

Abstract
Historically, serum therapy was previously used to combat infectious pathogens. However, serum sickness and anaphylaxis limited its broad application. The advancement of antibody engineering technologies has made it feasible to generate monoclonal antibodies. There are divergent methods for antibody engineering and optimization. In this chapter, we summarized the latest developments in engineering antibodies for infectious diseases.
Keywords: Engineering antibodies, Pathogens, Treatment
Introduction
The discovery of antibiotics has saved countless patients from pathogens. Thus, widespread availability, high efficacy and low costs make antibiotics a cornerstone of modern medicine. However, after many decades of empiric broad-spectrum antibiotics use and misuse, multiple antibiotic-resistant pathogens are emerging. These pathogens include, but not limited to, methicillin-resistant staphylococcus aureus, extreme drug-resistant Mycobacterium tuberculosis and plasmodium falciparum [17]. Antibiotic-resistant pathogens pose a threat to global health. Additionally, broad-spectrum antibiotics bring the risk of perturbing normal and beneficial microbiome in vivo [36]. The perturbations or dysbioses of normal or beneficial microbiome has been linked to a range of diseases including Clostridium difficile associated diarrhea, diabetes, obesity and immune defects [10]. These threats on public health worldwide are detrimental.
Clearly, there is a need to explore new weapons to combat infectious diseases. Among many alternatives, antibody-based therapy represents a major interest with the success in other diseases such as tumors (Opdivo and Keytruda), autoimmune diseases and inflammatory conditions (Remicade, Humira) [58]. With the advancement in antibody engineering, production and optimization, antibodies may hold great promise in combating infectious diseases. If successful, antibody-based therapy will provide precision weapons to destroy pathogens without comprising normal or beneficial microbiome.
Antibody-based therapy for infectious diseases is not new. As far as in the 1890s, serum immunoglobulin was used to neutralize bacterial virulence [7]. Later, serum therapy was applied to a wide range of other infectious diseases such as meningococcal meningitis, pneumococcal pneumonia, streptococcal scarlet fever, varicella, measles, the pandemic Spanish influenza [15] and Ebola virus disease as well as cytomegalovirus [14, 31]. However, adverse reactions such as serum sickness and anaphylaxis harmed the efficacy of serum therapy [9]. With the discovery of antibiotics, serum therapy in infectious pathogens quickly declined. Antibiotics therapy soon became a mainstay of bacterial infections treatment. However, the success did not last long with the emergence of antibiotic-resistant pathogens. The failure to develop new antibiotics with different mechanisms of actions again trapped patients by the super drug-resistant bacteria. Besides, for individuals with compromised immunity, antimicrobial chemotherapy provides a less effective treatment [28]. Obviously, antibiotics are ineffective at eliminating viruses. The emergence of antibiotics resistance calls for renewed efforts. Among the many options, monoclonal antibody (mAb) may be an alternative, reminiscent of the successful application of antitoxin in fighting against meningococcal meningitis in pre-antibiotic era. Indeed, Nebacumab, the first human therapeutic mAb reviewed by a regulatory agency, was engineered for the treatment of sepsis and gram-negative bacteremia [61]. However, the CHESS (Centocor: HA-1A Efficacy in Septic Shock) trial conducted in the USA which found non-statistically significant increase in mortality between the monoclonal anti-endotoxin antibody Nebacumab and placebo groups in patients with gram-negative bacteremia lead to the withdrawal of Nebacumab [20].Currently, only two humanized antibodies Palivizumab and Raxibacumab are approved for infectious diseases. Many other engineering antibodies against infectious pathogens are under development [2, 5, 16, 45, 67]. In this chapter, we summarize existing technologies for antibody engineering and optimization.
Serum Therapy in the Pre-antibiotics Era
Before the discovery of antibiotics, convalescent serum was the first effective strategy to combat infectious pathogens such as diphtheria, tetanus, hepatitis B, varicella, and cytomegalovirus et al. [13, 14]. It was later discovered that the mechanisms behind it were predominantly due to neutralizing activity and effector functions of immunoglobulins in serum. Antibodies derived from immune sera are polyclonal in nature. However, problems such as lot-to-lot variation and the immune response against animal-derived antibodies contributed to the reduced efficacy of serum therapy. Besides, the preparation of these serum products were expensive and labor-intensive. These complications together with the discovery of antibiotics limited the application of serum therapy in infectious pathogens after 1935. Nevertheless, this early attempt opened up the possibility of antibody-based therapies in infectious diseases.
Hybridoma Technology
The year 1975 witnessed the development of monoclonal antibody by mouse hybridoma technology [40]. This innovation makes it possible to produce large quantities of antibodies with a defined specificity in vitro, paving the way to use therapeutic monoclonal antibodies to treat different diseases. For example, E5 (XOMA Corp, Berkeley, Calif), an IgM antibody produced by hybridoma technology, was designed to neutralize endotoxin, the lipopolysaccharide component of the outer membrane of the J5 mutant of Echerichia coli [29]. The inconclusive results generated by clinical trials of E5 led to the withdrawal of this anti-endotoxin mAbs [29, p. 5].
Mouse monoclonal antibodies are limited by short-half life in circulation, inability to trigger human effector functions and the generation of human anti-mouse antibodies (the HAMA response). In an attempt to reduce the immunogenicity of therapeutic antibodies, efforts were directed to produce antibodies by using antibody engineering technology. Teng et al. generated a human monoclonal IgM antibody HA-1A by fusing human immunized B lymphocytes with heteromyeloma cells [61]. HA-1A was designed to fight against gram-negative bacteremia and shock by binding specifically to the lipid A domain of endotoxin. Clinical trials showed no overall benefit of HA-1A, but significant improvement in the survival rate in a subgroup of patients [76]. The common adverse events (hypersensitivity or allergic reactions) in children with meningococcal septic shock who were treated by HA-1A occurred at low rates (<2%) [26].
Approaches to Generate Humanized Antibodies
Traditional hybridoma technology generates antibodies with high immunogenicity. Such therapeutic antibodies in vivo invoke HAMA response. To attenuate the immunogenicity, several approaches are present to reduce non-human fragments in antibodies (Fig. 10.1).
Fig. 10.1.
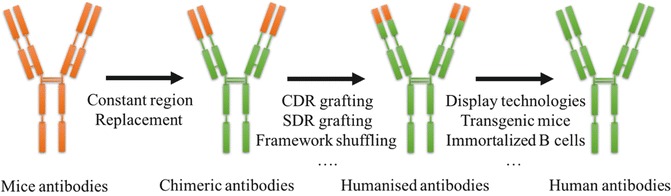
Chimeric antibodies and humanized antibodies
The orange segments originates from murine, the green segments are from human.
“CDR Grafting” Method
Many other strategies such as recombinant DNA methods are used to attenuate the human immune response, by reducing levels of non-human fragments in mAbs. Chimeric antibodies are produced by replacing mouse constant region with the human constant region. The generated antibodies with mouse variable regions and human constant regions were perceived as less immunogenic than mouse monoclonal antibodies . Nonetheless, human anti-mouse response still exists with the use of chimeric antibodies.
To further minimize the mouse component in the antibodies, murine-derived CDR loops responsible for antigen binding were grafted into the human variable-domain framework. Caution should be noted that such replacements often change the structural relationship between CDRs and framework regions (FRs). Additionally, the change of CDR loops may influence binding properties of humanized Abs [3]. Thus, additional molecule engineering is needed to restore the binding capacity of humanized antibody [53].
“CDR grafting” method are successfully used to generate “humanized” antibodies which are able to evoke immune effector functions such as ADCC and phagocytosis more effectively. However, the murine-derived CDR loops can still evoke the immune response in patients. To minimize the anti-idiotypic response in vivo, several strategies were exploited.
Specificity-Determining Residue (SDR) Grafting
In most cases, only part of the entire CDR residues are involved in antibody-antigen complexes. These residues located in the region of high variability are designated as specificity-determining residues (SDRs) [38]. Grafting the murine-derived SDRs onto the frameworks of human antibody generates humanized antibody with minimized immunogenicity. However, this strategy needs the identification of SDRs. Besides, the structure features of the target antibody should be preserved. Residues which are involved in VL-VH contact should also be preserved. Considerations should be taken that SDR grafting strategy usually generates antibodies with a reduced antigen-binding capacity. Thus, additional in vitro molecule engineering is needed to enhance antigen-binding affinity [23].
Framework Shuffling
“Framework shuffling” is another humanization approach by synthesizing a combinatorial library comprising CDRs of the non-human antibody fused in frame to pools of synthetic human germline frameworks. The corresponding libraries are then screened for antibodies with specific binding properties. This approach is simple, without requiring prior antibody structural knowledge, CDRs analysis or framework design [21, 22].
Human String Content Optimization (HSC)
Lazar et al. came up with a new paradigm termed “human string content” (HSC) for reducing immunogenic potential [43]. This approach is based on a metric of antibody humanness by comparing the non-human sequence with the human germline gene repertoire. The diversity of substitutions in a given variable heavy or light chain are scored as HSC. The targeted sequence is then humanized by maximizing its HSC.
Superhumanization
Superhumanization can be used to humanize mouse antibody. This method relies on comparison of CDRs. First, hypervariable loops of mice are compared with those of human. Variable sequences of human which are similar or identical to mice are then selected. Then, within the selected genes, those who share the highest homology to the mice sequence are used to construct a humanized antibody. Using this approach, antibodies with designed antigen-binding specificity and neutralizing activity can be generated [59].
Engineering Fully Human Monoclonal Antibodies
Although several humanized antibodies are approved for marketing, to some extent, humanized antibody still elicits human anti-human antibodies (HAHAs) response. For example, a patient after receiving adalimumab treatment (an effective treatment for rheumatoid arthritis) over a period of time, lost his initial response to adalimumab due to the formation of HAHAs [4].Besides, humanization of antibodies can be labor intensive, comprising complex procedures including sequence analysis, developing engineering technology, assessing binding properties, and evaluating HAHAs response. Therefore, developing fully human antibodies is a long pursuit. Several technologies including display technologies, transgenic animals, immortalized B cells and transchromosomal calves are available to generate fully human antibody.
Display Technologies
In vitro display technologies are among of the most commonly used methods for fully human antibody generation. Advantages of in vitro display technologies are overcoming immunological tolerance and generating antibodies against defined antigens. Specifically, phage and yeast display systems are best exemplified. Other display systems such as ribosomes, bacteria and virus display are also used to display repertoires of single-chain variable antibody fragments (scFvs) , antigen-binding fragments (Fabs) or domain antibodies (Dabs) on their surface.
These techniques mainly contained three steps:
Antibody Library Construction
Amplify human immunoglobulin variable regions derived from human B cells.
-
2.
Surface Display Vector Construction
The antibody library is cloned into the plasmid and transformed (transfected) in the host cell to construct surface display vector.
-
3.
Desired Antibody Screening
Desired antibodies are isolated by panning the library against the target antigen/epitope. Antibodies that don’t bind to the antigen are washed away and the binders are retained. The corresponding genes of variable regions are cloned into whole human IgG expression vectors and expressed in orthologous mammalian system to generate human mAbs.
Huse et al. first introduced a lambda phage-based system which expressed a collection of functional antibody fragments in Escherichia coli [32]. However, it was laborious to distribute the libraries on agar plates. Besides, this method only assessed repertoires of roughly 1 × 107, less than the natural antibody repertoire of 1 × 108 in each individual [58]. Later, progress was achieved in the area of display technologies by extending the library size and quantity. Theodore et al. produced mAbs against the West Nile virus (WNV) E protein by yeast surface display [47]. In vitro studies showed that one mAb could neutralize 10 different strains of WNV [47]. The animal experiment demonstrated the efficacy of the mAb against WNV by administering the mAb in a mouse model of WNV infection [47].
Different display systems may differ in antibody folding efficiency, post-translational modification and epitope accessibility which may generate antibodies with different binding properties [46]. For example, Bowley et al. compared yeast display and phage display by using the same HIV-1 immune scFv cDNA library and the same selecting antigen (HIV-1 gp120) [11]. Their results showed that yeast display system generated more full scFvs than phage display system [11]. Besides, yeast display system produced many more novel antibodies [11].
There are many display platforms, including phage display, ribosome display, yeast display et al. Among these display systems, phage display-based selections are most often preferred for the generation of fully human antibodies. Other display methods can be used as complementary methods. Display technologies have become a standard method to generate antibodies with high affinity. However, a drawback is that the antigen must be known prior, in order to select for antibodies with high affinity.
Transgenic Mice
Another strategy to generate human mAbs is by using human immunoglobulin transgenic mice [12]. With the development of embryonic stem cell and gene transfer methods, transgenic mice are generated to carry human immunoglobulin gene. These transgenic mice have a normal humoral immune response [34]. B cells that produce human antibodies are harvested from immunized transgenic mice, and then cloned either as hybridomas or in vitro combinatorial library. Antibodies obtained via transgenic mice are of good antigen-binding properties with low immunogenicity. There are three main transgenic mice platforms including XenoMouse, HuMab mouse and VelociMouse [8, 24].
However, flaws may exist when using transgenic mice method. Immune tolerance may arise when using immune antigens that are highly homologous to mouse. Thus, more immunizations or antibody screens are needed.
Transgenic mice are used to generate human antibodies against infectious pathogens including bacteria and virus. For example, Coughlin et al. generated a human monoclonal antibody against severe acute respiratory syndrome coronavirus (SARS-CoV) using XenoMouse [18]. Paul et al. generated a Human mAb (V2L2-MD) by immunizing humanized VelocImmune mice. The generated mAb is specific for P. aeruginosa PcrV, and can protect murine infection models from P. aeruginosa challenge [66].
Immortalized B Cells
Memory B cells are produced after infection or vaccination, and these antigen specific B cells could persist for a lifetime. Thus, memory B cells can be used as a source of human mAbs. After immortalization by Epstein Barr Virus (EBV), the antigen-specific B cells can be cultured to secrete a large amounts of antibodies. The surface immunoglobulins of EBV-immortalized B cells can be used to select antigen-specific B cells by fluorescence-activated cell sorting (FACS). Traggiai E et al. used this method to isolate 35 human monoclonal antibodies against SARS-CoV, and found one antibody exerted effective protection in vivo [64]. Selection of antibodies against H5N1, HCV, HIV and CMV by EBV-immortalized B cells were also reported [55, 57, 62, 68].
Producing human antibodies by EBV-immortalized B cells was limited by the low efficiency of EBV-immortalization. This limitation is overcome with the discovery that a TLR agonist can increase the efficiency of EBV B-cell immortalization and promote cloning of immortalized B cells [64]. Now, EBV-immortalized B cells has become an interesting shortcut to generate human monoclonal antibodies by isolating memory B cells from a donor who is infected or vaccinated by infectious pathogens.
Transchromosomal Calves
Transchromosomal calves are recently used to produce large-amounts of human mAbs by transferring mammalian artificial chromosome vectors carrying human Ig loci to bovine primary fetal fibroblasts to produce cloned transchromosomic (Tc) calves [42]. The cloned Tc calves will contain a high rate of artificial chromosome positive cells. The Ig loci transferred in the cells undergoes rearrangement, diversification and expression. Secreted immunoglobulins in circulation will then be detected [42]. Production of Tc cattle is limited by the mitotic instability of human microchromosomes and the difference in immunophysiology between cattle and humans [42].
Immunospot Array Assay on a Chip (ISAAC)
Immunospot array assay on a chip (ISAAC) is a rapid and efficient method to generate antibodies from single cells [37]. This method uses a microarray chip whose surface is coated with antibodies against immunoglobulins to trap antibody-secreting cells (ACSs) [37]. ACSs are cultured on a chip for several hours. The antibodies secreted by ACSs bind to labeled antigens to form circular spots which are easily identified [37]. Then, the antigen-specific ASCs are retrieved to obtain antibody cDNA. The obtained antibody cDNA is inserted into expression vectors, and is transfected into cells for expression. The secreted antibodies can be detected by ELISA assay [37]. Jin et al. applied this system to produce human mAbs against viruses within a week, demonstrating ISAAC to be an alternative strategy for human mAbs generation [37].
New Technologies for Monoclonal Screening and Discovery
Next-generation sequencing technologies
Traditional in vitro monoclonal antibodies engineering technologies are laborious which depends on high-throughput screening of immortalized B cells. Reddy et al . developed a new method for antibody isolation bypassing antibody screening [50]. They used next generation sequencing and bioinformatics analysis to analyse the variable-gene repertoires from bone marrow plasma cells. They found that the variable region of immunoglobulin derived from immunized mice became highly polarized. The high-throughput DNA sequencing enabled them to identify several abundant VL and VH gene sequences rapidly [50]. VL and VH genes with relative frequencies within the repertoire were paired and synthesized by oligonucleotide and PCR assembly. Single-chain variable fragments and full-length IgG were expressed in expression systems. Most of the antibodies produced by this method were antigen specific [50].
Next-generation sequencing is also applied to sequence single antibody-secreting B cells. DeKosky et al . used next-generation sequencing technologies to identify large numbers of VH and VL in a single B cell repertoire [25]. They first isolated single B cells from tetanus toxoid immunized human by depositing B cells in a high-density microwell plate. mRNA was then captured and reverse transcribed by RT-PCR. The sequence information of the transcripts was analyzed by next-generation sequencing technology [25].
-
2.
Mass spectrometry
Wine et al. combined next-generation sequencing technology with mass spectrometry to deconvolute the polyclonal serum response after immunization [69]. They sequenced cDNA derived from desired B lymphocyte to generate a database of unique V genes by Roche 454 sequencing. Serum IgGs were pepsin-digested to obtain F(ab)2. Antigen specific F(ab)2 fragments were purified by standard antigen-affinity chromatography and were analyzed by bottom-up, liquid chromatography-high-resolution tandem mass spectrometry. By mapping peptides marking unique VH CDRH3 sequences, this method can be used to identify a set of V-genes constituting the serum polyclonal responses [69].
-
3.
Fluorescence-activated cell sorting (FACS)
Single B cells from defined subpopulations can be isolated by FACS [63]. The full-length variable region gene transcripts were obtained and amplified by RT-PCR. In vitro antibody were subsequently generated by eukaryotic expression system [63].
Prolong Half-Life
Once the engineered antibodies enter into the circulation, they would be eliminated by proteolysis, renal elimination and hepatic elimination, as well as neonatal Fc receptor-mediated endocytosis. To prolong serum circulation of engineered antibodies, several strategies are proposed. Extending serum half-life of therapeutic antibodies could reduce of the number of applications and the doses.
PEGylation
Attaching highly flexible hydrophilic molecules such as polyethylene glycol (PEG) could increase the hydrodynamic volume of engineered antibodies [27]. The increased volume thus improves the serum half-life. Careful attention should be paid that the number and size of PEG labeled may decrease the activation or binding affinity of the engineered antibodies [41]. Different coupling methods including random and site-directed approaches exist. Site-directed approach may be a better method to conjugate a single PEG chain to the antibody.
Fusion to Human Serum Albumin
Fusing human serum albumin to engineered antibodies provides an alternative way to extend serum half-life of antibodies. The binding site of albumin on FcRn does not alter the affinity for antigen or the affinity for FcRn. Covalent linkage of albumin can be achieved either by chemical coupling or genetic engineering methods. Albumin and IgG taken up by cells through macropinocytosis will bind to the FcRn of the early endosome. This binding will protect IgG from degradation in the lysosomal compartment. Antibodies are then redirected to the plasma membrane and released back into the blood.
Affinity Optimization
Humanization may hamper the affinity of antibodies. Sometimes, the affinity of engineered antibodies may not satisfy actual needs. Affinity optimization is, therefore, desired. Most affinity optimization methods depend on optimizing the CDR residues [71]. Targeted or random mutagenesis is used to generate libraries of variants. Normally, libraries of individual CDRs are cloned and screened for improved binding to antigens. Yang et al. successfully generated high affinity human antibodies against human envelope glycoprotein gp120 of HIV-1 by saturation mutagenesis of CDRs [71]. The Fab fragments were displayed and selected for improved binding to the immobilized gp120 [71]. Among the CDR residues, residues in CDR3 are often concentrated for affinity optimization [52]. It is speculated that key variants in CDR3 regions play a major role in antigen affinity mainly for the reason that CDR3 regions are located in the center of the antibody combining site [52].
Fc Engineering
The therapeutic efficacy of the antibody is directly influenced by the affinity of antibodies to pathogens. However, effector functions mediated by the Fc domain also modulates the efficacy of these antibodies. Fc domain plays an important role in opsonophagocytic killing activity, toxin neutralization efficiency [1, 72]. Additionally, Fc domain is involved in mediating effector function, namely complement-dependent cytotoxicity (CDC), antibody-dependent cell-mediated cytotoxicity (ADCC) and antibody-dependent cell phagocytosis (ADCP).
Optimizing the affinity of the Fc domain for Fc receptors is an alternative to increase the efficacy of therapeutic antibodies [56]. Several approaches including alanine scanning, site-directed mutagenesis, computational structure-based design and selection-based method are available to improve the affinity for Fc receptor [49].
Besides, the N-linked glycans in the Fc region of IgG also influence the interaction of the Fc region with its receptors. IgG1 antibodies that are deficient in fucose attached to the Asn297-linked carbohydrate show improved binding affinities with FcγRIIIA receptor [54]. Several different methods can be used to modulate the fucose content on the IgGs. Different expression platforms may generate antibodies with different fucose content. For example, IgG produced by rat hybridomas technologies often has low fucose content [33]. Yeast-and plant-based systems produce antibodies with quite different glycosylation patterns [19, 44].
Antibodies of IgG isotype have a prolonged circulation through FcRn-mediated recycling. Research showed that some mutations in Fc domain may increase the affinity of Fc to its receptor at acidic pH instead of neutral pH. Incorporating such mutations in the Fc domain of IgG will extend the half-life of antibodies [75].
Immunoglobulin Isotype Selection
There are five major classes of immunoglobulin: IgG, IgA, IgD, IgE and IgM. Among these, the IgG class is the most preferred to develop therapeutic antibodies.
IgG contains four isotypes: IgG1, IgG2, IgG3 and IgG4. The main differences between IgG isotypes are located in the Fc domain. To date, most approved human mAbs are mainly of IgG1. Palivizumab (Synagis; MedImmune Inc) approved against RSV infections is a humanized IgG1 monoclonal antibody [30]. IgG1 is preferred for the reason that IgG1 can induce robust cell killing activities by activating various effector cells.
IgG2 is chosen as a therapeutic backbone when weak effector function is needed. Now, several therapeutic IgG2 antibodies are either on market or in clinical development [51]. However, IgG2 dimers occur in vivo naturally. A more in-depth analysis by cleaving recombinant IgG2 suggested that Cys residues in the hinge are involved in the formation of covalent dimers. Besides, the reduced environments in vivo facilitate the formation of IgG2 dimers [74]. The mechanism of dimerization and the exact structure of IgG2 dimers have yet to be investigated. The report of IgG2 disulfide linkages interconverting in vivo may shed some light on IgG2 dimer formation [74]. One consequence of IgG2 dimerization is to enhance antibody avidity, which may play a better protective role [51]. IgG2 dimers exist at low levels (<1%) in vivo, and their speculative roles should not be magnified [73].
IgG3 is less chosen as a backbone of therapeutic antibodies for several reasons. First of all, IgG3 accounts for a minor component of all IgGs in humans (~10%) [51]. Secondly, IgG3 is susceptibility to proteolysis and has a short half-life of ~7 days in vivo. Finally, IgG3 displays extensive polymorphism, within the constant domains [35].
IgG4 can’t activate the classical complement pathway effectively and has reduced effector function [65]. Besides, IgG4 antibodies exchange half-molecules in vivo to form undesired crosslink [70]. To abrogate half-molecule exchange of IgG4 and to eliminate the possible unseen adverse effects, Ser 228 to Pro (S228P) mutation is introduced by antibody engineers to optimize IgG4-based therapeutic antibodies. This mutation not only reduces half-molecule change to a large extent, but also extends serum half-life.
Bispecific Antibodies
Bispecific antibodies (BsAbs) have two different binding specificities, which enables them to simultaneously recognize two different mediators/pathways that exert important roles in pathogenesis [27]. This dual binding capacity will increase targeting specificity or redirect specific immune cells to pathogens or infected cells, thus enhancing pathogen elimination. Recently, bispecific antibodies BiS4αPa against Pseudomonas aeruginosa was generated [39]. BiS4αPa was designed to bind to PsI and PcrV. PsI is an extracellular polysaccharide which involves in immune evasion and biofilm formation. PcrV takes part in the secretion of virulence factors. Animal studies showed that binding to PsI and PcrV led to superior protective activity of BiS4αPa [39]. Now BiS4αPa is in clinical candidate for the treatment of P. aeruginosa [39]. Berg et al. constructed a bispecific antibody by linking one heavy/light chain pair from an antibody against CD3 on cytotoxic T cells to a heavy chain whose variable region was replaced by sequence from CD4 [6]. The constructed antibody has dual binding specificity with one arm binding to CD3 on cytotoxic T cells and the other arm which contained sequences from CD4 binding to viral envelope protein gp120 of HIV [6]. Thus, the bispecific antibody redirected cytotoxic T cells to HIV-infected cells whose surface express integral viral proteins to eliminate pathogens. Bispecific antibodies hold great promise in the elimination of HBV, bacteriophages and other pathogens [48, 60].
The formats of bispecific antibodies can be roughly divided into two categories: IgG-like molecules and non-IgG-like molecules (Fig. 10.2). Divergent approaches are used to engineer bispecific antibodies (Table 10.1). For details, please refer to Fan et al. [27].
Fig. 10.2.

Formats of bispecific antibodies
Table 10.1.
Methods to engineer bispecific antibodies
| Formats | IgG-like molecules | Non-IgG-like molecules |
|---|---|---|
| Methods | Quadromas | Tandem scFvs |
| Knobs-into-holes | Diabody format | |
| Dual-variable domains Ig (DVD-Ig) | Single-chain diabodies | |
| IgG-single-chain Fv (IgG-scFv) | Tandem diabodies (TandAbs) | |
| Dual action Fab (DAF) | Dual-affinity retargeting molecules (DARTs) | |
| Half-molecule exchange | Nanobodies | |
| κλ-bodies | Dock-and-lock (DNL) | |
| scFvs connected to other molecules to form bispecific |
Conclusion
In the era of antibiotic resistance, antibody-based therapy holds great promise in treating infectious pathogens. It is a holy grail to produce human antibodies with high therapeutic efficacy. The therapeutic potential of antibodies is derived from less immunogenicity, proper serum circulating time, high antigen-binding affinity, exquisite specificity and robust effector function. Advancement in antibody engineering technologies makes it possible to produce antibodies tailored to different infection pathogens. Over the past decades, the “magic bullet” has obtained fruitful success in a wide range of diseases including tumors, autoimmune diseases and inflammatory conditions. However, in the field of infections, there is a gap between preclinical researches and clinical applications. Many engineered antibodies are still making their way through preclinical/clinical tests, only few are approved for market. Here, we provided a review of technologies for antibody engineering and optimization. We are optimistic about the application of engineered antibodies to treat infectious pathogens. With the emergence of new infectious pathogens and multidrug resistant bacterial, engineered antibodies may offer another choice to combat these threats.
Contributor Information
Theam Soon Lim, Email: theamsoon@usm.my.
Jinming Li, Email: jmli@nccl.org.cn.
References
- 1.Abboud N, Chow S-K, Saylor C, Janda A, Ravetch JV, Scharff MD, Casadevall A. A requirement for FcγR in antibody-mediated bacterial toxin neutralization. J Exp Med. 2010;207:2395–2405. doi: 10.1084/jem.20100995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Babcock GJ, Iyer S, Smith HL, Wang Y, Rowley K, Ambrosino DM, Zamore PD, Pierce BG, et al. High-throughput sequencing analysis of post-liver transplantation HCV E2 glycoprotein evolution in the presence and absence of neutralizing monoclonal antibody. PLoS One. 2014;9:e100325. doi: 10.1371/journal.pone.0100325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baca M, Presta LG, O’Connor SJ, Wells JA. Antibody humanization using monovalent phage display. J Biol Chem. 1997;272:10678–10684. doi: 10.1074/jbc.272.16.10678. [DOI] [PubMed] [Google Scholar]
- 4.Bartelds GM, Wolbink GJ, Stapel S, Aarden L, Lems WF, Dijkmans BAC, Nurmohamed MT. High levels of human anti-human antibodies to adalimumab in a patient not responding to adalimumab treatment. Ann Rheum Dis. 2006;65:1249–1250. doi: 10.1136/ard.2005.049858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beigel JH, Nordstrom JL, Pillemer SR, Roncal C, Goldwater DR, Li H, Holland PC, Johnson S, et al. Safety and pharmacokinetics of single intravenous dose of MGAWN1, a novel monoclonal antibody to West Nile virus. Antimicrob Agents Chemother. 2010;54:2431–2436. doi: 10.1128/AAC.01178-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berg J, Lötscher E, Steimer KS, Capon DJ, Baenziger J, Jäck HM, Wabl M. Bispecific antibodies that mediate killing of cells infected with human immunodeficiency virus of any strain. Proc Natl Acad Sci. 1991;88:4723–4727. doi: 10.1073/pnas.88.11.4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berry JD, Gaudet RG. Antibodies in infectious diseases: polyclonals, monoclonals and niche biotechnology. New Biotechnol. 2011;28:489–501. doi: 10.1016/j.nbt.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhatt RR, Haurum JS, Davis CG. Technologies for the generation of human antibodies. In: Tabrizi MA, Bornstein GG, Klakamp SL, editors. Development of antibody-based therapeutics. New York: Springer; 2012. pp. 33–63. [Google Scholar]
- 9.Bielory L, Yancey KB, Young NS, Frank MM, Lawley TJ. Cutaneous manifestations of serum sickness in patients receiving antithymocyte globulin. J Am Acad Dermatol. 1985;13:411–417. doi: 10.1016/S0190-9622(85)70182-X. [DOI] [PubMed] [Google Scholar]
- 10.Bien J, Palagani V, Bozko P. The intestinal microbiota dysbiosis and Clostridium difficile infection: is there a relationship with inflammatory bowel disease? Ther Adv Gastroenterol. 2013;6:53–68. doi: 10.1177/1756283X12454590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bowley DR, Labrijn AF, Zwick MB, Burton DR. Antigen selection from an HIV-1 immune antibody library displayed on yeast yields many novel antibodies compared to selection from the same library displayed on phage. Protein Eng Des Sel PEDS. 2007;20:81–90. doi: 10.1093/protein/gzl057. [DOI] [PubMed] [Google Scholar]
- 12.Brüggemann M, Neuberger MS. Strategies for expressing human antibody repertoires in transgenic mice. Immunol Today. 1996;17:391–397. doi: 10.1016/0167-5699(96)10025-6. [DOI] [PubMed] [Google Scholar]
- 13.Casadevall A, Scharff MD. Serum therapy revisited: animal models of infection and development of passive antibody therapy. Antimicrob Agents Chemother. 1994;38:1695–1702. doi: 10.1128/AAC.38.8.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Casadevall A, Scharff MD. Return to the past: the case for antibody-based therapies in infectious diseases. Clin Infect Dis Off Publ Infect Dis Soc Am. 1995;21:150–161. doi: 10.1093/clinids/21.1.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Casadevall A, Dadachova E, Pirofski L. Passive antibody therapy for infectious diseases. Nat Rev Microbiol. 2004;2:695–703. doi: 10.1038/nrmicro974. [DOI] [PubMed] [Google Scholar]
- 16.Chung RT, Gordon FD, Curry MP, Schiano TD, Emre S, Corey K, Markmann J, et al. Human monoclonal antibody MBL-HCV1 delays HCV viral rebound following liver transplantation: a randomized controlled study. Am J Transplant Off J Am Soc Transplant Am Soc Transpl Surg. 2013;13:1047–1054. doi: 10.1111/ajt.12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen ML. Epidemiology of drug resistance: implications for a post-antimicrobial era. Science. 1992;257:1050–1055. doi: 10.1126/science.257.5073.1050. [DOI] [PubMed] [Google Scholar]
- 18.Coughlin M, Lou G, Martinez O, Masterman SK, Olsen OA, Moksa AA, Farzan M, et al. Generation and characterization of human monoclonal neutralizing antibodies with distinct binding and sequence features against SARS coronavirus using XenoMouse. Virology. 2007;361:93–102. doi: 10.1016/j.virol.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cox KM, Sterling JD, Regan JT, Gasdaska JR, Frantz KK, Peele CG, Black A, et al. Glycan optimization of a human monoclonal antibody in the aquatic plant Lemna minor. Nat Biotechnol. 2006;24:1591–1597. doi: 10.1038/nbt1260. [DOI] [PubMed] [Google Scholar]
- 20.Cross AS. Antiendotoxin antibodies: a dead end? Ann Intern Med. 1994;121:58–60. doi: 10.7326/0003-4819-121-1-199407010-00011. [DOI] [PubMed] [Google Scholar]
- 21.Dall’Acqua WF, Damschroder MM, Zhang J, Woods RM, Widjaja L, Yu J, Wu H. Antibody humanization by framework shuffling. Methods. 2005;36:43–60. doi: 10.1016/j.ymeth.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 22.Damschroder MM, Widjaja L, Gill PS, Krasnoperov V, Jiang W, Dall’Acqua WF, Wu H. Framework shuffling of antibodies to reduce immunogenicity and manipulate functional and biophysical properties. Mol Immunol. 2007;44:3049–3060. doi: 10.1016/j.molimm.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 23.De Pascalis R, Gonzales NR, Padlan EA, Schuck P, Batra SK, Schlom J, Kashmiri SVS. In vitro affinity maturation of a specificity-determining region-grafted humanized anticarcinoma antibody: isolation and characterization of minimally immunogenic high-affinity variants. Clin Cancer Res Off J Am Assoc Cancer Res. 2003;9:5521–5531. [PubMed] [Google Scholar]
- 24.DeChiara T, Poueymirou W, Auerbach W, Frendewey D, Yancopoulos G, Valenzuela D. VelociMouse: fully ES cell-derived F0-generation mice obtained from the injection of ES cells into eight-cell-stage embryos. In: Wurst W, Kühn R, editors. Gene knockout protocols, methods in molecular biology. New York: Humana Press; 2009. pp. 311–324. [DOI] [PubMed] [Google Scholar]
- 25.DeKosky BJ, Ippolito GC, Deschner RP, Lavinder JJ, Wine Y, Rawlings BM, Varadarajan N, et al. High-throughput sequencing of the paired human immunoglobulin heavy and light chain repertoire. Nat Biotechnol. 2013;31:166–169. doi: 10.1038/nbt.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Derkx B, Wittes J, McCloskey R. Randomized, placebo-controlled trial of HA-1A, a human monoclonal antibody to endotoxin, in children with meningococcal septic shock. European Pediatric Meningococcal Septic Shock Trial Study Group. Clin Infect Dis Off Publ Infect Dis Soc Am. 1999;28:770–777. doi: 10.1086/515184. [DOI] [PubMed] [Google Scholar]
- 27.Fan G, Wang Z, Hao M, Li J. Bispecific antibodies and their applications. J Hematol Oncol. 2015;8:1–14. doi: 10.1186/s13045-014-0099-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gould IM. A review of the role of antibiotic policies in the control of antibiotic resistance. J Antimicrob Chemother. 1999;43:459–465. doi: 10.1093/jac/43.4.459. [DOI] [PubMed] [Google Scholar]
- 29.Greenman RL, Schein RMH, Martin MA, Wenzel RP, Maclntyre NR, Emmanuel G, Chmel H, et al. A controlled clinical trial of E5 murine monoclonal IgM antibody to endotoxin in the treatment of gram-negative sepsis. JAMA. 1991;266:1097–1102. doi: 10.1001/jama.1991.03470080067031. [DOI] [PubMed] [Google Scholar]
- 30.Group*, T.Im.-R.S Palivizumab, a humanized respiratory syncytial virus monoclonal antibody, reduces hospitalization from respiratory syncytial virus infection in high-risk infants. Pediatrics. 1998;102:531–537. doi: 10.1542/peds.102.3.531. [DOI] [PubMed] [Google Scholar]
- 31.Gutfraind A, Meyers LA. Evaluating large-scale blood transfusion therapy for the current Ebola epidemic in Liberia. J Infect Dis. 2015;211:1262–1267. doi: 10.1093/infdis/jiv042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huse WD, Sastry L, Iverson SA, Kang AS, Alting-Mees M, Burton DR, Benkovic SJ, et al. Generation of a large combinatorial library of the immunoglobulin repertoire in phage lambda. Science. 1989;246:1275–1281. doi: 10.1126/science.2531466. [DOI] [PubMed] [Google Scholar]
- 33.Jain M, Kamal N, Batra SK. Engineering antibodies for clinical applications. Trends Biotechnol. 2007;25:307–316. doi: 10.1016/j.tibtech.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 34.Jakobovits A, Amado RG, Yang X, Roskos L, Schwab G. From XenoMouse technology to panitumumab, the first fully human antibody product from transgenic mice. Nat Biotechnol. 2007;25:1134–1143. doi: 10.1038/nbt1337. [DOI] [PubMed] [Google Scholar]
- 35.Jefferis R. Antibody therapeutics: isotype and glycoform selection. Expert Opin Biol Ther. 2007;7:1401–1413. doi: 10.1517/14712598.7.9.1401. [DOI] [PubMed] [Google Scholar]
- 36.Jernberg C, Löfmark S, Edlund C, Jansson JK. Long-term impacts of antibiotic exposure on the human intestinal microbiota. Microbiol Read Engl. 2010;156:3216–3223. doi: 10.1099/mic.0.040618-0. [DOI] [PubMed] [Google Scholar]
- 37.Jin A, Ozawa T, Tajiri K, Obata T, Kondo S, Kinoshita K, Kadowaki S, et al. A rapid and efficient single-cell manipulation method for screening antigen-specific antibody–secreting cells from human peripheral blood. Nat Med. 2009;15:1088–1092. doi: 10.1038/nm.1966. [DOI] [PubMed] [Google Scholar]
- 38.Kashmiri SVS, De Pascalis R, Gonzales NR, Schlom J. SDR grafting – a new approach to antibody humanization. Methods. 2005;36:25–34. doi: 10.1016/j.ymeth.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 39.Kingwell K. Infectious diseases: two-hit antibody tackles bacteria. Nat Rev Drug Discov. 2015;14:15–15. doi: 10.1038/nrd4525. [DOI] [PubMed] [Google Scholar]
- 40.Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 41.Kontermann RE. Strategies for extended serum half-life of protein therapeutics. Curr Opin Biotechnol. 2011;22:868–876. doi: 10.1016/j.copbio.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 42.Kuroiwa Y, Kasinathan P, Choi YJ, Naeem R, Tomizuka K, Sullivan EJ, Knott JG, et al. Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo. Nat Biotechnol. 2009;27:767–771. doi: 10.1038/nbt.1521. [DOI] [PubMed] [Google Scholar]
- 43.Lazar GA, Desjarlais JR, Jacinto J, Karki S, Hammond PW. A molecular immunology approach to antibody humanization and functional optimization. Mol Immunol. 2007;44:1986–1998. doi: 10.1016/j.molimm.2006.09.029. [DOI] [PubMed] [Google Scholar]
- 44.Li H, Sethuraman N, Stadheim TA, Zha D, Prinz B, Ballew N, Bobrowicz P, Choi B-K, et al. Optimization of humanized IgGs in glycoengineered Pichia pastoris. Nat Biotechnol. 2006;24:210–215. doi: 10.1038/nbt1178. [DOI] [PubMed] [Google Scholar]
- 45.Lowy I, Molrine DC, Leav BA, Blair BM, Baxter R, Gerding DN, Nichol G, et al. Treatment with monoclonal antibodies against Clostridium difficile toxins. N Engl J Med. 2010;362:197–205. doi: 10.1056/NEJMoa0907635. [DOI] [PubMed] [Google Scholar]
- 46.Marasco WA, Sui J. The growth and potential of human antiviral monoclonal antibody therapeutics. Nat Biotechnol. 2007;25:1421–1434. doi: 10.1038/nbt1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oliphant T, Engle M, Nybakken GE, Doane C, Johnson S, Huang L, Gorlatov S, et al. Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nat Med. 2005;11:522–530. doi: 10.1038/nm1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park SS, Ryu CJ, Kang YJ, Kashmiri SVS, Hong HJ. Generation and characterization of a novel tetravalent bispecific antibody that binds to hepatitis B virus surface antigens. Mol Immunol. 2000;37:1123–1130. doi: 10.1016/S0161-5890(01)00027-X. [DOI] [PubMed] [Google Scholar]
- 49.Presta LG. Molecular engineering and design of therapeutic antibodies. Curr Opin Immunol. 2008;20:460–470. doi: 10.1016/j.coi.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 50.Reddy ST, Ge X, Miklos AE, Hughes RA, Kang SH, Hoi KH, Chrysostomou C, et al. Monoclonal antibodies isolated without screening by analyzing the variable-gene repertoire of plasma cells. Nat Biotechnol. 2010;28:965–969. doi: 10.1038/nbt.1673. [DOI] [PubMed] [Google Scholar]
- 51.Salfeld JG. Isotype selection in antibody engineering. Nat Biotechnol. 2007;25:1369–1372. doi: 10.1038/nbt1207-1369. [DOI] [PubMed] [Google Scholar]
- 52.Schier R, McCall A, Adams GP, Marshall KW, Merritt H, Yim M, Crawford RS, et al. Isolation of picomolar affinity anti-c-erbB-2 single-chain Fv by molecular evolution of the complementarity determining regions in the center of the antibody binding site. J Mol Biol. 1996;263:551–567. doi: 10.1006/jmbi.1996.0598. [DOI] [PubMed] [Google Scholar]
- 53.Schlapschy M, Gruber H, Gresch O, Schäfer C, Renner C, Pfreundschuh M, Skerra A. Functional humanization of an anti-CD30 Fab fragment for the immunotherapy of Hodgkin’s lymphoma using an in vitro evolution approach. Protein Eng Des Sel PEDS. 2004;17:847–860. doi: 10.1093/protein/gzh098. [DOI] [PubMed] [Google Scholar]
- 54.Shields RL, Lai J, Keck R, O’Connell LY, Hong K, Meng YG, Weikert SHA, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human FcγRIII and antibody-dependent cellular toxicity. J Biol Chem. 2002;277:26733–26740. doi: 10.1074/jbc.M202069200. [DOI] [PubMed] [Google Scholar]
- 55.Stamataki Z, Shannon-Lowe C, Shaw J, Mutimer D, Rickinson AB, Gordon J, Adams DH, et al. Hepatitis C virus association with peripheral blood B lymphocytes potentiates viral infection of liver-derived hepatoma cells. Blood. 2009;113:585–593. doi: 10.1182/blood-2008-05-158824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stavenhagen JB, Gorlatov S, Tuaillon N, Rankin CT, Li H, Burke S, Huang L, et al. Fc optimization of therapeutic antibodies enhances their ability to kill tumor cells in vitro and controls tumor expansion in vivo via low-affinity activating Fcγ receptors. Cancer Res. 2007;67:8882–8890. doi: 10.1158/0008-5472.CAN-07-0696. [DOI] [PubMed] [Google Scholar]
- 57.Sun Z-YJ, Oh KJ, Kim M, Yu J, Brusic V, Song L, Qiao Z, et al. HIV-1 broadly neutralizing antibody extracts its epitope from a kinked gp41 ectodomain region on the viral membrane. Immunity. 2008;28:52–63. doi: 10.1016/j.immuni.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 58.Tabrizi MA, Bornstein GG, Klakamp SL, editors. Development of antibody-based therapeutics. New York: Springer; 2012. [Google Scholar]
- 59.Tan P, Mitchell DA, Buss TN, Holmes MA, Anasetti C, Foote J. “Superhumanized” antibodies: reduction of immunogenic potential by complementarity-determining region grafting with human germline sequences: application to an anti-CD28. J Immunol Baltim Md. 2002;1950(169):1119–1125. doi: 10.4049/jimmunol.169.2.1119. [DOI] [PubMed] [Google Scholar]
- 60.Taylor RP, Martin EN, Reinagel ML, Nardin A, Craig M, Choice Q, Schlimgen R, et al. Bispecific monoclonal antibody complexes facilitate erythrocyte binding and liver clearance of a prototype particulate pathogen in a monkey model. J Immunol. 1997;159:4035–4044. [PubMed] [Google Scholar]
- 61.Teng NN, Kaplan HS, Hebert JM, Moore C, Douglas H, Wunderlich A, Braude AI. Protection against gram-negative bacteremia and endotoxemia with human monoclonal IgM antibodies. Proc Natl Acad Sci U S A. 1985;82:1790–1794. doi: 10.1073/pnas.82.6.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Throsby M, van den BE, Jongeneelen M, Poon LLM, Alard P, Cornelissen L, Bakker A, Cox F, van DE, Guan Y, et al. Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS One. 2008;3:e3942. doi: 10.1371/journal.pone.0003942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tiller T, Busse CE, Wardemann H. Cloning and expression of murine Ig genes from single B cells. J Immunol Methods. 2009;350:183–193. doi: 10.1016/j.jim.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 64.Traggiai E, Becker S, Subbarao K, Kolesnikova L, Uematsu Y, Gismondo MR, Murphy BR, et al. An efficient method to make human monoclonal antibodies from memory B cells: potent neutralization of SARS coronavirus. Nat Med. 2004;10:871–875. doi: 10.1038/nm1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Van Der Zee JS, Van Swieten P, Aalberse RC. Serologic aspects of IgG4 antibodies. II. IgG4 antibodies form small, nonprecipitating immune complexes due to functional monovalency. J Immunol. 1986;137:3566–3571. [PubMed] [Google Scholar]
- 66.Warrener P, Varkey R, Bonnell JC, DiGiandomenico A, Camara M, Cook K, Peng L, et al. A novel anti-PcrV antibody providing enhanced protection against Pseudomonas aeruginosa in multiple animal infection models. Antimicrob Agents Chemother. 2014;58:4384–4391. doi: 10.1128/AAC.02643-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weisman LE, Thackray HM, Steinhorn RH, Walsh WF, Lassiter HA, Dhanireddy R, Brozanski BS, et al. A randomized study of a monoclonal antibody (Pagibaximab) to prevent staphylococcal sepsis. Pediatrics. 2011;128:271–279. doi: 10.1542/peds.2010-3081. [DOI] [PubMed] [Google Scholar]
- 68.Wiesner M, Zentz C, Hammer MH, Cobbold M, Kern F, Kolb H-J, Hammerschmidt W, et al. Selection of CMV-specific CD8+ and CD4+ T cells by mini-EBV-transformed B cell lines. Eur J Immunol. 2005;35:2110–2121. doi: 10.1002/eji.200425936. [DOI] [PubMed] [Google Scholar]
- 69.Wine Y, Boutz DR, Lavinder JJ, Miklos AE, Hughes RA, Hoi KH, Jung ST, et al. Molecular deconvolution of the monoclonal antibodies that comprise the polyclonal serum response. Proc Natl Acad Sci. 2013;110:2993–2998. doi: 10.1073/pnas.1213737110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang† YHJ, Ngo C, Yeh IN, Uemura Y. Antibody Fc functional activity of intravenous immunoglobulin preparations treated with solvent-detergent for virus inactivation. Vox Sang. 1994;67:337–344. doi: 10.1111/j.1423-0410.1994.tb01270.x. [DOI] [PubMed] [Google Scholar]
- 71.Yang WP, Green K, Pinz-Sweeney S, Briones AT, Burton DR, Barbas CF. CDR walking mutagenesis for the affinity maturation of a potent human anti-HIV-1 antibody into the picomolar range. J Mol Biol. 1995;254:392–403. doi: 10.1006/jmbi.1995.0626. [DOI] [PubMed] [Google Scholar]
- 72.Yang J, Goetze AM, Flynn GC. Assessment of naturally occurring covalent and total dimer levels in human IgG1 and IgG2. Mol Immunol. 2014;58:108–115. doi: 10.1016/j.molimm.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 73.Yang X, Zhang Y, Wang F, Wang LJ, Richardson D, Shameem M, Ambrogelly A. Analysis and purification of IgG4 bispecific antibodies by a mixed-mode chromatography. Anal Biochem. 2015;484:173–179. doi: 10.1016/j.ab.2015.06.014. [DOI] [PubMed] [Google Scholar]
- 74.Yoo EM, Wims LA, Chan LA, Morrison SL. Human IgG2 can form covalent dimers. J Immunol Baltim Md. 2003;1950(170):3134–3138. doi: 10.4049/jimmunol.170.6.3134. [DOI] [PubMed] [Google Scholar]
- 75.Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IWL, Sproule TJ, Lazar GA, et al. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol. 2010;28:157–159. doi: 10.1038/nbt.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ziegler EJ, Fisher CJ, Sprung CL, Straube RC, Sadoff JC, Foulke GE, Wortel CH, et al. Treatment of gram-negative bacteremia and septic shock with HA-1A human monoclonal antibody against endotoxin. A randomized, double-blind, placebo-controlled trial. The HA-1A Sepsis Study Group. N Engl J Med. 1991;324:429–436. doi: 10.1056/NEJM199102143240701. [DOI] [PubMed] [Google Scholar]