

Abstract
Disease recurrence causes significant mortality in B-progenitor acute lymphoblastic leukemia (B-ALL). Genomic analysis of matched diagnosis and relapse samples show relapse often arising from minor diagnosis subclones. However, why therapy eradicates some subclones while others survive and progress to relapse remains obscure. Elucidation of mechanisms underlying these differing fates requires functional analysis of isolated subclones. Here, large-scale limiting dilution xenografting of diagnosis and relapse samples, combined with targeted sequencing, identified and isolated minor diagnosis subclones that initiate evolutionary trajectory toward relapse (termed diagnosis Relapse Initiating clones, dRI). Compared to other diagnosis subclones, dRI were drug tolerant with distinct engraftment and metabolic properties. Transciptionally, dRI displayed enrichment for chromatin remodelling, mitochondrial metabolism, proteostasis programs and an increase in stemness pathways. The isolation and characterization of dRI subclones reveals new avenues for eradicating dRI cells by targeting their distinct metabolic and transcriptional pathways before further evolution renders them fully therapy resistant.
Keywords: B-ALL, relapse, clonality, metabolism, therapy
INTRODUCTION
Despite significant advancements in the treatment of acute lymphoblastic leukemia (ALL), the disease recurs in 15–20% of pediatric and 40–75% of adult patients, with the prognosis for patients who relapse being dismal(1–3). Analysis of paired diagnosis and relapse ALL samples with increasingly broader and deeper genomic sequencing methods has shown that classical Darwinian branching evolution of genomically distinct subclones is a hallmark of disease recurrence(4,5). At both diagnosis and relapse, a single neoplasm may contain multiple genetic subclones related to each other through complex evolutionary trajectories(4–6). While relapse may evolve from the predominant clone at diagnosis, in the majority of patients relapse arises from pre-existing minor subclones within the diagnosis sample or from a rare ancestral clone(4–6). Although the population-level genetic analyses upon which these conclusions are based rely on computational inference of their evolutionary relationships from analysis of bulk leukemic cells, resolution of leukemic subclones at clonal levels, either through isolation of subclones in xenografts or from single cell analysis, have largely substantiated these predictions(7,8). Functional studies to explain therapy failure have mainly been undertaken by comparing diagnosis cells with those from relapse and identifying drug resistance mechanisms present in relapse and absent at diagnosis. However, the properties of the relapse sample have been shaped by exposure to chemotherapy causing further evolution and mutagenesis. Thus two critical questions remain: what are the unique properties and mechanisms that contribute to the relapse-fate of a particular diagnosis subclone prior to full evolution to drug resistant relapse disease; and when does drug tolerance arise? Drug tolerance may arise stochastically through genetic or epigenetic mecahnisms prior to exposure to therapy, and be selected for by both cell autonomous and non-cell autonomous processes(9–12). Alternatively, therapy may induce genomic aberrations that are then selected for during disease progression, particularly if such alterations reduce leukemic cell fitness(13) during disease establishment and prior to the administration of therapy(14,15). Without isolation of the subclones that contribute to disease progression from diagnosis samples, it is not possible to answer these questions and uncover the cellular and molecular properties that explain their differing subclonal fates and drug tolerance.
Many therapy resistance mechanisms have been implicated in B-ALL including acquisition of stemness programs(16), dormancy, the protective role of the niche and the acquisition of resistance driver mutations(13,17–20). However, the ability to evade drug treatment is only one prerequisite of relapse; surviving cells must also possess significant clonal regenerative capacity in order to regrow or reproduce disease. For many human cancers, only rare fractions of malignant cells are capable of significant clonal propagation as detected by xenograft-based cancer or leukemia-initiating cell (L-IC) assays(9). Indeed, methods to propagate primary human leukemia samples were first undertaken in B-ALL with patient-derived xenografts (PDXs) recapitulating many features of the patient’s disease(21,22). Subsequent studies used a limiting dilution approach to generate xenografts, thereby tracking the growth properties and genetic identity of single L-ICs(8). Thus, depending on the cell dose transplanted, rare subclones with poorer competitive repopulation properties, compared to more aggressive or numerous L-IC within the sample, could be identified. By genetic analysis of such xenografts, evidence was found for the existence of ancestral and/or minor subclones proving that branching evolution and clonal diversity occur at the level of L-IC, however these studies did not test for relapse-fated subclones(7,8). Conceptually, L-ICs with the capacity for clonal propagation should serve as the units of selection during disease progression since only mutations accumulating in clonal propagating cells are relevant for further disease evolution. Thus the relationship between disease progression and properties of stemness is an active area of investigation(9,23). The pairing of xenografting assays with genomic studies provides a unique opportunity to enrich for the reservoirs of relapse. However, a direct test of this concept through paired diagnosis and relapse xenografting in B-ALL has been limited(22,24,25). Paired studies in AML and T-ALL have demonstrated that xenografts can capture distinct subclones present within the diagnosis samples of relapsing patients; some genetically more closely related to the predominant diagnosis subclone and others to a corresponding relapse sample(26–29). These results suggest that the latent relapse-initating subclones possess competitive growth advantages when assessed by xenografting, but remain suppressed by the predominant clone in the patient making them difficult to study.
Here, we undertook a combined functional and genomic analysis of 14 genetically distinct paired diagnosis and relapse B-progenitor ALL patient samples to isolate latent subclones within the diagnosis sample that initiate relapse. Isolation of subclones enabled functional analysis of their growth and drug response properties as well as molecular analysis of their transcriptomic profiles.
RESULTS
Isolation of relapse-initiating subclones in B-ALL
Whole exome sequencing (WES) and single nucleotide polymorphism (SNP) microarray analysis of 6 adult and 8 pediatric B-ALL patients of varying genetic subtypes were undertaken to identify somatic single nucleotide variants (SNV), insertion-deletion mutations (indels) and DNA copy number alterations (CNA). The patients encompassed a range of cytogenetic subtypes and varied in the length of their disease remission (range 5.88 – 94.8 months) (Supplementary Table S1). Diagnosis samples had a median of 24 somatic SNV/ indels (range 7–100) and 13.5 CNA (range 1–51), whereas relapse samples contained a median of 39.5 SNV/indels (range 22–405) and 16.5 CNA (range 2–58) (Supplementary Table S1). Leukemic variants were confirmed by targeted sequencing using a custom capture array of the variants identified by WES (Fig. 1a). Targeted sequencing and WES data were merged resulting in a coverage of ~350x. Computational analysis of the variant allele frequencies (VAFs) of leukemic variants comparing diagnosis and relapse samples predicted that the origin of relapse arose from a minor subclone present at diagnosis in 10 patients (patients 1,3–7,9 and 12–14) (Fig. 1b and Supplementary Table S1) and through further evolution of the major diagnostic subclone in 4 patients (patients 2,8,10 and 11) (Supplementary Table S1). Broadly, these findings of evolutionary origins are representative of the much larger analysis of 92 paired samples that reports the mutational landscape and patterns of clonal evolution of relapsed childhood ALL as described in Waanders et al.(30).
Figure 1: PDXs capture clonal diversity present in paired diagnosis and relapse B-ALL samples.
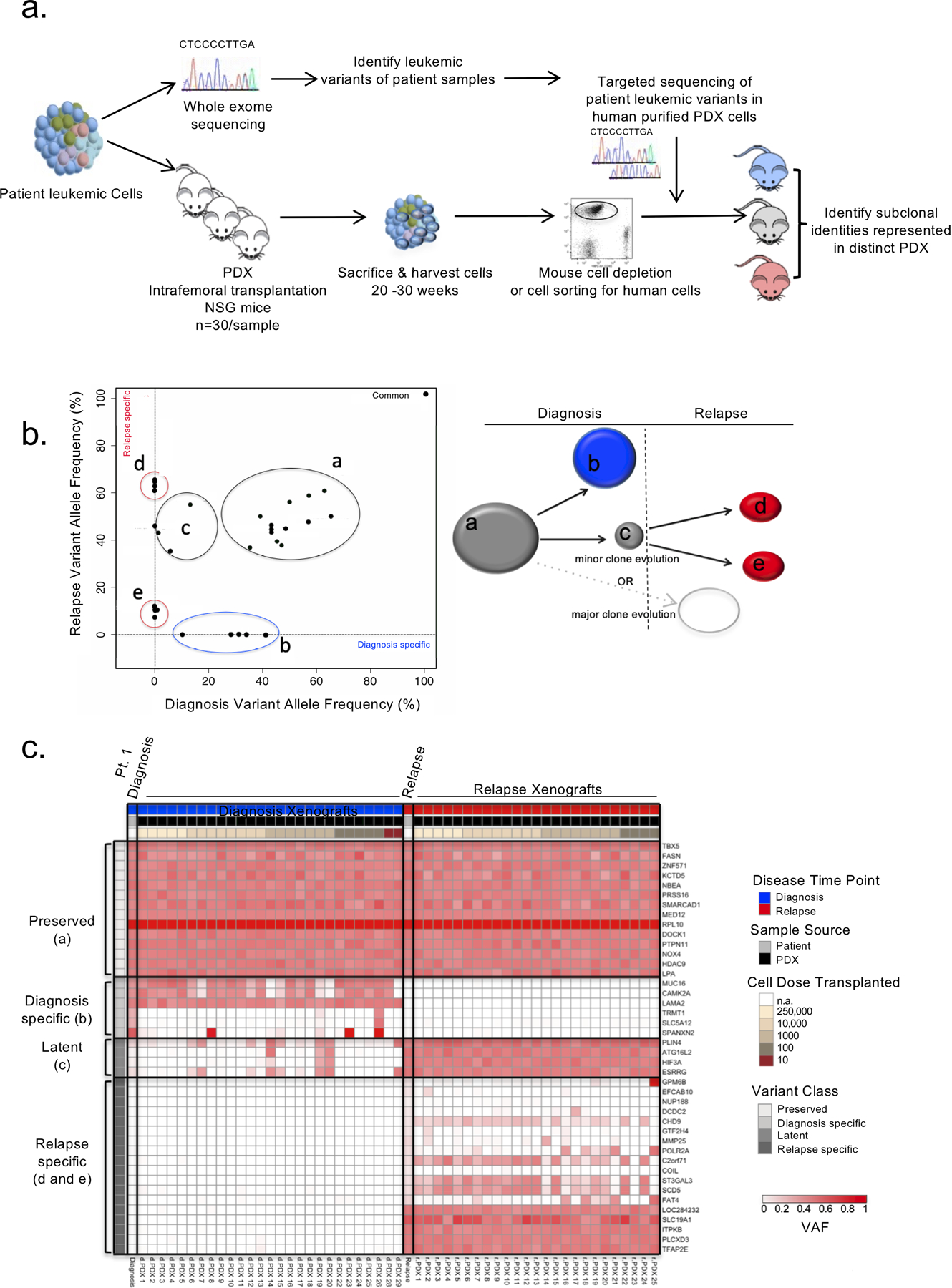
a. Experimental schematic. PDXs were generated for 6 adult and 8 pediatric B-ALL patient samples at diagnosis and relapse by intrafemoral transplantation of sorted leukemic blasts into 30 irradiated NSG mice in a limiting dilution assay. Mice were sacrificed 20–30 weeks post transplant and their engraftment was assessed by flow cytometry. Patient samples were also subjected to genomic analysis including whole exome sequencing (WES). Variants identified from the WES of patient samples at either time point were used to create custom capture baits for targeted sequencing at a deeper depth in the patient samples and their corresponding PDX. PDXs representing varying clones were identified. b. Schematic representation of the results obtanined by mutational clustering of variants based on the variant allele frequencies (VAF) at diagnosis (x-axis) and relapse (y-axis) of patient 1 in 2D VAF plots showing evolution from a minor subclone as depicted. Each dot represents a variant. Shared variants are shown in grey clusters (clusters a and c). Diagnosis and relapse specific variants are shown in the blue cluster (cluster b) and red clusters (clusters d and e) respectively. c. Heatmap of the variant allele frequencies of leukemic variants at diagnosis, relapse and in their corresponding PDXs for patient 1. Variant classes are labeled with their class and a letter corresponding to the clusters illustrated in b.
To gain insight into the genetic diversity at the level of leukemia initiating-cells (L-IC) and uncover rare and/or outcompeted clones, purified leukemic blasts from primary diagnosis and relapse samples were transplanted intrafemorally in a limiting dilution assay into 872 NOD.CB17-PrkdcscidIl2rgtm1Wjl/Szj (NSG) mice to generate primary patient derived xenografts (termed PDX). The frequency of L-IC varied widely between samples with L-IC ranges corresponding to those previously described(31,32), however paired analysis between diagnosis and relapse samples did not show a consistent trend in L-IC enrichment at either time point (Supplementary Table S2).This collection of PDXs (n=402, average 28 per patient (range 11–58), 13 per sample (range 0–27)) was used to further study the clonal landscape present in the patient samples. To determine if PDXs captured the clonal diversity and disease evolution present in the patient samples, human cells were isolated from the bone marrow (BM), spleen (SPL) and central nervous system (CNS) of engrafted mice and subjected to targeted sequencing using the custom capture array designed for the patient samples (Fig. 1a). PDXs with sufficient human engraftment for genotyping (>10% human chimerism, 372 PDX total, average 26 PDX per patient) were analyzed. Individual PDXs from the same patient sample were often found to vary in their clonal composition in terms of the presence and frequency of variants suggesting that the L-IC initiating the grafts derived from genetically diverse subclones often capturing the totality of the clonal diversity present in the diagnosis patient sample (Fig. 1c).
Leukemic variants were classified on the basis of the VAF of the variants in the bulk patient diagnosis and relapse samples from which the PDX were generated: preserved variants (VAF > 30% in both diagnosis and relapse samples, or preserved between samples); diagnosis-specific variants (present at diagnosis and absent at relapse); latent variants (present at diagnosis with VAF < 30% and increasing at relapse); relapse-specific variants (absent at diagnosis and present at relapse) (Supplementary Table S3). The limit of detection of our combined sequencing was a VAF of 1%. This analysis revealed three patterns of engraftment in PDX derived from diagnosis samples (termed dPDXs). In 10 of 13 engrafting diagnosis samples (76.9%, patients 1–7,9,12 and 14), latent variants were enriched in dPDXs (>10% increase in VAF in dPDX) as compared to the diagnosis patient sample demonstrating the regenerative potential of clones marked by these variants (Fig. 2a and Supplementary Fig. S1a–b). In 4 of 13 patients (patients 5,6,11 and 13), dPDXs were generated where leukemia cells bore relapse-specific variants establishing their existence within the diagnosis sample, despite being at levels below the detectable limit of the sequencing (Fig. 2b). Lastly, in one patient (patient 8), whose relapse was predicted to evolve from the major diagnosis clone based on analysis of the bulk patient samples, only diagnosis clones engrafted in dPDXs (Supplementary Fig. S1c). This patient carried an ETV6-RUNX1 translocation and had the longest remission, relapsing eight years after the initial ALL diagnosis. Our approach of generating PDXs with differing cell doses was instrumental in showing the existence of subclones at diagnosis that bear latent (patient 2) or relapse specific variants indicative of ancestral clones (patient 11) in two patients in which genetic analysis of bulk diagnosis and relapse samples had predicted evolution from the major diagnosis clone (Fig. 2b and Supplementary Table S3). Only one diagnosis patient sample (patient 10), a sample predicted to arise from evolution of the major diagnostic clone, did not engraft at the highest transplanted cell dose (250,000 cells). Therefore, xenografting added considerable new insight into the subclonal repertoire of L-ICs in these patients, and their evolutionary fates and patterns.
Figure 2: PDXs enrich for latent diagnosis clones.
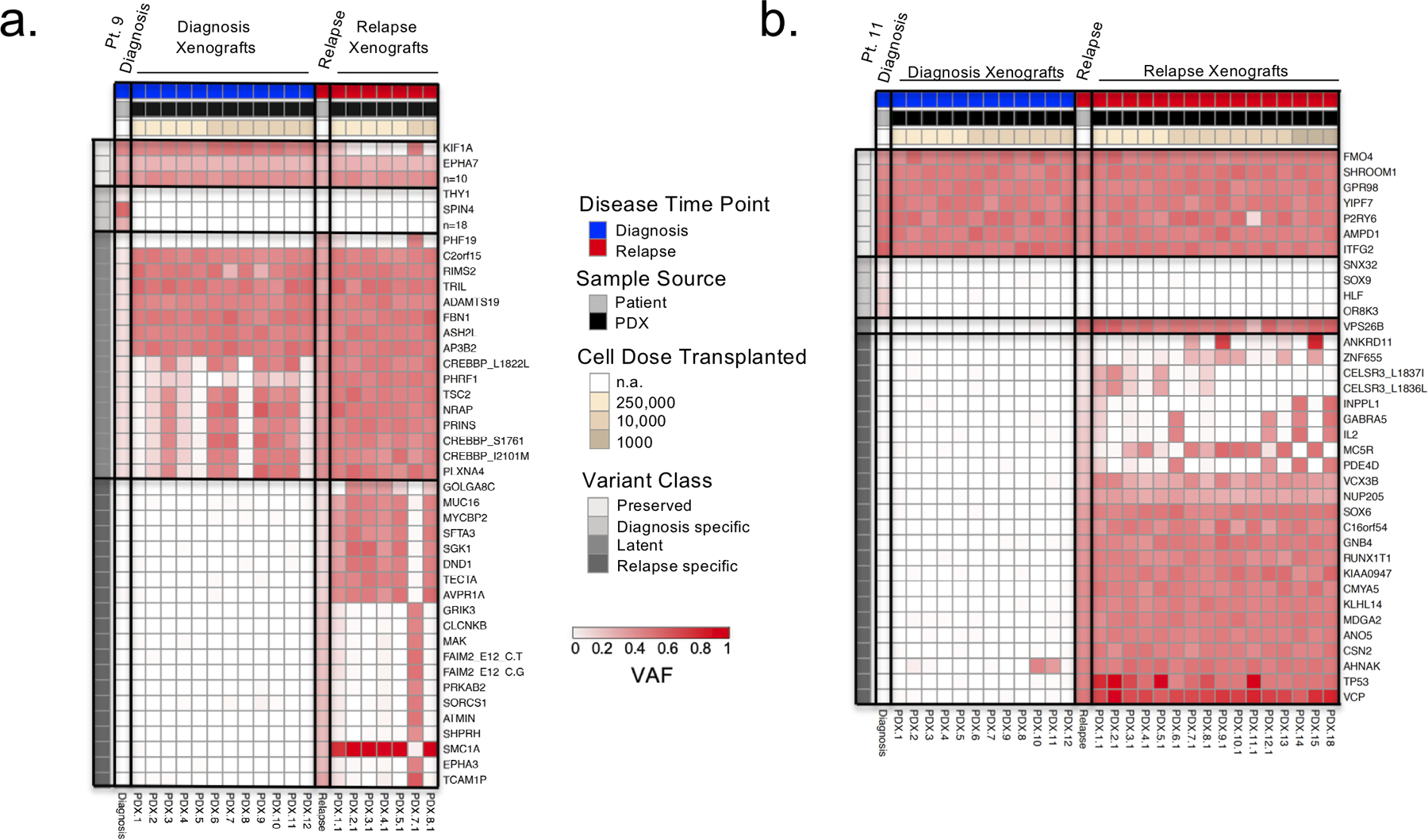
a. and b. Heatmaps of VAF of the SNV and indel leukemic variants identified by whole exome and targeted sequencing in diagnosis/relapse patient samples and PDX respectively. Variants are clustered as preserved (present in diagnosis and relapse patient samples), diagnosis specific (present in diagnosis patient sample and absent in relapse patient sample), latent (present in diagnosis patient sample with VAF < 0.3 and expanding in relapse sample), and relapse specific (present in relapse patient sample and absent in diagnosis patient sample). PDX are ordered in decreasing numbers of transplanted cell doses. a. Representative heatmap for the selection of latent variants in diagnosis PDX as observed in patient 9. b. Representative heatmap of patient 11 describing the identification of a relapse specific variant undetectable in the patient diagnosis sample but present in diagnosis PDX.
Genetic analysis of xenografts provides insight into the dynamics of subclone evolution
To gain insight into the evolutionary relationships and processes underlying the divergence of subclones, we undertook two approaches: population phylogenetic analysis to examine the genetic similarity between xenografts and patient samples; and generation of mutational trees to reconstruct the clonal hierarchies. We began by using a population genetics approach where phylogenetic analysis was performed for the patient samples and the clones isolated from their PDXs to depict the evolutionary relationships between xenografts and the patient samples (Fig. 3a–b and Supplementary Fig. S1d–g). These analyses demonstrate the ability of PDXs to capture diagnosis subclones more closely related to the diagnosis patient sample, as well as subclones present in the diagnosis patient sample that are on the trajectory to relapse. We term these partially evolved subclones that go on to cause relapse – diagnosis relapse-initiating (dRI) clones. dRI were identified in all engrafting diagnosis patient samples except for the patient with the longest time of remission (patient 8). Thus, our xenografting approach enabled the capture and isolation of subclones, including those on the evolutionary trajectory of relapse, for further functional analysis and to aid in the determination of mutational acquisition.
Figure 3: PDXs identify relapse-fated clones in diagnosis patient samples.
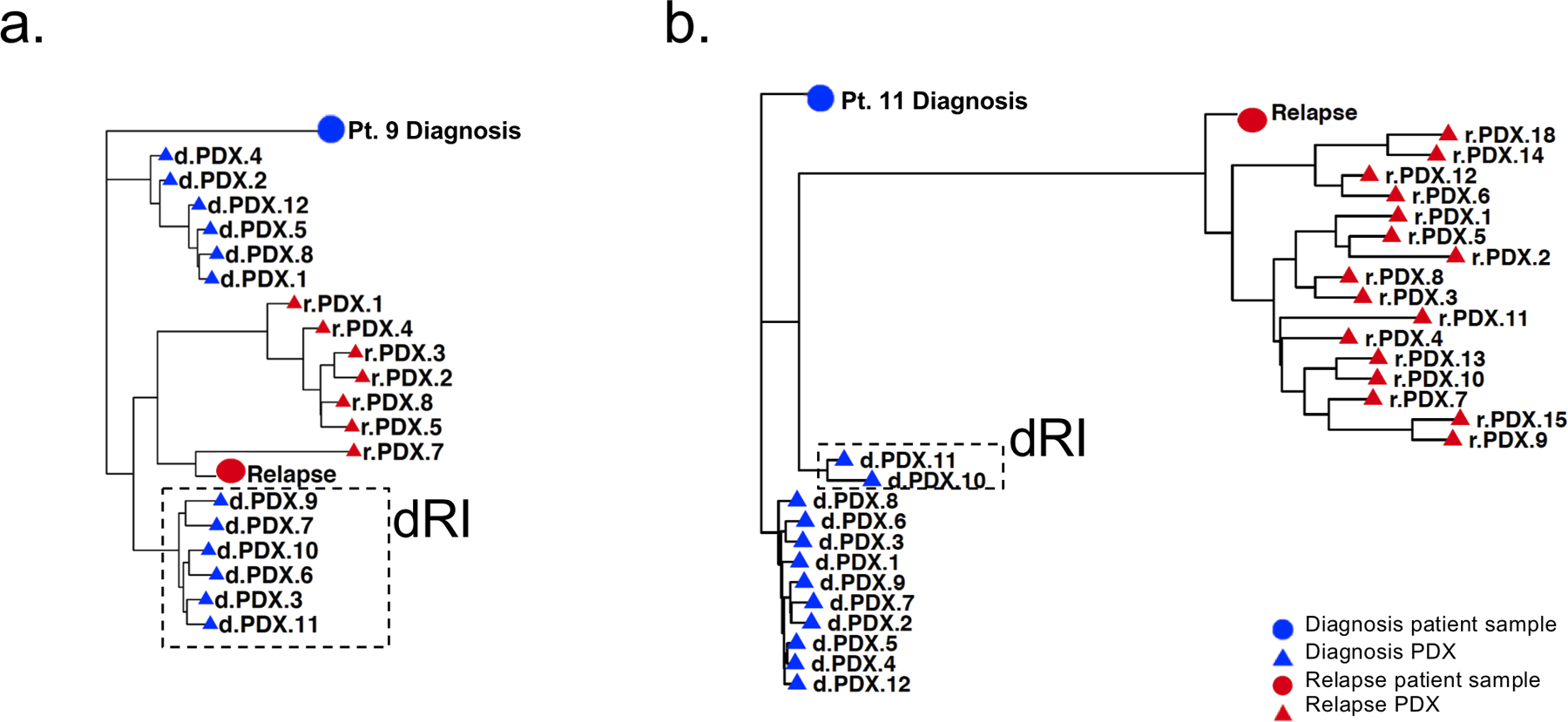
a. and b. Phylogenetic analysis showing relationship of patient 9 (a) and patient 11 (b) patient samples and xenografts, based on VAF of leukemic variants. The distance between symbols on the tree were estimated by a nearest neighbour joining method and represent the degrees of relation between them (Minkowski’s distance). Circles represent patient samples and triangles represent PDX; blue represents diagnosis and red represents relapse. Diagnosis clones on the trajectory to relapse were termed diagnosis relapse initiating clones or dRI and are indicated by a box with a hatched border.
For our second approach, we used the extensive mutational information garnered from the 372 PDXs to supplement the patient analysis. For each patient, computational analysis using PairTree (Wintersinger et al. in preparation) was undertaken to group leukemic variants with similar VAFs across numerous PDX and bulk samples to define genetically unique mutational populations. We then constructed mutational trees to describe the evolutionary history of each leukemia and the order of mutational acquisition (Fig. 4; Supplementary Fig. S2a–c; Supplementary Fig. S3a–c; Supplementary Fig. S4a–e; Supplementary Fig. 5a–c; Supplementary Table S3). In 7 of 14 patients, the PDXs helped seperate the mutational aquistion into multiple distinct populations that could not be resolved using the sequencing data of the primary patient samples alone. For instance, in patient 1, analysis of the xenografts revealed the existence of four distinct subclonal populations (populations 10,12,14 and 15) at relapse that, when only the patient samples were considered, had been predicted to be only a single subclone (population G) (Fig. 4a–d). This analysis further enabled comparisons of the state of the evolutionary trajectory of the subclones captured within each individual PDXs (denoted by a numbered PDX referring to a specific mouse) to the patient samples from which they were derived; such as dPDX7 in patient 1 whose clonal composition is more closely related to the relapse than the diagnosis sample (Fig. 4c–d). Additionally, the PDXs aided in subdividing single mutational populations into more than a single linearly related population or branch in 11 of 14 patients. For example, population G in patient 9 was expanded into two separate populations (populations 8 and 9) when PDXs were included (Fig. 4e–f; Supplementary Fig. S2a–c; Supplementary Fig. S3a–c; Supplementary Fig. S4a–e; Supplementary Fig. S5a–c).
Figure 4: Generation of mutational trees from the combined genomic data of xenografts and bulk diagnosis and relapse patient samples.
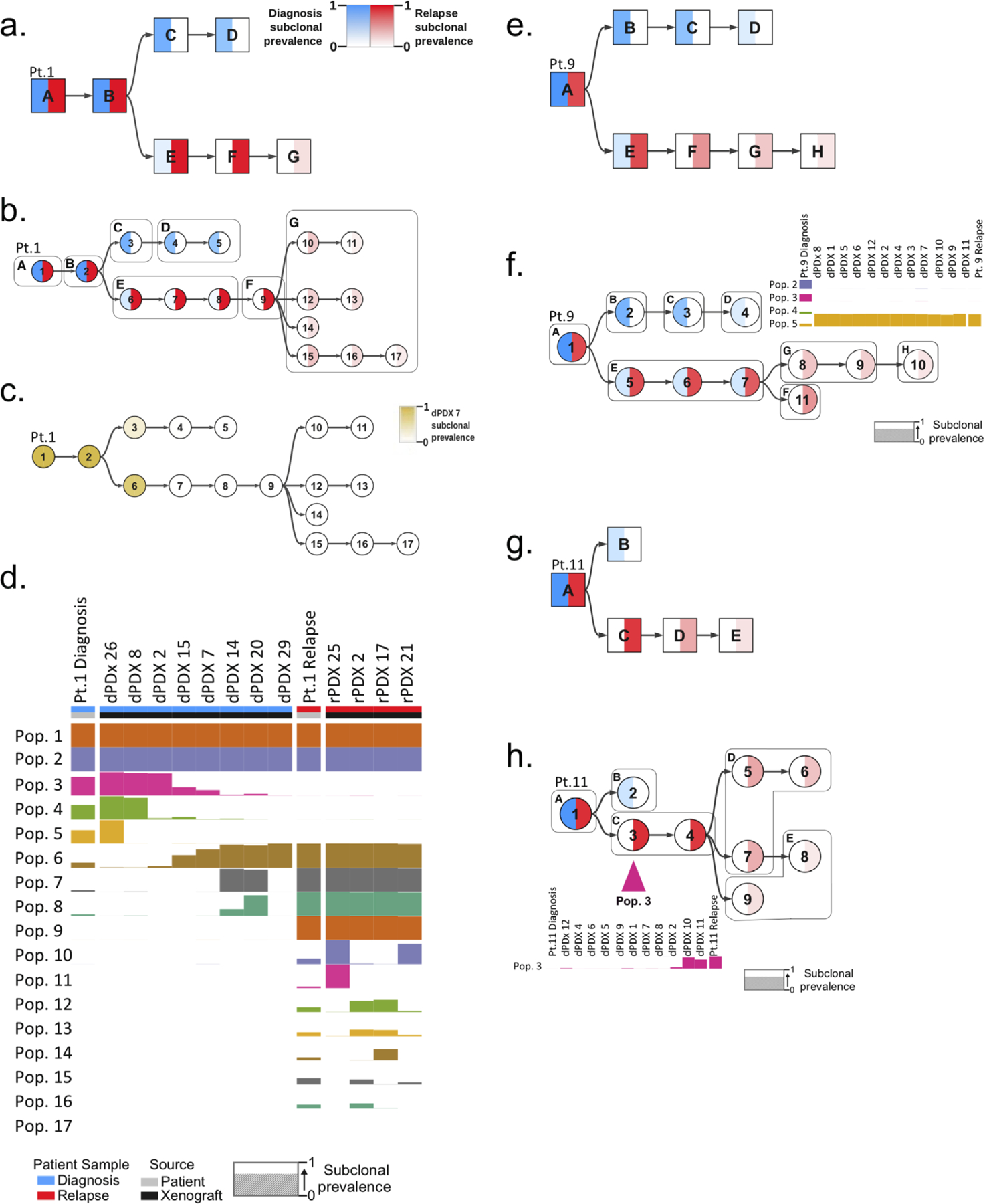
Mutational trees of variants clustered to form populations using the PairTree algorithm. Nodes in mutational trees are divided in half, with the intensity of blue in the left half indicating the frequency of the population’s variants at diagnosis, and the intensity of red in the right half showing the frequency of the population’s variants at relapse. Colour intensity shows subclonal prevalence as noted in the legend of a. and applicable to all other trees except c. a. Mutational tree of patient 1 derived by analysis of the patient samples (diagnosis and relapse) alone. Mutational populations identified from bulk patient samples alone are denoted by a square node labeled with with an alphabetical letter. b. Combined mutational tree derived from the variant analysis of both patient 1 patient samples and all their generated xenografts. Mutational populations derived from combined patient and xenograft analysis are represented by cicrcular nodes labeled with numerals. The mutational populations identified using the patient samples alone in a., are overlaid on the tree as boxes labeled with their corresponding alphabetical letter. This identifies instances where single populations in a correspond to multiple populations when xenografts are included (ie. mutational population G). c. Combined mutational tree of patient 1 shaded to indicate the prevalence of variants in dPDX 7 (instead of diagnosis and relapse) demonstrating that this PDX is composed primarily of variants of the relapse lineage. d. Presence of identified mutational populations in patient samples and representative xenografts. Mutational populations (Pop.) are displayed on the y-axis and individual patient samples or xenografts are displayed on the x-axis. The height of the population bar represents the prevalence of the lineage in the patient sample (Pt.) or xenograft. e. Mutational tree, similar to a, derived from patient samples alone of patient 9. f. Combined mutational tree, similar to b, derived jointly from patient 9 patient samples and xenografts. Subclonal prevalences of populations 2–5 are shown, indicating the absence of diagnosis populations 2–4 and presence of population 5 (the first node in the relapse lineage branch) in all dPDX. g. Mutational tree, similar to a, of patient 11 derived from patient samples alone. h. Combined mutational tree, similar to b, of patient 11 derived from patient samples and xenografts. The prevalence of mutational population 3 is displayed, highlighting its absence in the diagnosis patient and its detection in only two dPDX.
Genetically diverse subclones have differing xenograft repopulation kinetics
The mutational analysis of the xenografts performed using PairTree helped illuminate the competitive differences in xenograft repopulation kinetics of specific subclones. We compared the predominant diagnosis mutational populations in the patient sample to the mutational populations that engrafted in the xenografts. In all but four patient samples the dPDX captured the diversity of clones present in the diagnosis samples (Fig. 4; Supplementary Fig. S2a–c; Supplementary Fig. S3a–c; Supplementary Fig. S4a–e; Supplementary Fig. S5a–c). In two of these four patients (patients 7 and 11) minor diagnosis-specific mutational populations including a population harbouring missense NRAS mutations in patient 7 did not engraft in the dPDX mice (Supplementary Fig. S4a; Supplementary Table S3). In the other two patients (patients 9 and 12) all the dPDX were initiated from a minor population in the diagnosis sample ancestral to the relapse (Fig. 4; Supplementary Fig. S4c; Supplementary Fig. S5a). These data show the enhanced competitive repopulation properties of these dRI clones. In patient 9, all dPDX were initiated from mutational population 5 that corresponded to only 22% of the diagnosis leukemic cells (initially described as population E in the patient sample alone mutational tree) in comparison to the mutational population 2 lineage that represented 71% of the diagnosis leukemic cells (initially described in population B) (Fig. 4e–f). In the dPDX, population 5 displayed a strong selective advantage representing at least 93% of the leukemic cells, outcompeting the population 2 lineage (encompassing populations 2–4) and recapitulating the evolutionary dynamics of the patient relapse, where 98% of the leukemic cells came from the population 5 lineage (Fig. 4f). In contrast, in 5 patients (patients 1,4,6,7 and 11) the relapse-fated mutational population represented only a small percentage of the engrafting cells in most dPDX and only rose to predominance in rare mice. For example, in patient 11, relapse-fated mutational population 3, initially described in population C, only corresponded to 1% of leukemic cells at diagnosis and remained at similarly low levels in most dPDX. However, in two dPDX transplanted with a low, near limiting cell dose (10,000 cells), population 3 rose to 92% and 74% of leukemic cells distinguishing it from population 4 and highlighting the importance of the limiting dilution approach to capture rare subclones by isolating them away from other subclones with higher competitive capacity (Fig. 4g–h; Supplementary Fig. S4e). Population 3 was defined by a single variant in the 3’UTR of AHNAK, a gene whose expression has previously been implicated in disease relapse in T-ALL(33). Thus, the xenografting strategy permitted integration of functional and genomic information providing insight into the distinct competitive growth properties of functionally defined L-IC subclones. Taken together, the phylogenetic analysis and mutational evolutionary trees provide strong evidence that relapse-fated subclones were already present in the diagnosis sample confirming prior studies of several human acute leukemic diseases and in silico predictions(4,5,27,28).
Genetically diverse subclones have differing immunophenotypic and migratory properties
To further examine whether genetically distinct subclones also possessed variation in their immunophenotype or functional properties that might explain why some are fated to contribute to relapse and others are not, we examined their differentiation, growth, and migration properties. We first interrogated the differential properties of the diagnosis clones isolated from patient 9 in which the presence of latent variants including the known relapse driver CREBBP(20), was segregated between clones. Differences in immunophenotype were observed for dRI-PDX enriched for these latent variants which were present at low VAFs in the diagnosis patient sample (1–11%) (Fig. 5a–b). The dRI-PDX exhibited a CD45negCD34+ immunophenotype reflective of the relapse sample whereas others contained both a CD45negCD34+ and a CD45dim CD34+ population as observed in the diagnosis patient sample (Fig. 5a). Relapse PDX (termed rPDX), like the relapse sample itself were CD45neg (Fig. 5a). dPDX that were engrafted primarily with CD45dim or both immunophenotypic populations had a genotype that was more closely related to the diagnosis sample (Fig. 5b). The differences noted in immunophenotype appeared to reflect the rise of the relapse fated clone and aided in identifying different subclones present within a single xenograft. To validate that the difference in immunophenotype segregated the predominant diagnosis clone from the minor subclone that seeded the relapse, we isolated the two immunophenotypic poplulations by flow sorting cells from 6 dPDX and subjected the populations to targeted sequencing using the custom capture array designed for the patient samples. This analysis confirmed the enrichment of the dRI clone in the CD45neg population, whereas latent variants were absent or very rare (<6%) in the CD45dimCD34+ population (Fig. 5b). Of note, change in CD45 expression between diagnosis and relapse timepoints were identified in 5 of our patient samples. PDX from patient 9 showed markedly reduced leukemic dissemination from the injected femur to other hematopoietic sites (Supplementary Fig. S6a).
Figure 5: Competitive dRI-PDX clones identified in diagnosis PDX.
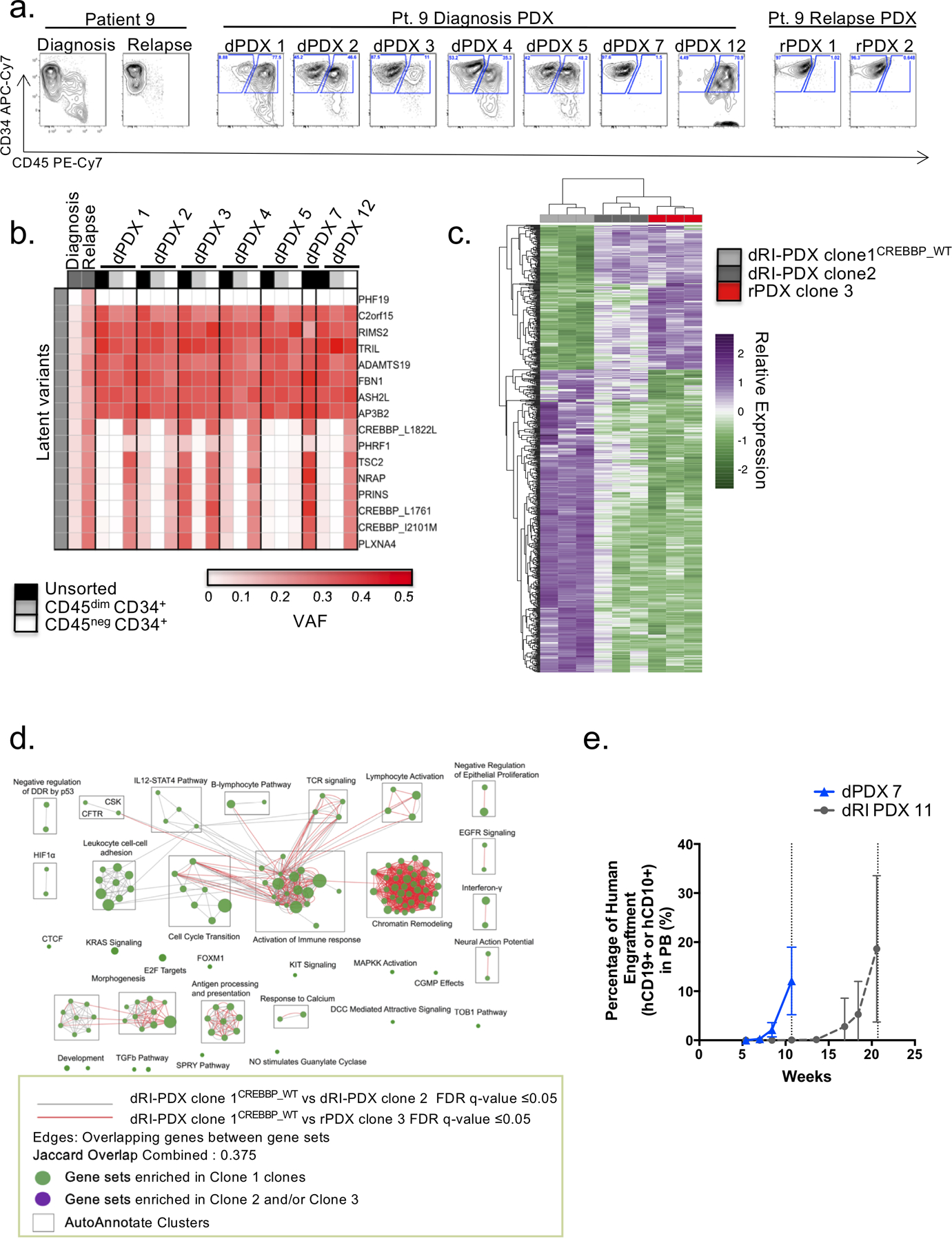
a.Flow cytometry analysis of patient samples and representative dPDX and rPDX of patient 9 display the presence of two different immunophenotypic populations: a CD45dimCD34+ and a CD45negCD34+. b.Targeted sequencing of the dPDX revealed variability in the VAF of latent variants that corresponded with the shift in immunophenotypic populations. Cell sorting for immunophenotypic populations followed by targeted sequencing revealed the isolation of the latent variants in the CD45negCD34+ population. c. RNA-sequencing analysis of differentially expressed genes (adjusted p-value of < 0.05 and absolute log2 fold change of > 1) between the two dPDX (dRI-PDX clone 2; dRI-PDX clone 1CREBBP_WT) vs rPDX for Patient 9. Relative expression was generated from variance stabilized normalized counts. d. Enrichment map of gene sets differentially enriched in patient 9 dRI-PDX clone 1CREBBP_WT vs dRI-PDX clone 2 (FDR q value ≤ 0.05) and dRI-PDX clone 1CREBBP_WT vs rPDX clone 3 (FDR q value ≤ 0.05). Node size is proportional to the number of genes included in each gene set (minimum 10 genes/gene set). Grey and Red edges indicate gene overlap. Green node: enrichment in dRI-PDX clone 1CREBBP_WT (positive NES). Purple node: enrichment in dRI-PDX clone 2 and/or rPDX clone 3 (negative NES). Autoannotate app in Cytoscape was used to automatically annotate clusters (black squares). NES: normalized enrichment score; DDR: DNA Damage Response; TCR: T-Cell Receptor; UPR: Unfolded Protein Response; cGMP: cyclic Guanosine Monophosphate; DCC: Deleted in Colorectal Cancer gene; NO: Nitric Oxide; CSK: C-terminal Src Kinase; CFTR: Cystic Fibrosis Transmembrane Conductance Regulator; SPRY: Sprouty gene. e. Human purified cells from primary dPDX and dRI-PDX from patient 4 were transplanted into secondary NSG recipients. Mice were monitored for peripheral blood human chimerism until mean blood levels reached greater than 10% revealing different kinetics of chimerism between dPDX 7 and dRI-PDX 11. Symbols represent the mean chimerism value of dPDX 7 (n= 20 mice) and dRI-PDX 11 (n= 16 mice) and bars represent standard deviation.
The ability to use differences in immunophenotype to segregate clones enabled us to select evolutionarily related dRI subclones for RNA-sequencing (RNA-seq) analysis from patient 9. We compared the expression profiles of two dRI clones, an ancestral and a daughter clone (clones 1 and 2 respectively), to a representative relapse clone (clone 3). The dRI clone most genetically similar to the relapse, bearing latent leukemic variants including 3 variants in CREBBP (2 missense and 1 silent) as well as variants in TCS2, NRAP, PLXNA4 and PRINS (dRI-PDX clone 2) had an expression profile very closely related to representative relapse clones with only 24 differentially expressed genes (Fig. 5c–d; Supplementary Table S4). In comparison, the ancestral clone not harbouring CREBBP (dRI-PDX clone 1CREBBP_WT) showed 479 differentially expressed genes compared to relapse clones suggesting that the majority of transcriptional changes that are seen at relapse had already occured in dRI-PDX clone 2 at diagnosis prior to exposure to chemotherapy (Supplementary Table S4). Broadly, the changes in gene expression of dRI-PDX clone 2 in relation to dRI-PDX clone 1CREBBP_WT centered on dysregulation of histone variants and inflammation-related genes as well as downregulation of genes involved in morphogenesis (TGFβ signaling, GATA3), the TCR and BCR pathways (CD19, CD79A, CD4, etc) and antigen processing and presentation pathway, while heat-shock response (HSR) and unfolded protein respone (UPR) pathways were enriched (Supplemental Table S4). Thus the early acquisition of additional leukemic variants in this relapse-fated subclone, including the CREBBP mutations, caused significant changes in gene expression. Interestingly, tyrosine kinases such as EPHB2, LTK and ERBB2 were upregulated after acquistion of CREBBP variants in both dRI-PDX clone 2 and the relapse clone suggesting possible vulnerabilities to tyrosine kinase inhibitors. Despite this striking change, the dRI subclone remained dormant within the patient for many years as this patient displayed a long remission (4 years).
PDX from the majority of samples showed extensive migration of leukemic cells to other hematopoietic sites and other tissues including the SPL, CNS and peripheral blood (Supplementary Fig. S6b–c). Since patients with B-ALL may present with leukemic dissemination to the CNS and testes where they can provide a sanctuary for relapse, we investigated whether there were differences in the dissemination of subclonal populations to each site. Targeted sequencing analysis showed that there was genetic discordance between CNS and bone marrow in 40% (44 of 111) of xenografts and between SPL and BM in 17.8 % (28 of 157) of xenografts (Supplementary Table S5). In one patient (patient 7), our analysis of the genetic discordance revaled the presence of a dRI clone engrafting in the CNS (VAFs > 39%) of a a xenograft transplanted with a high cell dose (250,000 cells), in which the clone was a minor clone barely detectable in the BM (VAFs of < 3%) and outcompeted by a more predominate diagnosis clone. (Supplementary Fig. S6b). The identification of the dRI subclone in the CNS of dPDX is consistent with the ability of relapse-fated cells to disseminate and cause disease recurrence in the CNS of B-ALL patients. We also noted the occurrence of a difference in peripheral blood dissemination of dRI in an additional patient (patient 4), with delayed mobilization of the dRI-PDX subclone as compared to the representative diagnosis clone (Fig. 5e). Collectively, the combined functional and genotyping studies show that individual subclones possess distinct immunophenotypic, competitive repopulation, proliferative, and migration properties and that dRI subclones can already possess distinct biological properties at diagnosis, before exposure to chemotherapeutic agents.
dRI subclones display differential response to chemotherapeutic agents
To directly test the functional properties of dRI subclones with their ability to survive and contribute to relapse, we compared the drug sensitivities of individual subclones for 5 of the genetically distinct patients. Multiple secondary PDX were generated from dPDX with known dRI clones (dRI-PDX), predominant diagnosis clones (dPDX), or representative relapse clones (rPDX). Treatment of B-ALL patients includes combination chemotherapy with supportive care, however, it is not possible to replicate human therapy precisely in xenografts. Our interest was to determine if there was any variation in the responsiveness of different subclones to individual drugs used in these chemotherapeutic protocols. Following engraftment, PDX received single agent treatments of dexamethasone, vincristine, L-asparaginase or saline for 4 weeks (Fig. 6a–b and Supplementary Fig. S7a–f). Differences in therapeutic responses to one or more drugs between dRI and representative diagnosis clones were observed for four patient samples. In three patient samples, (patient 1, 6, 7), dPDX harbouring a dRI clone (dRI-PDX) were compared to dPDX repopulated with the major diagnosis clone and demonstrated reduced sensitivity to 2 or 3 of the 3 chemotherapy agents tested (Fig. 6a–b and Supplementary Fig. S7a–d). Reduced sensitivity to a single chemotherapeutic agent was also observed in one additional patient (patient 4; dexamethasone, significant in injected femur and trend in BM and SPL) (Supplementary Fig. S7e). By contrast, there was no difference in the therapeutic sensitivity of two dRI-PDX from patient 11 defined by the presence of the AHNAK 3’UTR varaint. This patient has a strong driver KMT2A (MLL) translocation and the presence of the single relapse specific variant does not appear to confer sufficient evolution of the leukemia to alter the therapeutic sensitivity in this context (Supplementary Fig. S7f). Purification of human cells from the secondary PDXs post therapy and targeted sequencing confirmed their genotype and did not reveal the selection of any further relapse-specific variants (Supplementary Fig. S8a–c). The observed differences in drug response could not be accounted for by any consistent changes in L-IC frequency (Supplementary Table S6).
Figure 6: dRI subclones display decreased sensitivity to commonly used chemotherapeutic drugs.
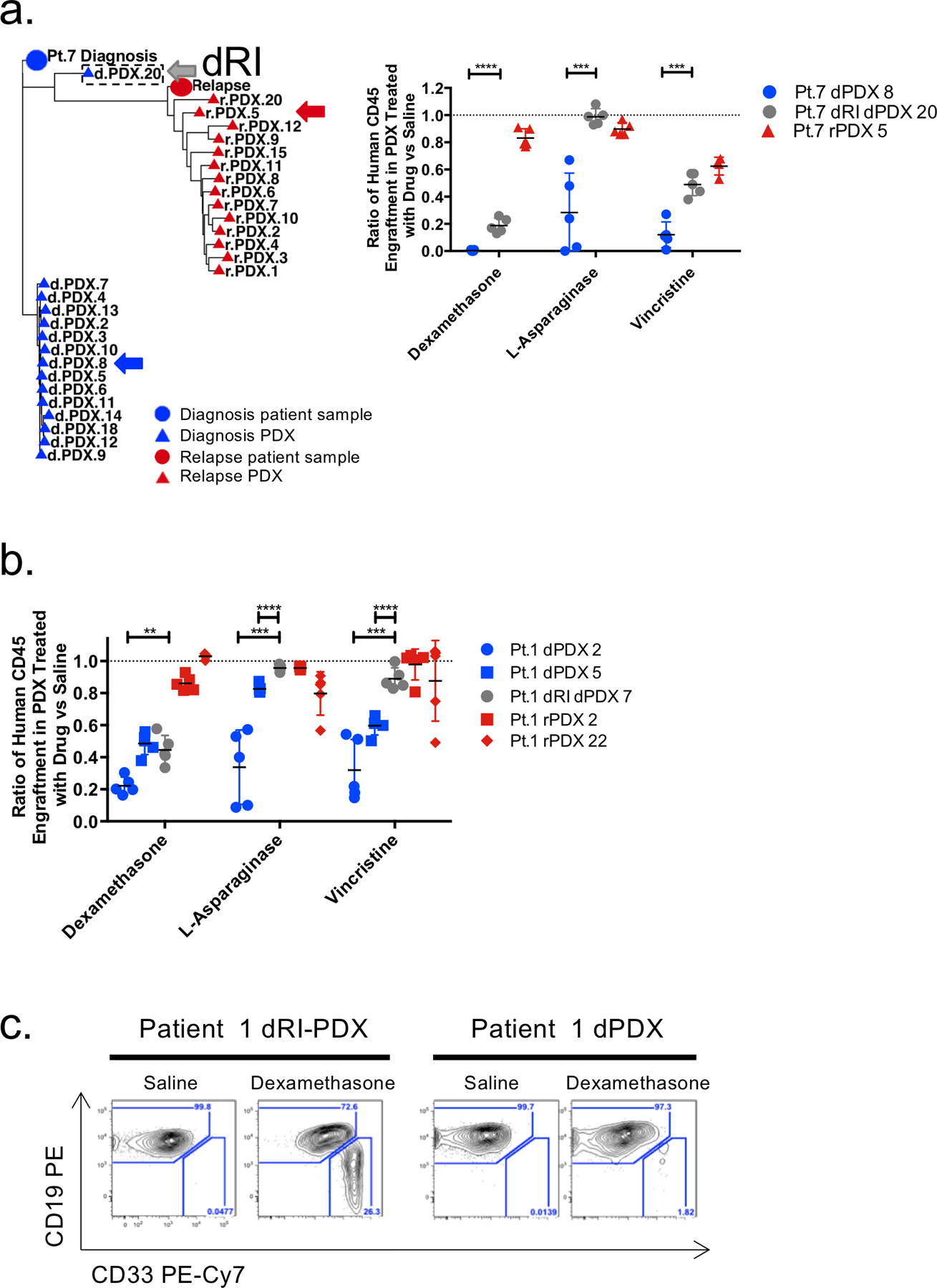
a. Phylogenetic analysis showing clonal relationships in patient 7 based on VAF of leukemic variants shows clear evidence of the isolation of a relapse-fated, dRI, clone in dPDX 20. The distance between symbols on the tree were estimated by a nearest neighbour joining method and represent the degrees of relation between them (Minkowski’s distance). Circles represent patient samples and triangles represent PDX; blue represents diagnosis and red represents relapse. dRI-PDX 20 is indicated by a hatched border box. Purified human cells from primary dPDX 7, dRI-PDX 20 and rPDX 5 (representative relapse genetics) xenografts were injected into secondary NSG mice and allowed to engraft. Mice were randomized into 4 groups (with 4–5 mice per group) and treated with either saline, dexamethasone, L-asparagine or vincristine. After 4 weeks of treatment mice were sacrificed and engraftment in the IF, BM and SPL were analyzed by flow cytometry. Ratio of human chimerism in the BM of drug treated mice in comparison to saline controls is shown. b..Ratio of human chimerism in the BM of drug treated mice in comparison to saline controls of dPDX, dRI-PDX and rPDX of patient 1. c. Representative flow plots of the CD19 and CD33 immunophenotype of dPDX and dRI-PDX dexamethasone treated mice from patient 1. Lines represent mean and standard deviation. Only significance between dPDX and dRI-PDX are shown. **p < 0.01, *** p < 0.001, **** p < 0.0001, unpaired two-sided t-tests.
Unexpectedly in two of five patient samples used for drug testing, we found that the dRI subclones exhibited immunophenotypic plasticity as compared to the predominant diagnosis clone when exposed to drugs. In the secondary recipients of dRI-PDX of patient 1 and dRI-PDX of patient 7, both patients harbouring DUX4 translocations at diagnosis, there was the emergence of a distinct CD33+CD19dim/neg population upon dexamethasone treatment but not in saline controls or upon treatment with the other two drugs (Fig. 6c) (Supplementary Fig. S9a–b). This population was very rare or not observed in the primary recipients. The CD33+CD19dim/neg cells resembled myeloid cells with respect to their cell surface marker expression, size and granularity (Supplementary Fig. S9c). An immunophenotypic shift towards the myeloid lineage especially with steroid challenge has previously been reported in ERG/DUX4 patients but our finding of a subclone specific switch is of interest(34). The emergence of CD33+CD19dim/neg cells was not due to the outgrowth of a different subclone as these cells were genetically identical to their CD19+ counterpart (Supplementary Fig. S9d–e). We speculate that this propensity for immunophenotypic plasticity might be linked to chromatin remodelling and the ability to evade therapy as other studies have shown(35–38). Overall, our data provide strong evidence that prior to exposure to chemotherapy, dRI subclones already possess distinct pre-existing functional properties including differential sensitivity to standard chemotherapy agents.
Transcriptional analysis of dRIs reveal metabolic rewiring and enrichment of a stem cell state in progression to relapse
To gain mechanistic insight into the molecular pathways present in dRIs, RNA-seq analysis was performed on cells from dRI-PDX, dPDX, or rPDX of four patients (Patients 1,4,6,7; n=14 dPDX, 15 dRI-PDX and 13 rPDX) (Supplementary Table S7). This analysis confirmed the placement of dRIs as intermediates between diagnosis and relapse, sharing transcriptional programs with both timepoints (Supplementary Fig. S10a). Given the distinct B-ALL subtypes/cytogenetics of the patient samples analyzed, as expected only few (n= 23) differentially expressed genes reached significance when comparing dPDX vs dRI-PDX across all samples (Supplementary Table S7). Suprisingly, one of the genes upregulated in the dRI-PDX is asparaginase (ASPG), a catalytic enzyme that hydrolyzes asparagine to aspartic acid, albeit non-robustly in human cells. The human ASPG protein can display cytotoxic activity in human leukemic cell lines(39) suggesting that the dRI-PDX cells may have altered their response to cytotoxic stress or metabolic requirements thereby explaining why some of the dRI clones are less sensitive to L-asparaginase treatment than dPDX clones.
As pathway enrichment analysis is more sensitive than differential gene expression for finding differences between populations we undertook this approach to uncover significantly differentially enriched pathways that were shared amongst all patients. Gene set enrichment analysis (GSEA) comparing the pathways present in dRI-PDX vs dPDX revealed that most pathways significantly enriched in dRI-PDX (FDR q-value ≤ 0.05) were also present or further enriched in the comparision of rPDX to dPDX; these are termed dRI-PDX/rPDX common pathways (Fig. 7a–b; Supplementary Fig. S10b–d; Supplementary Table S7). Network analysis revealed that these pathways were centered on a metabolic signature composed of genes involved in cellular and mitochondral metabolism including amino acid metabolism, TCA cycle, oxidative phosphorylation, mitochondrial translation and transport, and lipid metabolism (Fig. 7a–b; Supplementary Fig. S10b–d). In concordance with a previous study on ALL relapse(25), the central regulator of growth and metabolism, mTOR, was enriched in dRI and relapse clones (Supplementary Fig. S10b; Supplementary Table S7). Pathways identified as uniquely enriched in rPDX vs dPDX (rPDX-unique), included a large network of highly interconnected nodes involved in cell cycle regulation such as cell cycle checkpoints, DNA replication, DNA repair and microtubule organization (Fig. 7a; Supplementary Table S7).
Figure 7: dRI subclones share a common metabolic and stem cell profile.
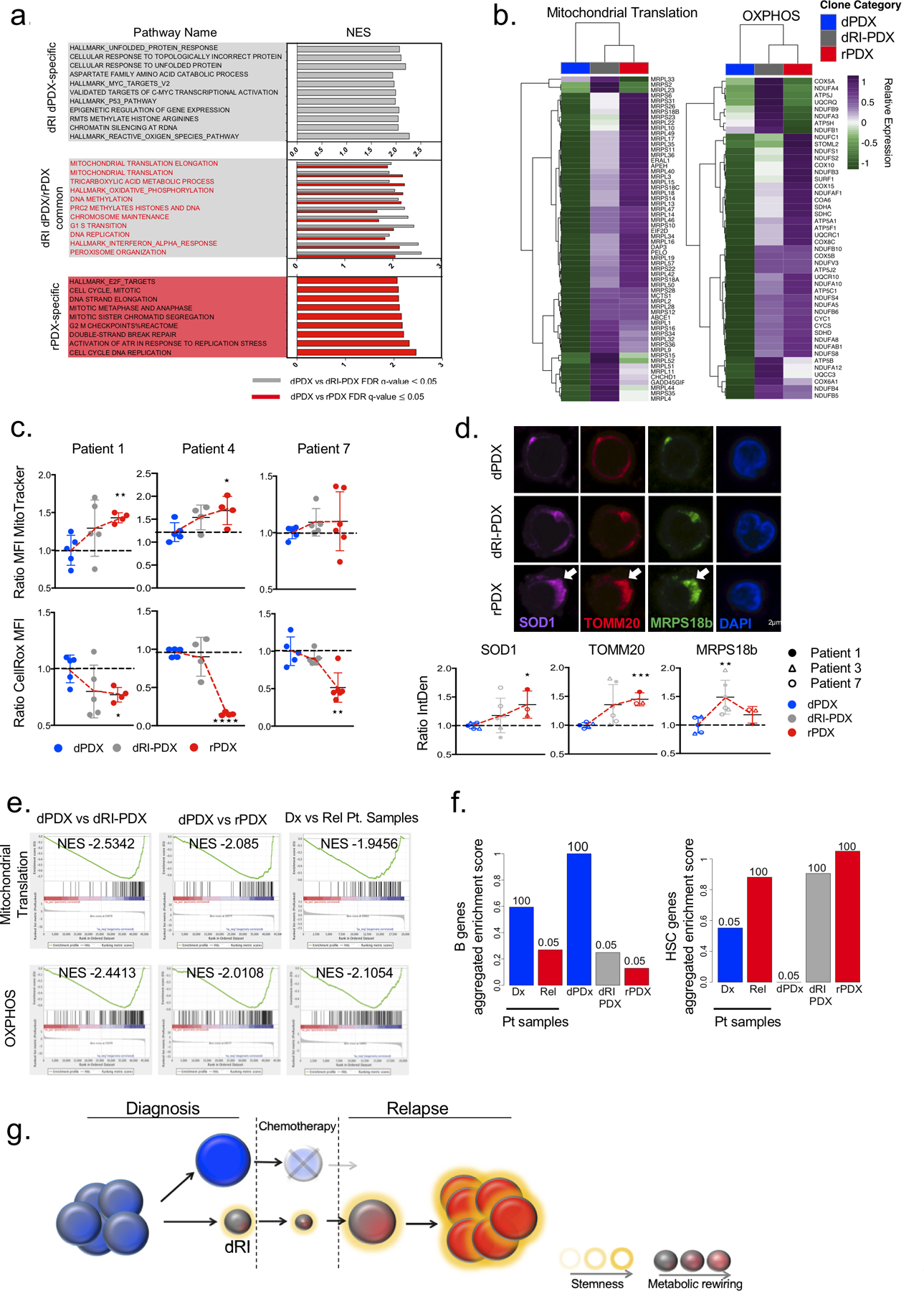
a. Plot showing the normalized enrichment score (NES) for the top differentially enriched gene sets (FDR q value ≤ 0.05) of dRI-PDX unique, dRI-PDX/rPDX common and rPDX unique groups from GSEA analysis of the following comparisons: dPDX vs dRI-PDX and dPDX vs rPDX. b. Heatmaps showing the expression of leading-edge genes (subset of genes found in the ranking at or just before the maximal enrichment score in GSEA analysis) for selected gene sets enriched in dRI-PDX and rPDX from enrichment map in (a). Relative expression was generated from variance stabilized normalized counts c. dPDX, dRI-PDX and rPDX from Patient 1, Patient 4 and Patient 7 were stained with MitoTracker and CellROX dyes and analyzed by flow cytometry. Mean fluorescence intensity (MFI) for each sample and dye is represented as ratio to dPDX for each patient (Patient 1: dPDX n=5, dRI-PDX n=5, rPDX=4; ; Patient 4: dPDX n=5, dRI-PDX n=4, rPDX=4; Patient 7: dPDX n=5, dRI-PDX n=5, rPDX=5). d. Immunostaining analysis for TOMM20, MRPS18B and SOD1 in dPDX, dRI-PDX and rPDX cells from Patient 1, 3 and 7. The Integrated Density (IntDen= Area* Mean Fluorescence Intensity) for 40–50 cells from each clone was analyzed using Fiji. The mean for each clone was normalized and calculated as a ratio to the dPDX for each patient separately. Representative images for Patient 7 are shown. (Patient 1: dPDX n=1, dRI-PDX n=2, rPDX=1; Patient 7: dPDX n=2, dRI-PDX n=2, rPDX=1; Patient 3: dPDX n=2, dRI-PDX n=2, rPDX=1). e. GSEA enrichment plots from the following comparisons (1) dPDX vs dRI-PDX (n=4 pts), (2) dPDX vs rPDX (n=4 pts) and (3) diagnosis vs relapse patient samples from our cohort, were generated for mitochondrial translation and OXPHOS gene sets. f. Barplot of the aggregated GSVA scores for B cell genes and HSC genes in each sample. GSVA scores for samples in each category were summed and scaled from 0 to 1. The numbers above the bars represent how many times the observed score was higher than random scores obtained in 1000 permutations using a list of 1000 random genes. g. Schematic diagram of dRI with altered metabolic and stem cell programs pre-exisiting in diagnosis patient samples that survive chemotherapy and seed the relapse disease. *p<0.05, **p < 0.01, ***p<0.001, unpaired two-sided t-test.
To validate the metabolic signature observed in dRIs, that was often further enhanced at relapse, we performed flow cytometry and immunostaining analysis in the PDX from five patient samples (Fig. 7c–d; Supplementary Fig. S10e–f). This analysis confirmed the convergence of similarities in metabolic rewiring of dRI-PDX/rPDX in comparison to dPDX in all 5 B-ALL patients. This functional validation showed that there was an increase in total mitochondrial mass, with individual mitochondria having similar mitochondria membrane potential in most of the patients analyzed. However, despite this increase in absolute mitochondria abundance, the levels of reactive oxygen species (ROS) were unexpectedly found to be lower in dRI-PDX and rPDX cells as compared to dPDX cells. The reduced ROS levels is suggestive of the presence of increased antioxidant defense upon progression toward relapse. This interpretation was concordant with our gene expression data that showed an enrichment of ROS-defense and peroxisomal activity genes (ie. catalase), which degrade toxic hydrogen peroxide and metabolize drugs, in the dRI-PDX and rPDX samples (Fig. 7a,c; Supplementary Fig. S10b,d–f; Supplementary Table S7). Significant enrichment for chromatin remodelling and cell stress response (such as the unfolded protein response (UPR)) were also identified as dRI-PDX/rPDX common pathways (Fig. 7a; Supplementary Fig. S10b–d). The chromatin remodelling pathways including expression of histone variants and isoforms were likely instrumental in the plasticity observed in the progression to relapse including the immunophenotypic plasticity we observed in dRI-PDX of patient 1,7 and 9 (Fig. 4a, Fig 5c), while the expression of the cell stress response pathways identified could contribute to enhanced survival of dRI subclones. To independently validate the results obtained from the PDX identified pathways, we directly evaluated bulk diagnosis and relapse patient samples. We found enrichment (FDR q-value ≤ 0.05) of pathways involved in metabolism, mitochondria regulation and cell cycle at relapse (Fig. 7e; Supplementary Fig.S10g; Supplementary Table S7). Furthermore, we found significant enrichment of a signature present at minimal residual disease (MRD)(17) in dRI-PDX and rPDX compared to dPDX lending further support for the relevance of the dRI-PDX/rPDX pathways for understanding the mechanisms of relapse disease initiation and their presence prior to chemotherapeutic challenge (Supplementary Fig. S10h). Collectively, our transcriptional analysis has for the first time uncovered the utilization of chromatin remodeling, stress responses and metabolic pathways in dRI subclones whose activity could serve to protect them during chemotherapy treatment and contribute to their ability to further progress to relapse disease. As such these newly identified pathways represent rich areas to investigate for new therapeutic strategies to target dRI specifically.
To investigate whether the broader cellular state of the dRIs contributes to their functional differences, we performed gene set variation analysis (GSVA) comparing the transcriptomic profile of representative clones to normal hematopoietic cell populations isolated from human umbilical cord blood using 2 independent datasets, one from our own newly generated data and the other previously published(40)(Supplementary Fig. S11a; Supplementary Table S7). dRI-PDX and rPDX exhibited a transcriptome profile significantly enriched for hematopoietic stem cell (HSC) genes and a slight reduction in B cell genes as compared to dPDX (Fig. 7f; Supplementary Fig. S11b–c). Depletion of B cell genes and enrichment of HSC genes at relapse were also observed in the bulk patient samples from our cohort (Fig. 7f). Furthermore, the leading-edge genes of common pathways enriched in both dRI-PDX and rPDX are upregulated in HSCs as compared to lymphoid cells (Supplementary Fig. S11d). The enrichment of mitochondrial metabolism and stemness at relapse were also validated in the larger Waanders et al. cohort of paired diagnosis-relapse patient B-ALL samples(30) encompassing several genetic subtypes (Supplementary Fig. S11e,f). These findings are in line with previous studies in the normal and leukemia stem cell fields(24,41,42) where several metabolic and stress response pathways such as those identified here (mitochondria metabolism, UPR, antioxidant defense) have been described to be crucial to the maintenance of stem cell homeostasis and function. Additionally, enrichment of stemness signatures are also a hallmark of high-risk B-ALL(43). Thus, our findings report a link for the acquisition of HSC stemness properties in combination with metabolic rewiring in dRI as part of progression to relapse in lymphoid B-ALL leukemia (Fig. 7g).
DISCUSSION
Our study provides new insight into the leukemogenic process of human B-ALL through a deep analysis of the functional and molecular properties of genetically diverse diagnosis subclones isolated through xenografting from pediatric and adult B-ALL patients. By combining xenografting with sequencing, broad clonal structures were unambiguously demonstrated and additional subclones are uncovered. We identify relapse-fated dRI subclones, prior to chemotherapeutic exposure, with the capacity for clonal propagation and leukemia initiation in B-ALL that are both genetically and transcriptionally related to the relapse. Our data extends prior B-ALL studies that relied on genetic analysis and computational methods of bulk leukemia samples without functional studies, and that could only infer the presence and survival of minor subclones that were ancestral to the relapse driving clone and could not speak to the functional properties of these cells at diagnosis(4,5). As shown in Waanders et al.(30), while our xenografting approach substantiates the predicted lineage relationships of the traditional genetic analysis of bulk tissues it also extends our understanding of leukemia evolution by revealing a richer diversity of branching evolution and additional subclones. Our functional studies of subclones analysed either alone or in the context of competitive repopulation demonstrate that dRI subclones can be found to display increased clonal repopulation kinetics, immunophenotypic plasticity and differences in leukemic dissemination such as dissemination to the CNS, a known disease sanctuary site in patients. Importantly, dRI showed an intrinsic difference in tolerance to standard induction chemotherapeutic agents. Moreover, dRI subclones were typically very minor latent subclones in the diagnostic sample suggesting that they are restrained within the patient in comparison to the predominant subclone perhaps due to dormancy, timing, subclonal cross-talk or slow cycling, although this was not directly measured in our studies. Our study resolved the theoretical question as to whether functional properties of relapse clones are present prior to chemotherapeutic challenge or are induced by chemotherapy. While some mutations not present at diagnosis are recurrently acquired after chemotherapy (e.g. USH2A, NT5C2), our study documents the pre-existence of dRI subclones with intrinsic tolerance to clinical drugs. This data provides direct evidence that the Luria-Delbrück principle occurs in human leukemia(44). We speculate that the dRI subclones arise stochastically and these partially evolved subclones then become selected for by chemotherapy. Gene expression and pathway analysis showed that even at this early stage where relapse fate arises, there was considerable metabolic rewiring within dRI subclones that was often further enhanced in relapse clones. Recurrent pathways were identified across a spectrum of different B-ALL patients with distinct genetic drivers suggestive of convergent evolution. Broadly, we found that adult and pediatric dRI had similar properties in terms of their functional xenografting properties, therapy response, transcriptional pathways and metabolic rewiring also supporting the concept of convergent evolution. Pathways regulating mitochondrial dynamics and proteostasis are particularly relevant since they are linked to survival under stress, processes that may underpin the ability of dRI cells to adapt and survive chemotherapy. Futher evolution and mutagenesis of such subclones during the treatment and/or remission phases ultimately results in disease recurrence. Expression of the dRI-PDX/rPDX common pathways converged on a HSC signature, shared by both dRI and relapse cells, analogous to recent studies we have undertaken from purified acute myeloid leukemia (AML) leukemia stem cell populations(23,45). The finding of stemness signatures already present in dRI subclones, together with the identified metabolic changes provides the molecular basis to explain why dRI subclones may both survive therapy and posses the regenerative capacity to intiate relapse disease after a period of dormancy. Future studies that explore the transcriptome of dRI subclones from across an even wider spectrum of B-ALL patient samples would give insight into how the timing of acquisition of these molecular pathways leads to relapse.
The ability to isolate and characterize dRI subclones provides an important first step in understanding the basis for therapy resistance and clonal propagation. Such subclones isolated in this way are not simply an in silico depiction, rather they can be viably preserved cells or serially propagated xenografts for future studies. Whole genome sequence, methylation and chromatin accessibility studies could be undertaken to build upon our transcriptomic analysis; thereby exploring more deeply the mechanisms that drive their functional properties. The signatures developed from isolated dRI clones could yield biomarkers to improve the classification of patients who are at increased risk of relapse and to better monitor residual disease. The immunophenotypic plasticity we linked to different genetic subclones points to the need to understand the breadth of cell surface phenotypes present to ensure that all leukemic subclones are properly tracked during flow cytometry based residual disease monitoring. Finally, further investagtion of the dRI transcriptomic profile and metabolic rewiring may be used to uncover the vulnerabilities of dRI subclones resulting in new therapeutic targets. Improved eradication of dRIs during early treatment phases before the subclones evolve would prevent progression to more aggressive, therapy resistant disease. Prior studies from us and others have shown that xenografting of a wide spectrum of primary patient samples provides a powerful tool to evaluate novel therapeutics and develop biomarkers(22,24,46,47). Our study suggests that extending the xenograft-based drug development paradigm by including genetic analysis to uncover subclonal responses to drug treatment will open up avenues to evaluate whether relapse fated clones are effectively targeted.
METHODS
Patient Samples
Patient samples were obtained at diagnosis and relapse from six adult and eight pediatric B-ALL patients according to pre-established guidelines approved by the Research Ethics Board of the University Health Network and the St. Jude Institutional Review Board respectively and were conducted in accordance with recognized ethical guidelines. Adult samples were collected at the Princess Margaret Cancer Centre and pediatric samples were collected at St. Jude Children’s Research Hospital. Written informed consent was obtained from all patients or patient families. All samples were frozen viably and stored long term at −150 °C. Samples were selected retrospectively based on sample and paired sample availability.
Patient Derived Xenograft Generation and Limiting Dilution Analysis
Twenty-nine clinical samples obtained from the 14 patients were stained with the following antibodies: anti-CD19 PE (BD, clone 4G7), anti-CD3 FITC (BD, clone SK7) or anti-CD3 APC (BC clone UCHT11), anti-CD45 APC (BD, clone 2D1) or anti-CD45 FITC (BD, clone 2D1) and anti-CD34 APC-Cy7 (clone 581). Each sample was sorted on a FACSAria III (BD Biosciences) for leukemic blasts (CD19+CD45dim/-) and T cells (CD3+, CD45hi). NOD.CB17-PrkdcscidIl2rgtm1Wjl/Szj (NSG) mice were bred according to protocols established and approved by the Animal Care Committee at the University Health Network. Eight-to-twelve-week-old mice were sublethally irradiated at 225cGy 24h before transplant. Only female mice were used in these studies. Intrafemoral injections of 10–250 000 sorted leukemic blasts were performed as previously described(48). Mice were sacrificed 20–30 weeks post-transplant or at the onset of disease symptoms. Human cell engraftment in the injected femur, bone marrow (non-injected bones, left tibia, right tibia, left femur), spleen and central nervous system were accessed using human specific antibodies for CD45 (PE-Cy7, BD, clone HI30; v500, BD, clone HI30), CD44 (PE, BD clone 515; FITC, BD clone L178), CD3 (APC, BD, clone UCHT1), CD19 (PE-Cy5, Beckman Coulter, clone J3–119), CD33 (PE-Cy7, BD, clone P67–6; APC, BD, P67–6) and CD34 (APC-Cy7, BD, clone 581) analyzed on an LSRII (BD Biosciences). Mice were considered to be engrafted when >0.1% of cells in the injected femur were positive for one or more human B-ALL specific cell surface markers (CD45, CD44, CD19, CD34). The confidence intervals for the frequency of leukemia-initiating cells was calculated using ELDA software(49).
Whole exome sequencing (WES)
DNA from the adult samples was extracted from sorted leukemic blasts (CD19+CD45dim/-, purity >90%) and sorted T cells (CD3+CD45hi, purity >90%) using the Gentra Puregene Cell Kit (Qiaqen). T cells from patients 2, 3 and 5 were whole genome amplified (REPLI-g Mini Kit, Qiagen) due to limited material. DNA from the pediatric samples with >90% leukemic blasts was extracted from diagnosis, remission and relapse samples using phenol-chloroform. Exomes were captured using the TruSeq Exome Library Prep kit (67Mb, 1μg DNA input) or the Nextera Rapid Capture Expanded Exome (62Mb, 50ng DNA input; Illumina, San Diego, CA, USA). Paired-end sequencing was performed with the HiSeq 2500 genome sequencer (Illumina). The data was mapped to human reference genome hg19 and variant calling was performed using the Bambino variant detector as previously described(50). Briefly, leukemia and germline files were combined by the program and aligned against the reference genome. Putative sequence variants including SNV and indels were detected by running the variation detection module of Bambino. The output contained detailed read counts for each variant with columns for tumour/normal status, allele and strand. Variants were not filtered for coverage prior to combination with targeted-sequencing results.
Copy Number Analysis
Patient copy number aberrations were determined using SNP6.0 microarrays according to manufacturer’s instructions (Affymetrix, Santa Clara, CA, USA). Data was analyzed as previously described(51) using optimal reference normalization(52) and circular binary(53,54) segmentation with Genotyping Console (Affymetrix) and dCHIP (build Apr 2010)(55). Detection of loss of heterozygosity (LOH) and allelic ratios were performed using Nexus 7.5.2 software (BioDiscovery Inc, Hawthorne, CA, USA). All segments were manually curated.
Recovery of Human Cells and DNA Isolation from Xenografts
Cells from the injected femur (IF), bone marrow (BM) and spleen (SPL) were frozen viably after sacrifice. The IF and BM of mice engrafted with > 10% human cells were combined. These cells as well as cells from diagnosis PDX spleens were depleted of mouse cells by using the Miltenyi Mouse Cell Depletion Kit (Miltenyi Biotech) (samples with >20% engraftment) or by cell sorting with human CD45 and human CD19 and or CD34 cell surface antibodies to a purity of >90% as determined by post-processing flow cytometry. CNS cells from mice with greater than 60% engraftment were used directly for DNA isolations. DNA was isolated using the QIAamp DNA Blood Mini or Micro Kit (Qiagen).
Targeted Sequencing
All somatic SNVs and indels identified by WES were validated in the patient samples using NimbleGen SeqCap Target Enrichment according to manufacturer’s instructions (Roche, NimbleGen). Library preparation was completed using 250–500ng of DNA using the NEXTflex DNA-SEQ Library Prep Kit (BiooScientific) with NEXTflex-96 DNA Barcodes (BiooScientific). Sequencing was performed on a HiSeq 2500 genome sequencer to a mean coverage >350x for patient samples and >200x for PDXs.
Targeted-sequencing data analysis
Final patient variant calls used the combined results of WES and capture validation. Variants were filtered out if the variant allele frequency (VAF) in the germline was greater than 10% or if there was a dbSNP frequency of greater than 1%. Variants were classified based on the VAFs in the bulk patient diagnosis and relapse samples as: preserved variants (VAF > 30% in both diagnosis and relapse samples, or preserved between samples); diagnosis-specific variants (present at diagnosis (> 1%) and absent at relapse (< 1%)); latent variants (present at diagnosis with VAF < 30% and increasing at relapse); relapse-specific variants (absent at diagnosis (<1%) and present at relapse (>1%)). For xenograft analysis, varaints with less than 5x coverage or unconvered in numerous xenografts were removed. Results were analyzed by visualization in heatmaps (ie. Figure 1C). Phylogenetic analysis showing genetic relationships of patient samples and xenografts were estimated using Minkowski’s distance calculated from the VAFs and represented visually using a nearest neighbour joining algorithm. Genetic concordance between different tissues of the same xenograft were determined by visual assessment by three independent and blinded individuals. Discordance was only called when all three investigators were in agreement.
Generation of Mutational Trees from Patient Samples and Xenografts
Two independent computational analyses were performed—first for patient-only tissue samples, and then patient samples augmented with xenografts (BM and SPL)—using an early version of the PairTree algorithm (Wintersinger et al., in preparation). PairTree uses variant read counts to estimate the posterior probability distribution over four possible ancestral relationships between every variant pair (A, B). The four ancestral relationships are as follows: variant A and variant B occurred in the same cells, such that no cell possessed one variant but not the other; variant A is ancestral to variant B, such that some cells have A but not B; variant B is ancestral to variant A, such that some cells have B but not A; or neither is the ancestor of the other, such that A and B are on different branches of the evolutionary tree.
To permit temporal ordering of mutations, the infinite sites assumption was made, such that variant A could never be the ancestor of variant B if A’s cancer cell frequency (CCF) was lower than B’s in any sample, after their CCFs were estimated from each mutation’s variant allele frequency (VAF). To simplify the estimation of CCF from VAF, variants were discarded if they lay in CNA-affected regions determined by SNP6.0 analysis, of the patient samples, or if their VAFs were suggestive of an uncalled CNA regions. X-chromosome variants in male patients had their VAFs halved when estimating CCF to correct for their haploid nature. Distinct mutational populations were defined by semi-automatically clustering variants, with variants clustered together when their mutual “A and B occurred in the same cell” probabilities were high.
For each cluster, a representative “supervariant” was created. To compute a precise estimate of supervariant CCF, we summed the read counts of all variants within the supervariant’s cluster, then computed the pairwise ancestral relationship probabilities between supervariants. These probabilities guided expert-driven tree construction, in which trees were built according to how well their implied pairwise relationships between clusters matched the computed pairwise probabilities. Confirmation that these expert-driven trees yielded data likelihoods as good as or better than fully automated tree reconstructions produced by a more mature version of the PairTree algorithm (Wintersinger et al., in preparation) was performed. The tree-constrained lineage frequencies of each population in each sample were then computed using PairTree. The variants included in the populations defined by the patient-only analysis were then compared to the variants included in the populations defined for the patient and xenograft combined analysis to reveal the overlap, differences and additional clonality described with inclusion of the xenograft samples.
RNA-Sequencing
For PDX:
Cell pellets for PDX RNA extraction were frozen in 1mL of TRIzol (Invitrogen) and kept at −80 °C. Total RNA was purified by phenol/chloroform and integrity and concentration were verified on a Bioanalyzer pico chip (Agilent Technologies). cDNA conversion was performed using SMART-Seq® v4 Ultra Low Input RNA Kit for Sequencing (Takara) (1ng total RNA input) and libraries were prepared using Nextera XT DNA Library Preparation Kit (Illumina) (1ng input of cDNA). Equimolar quantities of libraries were pooled and sequenced 4 cDNA libraries per lane on a High Throughput Run Mode Flowcell with v4 sequencing chemistry on the Illumina HiSeq 2500 following manufacturer’s protocol generating paired-end reads of 126-bp in length to reach depth of 65 million reads per sample.
For patient samples:
RNA was extracted from sorted leukemic blasts (CD19+CD45dim/-, purity >90%) using RNeasy Micro Kit (Qiagen) for adult patient samples. From these samples approximately 10ng of total RNA was processed using the SMART™ cDNA synthesis protocol including SMARTScribe Reverse Transcriptase (Clontech) as per manufactures instructions. The amplified cDNA was subject to automated Illumina paired-end library construction using the NEBNext paired-end DNA sample Prep Kit (NEB) and libraries were sequenced on HiSeq2000 (Illumina) to an average of approximately 161 million Chastity-passed paired reads of 75bp in length per sample. For pediatric samples RNA was extracted from patient samples with >90% leukemic blasts using TRIzol (Life Technologies) and quality and quantity assessed by Qubit (ThermoFisher) and RNA6000 Chip (Agilent). 1ug of RNA was used for library preparation with TruSeq RNA Library Prep Kit v2 (Illumina) and 2× 100bp paired-end sequencing was performed on a HiSeq 2500 (Illumina). Patient RNA-seq samples were aligned against the human genome (hg19) using STAR v2.3 with default parameters. All PDX samples were aligned with STAR v2.5.2b(56) against the human genome build version GRCh38 and the ensembl v90 gtf file. Default parameters were used except for; chimeric segments were screened with a minimum size 12, junction overlap 12 and segment reads gap maximum 3; splice junction overlap 10, aligned mates gap maximum 100000, aligned intron maximum 100000 and alignSJstitchMismatchNmax 5 −1 5 5. For both patient samples and PDX, transcript counts were obtained using HTSeq v0.7.2(57). Data was library size normalized using RLE, followed by a variance stabilizing transformation using DESeq2 v1.22.1(58). PCA plots were generated on a per sample basis using the top 1000 variable genes. For downstream visualization, differential expression and pathway analysis, the mean expression of each sample clone condition was utilized. On a per patient level, differentially expressed genes were identified. Genes with adjusted p-value of < 0.05 and absolute log2 fold change of > 1 were considered significant. All visualizations were generated using R v3.5.1 and the pheatmap v1.0.10 and lattice v0.20–38 packages and ggplot2 v3.1.0 packages.
Pathway enrichment analysis and visualization
Pathway enrichment analysis and visualization was performed as described previously(59). Briefly, a score to rank genes from top up-regulated to down-regulated was calculated using the formula -sign(logFC) * -log10(pvalue). The rank file from each comparison was used in GSEA analysis (http://software.broadinstitute.org/gsea/index.jsp) using 2000 permutations and default parameters against a pathway database containing Msigdb c2 and c3, NCI, IOB, NetPath, HumanCyc, GO BP, Reactome and Panther (http://baderlab.org/GeneSets, version June 2018). GSEA progressively examines genes from the top to the bottom of the ranked list, increasing the enrichment score if a gene is part of the pathway and decreasing the score otherwise. These running sum values are weighted, so that enrichment in the very top- (and bottom-) ranking genes is amplified, whereas enrichment in genes with more moderate ranks are not amplified. The ES score is calculated as the maximum value of the running sum and normalized relative to pathway size, resulting in a normalized enrichment score (NES) that reflects the enrichment of the pathway in the list. Positive and negative NES values represent enrichment at the top and bottom of the list, respectively. A permutation-based p-value is computed and corrected for multiple testing to produce a permutation-based FDR q-value that ranges from zero (highly significant) to one (not significant). EnrichmentMap version 3.1.0 in Cytoscape 3.7.0 was used to visualize enriched gene-sets at FDR q-value<=0.05 for each comparison with a Jaccard coefficient set to 0.375. The Enrichment Map software takes as input a text file containing pathway enrichment analysis results (from GSEA) and another text file containing the pathway gene sets used in the original enrichment analysis (http://baderlab.org/GeneSets, version June 2018). An enrichment map is a network representing overlaps among enriched pathways with pathways represented as circles (nodes) that are connected with lines (edges) to other pathways with overlapping genes. The network layout and clustering algorithm Autoannotate app in Cytoscape was used to automatically display and group similar pathways as major biological themes. Pathways differentially enriched in dPDX vs dRI-PDX and/or dPDX vs rPDX clones were classified as: dRI-PDX-unique (pathways significantly enriched (FDR q-value<=0.05) in dRI-PDX in comparison to dPDX), dRI-PDX/rPDX common (pathways significantly enriched (FDR q-value<=0.05) in both dRI-PDX and rPDX in comparison to dPDX) or rPDX-unique (pathways significantly enriched (FDR q-value<=0.05) in rPDX in comparison to dPDX).
Isolation of HSC and B cells from human cord blood samples
Human cord blood samples were obtained with informed consent from Trillium and Credit Valley Hospital according to procedures approved by the University Health Network Research Ethics Board. Mononuclear cells were obtained by centrifugation on Lymphoprep medium (Stem Cell Technologies) followed by red blood cell lysis using ammonium chloride (Stem Cell Technologies). Human CD34+ and CD34- CB cells were separated using CD34 Microbeads kit (Miltenyi Biotech) according to manufacturer’s protocol and stored at −150°C. Cells were stained with antibodies (all from BD, unless stated otherwise): FITC–anti-CD45RA (1:50, 555488), PE–anti-CD90 (1:50, 555596), PECy5–anti-CD49f (1:50, 551129), V450–anti-CD7 (1:33.3, 642916), PECy7–anti-CD38 (1:100, 335790), APC–anti-CD10 (1:50, 340923), APCCy7–anti-CD34 (1:200, custom made by BD) and sorted on FACS Aria III (Becton Dickinson), consistently yielding >95% purity. HSC were sorted based on the following markers: CD34+ CD38- CD45RA- CD90+ CD49f+ from CD34+ CB cells as previously described(60). B cells were sorted as CD34- CD38+CD19+CD33-CD3-CD56-.
Gene set variation analysis (GSVA)
Read counts from 5 B cell and 3 HSC sorted populations from normal human umilical cord blood were normalized using the trimmed of M values using the edgeR_3.24.3 R package and gene differential expression was calculated using the quasi likelihood ratio method and include cord blood batch correction in the experimental design. The top 1000 genes enriched in B cells (B cell genes) and the top 1000 genes enriched in HSC (HSC genes) were selected to be used as the reference signature. Two mixed profiles were used: dPDX, dRI-PDX and rPDX clones and 14 bulk diagnosis and relapse patient samples. The mixes profiles read counts were variance stabilized and library size normalized using DESeq_1.34.1). The gsva() function from the R GSVA_1.30.0 package was employed using a gaussian kernel to estimate enrichment of reference signatures in each sample of the mixed profile dataset. GSVA score for each patient and each category were plotted on a boxplot and a stripchart. Gsva score were summed for each mixed profile category and standardized from 0 to 1. 1000 permutations with a random gene list of size 1000 were performed on the mixed profile and percentages calculated to indicate how many times the observed score was higher than the random scores. These results were confirmed using the HSC and B cell expression profiles from Novershtern et al., Cell 2011(40) (Supplementary Table S7).
Transcriptomic Validation Experiments
Staining for mitochondria content, ROS, mitochondrial membrane potential and mitochondrial superoxide levels was performed by incubating thawed PDX cells at 37°C with: 1mM of MitoTrackerGreen (M7514) (dilution 1:10000); 5 μM CellROX deep red (C10422) (dilution 1:500); 1 μM TMRE (T668) (dilution 1:1000) or 5mM MitoSOX Red (M36008) (dilution 1:1000) following manufacturer’s instructions (ThermoFisher) and directly analyzed on a BD LSRII cytometer. Mean fluorescence intensity (MFI) for each sample and dye is represented as ratio to dPDX for each patient. Immunostaining analysis for TOMM20, MRPS18B and SOD1 were performed on PDX cells. Briefly, cells were spun onto Poly-L-Lysine (Sigma)-coated slides (Ibidi, 200 xg, 10 min), fixed with 4% paraformaldehyde (Sigma) and permeabilized with 0.5% Triton (Sigma) before blocking (PBS, 10% FBS, 5% BSA). Slides were incubated with primary antibodies (TOMM20: ab56783, dilution 1/200; SOD1: ab8866, dilution 1/100 and MRPS18B: ab191891, dilution 1/200) in blocking solution O/N at 4°C. Secondary anti-mouse AF568, anti-rabbit AF488 and anti-sheep AF647 (Invitrogen, 1:400) antibodies were added (PBS, 0.025% Tween, Sigma, 1.5 h, RT). After washing, nuclei were stained with 1 μg/mL DAPI (Invitrogen) and slides were mounted (Fluoromount G, Invitrogen). Images were captured by a Zeiss LSM700 Confocal (oil, 63x/1.4NA, Zen 2012) and analyzed with ImageJ/Fiji. The Integrated Density (IntDen= Area* Mean Fluorescence Intensity) for 40–50 cells from each clone was analyzed. The mean for each clone was normalized and calculated as a ratio to the dPDX for each patient separately.
Secondary Transplantations for Drug Assays and Limiting Dilution Assays
Human purified cells from the primary recipients were thawed and transplanted into eight-to-twelve-week-old female NSG mice sublethally irradiated as described for the primary recipients. The number of mice used for secondary transplantation experiements/drugs were determined by cell and mouse avalibility and feasibility. Intrafemoral injections of 26000 −100 000 leukemic blasts were performed for drug assays and a range of 10 – 100 000 leukemic blasts were injected for secondary limiting dilution assays. After 4 weeks mice were bled from the saphenous vein and human chimerism was evaluated by flow cytometry. Once human engraftment in the peripheral blood reached between 1–10%, or after 10–14 weeks for those samples in which leukemic blasts did not mobilize to the peripheral blood, mice were randomized and single agent treatments were started. Dexamethasone (15mg/kg), L-asparginase (1000kU/kg) and saline were given daily by intraperitoneal injection 5 days a week. Vincristine (0.5mg/kg) was given once a week by intraperitoneal injection. All four arms of the drug treatment were performed for 4 weeks and mice were sacrificed the following day (vincristine/dexamethasone/saline) or one week after the last treatment (vincristine). Analysis of the secondary limiting dilution assay was performed 16 weeks post-transplant. Human cell engraftment in the injected femur, bone marrow and spleen were assessed using human specific antibodies for CD45 (FITC, BD, clone 2D1; v500, BD, clone HI30), CD19 (BD, PE, clone 4G7), CD33 (PE-Cy7,BD, clone P67–6), CD3 (APC, BD, clone UCHT1), CD10 (Qdot605, BD, clone HI10A), CD38 (BV421, BioLegend, clone HIT2) and CD34 (APC-Cy7, BD, clone 581). Mice were considered to be engrafted when >0.1% of cells in the injected femur were positive for one or more human B-ALL specific cell surface markers (CD45, CD44, CD19, CD34). Respose to treatment was analyzed as a ratio of human engraftment of drug vs saline treated mice to eliminate interclonal differences in engraftment levels. Ratio of human engraftment in each individual drug treated PDX to the average engraftment level of all saline controls was calculated. Lineage stains were performed on xenografts expressing CD33 (APC, BD clone P67–6) and CD19 (PE, BD, clone 4G7) including CD14 (PE-Cy7, Beckman Coulter, clone 52), CD15 (v450, BD, clone MMA), CD10 (Qdot605, BioLegend, clone HIT2) and CD34 (APC-Cy7, BD, clone 581). The confidence intervals for the frequency of leukemia-initiating cells was calculated using ELDA software(49). Statistical analysis was performed using PRISM 6 (GraphPad Software).
Fluorescence-activated Cell Sorting from Xenografts
Leukemic cells from primary xenografts were sorted for immunophenotypic populations on a FACSARIAIII (BD Biosciences). Cells from the IF and BM were pooled and stained with CD19, CD34, CD45, CD10 and CD33 and collected at a sort purity of >99%.
Supplementary Material
STATEMENT OF SIGNIFCIANCE.
Isolation and characterization of subclones from diagnosis samples of B-ALL patients who relapsed, showed that relapse-fated subclones had increased drug tolerance and distinct metabolic and survival transcriptional programs compared to other diagnosis subclones. This study provides strategies to identify and target clinically relevant subclones before further evolution towards relapse.
ACKNOWLEDGEMENTS:
We thank the patients, their families and physicians who made this study possible. We also thank N. Simard, S. Laronde, L. Jaimeson, A. Khandani, T. Velanthapillai and S. Zhao at the UHN/SickKids Flow Cytometry Facility; P.Lo and R. Lopez from the UHN Animal Resource Centre; the Genome Sequencing Facility of the Hartwell Centre for Bioinformatics and Biotechnology, and the Biorepository of St. Jude Children’s Research Hospital. We thank J. Ho, E. Lechman, A. Mitchell, L. Jin, M. Doedens, J. Loo-Yong-Kee, K.L. Woo, M.C. Shoong, and J. Roth for their technical assistance; J.A. Kennedy for clinical annotation and K. Kaufmann for data analysis script.
Financial Support: This work was supported by funds to J.E.D. from the: Princess Margaret Cancer Centre Foundation, Ontario Institute for Cancer Research with funding from the Province of Ontario, Canadian Institutes for Health Research, Canadian Cancer Society Research Institute, Terry Fox Research Institute Program Project, Genome Canada through the Ontario Genomics Institute, and a Canada Research Chair; and to C.G.M. from the the American Lebanese Syrian Associated Charities of St. Jude Children’s Research Hospital, a St. Baldrick’s Foundation Robert J. Arceci Innovation Award, the Henry Schueler 41&9 Foundation, the National Cancer Institute grants P30 CA021765 (St. Jude Cancer Center Support Grant), and Outstanding Investigator Award R35 CA197695. Q.M. was supported by funds from the Natural Sciences and Engineering Research Council, a Compute the Cure award from the NVIDIA charitable foundation, and the Vector Institute. E.W. is funded by the Dutch Cancer Society (KUN2012-5366).
Footnotes
Data availability statement
The datasets generated during the current study are included in this published article (and its supplementary information files).
Code availability statement
Code used in this study is available at https://www.github.com/morrislab/pairtree
Conflict of Interest:
J.E.D. Celgene: research funding; Trillium Therapeutics: Advisory Board funding; Abbvie: research funding
C.G.M. Illumina: Advisory board, honorarium, sponsored travel; Pfizer: research
REFERENCES
- 1.Forman SJ, Rowe JM. The myth of the second remission of acute leukemia in the adult. Blood 2013;121(7):1077–82 doi 10.1182/blood-2012-08-234492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liew E, Atenafu EG, Schimmer AD, Yee KW, Schuh AC, Minden MD, et al. Outcomes of adult patients with relapsed acute lymphoblastic leukemia following frontline treatment with a pediatric regimen. Leuk Res 2012;36(12):1517–20 doi 10.1016/j.leukres.2012.08.019. [DOI] [PubMed] [Google Scholar]
- 3.Hunger SP, Mullighan CG. Acute Lymphoblastic Leukemia in Children. N Engl J Med 2015;373(16):1541–52 doi 10.1056/NEJMra1400972. [DOI] [PubMed] [Google Scholar]
- 4.Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 2008;322(5906):1377–80 doi 10.1126/science.1164266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma X, Edmonson M, Yergeau D, Muzny DM, Hampton OA, Rusch M, et al. Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat Commun 2015;6:6604 doi 10.1038/ncomms7604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oshima K, Khiabanian H, da Silva-Almeida AC, Tzoneva G, Abate F, Ambesi-Impiombato A, et al. Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 2016;113(40):11306–11 doi 10.1073/pnas.1608420113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 2011;469(7330):356–61 doi 10.1038/nature09650. [DOI] [PubMed] [Google Scholar]
- 8.Notta F, Mullighan CG, Wang JC, Poeppl A, Doulatov S, Phillips LA, et al. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature 2011;469(7330):362–7 doi 10.1038/nature09733. [DOI] [PubMed] [Google Scholar]
- 9.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell 2014;14(3):275–91 doi 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 10.Foo J, Michor F. Evolution of acquired resistance to anti-cancer therapy. J Theor Biol 2014;355:10–20 doi 10.1016/j.jtbi.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim C, Gao R, Sei E, Brandt R, Hartman J, Hatschek T, et al. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018;173(4):879–93 e13 doi 10.1016/j.cell.2018.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iqbal Z, Aleem A, Iqbal M, Naqvi MI, Gill A, Taj AS, et al. Sensitive detection of pre-existing BCR-ABL kinase domain mutations in CD34+ cells of newly diagnosed chronic-phase chronic myeloid leukemia patients is associated with imatinib resistance: implications in the post-imatinib era. PLoS One 2013;8(2):e55717 doi 10.1371/journal.pone.0055717. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Tzoneva G, Dieck CL, Oshima K, Ambesi-Impiombato A, Sanchez-Martin M, Madubata CJ, et al. Clonal evolution mechanisms in NT5C2 mutant-relapsed acute lymphoblastic leukaemia. Nature 2018;553(7689):511–4 doi 10.1038/nature25186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hunter C, Smith R, Cahill DP, Stephens P, Stevens C, Teague J, et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res 2006;66(8):3987–91 doi 10.1158/0008-5472.CAN-06-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500(7463):415–21 doi 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Churchman ML, Low J, Qu C, Paietta EM, Kasper LH, Chang Y, et al. Efficacy of Retinoids in IKZF1-Mutated BCR-ABL1 Acute Lymphoblastic Leukemia. Cancer Cell 2015;28(3):343–56 doi 10.1016/j.ccell.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ebinger S, Ozdemir EZ, Ziegenhain C, Tiedt S, Castro Alves C, Grunert M, et al. Characterization of Rare, Dormant, and Therapy-Resistant Cells in Acute Lymphoblastic Leukemia. Cancer Cell 2016;30(6):849–62 doi 10.1016/j.ccell.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Polak R, de Rooij B, Pieters R, den Boer ML. B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment. Blood 2015;126(21):2404–14 doi 10.1182/blood-2015-03-634238. [DOI] [PubMed] [Google Scholar]
- 19.Iwamoto S, Mihara K, Downing JR, Pui CH, Campana D. Mesenchymal cells regulate the response of acute lymphoblastic leukemia cells to asparaginase. J Clin Invest 2007;117(4):1049–57 doi 10.1172/JCI30235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mullighan CG, Zhang J, Kasper LH, Lerach S, Payne-Turner D, Phillips LA, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 2011;471(7337):235–9 doi 10.1038/nature09727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamel-Reid S, Letarte M, Sirard C, Doedens M, Grunberger T, Fulop G, et al. A model of human acute lymphoblastic leukemia in immune-deficient SCID mice. Science 1989;246(4937):1597–600. [DOI] [PubMed] [Google Scholar]
- 22.Lock RB, Liem N, Farnsworth ML, Milross CG, Xue C, Tajbakhsh M, et al. The nonobese diabetic/severe combined immunodeficient (NOD/SCID) mouse model of childhood acute lymphoblastic leukemia reveals intrinsic differences in biologic characteristics at diagnosis and relapse. Blood 2002;99(11):4100–8. [DOI] [PubMed] [Google Scholar]
- 23.Ng SW, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 2016;540(7633):433–7 doi 10.1038/nature20598. [DOI] [PubMed] [Google Scholar]
- 24.Liem NL, Papa RA, Milross CG, Schmid MA, Tajbakhsh M, Choi S, et al. Characterization of childhood acute lymphoblastic leukemia xenograft models for the preclinical evaluation of new therapies. Blood 2004;103(10):3905–14 doi 10.1182/blood-2003-08-2911. [DOI] [PubMed] [Google Scholar]
- 25.Meyer LH, Eckhoff SM, Queudeville M, Kraus JM, Giordan M, Stursberg J, et al. Early relapse in ALL is identified by time to leukemia in NOD/SCID mice and is characterized by a gene signature involving survival pathways. Cancer Cell 2011;19(2):206–17 doi 10.1016/j.ccr.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 26.Shlush LI, Mitchell A, Heisler L, Abelson S, Ng SWK, Trotman-Grant A, et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017;547(7661):104–8 doi 10.1038/nature22993. [DOI] [PubMed] [Google Scholar]
- 27.Clappier E, Gerby B, Sigaux F, Delord M, Touzri F, Hernandez L, et al. Clonal selection in xenografted human T cell acute lymphoblastic leukemia recapitulates gain of malignancy at relapse. J Exp Med 2011;208(4):653–61 doi 10.1084/jem.20110105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klco JM, Spencer DH, Miller CA, Griffith M, Lamprecht TL, O’Laughlin M, et al. Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell 2014;25(3):379–92 doi 10.1016/j.ccr.2014.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richter-Pechanska P, Kunz JB, Bornhauser B, von Knebel Doeberitz C, Rausch T, Erarslan-Uysal B, et al. PDX models recapitulate the genetic and epigenetic landscape of pediatric T-cell leukemia. EMBO Mol Med 2018;10(12) doi 10.15252/emmm.201809443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Waanders EGZ, Dobsom SM, Antic Z, Crawford JC, Ma X, et al. Mutational landscape and patterns of clonal evolution in relapsed pediatric acute lymphoblastic leukemia. Blood Cancer Discovery 2020;1:1–16 doi 10.1158/0008-5472.BCD-19-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rehe K, Wilson K, Bomken S, Williamson D, Irving J, den Boer ML, et al. Acute B lymphoblastic leukaemia-propagating cells are present at high frequency in diverse lymphoblast populations. EMBO Mol Med 2013;5(1):38–51 doi 10.1002/emmm.201201703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schinnerl D, Mejstrikova E, Schumich A, Zaliova M, Fortschegger K, Nebral K, et al. CD371 cell surface expression: a unique feature of DUX4-rearranged acute lymphoblastic leukemia. Haematologica 2019;104(8):e352–e5 doi 10.3324/haematol.2018.214353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chiaretti S, Li X, Gentleman R, Vitale A, Vignetti M, Mandelli F, et al. Gene expression profile of adult T-cell acute lymphocytic leukemia identifies distinct subsets of patients with different response to therapy and survival. Blood 2004;103(7):2771–8 doi 10.1182/blood-2003-09-3243. [DOI] [PubMed] [Google Scholar]
- 34.Slamova L, Starkova J, Fronkova E, Zaliova M, Reznickova L, van Delft FW, et al. CD2-positive B-cell precursor acute lymphoblastic leukemia with an early switch to the monocytic lineage. Leukemia 2014;28(3):609–20 doi 10.1038/leu.2013.354. [DOI] [PubMed] [Google Scholar]
- 35.O’Connell MP, Marchbank K, Webster MR, Valiga AA, Kaur A, Vultur A, et al. Hypoxia induces phenotypic plasticity and therapy resistance in melanoma via the tyrosine kinase receptors ROR1 and ROR2. Cancer Discov 2013;3(12):1378–93 doi 10.1158/2159-8290.CD-13-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kemper K, de Goeje PL, Peeper DS, van Amerongen R. Phenotype switching: tumor cell plasticity as a resistance mechanism and target for therapy. Cancer Res 2014;74(21):5937–41 doi 10.1158/0008-5472.CAN-14-1174. [DOI] [PubMed] [Google Scholar]
- 37.Chaidos A, Barnes CP, Cowan G, May PC, Melo V, Hatjiharissi E, et al. Clinical drug resistance linked to interconvertible phenotypic and functional states of tumor-propagating cells in multiple myeloma. Blood 2013;121(2):318–28 doi 10.1182/blood-2012-06-436220. [DOI] [PubMed] [Google Scholar]
- 38.Pisco AO, Huang S. Non-genetic cancer cell plasticity and therapy-induced stemness in tumour relapse: ‘What does not kill me strengthens me’. Br J Cancer 2015;112(11):1725–32 doi 10.1038/bjc.2015.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Belviso S, Iuliano R, Amato R, Perrotti N, Menniti M. The human asparaginase enzyme (ASPG) inhibits growth in leukemic cells. PLoS One 2017;12(5):e0178174 doi 10.1371/journal.pone.0178174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Novershtern N, Subramanian A, Lawton LN, Mak RH, Haining WN, McConkey ME, et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell 2011;144(2):296–309 doi 10.1016/j.cell.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones CL, Stevens BM, D’Alessandro A, Reisz JA, Culp-Hill R, Nemkov T, et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018;34(5):724–40 e4 doi 10.1016/j.ccell.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Galen P, Mbong N, Kreso A, Schoof EM, Wagenblast E, Ng SWK, et al. Integrated Stress Response Activity Marks Stem Cells in Normal Hematopoiesis and Leukemia. Cell Rep 2018;25(5):1109–17 e5 doi 10.1016/j.celrep.2018.10.021. [DOI] [PubMed] [Google Scholar]
- 43.Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med 2009;360(5):470–80 doi 10.1056/NEJMoa0808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luria SE, Delbruck M. Mutations of Bacteria from Virus Sensitivity to Virus Resistance. Genetics 1943;28(6):491–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med 2011;17(9):1086–93 doi 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- 46.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med 2006;12(10):1167–74 doi 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- 47.Chen WC, Yuan JS, Xing Y, Mitchell A, Mbong N, Popescu AC, et al. An Integrated Analysis of Heterogeneous Drug Responses in Acute Myeloid Leukemia That Enables the Discovery of Predictive Biomarkers. Cancer Res 2016;76(5):1214–24 doi 10.1158/0008-5472.CAN-15-2743. [DOI] [PubMed] [Google Scholar]
- 48.Mazurier F, Doedens M, Gan OI, Dick JE. Rapid myeloerythroid repopulation after intrafemoral transplantation of NOD-SCID mice reveals a new class of human stem cells. Nat Med 2003;9(7):959–63 doi 10.1038/nm886. [DOI] [PubMed] [Google Scholar]
- 49.Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 2009;347(1–2):70–8 doi 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 50.Edmonson MN, Zhang J, Yan C, Finney RP, Meerzaman DM, Buetow KH. Bambino: a variant detector and alignment viewer for next-generation sequencing data in the SAM/BAM format. Bioinformatics 2011;27(6):865–6 doi 10.1093/bioinformatics/btr032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mullighan CG. Single nucleotide polymorphism microarray analysis of genetic alterations in cancer. Methods Mol Biol 2011;730:235–58 doi 10.1007/978-1-61779-074-4_17. [DOI] [PubMed] [Google Scholar]
- 52.Pounds S, Cheng C, Mullighan C, Raimondi SC, Shurtleff S, Downing JR. Reference alignment of SNP microarray signals for copy number analysis of tumors. Bioinformatics 2009;25(3):315–21 doi 10.1093/bioinformatics/btn624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 2004;5(4):557–72 doi 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 54.Venkatraman ES, Olshen AB. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics 2007;23(6):657–63 doi 10.1093/bioinformatics/btl646. [DOI] [PubMed] [Google Scholar]
- 55.Lin M, Wei LJ, Sellers WR, Lieberfarb M, Wong WH, Li C. dChipSNP: significance curve and clustering of SNP-array-based loss-of-heterozygosity data. Bioinformatics 2004;20(8):1233–40 doi 10.1093/bioinformatics/bth069. [DOI] [PubMed] [Google Scholar]
- 56.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29(1):15–21 doi 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015;31(2):166–9 doi 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reimand J, Isserlin R, Voisin V, Kucera M, Tannus-Lopes C, Rostamianfar A, et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat Protoc 2019;14(2):482–517 doi 10.1038/s41596-018-0103-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Notta F, Doulatov S, Laurenti E, Poeppl A, Jurisica I, Dick JE. Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science 2011;333(6039):218–21 doi 10.1126/science.1201219. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.