

Abstract
Gene fusion technology is a key tool in facilitating gene function studies. Hybrid molecules in which all the components are joined precisely, without the presence of intervening and unwanted extraneous sequences, enable accurate studies of molecules and the characterization of individual components. This article reviews situations in which seamlessly fused genes and proteins are required or desired and describes molecular approaches that are available for generating these hybrid molecules.
Introduction
With the completion of various genome sequencing projects, functional analysis of gene products has taken the stage. Gene fusion technology has an important role in many aspects of gene function studies, including gene and protein tagging, reporter gene studies, domain swapping studies, mutagenesis studies and gene knock-in and/or knock-out experiments [1]. Classic gene fusion techniques involving type II restriction enzyme digestion and DNA ligation reactions (the so-called cut-and-paste reactions) have been used as standard procedures for generating hybrid genes. However, such procedures often leave behind operational sequences, such as restriction sites, at the junction. These unwanted sequences can change the spacing between the DNA elements and introduce extra amino acid residues at the junction, which could have undesired effects on the structure and activity of the fusion protein and could therefore interfere with accurate study of the fusion gene. This review discusses examples in which the use of precisely fused genes is required or desired and outlines approaches that can be used to achieve seamless gene fusion. Owing to the lack of reviews in this area some historical information together with recent advances will be discussed here.
Seamless gene fusion and applications
Seamless cloning and gene fusion are processes that allow two or more DNA fragments to be joined precisely so that no unwanted nucleotides are added at the junctions between DNA fragments. This is the ideal situation for generating hybrid genes and the following section highlights some examples that show the importance of seamless gene fusion.
Promoter and exon studies
Gene promoters contain arrays of regulatory elements to which transcriptional factors bind and interact with each other to regulate transcription. Promoter deletion analysis allows identification of these functional elements and provides crucial information on the mechanism of gene regulation. However, because spacing between various regulatory elements is often important, a linker of the same length is usually required to preserve the spacing and helical facing of the elements. Linker scanning analysis [2] (Box 1 , Case 1) of gene promoters requires seamless DNA fusion or sequence replacement. Molecular evolution approaches, such as exon and DNA shuffling, for producing proteins with desired biochemical and/or biophysical properties also require seamless splicing of various functional elements [3]. In eukaryotic cells, chimeric genes and/or proteins can be created by intron-mediated RNA splicing 4, 5. In these experiments synthesis of RNA substrates and/or exon-tagged ribozymes requires careful design and generation of the chimeric precursor genes. As long as the hybrid gene is generated correctly, seamless fusion can be achieved on splicing.
Box 1. Seamless gene fusion case studies.
Case 1. Scanning mutagenesis. Scanning mutagenesis experiments were designed to identify cis regulatory elements in gene promoters [2] or to study crucial amino acid residue(s) in protein structure and function. For a gene promoter, a linker scanning analysis is performed. A panel of DNA constructs is made and within each a segment of the promoter is replaced by a defined linker fragment of the same length to restore the spacing. For a protein, alanine scanning mutagenesis is often performed [65] in which a set of mutants are generated with each charged amino acid residue mutated to alanine. Traditionally, scanning mutagenesis constructs were generated by a laborious process involving the use of exonucleases, linkers and ligase [2] or by bacterial phage M13-based site-directed mutagenesis [20]. With the advent of PCR, the variant constructs can be easily generated by overlap PCR [66] or in vivo recombination 57, 58.
Case 2. The ubiquitin tagging system and applications. Ubiquitin is a highly conserved eukaryotic protein of 76 amino acid residues that is naturally expressed as polyubiquitin, which is then cleaved precisely in vivo at the C-terminus of the ubiquitin moiety by a family of ubiquitin processing proteases (Ubps). The amino acid residue at P1′ could be any residue except proline. The high specificity and the unique cleavage fashion of Ubps have made the ubiquitin–Ubp pair an ideal tagging system for gene expression in eukaryotic and prokaryotic cells [67]. To study the impact of the N-terminal amino acid residue on stability of proteins in yeast, Varshavsky and colleagues 21, 22 created a set of ubiquitin–X–βgal seamless fusion genes, in which X is a codon for one of the 20 amino acids. Upon expression in yeast, the nascent fusion proteins are de-ubiquitinated by Ubps in vivo, exposing the X–βgal proteins with various N-terminal residues. Stability studies of these X–βgal proteins in yeast led to the discovery of the N-end rule of protein stability in vivo [68]. Co-translational processing has also allowed protein production in eukaryotic cells 67, 69. In prokaryotic cells, ubiquitin fusion proteins are not processed in vivo owing to the lack of Ubps. After purification of the fusion protein, the ubiquitin tag can be removed with a purified Ubp in vitro [15]. Along the same line, the yeast small ubiquitin-like modifier (SUMO) protein, SMT3, has been used as a fusion tag for protein production in E. coli [14] (http://lifesensors.com/r_and_d/protein_expression.php3). This tag can be removed by treating with SUMO hydrolase (ubiquitin-like protease 1) in vitro. The ubiquitin–Ubp proteins thus provide a unique system for producing proteins with an authentic N-terminus, as long as the ubiquitin-ORF fusion is generated seamlessly.
Case 3. Viral genome assembly. In an effort to assemble a 31.5 kb full length infectious cDNA of a recombinant coronavirus, Yount and colleagues used a seamless gene fusion approach involving PCR and the use of a type IIS restriction enzyme [19]. To do so, the viral genome was PCR amplified as seven neighboring fragments using primers with an Esp3I cleavage site engineered at the ends. Esp3I digestion removes any unwanted nucleotide residues at the ends of the PCR fragments, leaving specific four base overhangs that are complementary for the neighboring segments. A stepwise ligation reaction allowed directional assembly of the full-length viral genome.
Protein functional studies and protein engineering
Proteins are composed of functional domains. To elucidate the function of a particular domain in a defined protein, mutant proteins with the domain deleted or replaced by a similar domain from a homologous protein are often needed. Such a domain deletion or swapping experiment would benefit greatly from seamless fusions of the relevant parts to eliminate potential negative effects caused by the presence of operational sequences. In more general terms, seamless fusions of various domains are essential for protein engineering, including the generation of novel hybrid molecules and antibody engineering. In the case of antibody engineering, grafting of the complementarity-determining region (CDR) from mouse antibodies to human frameworks (CDR-grafting) and site-directed mutagenesis (SDM) are routine procedures [6]. Chimeric proteins can also be generated via intein-mediated protein splicing and ligation [7]. As long as the intein-containing precursor proteins are generated seamlessly, precise protein fusions can be achieved.
Protein production
Protein production has been improved by the use of tags and fusion partners. They confer solubility and stability and facilitate subsequent affinity purification of the target protein [1]. In cases in which the activity of a particular protein is unaffected by the presence of its fusion partner or a tag, the entire fusion protein is used in subsequent applications. This is often the case for enzymatic studies and assay development. However, in many cases removal of the fusion partner is required or desired and can be achieved via an engineered protease cleavage site. This process requires a seamless junction between the protease cleavage site and the protein of interest. For structural studies, in some cases fusion proteins with a short tag (such as a hexa-Histidine tag) have been successfully crystallized and removal of the tag is unnecessary. However, for those fusion proteins with a tag that failed to crystallize, the impact of the tag is hard to assess and its removal is usually desirable.
In producing mature and active proteins, proteins with native amino acid residues are often required. This is exemplified by proteins such as RANTES (Regulated on Activation, Normal T Expressed and Secreted), interleukin-18 (IL-18), IL-1β and hirudin 8, 9, 10, 11. Addition of a methionine residue at the N-termini of these proteins significantly reduces activity. In the case of RANTES, Met-RANTES was found to behave as an antagonist for the authentic RANTES [8]. A practical approach for producing proteins with authentic N-terminal residues in E. coli is to express and purify them as fusion proteins with an N-terminal tag. Following purification, and refolding if necessary, the tag and any unwanted amino acid residues are then removed via a specifically engineered protease cleavage site. This process requires a seamless junction between the protease cleavage site and the protein of interest. Enterokinase [12], factor Xa [13] and ubiquitin-specific proteases (Ubps) 14, 15 that cleave at the C-terminal of their recognition sequences are often used for this purpose. The tobacco etch virus (TEV) protease [16] can also be used because it has a relaxed requirement on the amino acid residue immediately following the cleavage site (the P1′ position). The ubiquitin/Ubp system is probably the first tagging system that is used to generate proteins with native N-terminal residues both in vivo and in vitro (Box 1, Case 2). In producing mature human Apo A-I in E. coli, Moguilevsky and colleagues [17] found that the ubiquitin tagging system is one of the most straightforward methods.
Genome manipulation
Targeted gene knockout and knock-in technologies in model organisms provide powerful genetic tools for gene function analysis in vivo. This process often involves deletion of the entire open reading frame (ORF) or a part that encodes a particular domain, and in some cases replacement of these with sequences of a mutant allele or an orthologue. These studies require generation of knockout and knock-in constructs in which all the functional elements, including flanking regions, required for homologous recombination are precisely joined. Seamless gene fusion technologies would greatly facilitate construct generation for such studies. Link and colleagues [18] used the overlap PCR technology (see following section) to generate constructs with precise gene deletions for functional studies in the E. coli genome.
For molecular and pathogenesis studies of animal viruses and vaccine vector development, it is often necessary to synthesize the entire viral genome or to engineer hybrid viral vectors. Using a seamless gene fusion technique, Yount and colleagues [19] have successfully assembled a 31.5 Kb recombinant viral genome from multiple PCR fragments (Box 1, Case 3).
Methods for achieving seamless gene fusion
Before PCR was invented, seamless fusion of DNA fragments was achieved by complicated procedures routinely involving the use of bacterial phage M13-based site-directed mutagenesis, oligonucleotide primers and linkers and exonucleases, followed by plasmid propagation in E. coli. In some cases it was achieved using RecA-dependent homologous recombination. These processes required careful design of the target constructs and involved a laborious procedure. Given the difficulties with these classical approaches, only a few examples of seamless gene fusion using these methods can be found. One such example is the generation of a set of mutant constructs used for link-scanning analysis of gene promoters (Box 1, Case 1) 2, 20 and another for protein stability studies in yeast 21, 22 (Box 1, Case 2).
With the advent of PCR technology, seamless gene fusion has been enabled. Several methods for this purpose have been created, and many other approaches could be modified to achieve the purpose. These methods are discussed below and summarized in Table 1 . PCR is always involved, either to allow precise DNA manipulation or to modify the ends of the DNA elements for appropriate gene fusion. With these innovative approaches, our ability to manipulate DNA sequences has been taken to an unprecedented level. For example, in a study on the influence of P1′ amino acid residue on enterokinase (EK) cleavage in a fusion protein, a set of 20 variant GST–EK–X–calmodulin fusion genes (in which X is one of the 20 amino acid residues and GST is glutathione S-transferase) were created by a seamless gene fusion technique via PCR and ligation-independent cloning [12].
Table 1.
Methods for achieving seamless gene fusion
| Methods | Applications | Pros | Cons |
|---|---|---|---|
| Gene synthesis | Creation of hybrid or novel genes at will. Generation of DNA fragments with any desired changes. | Allow fusion gene designing with accuracy at base pair (bp) level. Allow codon optimization of any open reading frame. | Practical when the hybrid gene is <500 bp in length, although longer is possible. |
| Overlap PCR | Assembly of multiple DNA fragments. | Directional DNA assembly, versatile and efficient, independent of restriction sites. | Requires multiple PCR reactions. The longer the hybrid product is, the higher risk for PCR introduced errors. Reasonable for hybrids <10 Kb, although longer is possible. |
| Inverse PCR | Prepare vector backbones with desired terminal sequences for applications listed below. Can also be used to introduce point mutation, deletion, insertion and sequence replacement on a circular plasmid. | Ideal for preparing vectors with desired terminal sequences for seamless cloning. | The vector backbone might contain sequence errors introduced by PCR. The longer the vector backbone, the higher risk for PCR introduced errors. |
| QuickChange™ site-directed mutagenesis | Point mutation, deletion, insertion and sequence replacement on any plasmid backbones | Most widely used method with a series of kits commercially available. Multiple mutations in a single reaction possible. | Vector size >8 Kb may have a decreased efficiency, however, template up to 19 Kb has been used (www.stratagene.com). |
| Type IIS restriction enzyme-mediated gene fusion | Assembly of long multiple DNA fragments. | Directional ligation of multiple PCR fragments to assemble >15 Kb fusion genes or viral genomes. | Requires multiple PCR reactions and restriction digestion. For fusion of a PCR fragment with an existing fragment already contained in a vector, special vector is required. |
| Ligation-independent cloning | Fusion of a PCR fragment with a DNA element(s) contained in a vector. | Directional cloning, independent of restriction sites and the use of ligase. | Need a specially designed LIC vector. The insert and vector DNA fragments need to be treated to generate single-stranded overhangs. |
| In-Fusion™ cloning | Fusion of a PCR fragment with a DNA element(s) contained in a vector. | Directional cloning, independent of restriction sites and the use of ligase. | Mechanism of action not disclosed by the supplier. The In-Fusion™ enzyme is relatively expensive. Seamless only when the vector fragment doesn't contain extra sequence at the fusion junction. |
| RecA-dependent recombination | Allelic replacement in E. coli. Construction of recombinant adenoviral genomes in E. coli. | Intermolecular recombination between two circular molecules possible. | Work in recA+ strains and requires long (>1 Kb) homologous arms. A low frequency event, usually requires selection and/or conterselection. |
| RecA-independent recombination | Point mutation, deletion, insertion in E. coli. Linker scanning mutagenesis. Point mutation, deletion, insertion. | Work in commonly used E. coli strains such as DH5α and JM109 (recA strains), only requires >12 bp homologous arms. | Mechanism of action unclear, although independent of RecA. Most efficient if the origin of replication and a selectable marker is contained in a separate fragment. |
| Red/ET recombination | Point mutation, deletion, insertion. Subcloning from a complex source Allelic replacement in E. coli. | Requires 30–50 homologous arms. Vector fragments generated by inverse PCR have been used. | Work in recBC sbcA mutant strains such as JC8679 or in recBC+ strains overexpressing RedE/RedT/Redγ |
| Gap repair in yeast | Gene fusion and subcloning in yeast. | Requires >30 bp homologous arms, vector fragments can be generated by inverse PCR. | Requires a yeast origin of replication and a selectable marker in the backbone. |
Overlap PCR
Overlap PCR (Figure 1 ) was described shortly after the invention of PCR 23, 24. It is a robust process that is independent of any restriction sites; any two fragments can be freely joined at any predetermined sequence location, provided that the fragments can be faithfully amplified. With the same principle, multiple DNA fragments can be spliced together seamlessly [25] and recombinant fusion genes as long as 20 Kb have been obtained [26]. Overlap PCR has been used to introduce point mutations, insertions, deletions and replacements into any point of a gene in a seamless fashion 23, 24. The PCR generated fusion genes can subsequently be cloned into appropriate vectors for downstream applications. Note that a special case of overlap PCR is gene synthesis, in which overlapping oligonucleotides are used as PCR templates for gene assembly [27]. This approach can be readily used if there is no suitable template DNA that can be used for PCR amplification, and/or if the hybrid gene is relatively short (<500bp).
Figure 1.

Seamless gene fusion by overlap PCR. The diagram shows seamless fusion of DNA fragments X and Y. The two DNA fragments are PCR amplified individually. Primers P2 and P3 are designed so that the 5′-end 15 bases are complementary to each other. The PCR products are then used as templates for a second PCR amplification with primers P1 and P4. The complementary part of P2 and P3 could be part of fragment X or fragment Y. Note that to facilitate efficient PCR amplification, the melting temperature (Tm) for all primers should be made to be similar within the range of 55°C–75°C.
Site-specific mutagenesis
Site specific mutagenesis, including point mutations, insertions, deletions and replacements, is carried out on a circular plasmid template that contains the target gene. A variety of mutagenesis approaches are available for generating point mutations [28]. However, mutagenesis tasks involving deletion, insertion or replacement of a sequence are typically achieved by a PCR-based method. Inverse PCR uses two oppositely positioned primers to amplify a plasmid backbone, which allows generation of point mutations, deletions and insertions at any location of the plasmid [29]. However, the PCR step could potentially introduce sequence errors in the circular plasmid backbone. This issue was addressed by the QuickChange™ (www.stratagene.com) mutagenesis technology 30, 31. As illustrated in Figure 2 , the method relies on the use of two fully complementary mutagenic primers and Pfu DNA polymerase. Given that the primers are completely complementary to each other, and that Pfu lacks strand-displacement activity, the mutant strands are only linearly amplified from the original template DNA during the multiple thermal cycles. This design thus prevents amplification of any errors that occurred during DNA synthesis. The QuickChange™ procedure and improvements have been used to generate a DNA insertion, deletion or sequence replacement at any site of the template DNA 32, 33 and even to introduce multiple mutations into a template simultaneously [34]. The QuickChange™ mutagenesis kit thus allows easy manipulation of any circular plasmid DNA with high fidelity.
Figure 2.
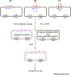
Seamless DNA manipulation by QuickChange™ site-directed mutagenesis. The diagram shows steps involved in site-directed mutagenesis for generating point mutations (a), insertions (b) or deletions (c). In all the cases, two complementary mutagenic primers (or megaprimers in case b) are used with each having >15 base homologous sequences flanking the mutagenic site. After primer extension cycles with Pfu polymerase, the undesired methylated template DNA and semi-methylated hybrids are fragmented by treating with restriction enzyme DpnI. The desired mutant circular duplexes are recovered in E. coli following transformation. The plasmid backbone contains an origin of replication (ori) and a selectable marker (sm).
The use of type IIS restriction enzymes
Type IIS restriction enzymes are a class of enzymes that cleave outside of their recognition sequences, for example SapI, BsaI and FokI [35]. They have been used to generate cohesive ends from PCR fragments for seamless assembly of genes and viral genomes 19, 36, 37 (Figure 3 a). In cases in which one of the two fragments is a generic one to which multiple partners will be fused, such as a purification tag (e.g. maltose binding protein) for protein production, the fragment is usually inserted into the expression vector first. The vector is then made to accept its fusion partner through seamless cloning 38, 39 (Figure 3b). The pCal.n.EK vector of Stratagene (www.stratagene.com) is an E. coli expression vector that contains the T7/lacO–CBP–EK–MCS expression cassette (CBP is calmodulin binding peptide and MCS is multiple cloning site). Cloning via the type IIS enzyme Eam1104 I allows seamless fusion of the target ORF immediately downstream of the CBP–EK ORF. After expression and purification of the CBP–EK–ORF fusion protein, removal of the CBP–EK polypeptide with enterokinase allows recovery of the target recombinant protein with native amino acid sequence [39]. The IMPACT™ vectors pTYB and pTWIN (www.neb.com) allow seamless fusion of ORFs for a target protein with an intein via SapI for protein production and subsequent protein splicing.
Figure 3.
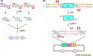
Seamless gene cloning and gene fusion via a type IIS restriction enzyme. SapI is used as an example. (a) Seamless assembly of fragments X, Y, and Z. The fragments are first individually PCR amplified, with primers containing a SapI site. The primers are so designed that upon SapI digestion specific cohesive ends are generated for each fragment. Following SapI digestion, ligation of the fragments results in seamless assembly of X, Y, and Z. The 5′-end of X and the 3′-end of Z are made to contain SapI (with ends incompatible to other ends of X, Y and Z) or any other restriction sites for further subcloning. (b) Seamless fusion of fragment X with fragments Y and Z contained in a vector. Fragment A is PCR amplified with primers containing a SapI site. The primers are so designed that upon digestion with SapI cohesive ends are generated. The vector fragment, which contains fragments Y and Z, was specially engineered and prepared by SapI digestion. Ligation of the SapI-treated fragment A with the vector fragment results in seamless fusion of X with Y and Z. The plasmid backbone contains an origin of replication (ori) and a selectable marker (sm). Note that the nucleotide bases comprising the cohesive ends can be part of the fragment X or its fusion partners, and can be chosen to make ligation of the fragments directional.
Type IIS restriction sites have also been used in preparing acceptor vectors for seamless cloning 38, 40 allowing a DNA fragment to be fused seamlessly with existing DNA fragments contained in the vector. This is achieved by inverse PCR of the plasmid backbone with a type IIS restriction site engineered in the primers. Subsequent restriction digestion generates the vector with desired compatible cohesive ends. It should be noted that the type IIS restriction enzyme-based approaches involve restriction enzyme digestion of both the vector and the inserted DNA fragments. In cases in which the DNA fragments contain internal sites for a particular type IIS enzyme in use, the methylation inhibition approach can be used to block digestion of the internal sites by the enzyme 38, 41 or a different type IIS enzyme is used to produce DNA fragments with desired cohesive ends, if possible. The vector and insert fragments can be assembled as illustrated (Figure 3a). Alternatively, the desired circular plasmid can be assembled from multiple PCR fragments using the strategy outlined in Figure 3a.
Ligation-independent cloning (LIC)
To circumvent limitations of the classical cut-and-paste methods for generating hybrid genes, LIC was developed to clone PCR fragments independent of restriction enzyme digestion and ligation [42]. The method relies on the use of DNA polymerases with 3′ to 5′ exonuclease activities (T4 or Pfu polymerases) to create 12 base 5′ overhangs on DNA fragments for annealing (Figure 4 ). With its unique properties, LIC has been used to achieve seamless cloning to join ORFs encoding protein domains for protein production 12, 39, 43, 44. Using chimeric PCR primers containing ribonucleotides, LIC ends with cohesive overhangs can also be generated by treating the PCR products with other reagents, such as uracil DNA glycosylase [45] or rare-earth metal ions [46]. Jarrell and colleagues [47] used primers with three consecutive ribonucleotide bases or a single 2′-O-methyl ribonucleotide to terminate DNA synthesis at the complementary strand, thus generating single-stranded overhangs during PCR. Given the simplicity, this method can be used to facilitate high-throughput cloning experiments.
Figure 4.

Seamless cloning by ligation-independent cloning (LIC). The diagram shows seamless fusion of fragment X with fragments Y and Z contained in a vector. Fragment X is first PCR amplified and purified. The primers are designed so that the 5′-end 12 bases are free of one specific nucleotide (e.g. dT as an example). The product is then treated with a DNA polymerase possessing 3′-5′ exonuclease activity (such as T4 DNA polymerase and Pfu) in the presence of dATP. The polymerase starts to remove nucleotides from 3′-ends of the fragment until a dA base is encountered and removed, which is subsequently added back by the enzyme's 5′-3′ polymerase activity. This reaction generates 12 base (or longer) overhangs on fragment X. The vector fragment, which contains fragments Y and Z was engineered and prepared similarly to yield 12 base (or longer) overhangs that are complementary to the insert fragment. The plasmid backbone contains an origin of replication (ori) and a selectable marker (sm). The LIC-ready vector fragment and the insert fragment are then annealed to form circular duplexes, which are recovered in E. coli following transformation 39, 42. Seamless fusion of X with Y and Z is achieved if the nucleotides comprising the LIC overhangs are made to be parts of X or its fusion partners.
The In-Fusion™ PCR cloning system (www.bdbiosciences.com) allows cloning of a PCR fragment into any linearized vector, as long as the PCR fragment contains 15 bp arms homologous to those in the vector fragment [48]. However, the exact components of the enzyme(s) involved in this reaction are not disclosed by the company. Because the terminal ends are crucial for this reaction, the vector fragment needs to be specially prepared to achieve seamless fusion of the insert with an existing ORF in the vector. The vector fragment can be prepared using inverse PCR, or by treating with a type IIS restriction enzyme to remove unwanted nucleotides [40]. The In-Fusion™ system is so far the most straightforward system for PCR cloning, it should enable automation of PCR cloning.
In vivo recombination
Homologous recombination allows exchange of genetic material between two molecules with homologous sequence regions. Because the location for homologous recombination can be freely chosen, it allows seamless DNA manipulation. Traditionally, this process is used in allelic replacement for gene function studies in E. coli and yeast cells. This is usually achieved by a two-step homologous recombination process 18, 49. A circular plasmid bearing a positive selectable marker and a negative selectable marker is first incorporated into a specific site of the target ORF on the chromosome or an episomal vector. Recombinants are selected through the positive selection marker. The selection fragment is subsequently replaced with another fragment containing a modified version of the ORF by counter selection against the negative selectable marker. This process allows precise and seamless gene manipulation on chromosome(s). In yeast, manipulation of genes carried on a plasmid is traditionally achieved by a one-step gap repair process, which requires only ∼30 bp homologous arms [50]. Using PCR technology, the hybrid constructs for such gap repair experiments can easily be generated to achieve deletion, insertion and sequence replacement. A yeast–E. coli–mammalian shuttle vector was used for generating hybrid fusion genes seamlessly in Saccharomyces cerevesiae followed by plasmid rescue in E. coli [51].
Homologous recombination in E. coli can be achieved by three different mechanisms: RecA-dependent, RecA-independent and Red/ET-dependent. RecA binds to single-stranded segments of DNA and promotes strand invasion and exchange between homologous sequences. RecA-mediated recombination requires a long homologous region (>1 Kb) and occurs at a low frequency [52]. It has been used to generate recombinant adenoviral genomes [53] and in the two-step allelic replacement experiment to modify genes carried on bacterial artificial chromosomes (BACs) and P1-derived artificial chromosomes (PACs) 54, 55. The RecA-independent pathway works in recA strains and requires the two DNA fragments to be recombined to have >12 bp homologies at the ends 56, 57, 58 (Figure 5 ). Co-transformation of the two linear DNA fragments allows recovery of circular plasmids. However, the exact mechanism for this reaction is unclear and the cloning efficiency can be low. A high cloning efficiency is obtained when the origin of replication and a selectable marker are contained in a separate fragment to enforce recombination 57, 58. This strategy has been used to generate a series of cysteine mutations of a protein in a high-throughput fashion [59]. The Red/ET system only requires 30–50 bp homologous regions between a linear vector fragment and a linear insert fragment for efficient recombination in E. coli (Figure 5). The technology relies on the function of the Redα/Redβ protein pair that of λ prophage, or their functionally equivalent RecE/RecT protein pair of Rac prophage, in which Redα and RecE are 5′-3′ exonucleases and Redβ and RecT are single-stranded DNA annealing proteins 52, 61. In recBC sbcA mutant strains, the RecBCD exonuclease activity is inactivated and the RedE/RedT proteins are expressed from a cryptic Rac prophage that is activated by the sbcA mutation, homologous recombination between linear fragments is possible 52, 60, 61. In recBC+ strains, homologous recombination between linear fragments can be enabled by over-expressing RedE/RedT proteins and the Redγ gene product, which inhibits RecBCD exonuclease activity 51, 60”.
Figure 5.
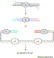
Seamless cloning by in vivo recombination. The diagram shows seamless fusion of fragment X with fragments Y and/or Z contained in a vector. Both the insert and the vector fragments are PCR amplified. The primers used in the reaction are designed so that the products contain a stretch of homologous sequences (15–40 bps) at the ends. The plasmid backbone contains an origin of replication (ori) and a selectable marker (sm). The two DNA fragments are then co-transformed into E. coli for in vivo recombination. Seamless fusion is achieved as long as the homologous sequences are parts of fragment X or its fusion partners.
The RecA-independent system and the Red/ET system are thus functionally comparable to the gap repair system in yeast cells. DNA fragments with the short arms can be generated by PCR and used directly in the in vivo recombination. These E. coli gap repair approaches have been used in construction generation for gene expression, cloning genes from complex sources and in achieving allelic replacement on chromosomes 52, 61.
Concluding remarks
Seamless cloning and gene fusion is the ideal situation for creating hybrid DNA molecules for all applications. As discussed here, in many cases it is imperative to ensure that no extra sequences are added during construction to enable straightforward data interpretation. In certain situations in which the presence of additional nucleotides or amino acid residues is unlikely to have a negative impact, making the hybrid construct free of operational sequences can only help assure the perfect design. However, although the presence of the operational sequence might not seem to have an undesired effect for one application of the hybrid molecule (for example in enzymatic assays) it might be problematic for another application, such as crystallization. Thus, being able to create the molecules exactly as desired adds value to data quality and reduces ambiguities in data interpretation.
Seamless cloning and gene fusions do not come without a price. Some procedures require special engineering or preparation of the insert and/or vector DNA fragments. For example, vectors for LIC must be free of extra nucleotide residues at the ends. All the methods described rely on the use of PCR to generate the correct ends on the insert and/or the vector fragments to enable seamless gene fusion or cloning. Although PCR fidelity was an issue for long PCRs in the late 1980s, it became less of a concern with the use of high fidelity DNA polymerases, such as Pfu DNA polymerase [62]. The use of high fidelity DNA polymerases or their blends has greatly facilitated the generation of long fusion genes and viral genomes 19, 26. Nevertheless sequencing confirmation of the fusion genes is always required. In addition, because the fusion junctions are free of operational sequences, such as restriction site(s) or site-specific recombination sequences, the fusion gene or part of it cannot be readily transferred to other vectors. This is in strong contrast to those hybrid genes with a seam, in which the DNA components can be transferred to other vectors by the ‘cut-and-paste’ approach or by site-specific recombination systems, such as the Gateway™ system (www.invitrogen.com) [63] or the Creator™ system [64] (www.bdbiosciences.com). Whenever possible, benefits of both the seamless system and the facile transfer system should be combined. For example, the hybrid gene can be assembled seamlessly first by overlap PCR and then subcloned into a Gateway™ entry vector to facilitate facile transfer of the hybrid ORF to other destination vectors.
With the accuracy of seamless fusion for hybrid genes and the flexibility of the facile transfer systems, our ability to manipulate genes and DNA elements has been enhanced. These technologies will enable construct generation for gene function studies and drug discovery at all levels, including gene knockout and knock-in studies in animal models or in cells, mutagenesis and allelic replacement studies, heterologous protein production and assay development.
Acknowledgements
Q.L. thanks Robert Ames, Thomas Kost, Danuta Mossakowska, Robert Kirkpatrick, Kyung Johanson, Christopher Jones and Abby Sukman at GlaxoSmithKline for critical reading of the manuscript and for helpful discussions on the topic.
References
- 1.Lu Q. Plasmid vectors for gene cloning and expression. In: Funnell B.E., Phillips G.J., editors. Plasmid Biology. ASM Press; 2004. pp. 545–566. [Google Scholar]
- 2.McKnight S.L., Kingsbury R. Transcriptional control signals of a eukaryotic protein-coding gene. Science. 1982;217:316–324. doi: 10.1126/science.6283634. [DOI] [PubMed] [Google Scholar]
- 3.Patten P.A. Applications of DNA shuffling to pharmaceuticals and vaccines. Curr. Opin. Biotechnol. 1997;8:724–733. doi: 10.1016/s0958-1669(97)80127-9. [DOI] [PubMed] [Google Scholar]
- 4.Mikheeva S., Jarrell K.A. Use of engineered ribozymes to catalyze chimeric gene assembly. Proc. Natl. Acad. Sci. U. S. A. 1996;93:7486–7490. doi: 10.1073/pnas.93.15.7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ayre B.G. Design of highly specific cytotoxins by using trans-splicing ribozymes. Proc. Natl. Acad. Sci. U. S. A. 1999;96:3507–3512. doi: 10.1073/pnas.96.7.3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marshall S.A. Rational design and engineering of therapeutic proteins. Drug Discov. Today. 2003;8:212–221. doi: 10.1016/s1359-6446(03)02610-2. [DOI] [PubMed] [Google Scholar]
- 7.Muir T.W. Semisynthesis of proteins by expressed protein ligation. Annu. Rev. Biochem. 2003;72:249–289. doi: 10.1146/annurev.biochem.72.121801.161900. [DOI] [PubMed] [Google Scholar]
- 8.Proudfoot A.E. Extension of recombinant human RANTES by the retention of the initiation methionine produces a potent antagonist. J. Biol. Chem. 1996;271:2599–2603. doi: 10.1074/jbc.271.5.2599. [DOI] [PubMed] [Google Scholar]
- 9.Kirkpatrick R.B. A bicistronic expression system for bacterial production of authentic human interleukin-18. Protein Expr. Purif. 2003;27:279–292. doi: 10.1016/s1046-5928(02)00606-x. [DOI] [PubMed] [Google Scholar]
- 10.Wingfield P. N-terminal-methionylated interleukin-1β has reduced receptor-binding affinity. FEBS Lett. 1987;215:160–164. doi: 10.1016/0014-5793(87)80133-3. [DOI] [PubMed] [Google Scholar]
- 11.Loison G. Expression and secretion in S. cerevisiae of biologically active leech hirudin. Biotechnology. 1988;6:72–77. [Google Scholar]
- 12.Hosfield T., Lu Q. Influence of the amino acid residue downstream of (Asp)4Lys on enterokinase cleavage of a fusion protein. Anal. Biochem. 1999;269:10–16. doi: 10.1006/abio.1998.3084. [DOI] [PubMed] [Google Scholar]
- 13.Jenny R.J. A critical review of the methods for cleavage of fusion proteins with thrombin and factor Xa. Protein Expr. Purif. 2003;31:1–11. doi: 10.1016/s1046-5928(03)00168-2. [DOI] [PubMed] [Google Scholar]
- 14.Li S-J., Hochstrasser M. A new protease required for cell-cycle progression in yeast. Nature. 1999;398:246–251. doi: 10.1038/18457. [DOI] [PubMed] [Google Scholar]
- 15.Wang T. Biotin-ubiquitin tagging of mammalian proteins in Escherichia coli. Protein Expr. Purif. 2003;30:140–149. doi: 10.1016/s1046-5928(03)00098-6. [DOI] [PubMed] [Google Scholar]
- 16.Kapust R.B. The P1′ specificity of tobacco etch virus protease. Biochem. Biophys. Res. Commun. 2002;294:949–955. doi: 10.1016/S0006-291X(02)00574-0. [DOI] [PubMed] [Google Scholar]
- 17.Maguilevsky N. Production of authentic human proapolipoprotein A-I in Escherichia coli: Strategies for the removal of the amino-terminal methionine. J. Biotechnol. 1993;27:159–172. doi: 10.1016/0168-1656(93)90105-v. [DOI] [PubMed] [Google Scholar]
- 18.Link A.J. Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: application to open reading frame characterization. J. Bacteriol. 1997;179:6228–6237. doi: 10.1128/jb.179.20.6228-6237.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yount B. Systematic assembly of a full-length infectious cDNA of mouse hepatitis virus strain A59. J. Virol. 2002;76:11065–11078. doi: 10.1128/JVI.76.21.11065-11078.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sudhof T.C. Three direct repeats and a TATA-like sequence are required for regulated expression of the human low density lipoprotein receptor gene. J. Biol. Chem. 1987;262:10773–10779. [PubMed] [Google Scholar]
- 21.Bachmair A. In vivo half-life of a protein is a function of its amino-terminal residue. Science. 1986;234:179–186. doi: 10.1126/science.3018930. [DOI] [PubMed] [Google Scholar]
- 22.Bachmair A., Varshavsky A. The degradation signal in a short-lived protein. Cell. 1989;56:1019–1032. doi: 10.1016/0092-8674(89)90635-1. [DOI] [PubMed] [Google Scholar]
- 23.Higuchi R. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 1988;16:7351–7367. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horton R.M. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 25.Gao X. Thermodynamically balanced inside-out (TBIO) PCR-based gene synthesis: a novel method of primer design for high-fidelity assembly of longer gene sequences. Nucleic Acids Res. 2003;31:e143. doi: 10.1093/nar/gng143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shevchuk N.A. Construction of long DNA molecules using long PCR-based fusion of several fragments simultaneously. Nucleic Acids Res. 2004;32:e19. doi: 10.1093/nar/gnh014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Casimiro D.R. PCR-based gene synthesis and protein NMR Spectroscopy. Structure. 1997;5:1401–1412. doi: 10.1016/s0969-2126(97)00291-8. [DOI] [PubMed] [Google Scholar]
- 28.Braman J., editor. 2nd edn. Vol. 182. Humana Press; 2002. (In Vitro Mutagenesis Protocols, Methods in Molecular Biology). [Google Scholar]
- 29.Weiner M.P. Site-directed mutagenesis of double-stranded DNA by the polymerase chain reaction. Gene. 1994;151:119–123. doi: 10.1016/0378-1119(94)90641-6. [DOI] [PubMed] [Google Scholar]
- 30.Papworth C. Site-directed mutagenesis in one day with >80% efficiency. Strategies. 1996;9:3–4. [Google Scholar]
- 31.Bauer, J.C. et al. Stratagene (1998) Circular site-directed mutagenesis. US patent #5789166 and #6391548
- 32.Wang W., Malcolm B.A. Two stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuickChange™ site-directed mutagenesis. Biotechniques. 1999;26:680–682. doi: 10.2144/99264st03. [DOI] [PubMed] [Google Scholar]
- 33.Geiser M. Integration of PCR fragments at any specific sites within cloning vectors without the use of restriction enzymes and DNA ligase. Biotechniques. 2001;31:88–92. doi: 10.2144/01311st05. [DOI] [PubMed] [Google Scholar]
- 34.Hogrefe H.H. Creating randomized amino acid libraries with the QuickChange™ multi site-directed mutagenesis kit. Biotechniques. 2002;33:1158–1165. doi: 10.2144/02335pf01. [DOI] [PubMed] [Google Scholar]
- 35.Szybalski W. Class-IIS restriction enzymes – a review. Gene. 1991;100:13–26. doi: 10.1016/0378-1119(91)90345-c. [DOI] [PubMed] [Google Scholar]
- 36.Lebedenko E.N. Method of artificial DNA splicing by directed ligation. Nucleic Acids Res. 1991;19:6757–6761. doi: 10.1093/nar/19.24.6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rebatchouk D. NOMAD: A versatile strategy for in vitro DNA manipulation applied to promoter analysis and vector design. Proc. Natl. Acad. Sci. U. S. A. 1996;93:10891–10896. doi: 10.1073/pnas.93.20.10891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Padgett K.A., Sorge J.A. Creating seamless junctions independent of restriction sites in PCR cloning. Gene. 1996;168:31–35. doi: 10.1016/0378-1119(95)00731-8. [DOI] [PubMed] [Google Scholar]
- 39.Wyborski D.L. An Escherichia coli expression vector that allows recovery of proteins with native N-termini from purified calmodulin-binding peptide fusions. Protein Expr. Purif. 1999;16:1–10. doi: 10.1006/prep.1999.1064. [DOI] [PubMed] [Google Scholar]
- 40.Stemmer W.P., Morris S.K. Enzymatic inverse PCR: a restriction site independent, single-fragment method for high-efficiency, site-directed mutagenesis. Biotechniques. 1992;13:214–220. [PubMed] [Google Scholar]
- 41.McClelland M. Effect of site-specific modification on restriction endonucleases and DNA modification methyltransferases. Nucleic Acids Res. 1994;22:3640–3659. doi: 10.1093/nar/22.17.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aslanidis C., de Jone P.J. Ligation-independent cloning of PCR products (LIC-PCR) Nucleic Acids Res. 1990;18:6069–6074. doi: 10.1093/nar/18.20.6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu Q. Using Schizosaccharomyces pombe as a host for expression and purification of eukaryotic proteins. Gene. 1997;200:135–144. doi: 10.1016/s0378-1119(97)00393-4. [DOI] [PubMed] [Google Scholar]
- 44.Hosfield T., Lu Q. S. pombe expression vector with 6x(His) tag for protein purification and potential for ligation-independent cloning. Biotechniques. 1999;27:58–60. doi: 10.2144/99271bm10. [DOI] [PubMed] [Google Scholar]
- 45.Rashtchian A. Uracil DNA glycosylase-mediated cloning of PCR-amplified DNA: application to genomic and cDNA cloning. Anal. Biochem. 1992;206:91–97. doi: 10.1016/s0003-2697(05)80015-6. [DOI] [PubMed] [Google Scholar]
- 46.Chen G.J. Universal restriction site-free cloning method using chimeric primers. Biotechniques. 2002;32:516–520. doi: 10.2144/02323st02. [DOI] [PubMed] [Google Scholar]
- 47.Donahue W.F. Rapid gene cloning using terminator primers and modular vectors. Nucleic Acids Res. 2002;30:e95. doi: 10.1093/nar/gnf094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clontechniques BD In-Fusion cloning kit. Precise, directional cloning of PCR products without restriction enzymes. CLONTECHniques. 2002;XVII:10–11. [Google Scholar]
- 49.Rothstein R. Targeting, discruption, replacement, and allele rescue: integrative DNA transformation in yeast. Methods Enzymol. 1991;194:281–301. doi: 10.1016/0076-6879(91)94022-5. [DOI] [PubMed] [Google Scholar]
- 50.Hua S-B. Minimum length of sequence homology required for in vivo cloning by homologous recombination in yeast. Plasmid. 1997;38:91–96. doi: 10.1006/plas.1997.1305. [DOI] [PubMed] [Google Scholar]
- 51.DeMarini D.J. Oligonucleotide-mediated, PCR-independent cloning by homologous recombination. Biotechniques. 2001;30:520–523. doi: 10.2144/01303st02. [DOI] [PubMed] [Google Scholar]
- 52.Muyrers J.P.P. Techniques: recombinogenic engineering – new options for cloning and manipulating DNA. Trends Biochem. Sci. 2001;26:325–331. doi: 10.1016/s0968-0004(00)01757-6. [DOI] [PubMed] [Google Scholar]
- 53.Danthinne X., Imperiale M.J. Production of first generation adenovirus vectors: a review. Gene Ther. 2000;7:1707–1714. doi: 10.1038/sj.gt.3301301. [DOI] [PubMed] [Google Scholar]
- 54.Yang X.W. Homologous recombination based modification in Escherichia coli and germline transmission in transgenic mice of a bacterial artificial chromosome. Nat. Biotechnol. 1997;15:859–865. doi: 10.1038/nbt0997-859. [DOI] [PubMed] [Google Scholar]
- 55.Imam A.M.A. Modification of human β-globin locus PAC clones by homologous recombination in Escherichia coli. Nucleic Acids Res. 2000;28:e65. doi: 10.1093/nar/28.12.e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bubeck P. Rapid cloning by homologous recombination in vivo. Nucleic Acids Res. 1993;21:3601–3602. doi: 10.1093/nar/21.15.3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Howorka S., Bayley H. Improved protocol for high-throughput cysteine scanning mutagenesis. Biotechniques. 1998;25:764–772. doi: 10.2144/98255bm03. [DOI] [PubMed] [Google Scholar]
- 58.Howorka, S. and Bayley, H. (2002) High-throughput scanning mutagenesis by recombination polymerase chain reaction. In In Vitro Mutagenesis Protocols, (2nd edn), Methods in Molecular Biology, (Vol. 182), pp 139–147 [DOI] [PubMed]
- 59.Howorka S. Surface-accessible residues in the monomeric and assembled forms of a bacterial surface layer protein. J. Biol. Chem. 2000;275:37876–37886. doi: 10.1074/jbc.M003838200. [DOI] [PubMed] [Google Scholar]
- 60.Court D.L. Genetic engineering using homologous recombination. Annu. Rev. Genet. 2002;36:361–388. doi: 10.1146/annurev.genet.36.061102.093104. [DOI] [PubMed] [Google Scholar]
- 61.Zhang Y. A new logic for DNA engineering using recombination in Escherichia coli. Nature Genet. 1998;20:123–128. doi: 10.1038/2417. [DOI] [PubMed] [Google Scholar]
- 62.Cline J. PCR fidelity of Pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res. 1996;24:3546–3551. doi: 10.1093/nar/24.18.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hartley J.L. DNA cloning using in vitro site-specific recombination. Genome Res. 2000;10:1788–1795. doi: 10.1101/gr.143000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Farmer, A.A. Clontech Laboratories Inc. (2002) Recombinase-based methods for producing expression vectors and compositions for use in practicing the same. US patent #6410317
- 65.Cunningham B.C., Wells J.A. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science. 1989;244:1081–1085. doi: 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- 66.Harlow P.P. Construction of linker-scanning mutations using the polymerase chain reaction. Methods Mol. Biol. 1994;31:87–96. doi: 10.1385/0-89603-258-2:87. [DOI] [PubMed] [Google Scholar]
- 67.Varshavsky A. Ubiquitin fusion technique and its descendants. Methods Enzymol. 2000;327:578–593. doi: 10.1016/s0076-6879(00)27303-5. [DOI] [PubMed] [Google Scholar]
- 68.Varshavsky A. The N-end rule: functions, mysteries, uses. Proc. Natl. Acad. Sci. U. S. A. 1996;93:12142–12149. doi: 10.1073/pnas.93.22.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ecker D.J. Increasing gene expression in yeast by fusion to ubiquitin. J. Biol. Chem. 1989;264:7715–7719. [PubMed] [Google Scholar]