

Abstract
The sequence of the S gene of a field canine coronavirus (CCoV), strain Elmo/02, revealed low nucleotide (61%) and amino acid (54%) identity to reference CCoV strains. The highest correlation (77% nt and 81.7% aa) was found with feline coronavirus type I. A PCR assay for the S gene of strain Elmo/02 detected analogous CCoVs of different geographic origin, all which exhibited at least 92–96% nucleotide identity to each other and to strain Elmo/02. The evident genetic divergence between the reference CCoV strains and the newly identified Elmo/02-like CCoVs strongly suggests that a novel genotype of CCoV is widespread in the dog population.
Keywords: Coronavirus, Dog, Genotype, Diversity
1. Introduction
Canine coronavirus (CCoV) is an enveloped, positive stranded RNA virus of dogs associated with moderate to severe enteritis in young pups. The genome contains two large open reading frames (ORFs), 1a and 1b, encoding two polyproteins leading to the viral replicase formation. Downstream to the ORF1b, there are 8–10 smaller ORFs encoding for the structural proteins S (ORF2), E (ORF4), M (ORF5) and the nucleocapsid (N) protein (Enjuanes et al., 2000).
The small membrane protein (E) has been found recently to be important for viral envelope assembling (Raamsman et al., 2000). The M protein is a type III glycoprotein consisting of a short amino-terminal ectodomain, a triple-spanning transmembrane domain, and a long carboxyl-terminal inner domain (Rottier, 1995). The ORF2 encodes for a glycosilated protein (S) ranging from 1160 to 1452 amino acids (aa) in length (Enjuanes et al., 2000), constituting the large, petal-shaped spikes on the surface of the virion. This large protein can be divided into three structural domains. The large external domain at the N-terminus is divided further into two subdomains S1 and S2. The S1 sub-domain includes the N-terminal half of the molecule and forms the globular portion of the spikes. It contains sequences that are responsible for binding to specific receptors on the membrane of susceptible cells. S1 sequences are variable, containing various degrees of deletions and substitutions in different coronavirus strains or isolates. Mutations in the S1 region have been associated with altered antigenicity and pathogenicity. In contrast, S2 sequences are more conserved and contain two heptad repeat motifs that suggest a coiled-coil structure (Lai and Holmes, 2001).
On the basis of phylogenetic analysis and antigenic cross reactivity, three groups can be distinguished in the Coronaviridae family. Group I includes CCoV, the transmissible gastroenteritis virus of swine (TGEV), the porcine epidemic diarrhoea virus (PEDV), the porcine respiratory coronavirus (PRCoV), the feline coronaviruses (FCoVs) and the human coronavirus 229E (HCoV 229E). FCoVs can be distinguished into two serotypes, I and II, on the basis of a virus neutralization assay in vitro using both type-specific feline sera and monoclonal antibodies directed against the S protein (Herrewegh et al., 1998). In the field, FCoVs type I are predominant and FCoVs type II are detected only sporadically. Differences in the S gene of FCoVs type I and that of FCoVs type II may also account for the different properties observed in vitro, as indeed FCoVs type I grow poorly in tissue culture cells (Pedersen et al., 1984) while type II strains grow well.
In a previous study, sequence analysis of CCoVs detected in faecal samples collected from dogs with diarrhoea revealed multiple nucleotide substitutions accumulating over a fragment of the M gene (Pratelli et al., 2001). A genetic drift to FCoV type II was also observed in the sequence of CCoVs detected in the faeces of two pups infected naturally during the late stages of long-term viral shedding. It was thus hypothesized that (i) the dogs might have been infected by a mixed population of genetically different CCoVs, or (ii) the viruses detected in both the pups were the result of mutation/recombination events (Pratelli et al., 2002b).
Subsequently, extensive sequence analysis on multiple regions of the viral genome, including ORF1a, ORF1b and ORF5, of several CCoV positive faecal samples provided strong evidence for the existence of two separate genetic clusters of CCoV. The first cluster includes CCoVs intermingled with reference CCoV strains, such as Insavc-1 and K378, while the second cluster segregates separately from CCoVs and, presumably, represents a genetic outlier referred to as FCoV-like CCoV (Pratelli et al., 2003).
The aim of the present study was to evaluate the genetic differences between the FCoV-like and the ‘typical’ CCoVs in the sequence of the gene encoding for the S protein.
2. Materials and methods
2.1. Faecal samples
Twenty faecal samples, collected in four kennels in Southern Italy from 2–6 month-old pups affected with diarrhoea, were tested. Three of the kennels were sited in different areas of Puglia, about 50 km from each other, while the fourth shelter was located in Abruzzo, more than 400 km far from the other three. The faecal samples were stored at −20 °C until tested. All the samples were negative by a haemoagglutination test for canine parvovirus and positive for CCoV when submitted to a PCR assay targeting a fragment of the M gene (primers CCoV1–CCoV2) (Pratelli et al., 1999). The presence of FCoV-like CCoVs in the same samples was detected by means of a differential PCR assay, using primers (CCoV1a–CCoV2) able to recognise nucleotide substitutions conserved in the M gene across all the FCoV-like CCoVs (Pratelli et al., 2002a). The sequence of the primers and their positions in the M gene are shown in Table 1 .
Table 1.
Primers used for PCR amplification and sequence analysis
| Primer | Gene | Virusesa | Sequence 5′ to 3′ | Sense | Position | Amplicon size |
|---|---|---|---|---|---|---|
| CCoV1b | M | CCoV, FCoV-like CCoV, FCoV type II | TCCAGATATGTAATGTTCGG | + | 6729–6748d | 409 bp |
| CCoV2b | TCTGTTGAGTAATCACCAGCT | − | 7138–7118d | |||
| CCoV1ac | M | FCoV-like CCoV, FCoV type II | GTGCTTCCTCTTGAAGGTACA | + | 6900–6920d | 239 bp |
| CCoV2b | TCTGTTGAGTAATCACCAGCT | − | 7138–7118d | |||
| UCD1F | S | FCoV-like CCoV | CAGAATGGGAAGAAGTGACG | + | 3704–3723e | 502 bp |
| UCD1R | CACACATACCAAGGCCATTTT | − | 4185–4205e | |||
| V1F | S | FCoV-like CCoV | AAGGACGAGTGCACCGACTAYAAYATHTA | + | 2092–2120e | 1698 bp |
| V1R | TGCATACGTGTCATTAACACAA | − | 3738–3759e | |||
| V2F | S | FCoV-like CCoV | GACGGCTTCTCCTTCAACAAYTGGTTYHT | + | 868–896e | 1420 bp |
| V2R | CAGCAGCTTGAGCAGTTAAATC | − | 2257–2278e | |||
| V3F | S | FCoV-like CCoV | GTTTCTGATGCTATTAGTACTGTTTCC | + | 3274–3300e | 744 bp |
| V3R | ACCTTCAGTAAAATCTGGAATTGTG | − | 3993–4017e |
2.2. PCR on the S gene
Comparative sequence analysis of the S gene have revealed a higher degree of variation at the N-terminus rather than at the C-terminus of the S protein (Jacobs et al., 1987, Motokawa et al., 1996, Horsburgh and Brown, 1995, Wesley, 1999). Taking into account the sequence drift to FCoVs observed in the M gene, we designed a pair of primers, UCD1F–UCD1R, amplifying a 502 bp fragment at the very 3′ end of the sequence of the S gene of FCoVs type I (strains UCD1, KU-2 and Black), which encodes for the highly conserved C-terminus of the spike protein. All the 20 canine samples were tested with this primer pair and yielded an amplicon of the expected size. The sequence of the amplicons obtained was determined by direct sequencing of the PCR products and displayed 81–82% nucleotide identity to FCoV type I strains.
2.3. Determination of the sequence of the S gene of FCoV-like CCoV Elmo/02
To verify the extent of genetic variation between the two clusters of CCoV in the S gene, we determined the nearly-full length sequence of the ORF2 of one (Elmo/02) of the samples that had tested positive to FCoV-like CCoV.
Degenerate primers were designed with the CODEHOP strategy (Rose et al., 1998), using a wide selection of coronaviruses belonging to group I of the Coronaviridae. This strategy is based on the identification of blocks of homology in the amino acid sequences of distantly related organisms. Hybrid oligonucleotides, with a short 3′ degenerate core region and a longer 5′ consensus clamp region, are selected by retro-translation on the blocks individuated. CODEHOP sense primers with low degeneracy index were selected to amplify overlapping fragments of ORF2. One-step reverse transcription and PCR amplification were carried out using SuperScript™ One-Step RT-PCR for Long Templates (Life Technologies, Invitrogen. Milan, Italy). To select against any CCoV-like virus during PCR amplification, reverse primers specific for the S gene of FCoV-like CCoV Elmo/02 were used in combination with the forward degenerate primers. Reference CCoV strains, S378, K378, 45/93 (Buonavoglia et al., 1994), USDA, 1/71 and SE, were used as controls in the PCR reactions, to verify that no CCoV was amplified by the primers. The amplicons were cloned into pCR®2.1-TOPO® vectors (TOPO TA Cloning ® , Invitrogen, Milan, Italy) and the recombinant clones individuated by blue/white screening. Plasmid DNA was extracted and subjected to sequence analysis (Genome Express: Labo Grenoble, France). Following this strategy, the fragments of the FCoV-like virus Elmo/02 were inserted into the clones. A consensus of the sequences obtained was determined and the overlapping fragments were manually edited. Alignments and sequence analysis were performed using the bioedit software package (Hall, 1999). The guidelines of the strategy used to determine the sequence of the S gene of the FCoV-like CCoV are schematised in Fig. 1 . The position and sequence of the degenerate primers are reported in Table 1. The nucleotide sequence of Elmo/02 will appear in the DDBJ/EMBL/GenBank databases under accession no. AY170345. The amino acid sequence of the FCoV-like CCoV Elmo/02 was inferred and aligned with a selection of coronaviruses of the group I. Phylogenetic and molecular evolutionary analyses were conducted using MEGA version 2.1 (Kumar et al. 2001) and PAUP version 4.0b (Swofford, 1998). A parsimony tree was elaborated using a heuristic algorithm and supplying statistical support by bootstrapping over 100 replicates.
Fig. 1.
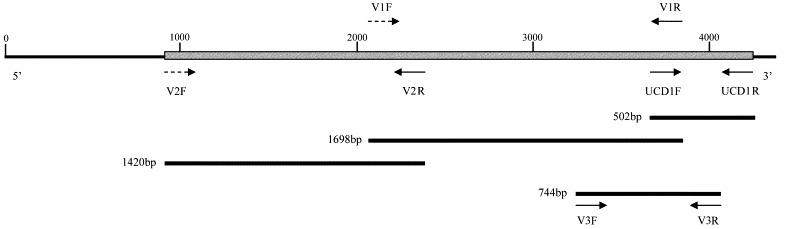
Outlines of the strategy followed to determine the sequence of ORF2 of strain Elmo/02. Dashed arrows indicate the degenerate primers. The position of the other primer pair used in this study, V3F–V3R, is also reported.
2.4. Analysis of field samples with a PCR specific for the S gene of FCoV-like CCoVs
The primer pair, V3F–V3R, designed on the sequence of virus Elmo/02, was chosen to selectively amplify FCoV-like CCoVs. The sequences and positions of the primers are shown in Table 1. All the samples previously characterised as FCoV-like by two separate primer pairs targeted to the M gene (CCoV1a–CCoV2) (Pratelli et al., 2002a) and to the S gene (UCD1F–UCD1R) were screened with the new primers specific for virus Elmo/02. The RNA was reverse transcribed with random hexamers using MuLV Reverse Transcriptase (Applied Biosystems, Roma, Italy) and then amplified with AmpliTaq DNA polymerase (Applied Biosystems, Roma, Italy), by 40 cycles at 94 °C for 1 min, 55 °C for 1 min and 72 °C for 1 min.
To assess the intra-genotypic variability in the S gene of FCoV-like CCoVs, four strains, each representative of a different geographical area, were selected and subjected to sequence analysis.
3. Results
All the 20 faecal samples characterised previously as FCoV-like CCoVs were recognised by the primer pair UCD1F–UCD1R, yielding an amplicon of the expected size of 502 bp.
About 80% (3347 nucleotides) of the ORF encoding for the S protein of strain Elmo/02 was determined. Using the ORF2 of strain UCD1 as a reference sequence, the fragment sequences between nt 868 and 4205 and between aa 300 and 1401 may be approximately localised.
The highest nucleotide identity was to FCoV type I strains KU-2, UCD1 and Black (∼77%), whereas identity to FCoVs type II and CCoVs was about 61%. Comparison of the inferred amino acid sequences revealed 80.81–81.76% identity to FCoVs type I, 53.88–54.31% to FCoVs type II and 54.31% to reference CCoV strains (Table 2 ). In accordance with previous observations, the sequence of the S protein was much more conserved at the C-terminus rather than at the N-terminus. For instance, amino acid identity to the best-matching sequence (strain KU-2) ranged from 73.39 to 88.4% and to strain Insavc-1 from 41.4 to 65.51% in the N- and C-terminus, respectively.
Table 2.
Amino acid identities between the spike protein of group I coronaviruses
| Elmo/02 | KU-2 | UCD1 | 79-1146 | 79-1683 | Insavc | K378 | Purdue | RM-4 | CV777 | VNot I-tk | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Elmo/02 | / | ||||||||||
| KU-2 | 80.81 | / | |||||||||
| UCD1 | 81.76 | 91.57 | / | ||||||||
| 79-1146 | 54.31 | 53.71 | 53.62 | / | |||||||
| 79-1683 | 53.88 | 53.19 | 53.02 | 97.34 | / | ||||||
| Insavc | 54.31 | 53.62 | 53.45 | 94.93 | 95.44 | / | |||||
| K378 | 54.31 | 53.45 | 53.36 | 95.62 | 95.96 | 95.36 | / | ||||
| Purdue | 54.05 | 53.71 | 53.45 | 93.98 | 94.24 | 92.52 | 93.38 | / | |||
| RM-4 | 53.62 | 53.28 | 53.1 | 92.6 | 92.52 | 91.23 | 91.92 | 96.56 | / | ||
| CV777 | 52.67 | 52.93 | 53.88 | 54.25 | 53.24 | 54.39 | 53.88 | 54.65 | 54.91 | / | |
| VNot I-tk | 50.69 | 51.38 | 51.98 | 53.28 | 54.31 | 52.87 | 53.19 | 53.19 | 53.28 | 55.68 | / |
Values indicate the arithmetic average×extrapolated from the amino acid matrix of comparison and are expressed in percentage.
The inferred amino acid sequence of the S protein of strain Elmo/02 is shown in Fig. 2 . Similar to other coronaviruses, there were several potential N-glycosilation sites, Asn–X–Ser (NXS) or Asn–X–Thr (NXT). Most of the glycosilation sites were conserved between strain Elmo/02 and feline/CCoVs, in particular with respect to the most closely related FCoVs type I. Interestingly, a potential cleavage site, the stretch of basic amino acids, Arg–Arg–Ala–Arg–Arg (RRARR), was found. The basic stretch is about at the same position as in the S protein of group II and group III coronaviruses, but it is absent in the S protein of all the other group I coronaviruses.
Fig. 2.
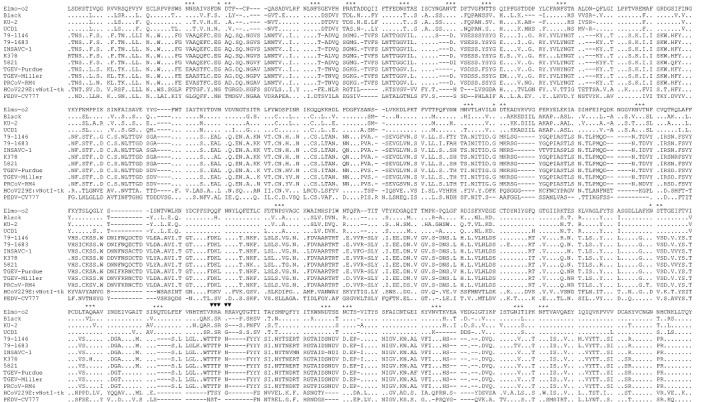
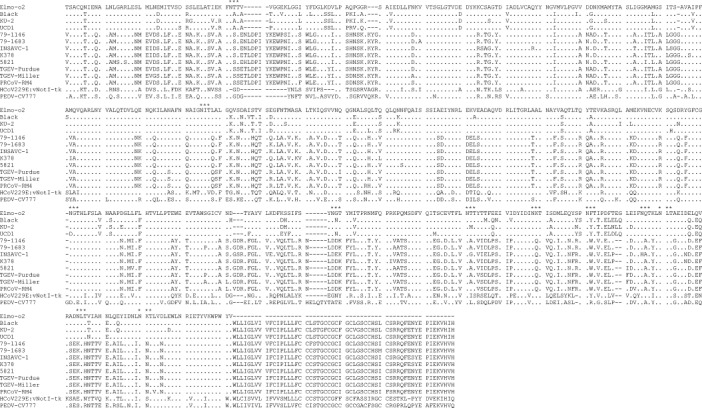
Alignment of the deduced amino acid sequence of the S protein of the FCoV-like strain Elmo/02 with reference CCoVs (Insavc-1, K378 and 5821), FCoVs type I (UCD1, Black and KU-2), FCoVs type II (79-1146 and 79-1683), TGEVs (Purdue and Miller), PRCoV (RM4), PEDV (CV777) and HCoV-229 (vNotI-tk). The potential glycosilation sites (*) and the putative cleavage site (▾) are indicated.
Parsimony analysis on the S protein of group I coronaviruses revealed that strain Elmo/02 is much more related to FCoVs type I rather than to typical CCoVs. Conversely, typical CCoVs tightly segregate with FCoVs type II and the porcine coronaviruses TGEV and PRCoV (Fig. 3 ).
Fig. 3.
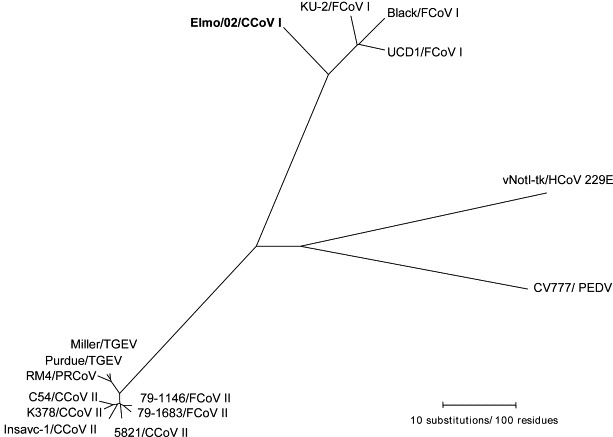
Parsimony phylogenetic tree of the S protein of group I coronaviruses. The tree is drawn to scale and rooted using the vaccinal strain vNot-tk of HCoV-229. Sequence for the coronavirus sequences reported is available from Gen Bank under the following accession numbers. CCoVs: Insavc-1, D13096; K378, X77047; C54, A22886; 5821, AB017789. FCoVs type II: 79-1146, X06170; 79-1683, X80799. FCoVs type I: KU-2: D32044; Black, AB088223; UCD1, AB088222. TGEV: Miller, S51223; Purdue, X05695. PRCoV: RM4, Z24675. PEDV: CV777, NC_00346. HCoV: vNotI-tk, NC_002645.1.
The new pair of primers, V3F–V3R, specific for strain Elmo/02, successfully amplified all the 20 samples tested, yielding an expected PCR product of 744 bp. The sequence of the V3F–V3R amplicon of four strains representative of different geographical areas was determined, revealing a nucleotide variability of 4–8%.
4. Discussion
Genetic divergence within the coronavirus group I is accounted for by linear evolution as well as by a sudden, dramatic shift generated by RNA deletions or recombination. For instance, the S protein of PEDV occupies an intermediate position between HCoV 229E and TGEV (Kocherhans et al., 2001), while the S protein of PRCoV is closely related to TGEV but has a large deletion in the N-terminus (more than 200 aa) that may explain the change in the pathobiology of the virus (Vaughn et al., 1994).
Comparative sequence analysis of the genome of FCoVs type I and type II and CCoV has demonstrated that FCoV type II has arisen from a template switch between FCoV type I and CCoV, which took place between the S and M genes. An additional template switch has been mapped in the ORF1b region for strain FCoV 79-1146 and in the ORF1ab region for strain FCoV 79-1683. The double recombination event determined the introduction of a large genome fragment, encompassing the CCoV-like S-encoding gene, into the background of a FCoV genome (Herrewegh et al., 1998).
The S gene of CCoV is closely related to FCoVs type II, TGEVs and PRCoVs, and is more divergent from FCoVs type I, PEDVs and HCoV 229E (Wesseling et al., 1994). So far, little evidence has been provided for genetic drifts or shifts affecting CCoV. Wesley (1999) has described a canine strain displaying a higher sequence identity to TGEV in the N-terminus of the S protein, explained as a possible recombination between CCoV and TGEV, and related to improved growth in swine testicular cells. The findings in the present study clearly indicate that a novel CCoV type, highly divergent from the reference CCoV strains, and more closely related to FCoVs type I, circulates among dogs. Indeed, by means of RT-PCR, Elmo/02-like strains were successfully detected in all the samples tested. All the samples had been characterised as ‘atypical’ CCoVs when screened with a RT-PCR targeted to the M gene and able to distinguish between the two genetic lineages previously identified (Pratelli et al., 2002a). Extensive sequence analysis of multiple regions in the ORF1a and 1b, as well in the M-encoding gene, has confirmed the existence of a distinct genetic lineage of CCoV, evolutionarily localised between CCoV and FCoV. Many amino acid residues observed in the M protein of FCoV-like CCoVs are the same as in FCoVs and presumably represent a retention of the sequence of an ancestral virus (Pratelli et al., 2003). Re-considering these data in the light of the findings of the present study and considering the analogies with closely related viruses, we have concluded that the extent of genetic variation observed within the CCoVs is limited in the ORF1a and slightly greater in the ORF1b and ORF5, though it still accounts for a clear pattern of segregation into a distinct genetic lineage. The two genotypes of CCoV diverge dramatically in the ORF2, where there is more than 38.4% nucleotide and about 45.5% amino acid variation from reference CCoVs. Analysis of the S gene of the Elmo/02-like CCoVs revealed a little degree of variation (4–8%), which may be explained by their different geographical origin. The majority of the sequence changes observed are conservative, demonstrating that there is some heterogeneity in the ORF2 of Elmo/02-like CCoVs. In conclusion, the findings suggest that the two canine genotypes underwent a linear evolution rather than a sudden shift originating from a recombinant event analogous to those leading to the appearance of FCoVs type II. Finally, recombination with an ancestral coronavirus from which FCoVs type I and Elmo/02-like CCoVs directly evolved may not be excluded.
Whether the Elmo/02-like CCoVs have phenotypic properties different from those of typical CCoVs, similarly to FCoVs type I and II, will be interesting to evaluate. The high divergence in the amino acid composition and the loss and gain of potential glycosilation sites, compared to the most closely related coronaviruses (FCoV type I, FCoV type II and typical CCoV), strongly suggest that the Elmo/02 strain is poorly correlated antigenically with the other coronaviruses of dogs and cats. Moreover, the presence of the stretch of basic residues RRARR is indicative of a potential cleavage of the protein (Wesseling et al., 1994). A similar basic motive is present, approximately in the same position, in all the coronaviruses identified to date of both group II and III, but it is absent in all the coronaviruses of group I. Cleavage of the S protein of coronaviruses has been correlated to cell-fusion activity in vitro (Hingley et al., 1998) but the potential implications in viral pathobiology have not been determined.
On the basis of the significant genetic differences between the reference and the Elmo/02-like CCoVs our tentative proposal is to designate the new genotype identified as CCoV type I, and to designate the reference strains, such as Insavc-1 and K378, as CCoVs type II. This new designation does not take into account the order of discovery of the viruses, but it is based on the genetic similarity between CCoVs type II and FCoVs type II and between CCoV type I and FCoV type I.
Acknowledgments
This study was supported by grants from CEGBA (Centro di Eccellenza di Genomica in Campo Biomedico e Agrario) and from Ministry of University, Italy (project: Enteriti virali del cane).
References
- Buonavoglia, C., Marsilio, F., Cavalli, A., Tiscar, P.G., 1994. L'infezione da coronavirus del cane: indagine sulla presenza del virus in Italia. Not. Farm. Vet. Nr. 2/94, ed. SCIVAC.
- Enjuanes L., Brian D., Cavanagh D., Holmes K., Lai M.M.C., Laude H., Masters P., Rottier P., Siddell S., Spaan W.J.M., Taguchi F., Talbot P. Coronaviridae. In: van Regenmortel M.H.V., Fauquet C.M., Bishop D.H.L., Carstens E.B., Estes M.K., Lemon S.M., Maniloff J., Mayo M.A., McGeoch D.J., Pringle C.R., Wickner R.B., editors. Virus Taxonomy, Classification and Nomenclature of Viruses. Academic Press; New York: 2000. pp. 835–849. [Google Scholar]
- Hall T.A. bioedit: a user-friendly biological sequence alignment and analysis program for Windows 95/98/NT. Nucl. Acids Symp. Ser. 1999;41:95–98. [Google Scholar]
- Herrewegh A.A.P.M., Smeenk I., Horzinek M.C., Rottier P.J.M., de Groot R.J. Feline coronavirus type II strains 79-1683 and 79-1146 originate from a double recombination between feline coronavirus type I and canine coronavirus. J. Virol. 1998;72:4508–4514. doi: 10.1128/jvi.72.5.4508-4514.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingley S.T., Leparc-Goffart I., Weiss R.S. The spike protein of murine coronavirus mouse hepatitis virus strain A59 is not cleaved in primary glial cells and primary hepatocytes. J. Virol. 1998;72:1606–1609. doi: 10.1128/jvi.72.2.1606-1609.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsburgh B.C., Brown T.D.K. Cloning, sequencing and expression of the S protein gene from two geographically distinct strains of canine coronavirus. Virus Res. 1995;39:63–74. doi: 10.1016/S0168-1702(95)00068-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs L., de Groot R., van der Zeijst B.A.M., Horzinek M.C., Spaan W. The nucleotide sequence of the peplomer gene of porcine transmissible gastroenteritis virus (TGEV): comparison with the sequence of the peplomer protein of feline infectious peritonitis virus (FIPV) Virus Res. 1987;8:363–371. doi: 10.1016/0168-1702(87)90008-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocherhans R., Bridgen A., Ackermann M., Tobler K. Completion of the porcine epidemic diarrhoea coronavirus (PEDV) genome sequence. Virus Genes. 2001;23:137–144. doi: 10.1023/A:1011831902219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Tamura K., Jakobsen I.B., Nei M. mega2: molecular evolutionary genetics analysis software. Bioinformatics. 2001;17:1244–1245. doi: 10.1093/bioinformatics/17.12.1244. [DOI] [PubMed] [Google Scholar]
- Lai M.M.C., Holmes K.V. Coronaviridae: the viruses and their replication. In: Knipe D.M., Howley P.M., Griffin D.E., Lamb R.A., Martin M.A., Roizman B., Strais S.E., editors. Vol. 1. Lippincott Williams and Wilkins; Philadelphia: 2001. pp. 1163–1185. (Fields Virology). [Google Scholar]
- Motokawa K., Hohdatsu T., Hashimoto H., Koyama H. Comparison of the amino acid sequence and phylogenetic analysis of the peplomer, integral membrane and nucleocapsid proteins of feline canine and porcine coronaviruses. Microbiol. Immunol. 1996;40:425–433. doi: 10.1111/j.1348-0421.1996.tb01089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen N.C., Black J.W., Boyle J.F., Evermann J.F., McKeiman A.J., Ott R.L. Pathogenic differences between various feline coronavirus isolates. In: Rottier P.J.M., van der Zeijst B.A.M., Spaan W.J.M., Horzinek M.C., editors. Molecular Biology and Pathogenesis of Coronaviruses. Plenum Press; New York: 1984. pp. 365–380. [Google Scholar]
- Pratelli A., Tempesta M., Greco G., Martella V., Buonavoglia C. Development of a nested PCR for the detection of canine coronavirus. J. Virol. Meth. 1999;80:11–15. doi: 10.1016/S0166-0934(99)00017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratelli A., Martella V., Elia G., Decaro N., Aliberti A., Buonavoglia D., Tempesta M., Buonavoglia C. Variation of the sequence in the gene encoding for transmembrane protein M of canine coronavirus (CCV) Mol. Cell. Probes. 2001;15:229–233. doi: 10.1006/mcpr.2001.0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratelli A., Tinelli A., Decaro N., Camero M., Elia G., Gentile A., Buonavoglia C. PCR assay for the detection and the identification of atypical canine coronavirus in dogs. J. Virol. Meth. 2002;106:209–213. doi: 10.1016/S0166-0934(02)00165-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratelli A., Elia G., Martella V., Tinelli A., Decaro N., Marsilio F., Buonavoglia D., Tempesta M., Buonavoglia C. M gene evolution of canine coronavirus in naturally infected dogs. Vet. Rec. 2002;151:758–761. [PubMed] [Google Scholar]
- Pratelli A., Martella V., Pistello M., Elia G., Decaro N., Buonavoglia D., Camero M., Tempesta M., Buonavoglia C. Identification of coronaviruses in dogs that segregate separately from the canine coronavirus genotype. J. Virol. Meth. 2003;107:213–222. doi: 10.1016/S0166-0934(02)00246-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raamsman M.J.B., Locker J.K., de Hooge A., de Vries A.A.F., Griffiths G., Vennema H., Rottier P.J.M. Characterization of the coronavirus mouse hepatitis virus strain A59 small membrane protein E. J. Virol. 2000;74:2333–2342. doi: 10.1128/jvi.74.5.2333-2342.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose T.M., Schultz E.R., Henikoff J.G., Pietrokovski S., McCallum C.M., Henikoff S. Consensus-degenerate hybrid oligonucleotide primers for amplification of distantly related sequences. NAR. 1998;26:1628–1635. doi: 10.1093/nar/26.7.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottier P.J.M. The coronavirus membrane protein. In: Siddell S.G., editor. The Coronaviridae. Plenum Press; New York: 1995. pp. 115–139. [Google Scholar]
- Swofford, D.L., 1998. PAUP*, phylogenetic analysis using parsimony (*and other methods), version 4.0b8. Sinauer, Sunderland, Mass.
- Vaughn E.M., Halbur P.G., Paul P.S. Three new isolates of porcine respiratory coronavirus with various pathogenicities and spike (S) gene deletions. J. Clin. Microbiol. 1994;32:1809–1812. doi: 10.1128/jcm.32.7.1809-1812.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesley R.D. The S gene of canine coronavirus, strain UCD-1, is more closely related to S gene of transmissible gastroenteritis virus than to that of feline infectious peritonitis virus. Virus Res. 1999;61:145–152. doi: 10.1016/S0168-1702(99)00032-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesseling J.G., Vennema H., Godeke G., Horzinek M.C., Rottier P.J.M. Nucleotide sequence and expression of the spike (S) gene of canine coronavirus and comparison with the S proteins of feline and porcine coronaviruses. J. Gen. Virol. 1994;75:1789–1794. doi: 10.1099/0022-1317-75-7-1789. [DOI] [PubMed] [Google Scholar]