) tail had been previously reported (Spagnolo and Hogue, 2000). Nevertheless, our studies report the first direct evidence of the effect of this protein in CoV RNA synthesis. CoV genomes mimic cellular mRNAs and are presumed to translate their genomes in a cap and poly(A)-dependent manner. The binding of cap-recognizing factors to the 5′ end and the interaction of the PABP with the poly(A) tail, could circularize the viral genome for translation. This mechanism would favor the recycling of ribosomes, the selection of complete molecules as templates and the controlled regulation of the switch between translation and RNA synthesis that occurs in opposite directions.
A novel finding of this study is the preferential association of EPRS protein to the 3′ end of the TGEV genome and the reduction in TGEV RNA synthesis and virus titers after silencing the expression of this protein. Aminoacyl-tRNA synthetases are ancient enzymes that catalyze the amino acid addition to cognate tRNAs (Ibba and Soll, 2000). In vertebrates, these enzymes have acquired features that are absent in ancestral forms including additional functions unrelated to aminoacylation (Jia et al., 2008, Sampath et al., 2004). Aminoacyl-tRNA synthetases play key roles in different viral systems, such as the packaging of primer tRNAs in retroviruses (Cen et al., 2002) or catalyzing the aminoacylation of tRNA-like domains at the 3′ end of some plant RNA viruses to control plant virus replication (Dreher, 2008, Kohl and Hall, 1974). With our data, it is possible to speculate that the binding of EPRS to a specific RNA motif at the 3′ termini of the TGEV genome could be a strategy to efficiently compete with cellular mRNAs for the translation machinery by recruiting a key enzyme and providing the substrate for protein synthesis.
Silencing of GAPDH led to a positive effect on TGEV RNA synthesis and virus production. Furthermore, an interaction of GAPDH with at least the 5′ end of the TGEV genome has been identified (data not shown), suggesting the possibility that the effect observed on RNA synthesis and virus production could be mediated by this interaction. GAPDH is a glycolytic enzyme that also has RNA-binding properties and plays different roles by interacting with the RNA genome of different viruses. GAPDH has been involved in suppression of cap-independent translation in picornaviruses by destabilization of the IRES structure (Yi et al., 2000), and is a member of the replicase complex of tombusviruses, where it may regulate viral asymmetric RNA synthesis (Wang and Nagy, 2008). Interestingly, a reduction in GAPDH gene expression has been found in SARS-CoV infected cells and in cells transfected with nsp1 (Kamitani et al., 2006, Wathelet et al., 2007). In our study the positive effect on TGEV RNA synthesis and virus production after GAPDH silencing is in agreement with a possible role of GAPDH in counteracting viral infection.
The effects of silencing the cellular genes described in this work on TGEV replicon activity were reproducible and statistically significant with two different siRNA sequences per gene and were proportional to the silencing potency of each siRNA sequence. The phenotypes observed in the TGEV replicon system were also corroborated with the infectious virus, confirming that the effects observed were reproducible, irrespective of the different experimental systems and cell types. Furthermore, although the differences in viral RNA synthesis and titers observed after the silencing of the host candidate genes could be considered moderate, similar quantitative results have been reported in other viral systems (Hara et al., 2009, Pettit Kneller et al., 2009). Overall, our data suggest an effect of GAPDH, PABP, hnRNP Q, and EPRS on TGEV RNA synthesis and virus production.
Materials and methods
Cells and viruses
Baby hamster kidney cells stably transformed with the porcine aminopeptidase N gene (BHK-pAPN) to make them susceptible to TGEV (Delmas et al., 1993) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% fetal bovine serum (FBS) (Biowhittaker, Berviers, Belgium) and geneticin (1.5 mg/ml) as a selection agent. Swine testis (ST) cells (McClurkin and Norman, 1966) were grown in DMEM supplemented with 10% FBS. Human liver derived Huh-7 cells were kindly provided by R. Bartenschlager (University of Heidelberg, Germany) and were grown in DMEM supplemented with 10% of heat-inactivated FBS. Human 293T cells were grown in DMEM supplemented with 5% FBS. TGEV PUR46-MAD strain (Sánchez et al., 1990) was used to infect BHK-pAPN and ST cells and TGEV PUR46-C11 strain (Sánchez et al., 1999) to infect Huh-7 cells. Virus titration was performed on ST cell monolayers as previously described (Jiménez et al., 1986).
DNA constructs
To generate a DNA template for the in vitro transcription of a precise TGEV 5′ end, the plasmid pM33L-637G (Galán et al., 2005) that contains the M33L minigenome was used as template to amplify the first 504 nt of the TGEV genome. The PCR was performed with the oligonucleotides XmaI-SP6p-G-5′TGEV-VS that includes the SP6 promoter and 5′TGEV-504-RS (Table 3 ). The resulting amplicon was purified with the QIAquick PCR Purification kit (QIAGEN), precipitated with 0.1 vol of sodium acetate 3M pH 5.2 and 2.5 vol of cold 100% ethanol (sodium acetate-ethanol precipitation), resuspended in Tris–HCl 10 mM pH 7.5, and then used as template for the in vitro transcription reactions. The last 493 nt at the 3′ end of TGEV genome were amplified from the plasmid pM33L-637G using the oligonucleotides XmaI-T7p-GGG-ORF7-VS (Table 3) that introduces the T7 promoter preceding the viral sequence and HindIII-poly(A)-RS (Table 3) that includes a 25 nt poly(A) tail. The PCR amplicon was digested with XmaI and HindIII and cloned into the same restriction sites of the plasmid pM33L-637G to generate the plasmid pM33L-3′-template. The plasmid pM33L-3′-template was linearized with HindIII, deproteinized by phenol chloroform extraction, precipitated by sodium acetate-ethanol precipitation, and resuspended in the reaction buffer of the Mung Bean nuclease (New England Biolabs). In order to generate a precise TGEV genome 3′ end, the linearized plasmid pM33L-3′-template was then treated with Mung Bean nuclease to degrade the three-nucleotide single-strand extension generated after the HindIII digestion, following the manufacturer's conditions. The template was deproteinized by phenol chloroform extraction, precipitated by sodium acetate-ethanol precipitation, resuspended in Tris–HCl 10 mM pH 7.5, and then used for the in vitro transcription reactions. In all cases the PCR reactions were performed with the Platinum Pfx DNA polymerase (Invitrogen) following the manufacturer's recommended conditions. All cloning steps were checked by sequencing the PCR-amplified fragments and cloning junctions.
Table 3.
Oligonucleotides used for PCR amplifications and Q-RT-PCR analysis.
| Oligonucleotidea | Methodb | Oligonucleotide sequence 5′ → 3′c | Amplicon |
|---|---|---|---|
| XmaI-SP6p-G-5′TGEV-VS | PCR 5′TGEV | cgcgCCCGGGATTTAGGTGACACTATA(G)ACTTTTAAAGTAAAGTGAGTGTAGC | 557 bp |
| 5′TGEV-504-RS | PCR 5′TGEV | CACCAATGACGTAGTGATCCTTACG | |
| XmaI-T7p-GGG-ORF7-VS | PCR 3′TGEV | cgcgCCCGGGTAATACGACTCACTATA(G)GGATGCTGTATTTATTACAGTTTTAATC | 545 bp |
| HindIII-poly(A)-RS | PCR 3′TGEV | gccgAAGCTTTTTTTTTTTTTTTTTTTTTTTTTTGTATCACTATC | |
| TGEV-Leader-VS | Q-RT-PCR mRNA 7 | CGTGGCTATATCTCTTCTTTTACTTTAACTAG | |
| TGEV-7(38)-RS | Q-RT-PCR mRNA 7 | AAAACTGTAATAAATACAGCATGGAGGAA | |
| TGEV mRNA 7 (TaqMan MGB probe) | Q-RT-PCR mRNA 7 | FAM-CGAATCAAACGAGATGCT-MGB |
VS, virus sense. RS, reverse sense. MGB, minor groove binder group.
PCR 5′ (or 3′) TGEV, PCR for amplification of the TGEV genome ends to generate transcription templates. Q-RT-PCR mRNA 7, primers and probe used for the quantification of the viral subgenomic mRNA 7.
Restriction endonuclease sites used for cloning are in italics. Additional sequences at the 5′ of the restriction site are in lower case. Transcription promoters are in bold. Transcription initiation sites are in brackets. Template sequence corresponding to the poly(A) is underlined.
The generation of the TGEV-derived replicon REP 2 has been previously described (Almazán et al., 2004).
In vitro transcription
In vitro transcription reactions were performed from 1 μg of linearized pM33L-3′-template or 200 ng of the PCR amplicon containing the 5′TGEV end using the SP6/T7 Transcription kit (Roche). Biotin-14-CTP (Invitrogen) was added at a final concentration of 0.16 mM in a 1:6.25 ratio to unlabeled CTP. The transcription reactions were incubated 2 h at 37 °C and treated with 10 U of DNaseI during 30 min at 37 °C. The resulting transcripts were purified with the RNeasy kit (QIAGEN) following the RNA cleanup protocol, analyzed by denaturing electrophoresis in 1% (w/vol) agarose-2.2 M formaldehyde gels and spectrophotometrically quantified.
Cell extracts
ST, BHK-pAPN and Huh-7 cells were grown in 15-cm diameter dishes to confluence and infected at a MOI of 10 with TGEV PUR46-MAD (for ST and BHK-pAPN cells) or at a MOI of 5 with TGEV PUR46-C11 (for Huh-7 cells). After an adsorption period of 1 h the inoculum medium was replaced by fresh medium and the cell extracts were prepared at 7 and 36 h postinfection with the TGEV PUR46-MAD and PUR46-C11 viruses, respectively. The cells were then washed with cold PBS, scrapped off the plates, centrifuged at 1500 ×g for 10 min at 4 °C, and the cell pellets were resuspended in cold PBS (4 × 107 cells/ml) and stored at − 80 °C. To generate cytoplasmic extracts, 4 × 107 cells were pelleted and resuspended in 400 μl of the extraction buffer [10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 1 mM DTT, protease inhibitor cocktail (Roche)] at 4 °C by gentle pipetting, incubated at 4 °C for 15 min, and after addition of 25 μl of 10% NP-40, vortexed vigorously for 10 s and centrifuged at 13,000 ×g for 30 s. The supernatant was collected and stored in 10% glycerol at − 80 °C. Total protein concentration was determined with the Coomassie Plus Protein Assay (Pierce) and the extracts were aliquoted to avoid repeated freeze/thaw cycles.
RNA affinity chromatography
Cell extracts (600 μg) were diluted 1:1 in binding-washing (BW) buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 5% glycerol, 0.01% NP-40) and pre-cleared three times with 20 μl of Streptavidin Sepharose beads (Streptavidin Sepharose High Performance, Amersham) during 4 h. All the incubations were performed at 4 °C in an orbital shaker and the beads were collected by centrifugation at 440 ×g. Biotinylated RNA (10 μg) was diluted in 200 μl of RNA-binding buffer (5 mM Tris–HCl pH 7.5, 0.5 mM EDTA, 1 M NaCl) and incubated with 20 μl of fresh Streptavidin Sepharose beads during 1 h. The immobilized RNA was washed three times for 15 min with 200 μl of BW buffer and then incubated with the pre-cleared protein extract overnight. The RNA–protein complexes were washed three times for 15 min with 200 μl of BW buffer, pulled down and resuspended in 30 μl of NuPage Sample buffer (Invitrogen) by incubation for 10 min at 65 °C. The beads were centrifuged and the supernatant containing RNA interacting proteins was recovered and analyzed by denaturing electrophoresis using NuPAGE 4–12% Bis-Tris gels and MOPS SDS Running buffer (Invitrogen). The gels were washed three times in deionized water, stained with Coomasie blue Simply Blue Safe Stain (Invitrogen), and the protein bands of interest were excised from the gels for their identification by mass spectrometry.
Identification of proteins by mass spectrometry
Excised protein bands were in-gel digested with sequencing grade modified porcine trypsin (Promega). Peptides were extracted from gel pieces with 100 μl of 50% acetonitrile in 0.1% formic acid, dried by speed vacuum centrifugation and frozen. Peptides were dissolved in 7 μl of 0.1% formic acid and analyzed on a Micromass Q-TOF Micro mass spectrometer (Manchester, UK) coupled to a CAPLC HPLC system at the Oxford Central Proteomics Facility of the Sir William Dunn School of Pathology of the University of Oxford (UK). Alternatively, protein digestions were eluted in 0.5% of trifluoroacetic acid, dried by speed vacuum centrifugation and resuspended in 4 μl of MALDI solution. A 0.8 μl aliquot of each peptide mixture was deposited and dried onto a 386-well OptiTOF™ plate (Applied Biosystems) and co-crystallized with 0.8 μl of matrix solution (3 mg/ml CHCA in MALDI solution). Samples were analyzed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry in an ABI 4800 MALDI TOF/TOF mass spectrometer (Applied Biosystems) at the Proteomics Facility of the Biotechnology National Centre (CNB-CSIC) of Madrid (Spain). Data were analyzed using MassLynx 4.0 (Micromass, Manchester, UK) or the ABI 4000 series explorer Software v3.6 and searches were performed with the MASCOT software v2.1 (Matrix Science) against the MSDB from the non-redundant NCBI protein database with a mass tolerance of 100 and 200 ppm. No restrictions were imposed for protein molecular weight, although in some cases taxonomy restrictions for the human or mammalian databases were included.
Silencing analysis using the TGEV replicon system
The transfection and silencing efficiencies of siRNAs were optimized in human 293T cells using a Cy3-labeled positive control siRNA targeting the human GAPDH gene (Table 2). In the optimized conditions, 293T cells were grown to 60% confluence in 35-mm-diameter plates and transfected with 15 nM of Silencer Select siRNAs (Ambion, Table 2) and 7.2 μl of RNAiMax (Invitrogen) according to the manufacturer's specifications. Cells were incubated at 37 °C for 24 h and then trypsinized and seeded on 96-well plates treated with poly-lysine at a confluence of 4 × 104 cells per well. The cells were re-transfected with 10 nM of siRNAs at 48 h after the first transfection and incubated for 5 h at 37 °C. In the case of EPRS siRNA #1 (Table 2), doses of 50 and 25 nM (for the first and second siRNA transfections, respectively) were required to achieve an efficient silencing and a significant effect on the TGEV replicon activity. Finally, the transfection medium was discarded and the cells were transfected with 200 ng of the TGEV-derived replicon REP 2 and 0.48 μl of Lipofectamine 2000 (Invitrogen) as previously described (Almazán et al., 2004). Total RNA and protein were collected at 36 and 48 h after the replicon transfection for further analysis.
Silencing analysis with the infectious TGEV
Human Huh-7 cells, susceptible to TGEV PUR46-C11 strain, were selected to analyze the effect of silencing host candidate genes on TGEV infection. After optimization of the transfection and silencing using a Cy3-labeled positive control siRNA targeting the human GAPDH gene (Table 2), the reverse transfection method was chosen. Briefly, for each well of a 24-well plate, 4.8 × 104 cells were incubated in suspension with 10 nM of Silencer Select siRNAs (Ambion, Table 2) and 2 μl of siPORT Amine (Ambion) diluted in 25 μl of Opti-MEM I Reduced Serum Medium (GibcoBRL-Invitrogen). Then, cells were plated onto each well using DMEM medium with 10% of heat-inactivated FBS. Cells were incubated at 37 °C for 48 h and then infected with the TGEV PUR46-C11 at a MOI of 5. Total RNA, protein and cell supernatants were collected at 36 and 72 h postinfection for further analysis.
Analysis of cellular gene expression and viral RNA synthesis
Cellular gene expression and viral RNA synthesis were quantified by Q-RT-PCR. Total cDNA was synthesized with random hexamers from 100 ng of total RNA using the High Capacity cDNA Transcription kit (Applied Biosystems). Cellular gene expression was analyzed using TaqMan gene expression assays (Applied Biosystems) specific for human hnRNP Q (Hs00749309_s1 synaptotagmin binding, cytoplasmic RNA interacting protein), EPRS (Hs00270083_m1 Inventoried EPRS, glutamyl-prolyl-tRNA synthetase), PABP (Hs00743792_s1 Inventoried PABPC1, poly(A)-binding protein, cytoplasmic 1) or GAPDH (Hs99999905_m1 Inventoried glyceraldehyde-3-phosphate dehydrogenase) genes. To analyze viral RNA synthesis a custom TaqMan assay (Applied Biosystems) specific for the TGEV mRNA 7 (Table 3) was used. Data were acquired with an ABI PRISM 7000 sequence detection system (Applied Biosystems) and analyzed with ABI PRISM 7000 SDS version 1.0 software. Relative gene expression referred to cells treated with a validated negative control siRNA (Ambion, Table 2). The data represent the average of biological triplicates.
Western-blot analysis
Cell lysates or pulled down RNA-binding proteins were analyzed by denaturing electrophoresis in NuPAGE 4–12% Bis-Tris gels with MOPS SDS Running buffer (Invitrogen). Proteins were transferred to a nitrocellulose membrane (Hybond-C Extra Nitrocellulose, Amersham Biosciences) with a Bio-Rad Mini protean II electroblotting apparatus at 100 V for 1 h in Bis-Tris transfer buffer (25 mM Bis-Tris, 25 mM Bicine, 1 mM EDTA) containing 20% methanol. Membranes were blocked for 1 h with 5% dried skimmed milk in TBS (20 mM Tris–HCl pH 7.5, 150 mM NaCl) and then probed with specific antibodies. Primary antibodies for hnRNP A1 (rabbit polyclonal E17), hnRNP Q (mouse mAb I8E4:sc-56703), hnRNP A2B1 (goat polyclonal G-16:sc-10036), and hnRNP A0 (goat polyclonal G-17:sc-16509) were purchased from Santa Cruz Biotechnology. Antibodies for hnRNP U (rabbit polyclonal ab20666), EPRS (rabbit polyclonal ab31531), GAPDH (rabbit polyclonal ab9485), and β-actin (mouse mAb ab8226) were purchased from Abcam. The hybridoma BB7 against PTB and the p100 transcriptional co-activator guinea pig polyclonal antiserum GP25 were purchased from the ATCC and Progen Biotechnik, respectively. The PABP rabbit polyclonal antiserum was kindly provided by Dra. Amelia Nieto (CNB-CSIC, Madrid). To detect the TGEV N protein the mouse mAb 3DC10 was used (Martín Alonso et al., 1992). Bound primary antibodies were detected with horseradish peroxidase-conjugated antibodies against the different species and the Immobilon Western chemiluminescent substrate (Millipore), following the manufacturer's recommendations.
Acknowledgments
We thank S. Zúñiga for critically reading the manuscript and helpful discussions. This work was supported by grants from the Ministry of Science and Innovation of Spain (BIO2007-60978 and CIT-01000-2007-8), the Community of Madrid (S-SAL-0185-2006), the European Community (Frame VII, RiViGene project, SSPE-CT-2005-022639) and Fort Dodge Veterinaria. C. G. received a contract supported by grants from the Ministry of Science and Innovation of Spain (CIT-01000-2007-8) and the Community of Madrid (S-SAL-0185-2006).
References
- Ahlquist P., Noueiry A.O., Lee W.M., Kushner D.B., Dye B.T. Host factors in positive-strand RNA virus genome replication. J. Virol. 2003;77(15):8181–8186. doi: 10.1128/JVI.77.15.8181-8186.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almazán F., Galán C., Enjuanes L. The nucleoprotein is required for efficient coronavirus genome replication. J. Virol. 2004;78(22):12683–12688. doi: 10.1128/JVI.78.22.12683-12688.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley I., Digard P., Inglis S.C. Characterization of an efficient coronavirus ribosomal frameshifting signal: requirement for an RNA pseudoknot. Cell. 1989;57(4):537–547. doi: 10.1016/0092-8674(89)90124-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudy A.A., Ketting R.F., Hammond S.M., Denli A.M., Bathoorn A.M., Tops B.B., Silva J.M., Myers M.M., Hannon G.J., Plasterk R.H. A micrococcal nuclease homologue in RNAi effector complexes. Nature. 2003;425(6956):411–414. doi: 10.1038/nature01956. [DOI] [PubMed] [Google Scholar]
- Cen S., Javanbakht H., Kim S., Shiba K., Craven R., Rein A., Ewalt K., Schimmel P., Musier-Forsyth K., Kleiman L. Retrovirus-specific packaging of aminoacyl-tRNA synthetases with cognate primer tRNAs. J. Virol. 2002;76(24):13111–13115. doi: 10.1128/JVI.76.24.13111-13115.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K.S., Huang P., Lai M.M. Polypyrimidine-tract-binding protein affects transcription but not translation of mouse hepatitis virus RNA. Virology. 2002;303(1):58–68. doi: 10.1006/viro.2002.1675. [DOI] [PubMed] [Google Scholar]
- Choi K.S., Mizutani A., Lai M.M. SYNCRIP, a member of the heterogeneous nuclear ribonucleoprotein family, is involved in mouse hepatitis virus RNA synthesis. J. Virol. 2004;78(23):13153–13162. doi: 10.1128/JVI.78.23.13153-13162.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas B., Gelfi J., Sjöström H., Noren O., Laude H. Further characterization of aminopeptidase-N as a receptor for coronaviruses. Adv. Exp. Med. Biol. 1993;342:293–298. doi: 10.1007/978-1-4615-2996-5_45. [DOI] [PubMed] [Google Scholar]
- Dreher T.W. Role of tRNA-like structures in controlling plant virus replication. Virus Res. 2008;139(2):217–298. doi: 10.1016/j.virusres.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten C., Günther S., Preiser W., van der Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A.M., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.C., Muller S., Rickerts W., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D.M.E. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348(20):1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- Elmen J., Thonberg H., Ljungberg K., Frieden M., Westergaard M., Xu Y., Wahren B., Liang Z., Orum H., Koch T., Wahlestedt C. Locked nucleic acid (LNA) mediated improvements in siRNA stability and functionality. Nucleic Acids Res. 2005;33(1):439–447. doi: 10.1093/nar/gki193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enjuanes L., Almazán F., Sola I., Zúñiga S. Biochemical aspects of coronavirus replication and virus–host interaction. Annu. Rev. Microbiol. 2006;60:211–230. doi: 10.1146/annurev.micro.60.080805.142157. [DOI] [PubMed] [Google Scholar]
- Enjuanes L., Gorbalenya A.E., de Groot R.J., Cowley J.A., Ziebuhr J., Snijder E.J. The Nidovirales. In: Mahy B.W.J., Van Regenmortel M., Walker P., Majumder-Russell D., editors. Encyclopedia of Virology. 3rd ed. Elsevier Ltd.; Oxford: 2008. pp. 419–430. [Google Scholar]
- Enjuanes L., Spaan W., Snijder E., Cavanagh D. In: Virus Taxonomy. Classification and Nomenclature of Viruses. van Regenmortel M.H.V., Fauquet C.M., Bishop D.H.L., Carsten E.B., Estes M.K., Lemon S.M., McGeoch D.J., Maniloff J., Mayo M.A., Pringle C.R., Wickner R.B., editors. Academic Press; San Diego, California: 2000. pp. 827–834. [Google Scholar]
- Escors D., Izeta A., Capiscol C., Enjuanes L. Transmissible gastroenteritis coronavirus packaging signal is located at the 5′ end of the virus genome. J. Virol. 2003;77(14):7890–7902. doi: 10.1128/JVI.77.14.7890-7902.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galán C., Enjuanes L., Almazán F. A point mutation within the replicase gene differentially affects coronavirus genome versus minigenome replication. J. Virol. 2005;79(24):15016–15026. doi: 10.1128/JVI.79.24.15016-15026.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghetti A., Pinol-Roma S., Michael W.M., Morandi C., Dreyfuss G. hnRNP I, the polypyrimidine tract-binding protein: distinct nuclear localization and association with hnRNAs. Nucleic Acids Res. 1992;20(14):3671–3678. doi: 10.1093/nar/20.14.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara H., Aizaki H., Matsuda M., Shinkai-Ouchi F., Inoue Y., Murakami K., Shoji I., Kawakami H., Matsuura Y., Lai M.M.C., Miyamura T., Wakita T., Suzuki T. Involvement of creatine kinase B in hepatitis C virus genome replication through interaction with the viral NS4A protein. J. Virol. 2009;83(10):5137–5147. doi: 10.1128/JVI.02179-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibba M., Soll D. Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 2000;69:617–650. doi: 10.1146/annurev.biochem.69.1.617. [DOI] [PubMed] [Google Scholar]
- Imbert I., Guillemot J.C., Bourhis J.M., Bussetta C., Coutard B., Egloff M.P., Ferron F., Gorbalenya A.E., Canard B. A second, non-canonical RNA-dependent RNA polymerase in SARS coronavirus. EMBO J. 2006;25(20):4933–4942. doi: 10.1038/sj.emboj.7601368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izeta A., Smerdou C., Alonso S., Penzes Z., Méndez A., Plana-Durán J., Enjuanes L. Replication and packaging of transmissible gastroenteritis coronavirus-derived synthetic minigenomes. J. Virol. 1999;73(2):1535–1545. doi: 10.1128/jvi.73.2.1535-1545.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J., Arif A., Ray P.S., Fox P.L. WHEP domains direct noncanonical function of glutamyl-prolyl tRNA synthetase in translational control of gene expression. Mol. Cell. 2008;29(6):679–690. doi: 10.1016/j.molcel.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez G., Correa I., Melgosa M.P., Bullido M.J., Enjuanes L. Critical epitopes in transmissible gastroenteritis virus neutralization. J. Virol. 1986;60(1):131–139. doi: 10.1128/jvi.60.1.131-139.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani W., Narayanan K., Huang C., Lokugamage K., Ikegami T., Ito N., Kubo H., Makino S. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc. Natl. Acad. Sci. U. S. A. 2006;103(34):12885–12890. doi: 10.1073/pnas.0603144103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl R.J., Hall T.C. Aminoacylation of RNA from several viruses: amino acid specificity and differential activity of plant, yeast and bacterial synthetases. J. Gen. Virol. 1974;25(2):257–261. doi: 10.1099/0022-1317-25-2-257. [DOI] [PubMed] [Google Scholar]
- Lai M.M.C. Cellular factors in the transcription and replication of viral RNA genomes: a parallel to DNA-dependent RNA transcription. Virology. 1998;244(1):1–12. doi: 10.1006/viro.1998.9098. [DOI] [PubMed] [Google Scholar]
- Li H.P., Zhang X., Duncan R., Comai L., Lai M.M.C. Heterogeneous nuclear ribonucleoprotein A1 binds to the transcription-regulatory region of mouse hepatitis virus RNA. Proc. Natl. Acad. Sci. U. S. A. 1997;94(18):9544–9549. doi: 10.1073/pnas.94.18.9544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.P., Huang P., Park S., Lai M.M.C. Polypyrimidine tract-binding protein binds to the leader RNA of mouse hepatitis virus and serves as a regulator of viral transcription. J. Virol. 1999;73(1):772–777. doi: 10.1128/jvi.73.1.772-777.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín Alonso J.M., Balbin M., Garwes D.J., Enjuanes L., Gascon S., Parra F. Antigenic structure of transmissible gastroenteritis virus nucleoprotein. Virology. 1992;188(1):168–174. doi: 10.1016/0042-6822(92)90746-C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006;66:193–292. doi: 10.1016/S0065-3527(06)66005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClurkin A.W., Norman J.O. Studies on transmissible gastroenteritis of swine. II. Selected characteristics of a cytopathogenic virus common to five isolates from transmissible gastroenteritis. Can. J. Comp. Med. Vet. Sci. 1966;30(7):190–198. [PMC free article] [PubMed] [Google Scholar]
- Mizutani A., Fukuda M., Ibata K., Shiraishi Y., Mikoshiba K. SYNCRIP, a cytoplasmic counterpart of heterogeneous nuclear ribonucleoprotein R, interacts with ubiquitous synaptotagmin isoforms. J. Biol. Chem. 2000;275(13):9823–9831. doi: 10.1074/jbc.275.13.9823. [DOI] [PubMed] [Google Scholar]
- Myer V.E., Steitz J.A. Isolation and characterization of a novel, low abundance hnRNP protein: A0. RNA. 1995;1(2):171–182. [PMC free article] [PubMed] [Google Scholar]
- Nanda S.K., Leibowitz J.L. Mitochondrial aconitase binds to the 3′ untranslated region of the mouse hepatitis virus genome. J. Virol. 2001;75(7):3352–3362. doi: 10.1128/JVI.75.7.3352-3362.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman S., Lane T.E., Buchmeier M.J. Coronavirus: hepatitis, peritonitis, and central nervous system disease. In: Cunningham M.W., Fujinami R.S., editors. Coronavirus: Hepatitis, Peritonitis, and Central Nervous System Disease. Lippincott Williams and Wilkins; Philadelphia: 2000. pp. 331–348. [Google Scholar]
- Pettit Kneller E.L., Connor J.H., Lyles D.S. hnRNPs relocalize to the cytoplasm following infection with vesicular stomatitis virus. J. Virol. 2009;83(2):770–780. doi: 10.1128/JVI.01279-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinol-Roma S., Dreyfuss G. hnRNP proteins: localization and transport between the nucleus and the cytoplasm. Trends Cell. Biol. 1993;3(5):151–155. doi: 10.1016/0962-8924(93)90135-n. [DOI] [PubMed] [Google Scholar]
- Sampath P., Mazumder B., Seshadri V., Gerber C.A., Chavatte L., Kinter M., Ting S.M., Dignam J.D., Kim S., Driscoll D.M., Fox P.L. Noncanonical function of glutamyl-prolyl-tRNA synthetase: gene-specific silencing of translation. Cell. 2004;119(2):195–208. doi: 10.1016/j.cell.2004.09.030. [DOI] [PubMed] [Google Scholar]
- Sánchez C.M., Jiménez G., Laviada M.D., Correa I., Suñé C., Bullido M.J., Gebauer F., Smerdou C., Callebaut P., Escribano J.M., Enjuanes L. Antigenic homology among coronaviruses related to transmissible gastroenteritis virus. Virology. 1990;174(2):410–417. doi: 10.1016/0042-6822(90)90094-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez C.M., Izeta A., Sanchez-Morgado J.M., Alonso S., Sola I., Balasch M., Plana-Durán J., Enjuanes L. Targeted recombination demonstrates that the spike gene of transmissible gastroenteritis coronavirus is a determinant of its enteric tropism and virulence. J. Virol. 1999;73(9):7607–7618. doi: 10.1128/jvi.73.9.7607-7618.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schelle B., Karl N., Ludewig B., Siddell S.G., Thiel V. Selective replication of coronavirus genomes that express nucleocapsid protein. J. Virol. 2005;79(11):6620–6630. doi: 10.1128/JVI.79.11.6620-6630.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X., Masters P.S. Evaluation of the role of heterogeneous nuclear ribonucleoprotein A1 as a host factor in murine coronavirus discontinuous transcription and genome replication. Proc. Natl. Acad. Sci. U. S. A. 2001;98(5):2717–2722. doi: 10.1073/pnas.031424298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S.T., Lai M.M. Viral and cellular proteins involved in coronavirus replication. Curr. Top. Microbiol. Immunol. 2005;287:95–131. doi: 10.1007/3-540-26765-4_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S.T., Huang P., Li H.P., Lai M.M. Heterogeneous nuclear ribonucleoprotein A1 regulates RNA synthesis of a cytoplasmic virus. EMBO J. 2000;19(17):4701–4711. doi: 10.1093/emboj/19.17.4701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S.T., Yu G.Y., Lai M.M. Multiple type A/B heterogeneous nuclear ribonucleoproteins (hnRNPs) can replace hnRNP A1 in mouse hepatitis virus RNA synthesis. J. Virol. 2003;77(19):10584–10593. doi: 10.1128/JVI.77.19.10584-10593.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L.M., Guan Y., Rozanov M., Spaan W.J.M., Gorbalenya A.E. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003;331(5):991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spagnolo J.F., Hogue B.G. Host protein interactions with the 3′ end of bovine coronavirus RNA and the requirement of the poly(A) tail for coronavirus defective genome replication. J. Virol. 2000;74(11):5053–5065. doi: 10.1128/jvi.74.11.5053-5065.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tijms M.A., Snijder E.J. Equine arteritis virus non-structural protein 1, an essential factor for viral subgenomic mRNA synthesis, interacts with the cellular transcription co-factor p100. J. Gen. Virol. 2003;84(Pt. 9):2317–2322. doi: 10.1099/vir.0.19297-0. [DOI] [PubMed] [Google Scholar]
- Vijaykrishna D., Smith G.J., Zhang J.X., Peiris J.S., Chen H., Guan Y. Evolutionary insights into the ecology of coronaviruses. J. Virol. 2007;81(8):4012–4020. doi: 10.1128/JVI.02605-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.Y., Nagy P.D. Tomato bushy stunt virus co-opts the RNA-binding function of a host metabolic enzyme for viral genomic RNA synthesis. Cell Host Microbe. 2008;3(3):178–187. doi: 10.1016/j.chom.2008.02.005. [DOI] [PubMed] [Google Scholar]
- Wathelet M.G., Orr M., Frieman M.B., Baric R.S. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J. Virol. 2007;81(21):11620–11633. doi: 10.1128/JVI.00702-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weighardt F., Biamonti G., Riva S. Nucleo-cytoplasmic distribution of human hnRNP proteins: a search for the targeting domains in hnRNP A1. J. Cell Sci. 1995;108(Pt. 2):545–555. doi: 10.1242/jcs.108.2.545. [DOI] [PubMed] [Google Scholar]
- Yi M., Schultz D.E., Lemon S.M. Functional significance of the interaction of hepatitis A virus RNA with glyceraldehyde 3-phosphate dehydrogenase (GAPDH): opposing effects of GAPDH and polypyrimidine tract binding protein on internal ribosome entry site function. J. Virol. 2000;74(14):6459–6468. doi: 10.1128/jvi.74.14.6459-6468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W., Leibowitz J.L. Specific binding of host cellular proteins to multiple sites within the 3′ end of mouse hepatitis virus genomic RNA. J. Virol. 1995;69(4):2016–2023. doi: 10.1128/jvi.69.4.2016-2023.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr J. The coronavirus replicase. Curr. Top. Microbiol. Immunol. 2005;287:57–94. doi: 10.1007/3-540-26765-4_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr J., Snijder E.J., Gorbalenya A.E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000;81(Pt. 4):853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]