

Abstract
Selenium (Se) is an essential trace element that functions in the form of the 21st amino acid, selenocysteine (Sec) in a defined set of proteins. Se deficiency is associated with pathological conditions in humans and animals, where incorporation of Sec into selenoproteins is reduced along with their expression and catalytic activity. Supplementation of Se-deficient population with Se has shown health benefits suggesting the importance of Se in physiology. An interesting paradigm to explain, in part, the health benefits of Se stems from the observations that selenoprotein-dependent modulation of inflammation and efficient resolution of inflammation relies on mechanisms involving a group of bioactive lipid mediators, prostanoids, which orchestrate a concerted action towards maintenance and restoration of homeostatic immune responses. Such an effect involves the interaction of various immune cells with these lipid mediators where cellular redox gatekeeper functions of selenoproteins further aid in not only dampening inflammation, but also initiating an effective and active resolution process. Here we have summarized the current literature on the multifaceted roles of Se/selenoproteins in the regulation of these bioactive lipid mediators and their immunomodulatory effects.
Keywords: Cyclooxygenase, prostaglandin, thromboxane, immune cell, infection
I. Introduction
Selenium (Se) is an essential trace element discovered by Jöns Jacob Berzelius in 1817, named after the Greek goddess of the moon, Selḗnē. In the periodic table of the elements, Se is in the chalcogen group, which consists of oxygen (O), sulfur (S), Se, tellurium (Te), and polonium (Po). Thus, Se possesses some of the properties of S and Te. One of the most distinguishing features of Se is its biological functions that occur via incorporation into selenoproteins through a specialized evolutionary machinery involving multiple components. Co-translational insertion of selenocysteine (Sec) into proteins endows them the enzymatic efficiency when compared to their cysteine (Cys) counterparts (Gladyshev and Hatfield 1999; Hondal et al. 2001; Arner 2010). Even though some organisms such as yeast, fungi, or higher plants cannot synthesize selenoproteins, humans and other eukaryotes utilize Sec for incorporation into a diverse array of cellular redox gatekeepers and other enzymes that play a key role in physiological and pathological conditions. In particular, different components of immune system have been reported to be modulated by the levels and forms of dietary Se and corresponding augmentation of selenoproteins biosynthesis in the body (Brown et al. 2000; Shrimali et al. 2008). This is particularly intriguing given the relationship between Se status and prostanoid metabolism and how the modulation of the latter impacts the immune system.
Here, we discuss the impact of Se and selenoproteins in health and disease with an emphasis on the immune system and its modulation via the regulation of prostanoid metabolism. Relevant studies that have dealt with infections (bacterial, viral, and parasitic), cancer immunity, immunodeficiency (acquired immunodeficiency syndrome, AIDS), and allergic asthma are discussed. Although prostanoids are of considerable importance in cardiovascular, pulmonary, and gastrointestinal systems, this review focuses on Se and/or selenoproteins and prostanoid metabolism in relation to immunity.
II. Se and Selenoproteins
Diet is the primary source of Se in animals and humans. Inorganic Se (mainly selenate and selenite) from soil is converted to organic forms including selenomethionine (SeMet) and methylselenocysteine, and Sec in plants. In mammals, different forms of dietary Se are taken up and metabolized into various intermediary metabolites including selenide (HSe−), which functions as a Se donor for Sec biosynthesis. Se in the form of Sec was first discovered in the redox-active enzyme, glutathione peroxidase 1 (GPX1) in which Sec is located in the active site (Rotruck et al. 1973). Later, it was recognized that the 21st amino acid was incorporated into GPXs via decoding the stop codon UGA (Chambers et al. 1986; Zinoni et al. 1986). This is an evolutionarily conserved synthetic mechanism shared by all selenoproteins (Salinas G. 2006), which entails a dedicated set of factors as described below. Dietary Se in the form of selenate or selenite is metabolized to HSe− followed by conversion to selenophosphate by selenophosphate sythetase-2 (SPS2) (Low et al. 1995; Guimaraes et al. 1996). Selenophosphate is further utilized to convert the phosphoseryl-tRNA[Ser]Sec to form Sec-tRNA[Ser]Sec (Diamond et al. 1981; Leinfelder et al. 1988). Sec-tRNA[Ser]Sec controls the expression of selenoproteins by incorporating Sec into a growing polypeptide chain via decoding the stop codon UGA in response to the Sec insertion sequence (SECIS) cis element in the 3’-untranslated region (3’-UTR) (Berry et al. 1991). Other factors including SECIS binding protein 2 (SBP2) (Copeland et al. 2000) and Sec-specific elongation factors (EFSecs) (Tujebajeva et al. 2000) serve as key components of the translational machinery for Sec incorporation with many key mechanistic questions that still remain unanswered (Howard and Copeland 2019). A schematic of Sec formation and incorporation into selenoproteins is presented in Fig. 1.
Figure 1. Sec formation and incorporation into selenoproteins.
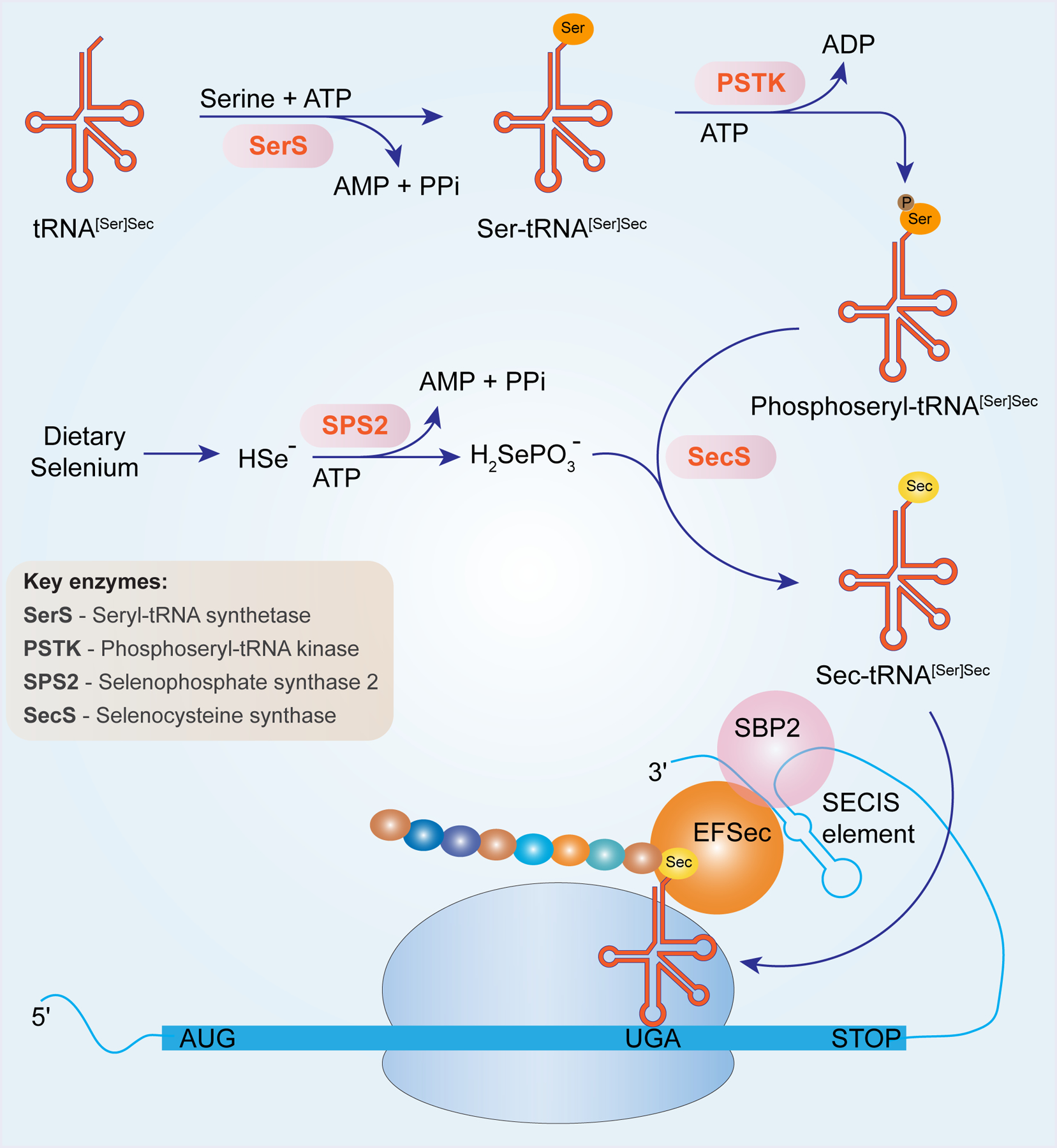
Dietary Se including selenite and selenate is converted to selenide (HSe−). The generation of selenophosphate (H2SePO3−) is catalyzed by SPS2 from selenide and ATP. Selenophosphate is charged on to tRNA[Ser]Sec by seryl-tRNA synthase (SerS). Phosphorylation of seryl-tRNA[Ser]Sec by phosphoseryl-tRNA kinase (PSTK) forms an intermediate, phosphoseryl-tRNA[Ser]Sec, which accepts selenophosphate to phosphoseryl-tRNA[Ser]Sec to generate Sec-tRNA[Ser]Sec. When SECIS is present in the 3’-UTR of mRNA, Sec incorporation is efficiently carried out by decoding UGA. Several factors such as SBP2, EFSec are also recruited to aid in the translation process.
Use of bioinformatic methods led to the identification of 25 and 24 selenoproteins in humans and rodents, respectively (Kryukov et al. 2003). Thus, the similarity of the selenoproteome between humans and rodents has enabled the use of rodent models to elucidate the physiological function of the selenoproteome as a whole and individual selenoproteins. However, the expression of selenoproteins in these and other higher mammals is tightly controlled in a tissue and cell-specific manner by hierarchical regulation dictated by developmental stages at both organismal and cellular levels, availability of Se, and sexual dimorphisms (Schomburg and Schweizer 2009). A commonly used model to ablate the selenoproteome utilizes the targeted deletion of Sec-tRNA[Ser]Sec gene (Trsp) (McBride et al. 1987; Bosl et al. 1995). Trsp knockout mice lack the capacity to synthesize selenoproteins, and thus, present an important tool to distinguish the physiological/pharmacological functions between selenoproteins and supplemented Se compounds that cannot be utilized for selenoprotein biosynthesis in the knockout mice. However, caution should be taken when extrapolating the results from murine models of selenoprotein deletion to humans. More recently, differences in the tolerance to selenoprotein loss between mouse and human were compiled by comparing human loss-of-function variants from a genome aggregation database (gnomAD) with selenoprotein knockout mouse models from large-scale projects IMPC (Santesmasses et al. 2019), suggesting the importance to differentiate the physiological effects of selenoproteins between mouse and human. Table 1 summarizes the currently known selenoproteins and their distributions within the cells of the immune system. A more detailed discussion of the functions of selenoproteins in immunity with emphasis on their involvement in inflammatory pathologies is presented in the following sections.
Table 1.
Summary of the distribution and functions of human selenoproteins
| Selenoprotein | Subcellular location | †RNA expression in immune cells | †Expression in tissues and organs | Function |
|---|---|---|---|---|
| *GPX1 (cGPX) | cytoplasm; mitochondrial membrane |
unclear | ubiquitous expression; high in kidney and liver | antioxidant; cardioprotective and anti-angiogenic (Galasso et al. 2006; Thu et al. 2010) |
| GPX2 (GI-GPX) | cytoplasm | NK cell > intermediate monocyte > neutrophil > naïve B cell > classical monocyte | gastrointestinal epithelium | antioxidant; dual role in carcinogenesis ((Brigelius-Flohe and Kipp 2012; Krehl et al. 2012; Muller MF et al. 2013; Emmink et al. 2014) |
| GPX3 (pGPX) | secreted abundantly in plasma | neutrophil > classical monocyte > myeloid DC > intermediate monocyte > total PBMC | plasma, kidney, thyroid gland | antioxidant; tumor suppressor (Barrett et al. 2013) |
| **GPX4 (PHGPX) | cytoplasm mitochondria and nuclei |
total PBMC > eosinophil > classical monocyte > basophil > non-classical monocyte | ubiquitous expression | antioxidant; regulation of ferroptosis and phospholipid hydroperoxide levels (Seiler et al. 2008); involved in spermiogenesis (Ursini et al. 1999) |
| GPX6 | Secreted protein | neutrophil > eosinophil > non-classical monocyte > intermediate monocyte | olfactory epithelium and embryonic tissues; secreted in male reproductive system |
antioxidant (Kryukov et al. 2003) |
| **Txnrd1 (TrxR1) | cytoplasm nucleoplasm |
neutrophil > basophil > eosinophil > non-classical monocyte > myeloid DC | ubiquitous expression | antioxidant; embryonic development (Conrad et al. 2004; Jakupoglu et al. 2005) |
| Txnrd2 (TrxR3) | cytoplasm Mitochondrial membrane |
classical monocyte > intermediate monocyte > myeloid DC > non-classical monocyte > eosinophil | ubiquitous expression | antioxidant; embryonic development (Conrad et al. 2004; Jakupoglu et al. 2005) |
| **Txnrd3 (TGR/TrxR2) | cytoplasm nucleoplasm |
naïve CD4 T cell > naïve CD8 T cell > classical monocyte > NK cell > intermediate monocyte | testis-specific expression | antioxidant; involved in spermiogenesis (Su et al. 2005) |
| DIO1 | plasma membrane | myeloid DC > classical monocyte > naïve CD4 T cell > intermediate monocyte > memory CD4 T-cell | thyroid gland, kidney, liver | regulation of the circulating thyroid hormone levels (St Germain and Galton 1997) |
| DIO2 | ER membrane | neutrophil > basophil > classical monocyte > plasmacytoid DC > eosinophil | thyroid gland, cervix, uterine | regulation of muscular thyroid hormone levels (St Germain and Galton 1997) |
| DIO3 | plasma membrane | not detected | thyroid gland, cervix, uterine, placenta | Inactivating thyroid hormone (St Germain and Galton 1997) |
| *SelenoH | nucleoplasm and nucleoli | plasmacytoid DC > classical monocyte > total PBMC > myeloid DC > intermediate monocyte | ubiquitous expression | antioxidant (Martin-Romero et al. 2001; Kryukov et al. 2003) |
| SelenoI | plasma membrane | memory B cell > naïve CD8 T cell > Treg > naïve CD4 T cell > plasmacytoid DC | ubiquitous expression | catalyzing the synthesis of phosphatidylethanolamine (Kryukov et al. 2003) |
| SelenoK | ER membrane | basophil > eosinophil > neutrophil > total PBMC > γδT cell | mainly expressed in heart and skeletal muscle; also present in other organs such as pancreas, liver, placenta | involved in ERAD and protein palmitoylation (Shchedrina et al. 2011; Fredericks et al. 2014) |
| SelenoM | ER membrane | naïve CD4 T cell > memory CD4 T cell > Treg > memory B cell > naïve CD8 T cell | expression is the highest in brain; also present in thyroid, heart, lung, kidney, uterus, placenta | the exact function remains unknown; possibly involved in neurodegeneration (Korotkov et al. 2002) |
| SelenoN | ER membrane | basophil > eosinophil > classical monocyte > intermediate monocyte > naïve CD4 T cell | ubiquitous expression | regulating the muscular regeneration (Moghadaszadeh et al. 2001) |
| SelenoO | mitochondrial membrane | eosinophil > classical monocyte > myeloid DC >MAIT cell > memory CD8 T cell | ubiquitous expression | the exact function remains unknow; possibly involved in redox regulation (Kryukov et al. 2003) |
| SelenoP (SEPP1) | secreted protein in plasma | plasmacytoid DC > NK cell > MAIT cell > naïve CD4 T cell > neutrophil | secreted to plasma by liver; but also seen in all tissues (Hill KE et al. 2003; Schomburg et al. 2003) | transporting Se to peripheral tissues |
| *SelenoR (MsrB1) | cytoplasm nucleoplasm |
neutrophil > eosinophil > classical monocyte > basophil > total PBMC | ubiquitous, mainly enriched in liver and kidney | antioxidant; regulation of actin polymerization (Lee et al. 2013) |
| SelenoS (SEPS1) | ER membrane | plasmacytoid DC > basophil > memory CD8 T cell > γδT cell > MAIT cell | ubiquitous expression | involved in ERAD and inflammation as well as immune response (Turanov et al. 2014) |
| SelenoT | ER membrane Golgi complex |
total PBMC > neutrophil > classical monocyte > MAIT cell > basophil | ubiquitous expression; abundant in brain at embryonic stages; but undetectable in the adult; persistently high expression in adult endocrine organs | possibly involved in redox regulation (Ursini et al. 1999) |
| SelenoV | unclear | neutrophil > classical monocyte > MAIT cell > naïve CD4 T cell > plasmacytoid DC | testes-specific expression | possibly involved in redox regulation (Kryukov et al. 2003) |
| *SelenoW | cytoplasm, a small fraction is bound to the cell membrane | eosinophil > Treg > total PBMC > naïve CD4 T cell > memory CD4 T cell | highly expressed in skeletal muscle, heart, and brain | antioxidant; possibly involved in muscle growth and differentiation (Vendeland et al. 1995) |
| SPS2 | nucleoplasm | NK cell > eosinophil > naïve CD4 T cell > MAIT cell > memory CD8 T cell | ubiquitous expression | autoregulation and synthesis of selenoproteins (Low et al. 1995; Guimaraes et al. 1996) |
| *Sep15 (SelenoF) | ER membrane | naïve B cell > plasmacytoid DC > memory B cell > total PBMC > basophil | Brain, prostate, testis, liver, and kidney | possibly implicated in cancer etiology (Kumaraswamy et al. 2000; Ferguson et al. 2006) |
Stress-responsive selenoproteins;
Housekeeping selenoproteins;
Data were extracted from “The Human Protein Atlas” (https://www.proteinatlas.org)
ER: endoplasmic reticulum; ERAD: ER-associated degradation; GI: gastrointestinal; MAIT: mucosal associated invariant T cell; PBMC: peripheral blood mononuclear cell; PHGPX: phospholipid hydroperoxide GPX
A family of lipid mediators that regulate various facets of inflammation, as well as its resolution, include cyclooxygenase (COX)-mediated synthesis of prostanoids. Esterified long-chain fatty acids as precursors of prostanoids are prone to oxidation, specifically during inflammation-associated activation of immune cells, including macrophages and neutrophils. This activation process involves physiologically regulated generation of reactive oxygen and nitrogen species. These reactive molecules can oxidize biomolecules, including unsaturated fatty acids, and can directly influence inflammatory pathways when cellular redox-regulatory pathways involving selenoproteins are dysregulated. Among selenoproteins, GPX4 plays a critical role in the reduction of lipid hydroperoxides that regulate the catalytic activity of COXs apart from playing a critical role in ferroptotic cell death (Ursini et al. 1982; Kulmacz and Lands 1983; Dixon et al. 2012; Sengupta et al. 2013). On the other hand, several key redox-sensitive transcription factors, including nuclear factor (NF)-κB, play pivotal roles in the progression and resolution of inflammation by modulating prostaglandin (PG) metabolism at different levels. Herein, we critically evaluate the direct association and plausible connections between redox-regulatory mechanisms involving Se/selenoproteins and inflammation and immunity in the context of PG metabolism.
III. Se/Selenoproteins in Prostanoid Metabolism
A. COX-mediated synthesis of prostaglandin H2 (PGH2)
Prostanoids are a class of lipid metabolites generated from the 20-carbon polyunsaturated fatty acids (PUFA), arachidonic acid (ARA 20:4, Δ5,8,11,14) or eicosapentaenoic acid (EPA 20:5, Δ5,8,11,14, 17), upon dioxygenation by COXs, which exist in two isoforms COX-1 [prostaglandin endoperoxide synthase 1 (Ptgs1)] and COX-2 (Ptgs2) (Vane et al. 1998). These lipid mediators consist of PGs (PGD2, PGE2, and PGF2α), prostacyclin (PGI2), and thromboxanes (TXs, TXA2 and TXB2) from ARA or their 3-series counterparts (PGD3, PGE3, PGF3α, PGI3, TXA3, and TXB3) from EPA.
Generally, ARA is released from plasma membrane phospholipids upon hydrolysis by phospholipase A2 (PLA2) (Dennis et al. 2011; Leslie 2015). ARA is abundant in mammalian tissues and serves as a precursor for various bioactive eicosanoids and their metabolites (Fig. 2). Specifically, ARA is oxidized by either COX-1 or COX-2 to produce prostaglandin G2 (PGG2) hydroperoxide that is further reduced to its corresponding alcohol, PGH2, by the peroxidase activity of COX enzymes or by GPX1 (Hong et al. 1989). COX-1 and COX-2 exist as homodimers in the microsomes and nuclear membranes (Rollins and Smith 1980). COX-1 is a constitutively expressed enzyme maintaining tissue homeostasis by providing the physiological levels of PGs in most tissues. However, cytokines such as interleukin (IL)-4 increase the expression of COX-1 via post-transcriptional and translational mechanisms that appears to be important in innate immune responses challenging the long-standing paradigm that COX-1 is devoid of any regulation (Shay et al. 2017). Apart from a few exceptions of its constitutive expression, COX-2 is mainly elevated in inflammation, including pathological conditions such as infections and cancers. COX-2 is expressed upon stimulation by a variety of triggers including cytokines (tumor necrosis factor (TNF)-α, IL-1β) (Feng et al. 1995), hormones (adiponectin) (Ouchi and Walsh 2007), toxins (lipopolysaccharide (LPS)) (Brock and Peters-Golden 2007), oxidative stress (hydrogen peroxide (H2O2)) (Feng et al. 1995), and physical stress (shear stress, forced-exercise) (Doroudi et al. 2000; Bilski et al. 2019).
Figure 2. Pathways of prostaglandin metabolism.
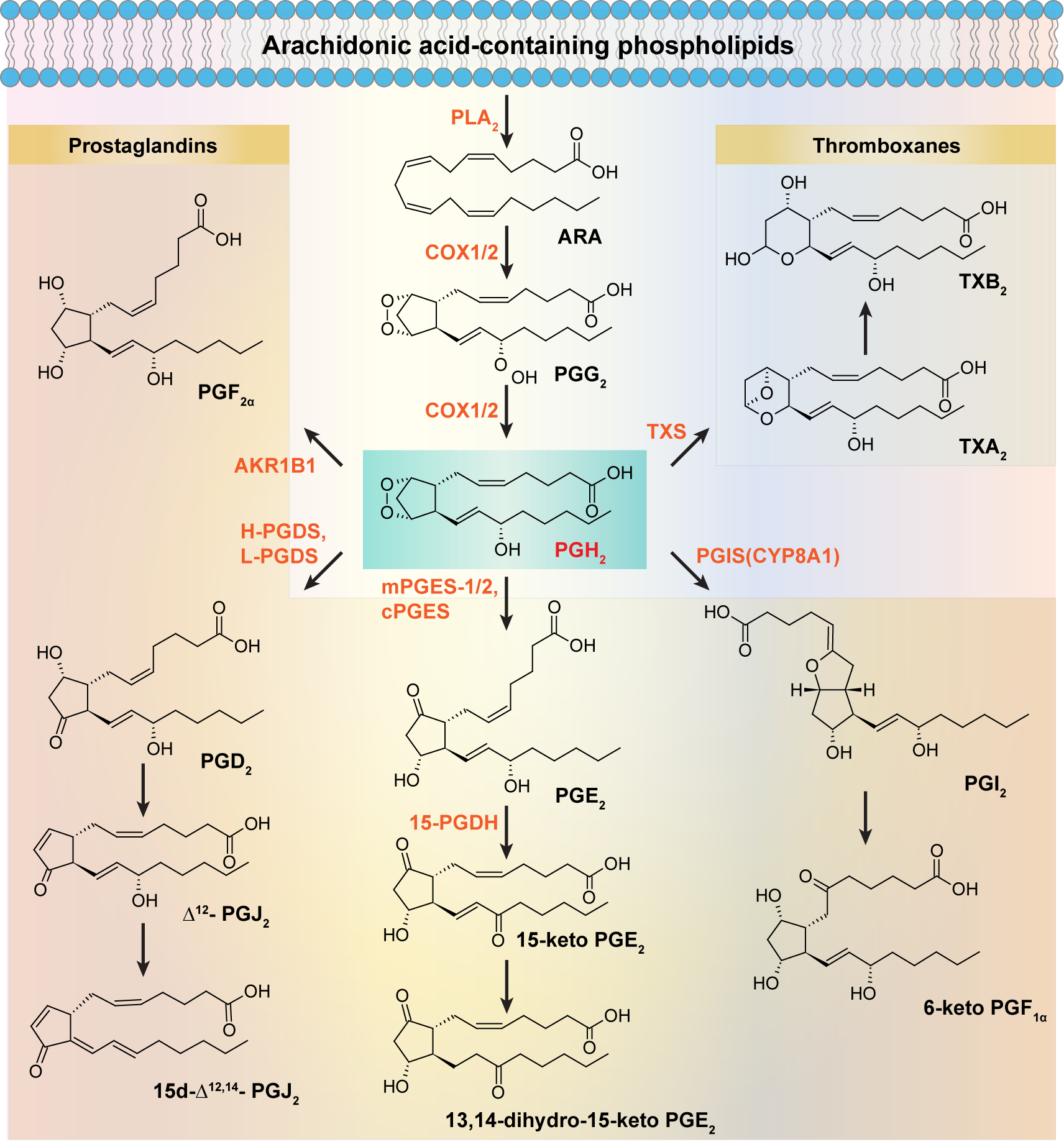
Upon stimulation, phospholipids in the plasma membrane are acted upon by PLA2 to release ARA, which is further converted to PGG2/PGH2 by COX1/2. PGH2 is the common precursor for D-series (PGD2, Δ13-PGJ2, Δ12-PGJ2, 15d-PGJ2), E-series (PGE2), prostacyclin (PGI2), PGF2α, and TXs (TXA2, TXB2) that is catalyzed by specific synthases.
1. Role of COXs in immunity
COX-1 and COX-2 vary in distribution and expression patterns. They modulate the production of different PGs via functional coupling with downstream synthases leading to both inflammation as well as resolution of inflammation. Given the enhanced expression of COX-2 in various inflammatory disorders, selective or conventional inhibitors in the form of non-steroidal anti-inflammatory drugs (NSAIDs) have been highly pursued since 1897 following the synthesis of aspirin (Green 2001). Furthermore, due to the role of COX-2 as well as its metabolites in the regulation of immunity, drug-candidates selectively inhibiting COX-2 have been used to treat immune system malfunction-associated morbidities. For example, celecoxib, a selective COX-2 inhibitor, is used to treat melanoma and breast cancer synergistically with programmed death 1 (PD-1) monoclonal antibody (mAb) in rodent models, which suppressed intra-tumoral expression of COX-2 (Li Y et al. 2016). A combination treatment of celecoxib with sunitinib (a multitargeted receptor tyrosine kinase inhibitor) in renal cell carcinoma increased CD4+ interferon (IFN)-γ+ and CD8+IFN-γ+ T cells, while reducing the CD4+FoxP3+ regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) (Wang X et al. 2013; Zhao et al. 2017). Similarly, in a murine model of Mycobacterium tuberculosis infection, celecoxib impaired T helper type 1 (Th1) response and exacerbated the infection (Mortensen et al. 2019), further confirming the dichotomous role of COX-2 in immune response. While celecoxib has shown promise in oncology, the cardiovascular side effects of this (and similar) drug often preclude its long-term use necessitating alternative methods to “fine tune” expression of this enzyme (Dogne et al. 2006; White et al. 2007). These studies suggest a unique role for COX-2 in immunity and provide an alternative candidate for the treatment of cancers based on the immunomodulatory mechanism that involves COX-2.
2. Regulation of COXs by Se/selenoprotein(s)
Se supplementation has been shown to downregulate the transcript levels of COX-2 in both bone marrow-derived macrophages (BMDMs) and RAW264.7 macrophage-like cell line that were stimulated with bacterial endotoxin LPS (Kalantari et al. 2008; Gandhi et al. 2011). For example, RAW264.7 macrophages cultured in the presence of sodium selenite (Na2SeO3) (0.1–1.5 μM) showed a decreased expression of COX-2 on both mRNA and protein levels comparing to the cells in Se deficient group (6 pmol/ml) upon stimulation with LPS. In vivo studies with low Se diet (0.033 mg/kg, Na2SeO3) increased the mRNA expression of COX-2 in the kidneys of broiler chicks, while a Se adequate diet (0.2 mg/kg, Na2SeO3) dampened the expression of COX-2 (Zhang JL et al. 2016). Other studies also identified the inhibitory effect of Se on the expression of COX-2 in colon cancer cells (Hwang et al. 2006), vascular endothelial cells (Li YB et al. 2011), and chicken neutrophils (Chen J et al. 2017). Taken together, in vitro and in vivo studies suggest a clear benefit of increased Se on COX-2 expression in experimental models.
These studies bring up an important question whether selenoproteins mediate the effect of Se on the inhibition of COX activity and/or expression. Early work found that GPX1 suppressed the activity of COX from sheep seminal vesicles (Smith WL and Lands 1972); and human plasma glutathione peroxidase activity (presumably GPX2) served as an important regulator of plasma hydroperoxide levels (Maddipati and Marnett 1987), which regulates the “peroxy tone” to modulate the catalytic activity of PG synthases in addition to COX enzymes (Kulmacz 1986; Kulmacz R.J. 1997). Furthermore, increased expression and activity of GPX1 were inversely associated with decreased expression and activity of COX-2 in Se supplemented macrophages (Zamamiri-Davis, Lu et al. 2002).
Phospholipid hydroperoxidase, GPX4, deserves a special attention in relation to COX-2 activity. Overexpression of Gpx4 in rat basophilic leukemia (RBL-2H3) resulted in decreased levels of PGD2 which was associated with inhibition of production of PGH2 from ARA by Cox-2 (Sakamoto et al. 2000). GPX4-overexpressing cells exhibited lower levels of lipid hydroperoxides, the availability of which plays a key role in the activation of Cox-2. Use of a chemical GPX4 allosteric activator (compound 102) reduced lipid hydroperoxides and intracellular ROS levels, inhibited ferroptosis, and most importanly, decreased NF-κB activation (Li C et al. 2018). Use of the GPX4 allosteric activator with zileuton, a 5-lipoxygenase (LOX) inhibitor, increased ARA metabolism through 12-LOX and 15-LOX leading to the enhanced production of proresolving metabolites in neutrophils (Li C et al. 2018). Keratinocyte-specific Gpx4 knockout mouse pups showed an impaired hair follicle morphogenesis, which was accompanied with an increased COX-2 expression, thereby suggesting an inverse relationship between Gpx4 and Cox-2 expression. This abnormal phenotype was partially reversed by feeding the nursing dams with a COX-2 specific inhibitor, celecoxib (Sengupta et al. 2013). Furthermore, in a dextran sodium sulfate (DSS)-induced colitis murine model, mice with monocyte/macrophage-specific knockout of selenoproteins (Trspfl/flLysMCre) showed a significantly increased expression of COX-2 and exacerbated inflammation in colons compared with their wild type (WT) littermates in the presence of Se supplementation (0.4 ppm, Na2SeO3) (Kaushal et al. 2014). Taken together, the incorporation of Se into selenoproteins was found to be essential in the control of COX-2.
The regulation of COX-2 expression by Se or selenoproteins occurs at multiple levels. For example, the regulation of expression of COX-2 depends on the activation and subsequent nuclear translocation of NF-κB. Cysteine 62 in the DNA-binding motif of p50 subunit of NF-κB is sensitive to oxidation and is necessary to be in the reduced state to mediate DNA binding (Hayashi et al. 1993). Activation of toll-like-receptor 4 (TLR4) by LPS in macrophages, further recruits other proteins, myeloid differentiation factor 88 (MyD88), IL-1 receptor-activated kinases, IRAK4 and IRAK1, followed by TNF receptor-associated factor 6 (TRAF6) [reviewed in (Lu et al. 2008)]. Ubiquitylated TRAF6 binds to two adaptors, transforming growth factor (TGF)β-activated kinase 1 (TAK1) and its binding partners, TAB1–3, which phosphorylate IκB kinases (IKK) in addition to other kinases such as p38 and c-Jun N-terminal kinase (JNK) (Ajibade et al. 2013). Several reports indicate TAK1 functions as a potential upstream redox sensor to maintain a steady state level of the master regulator of antioxidant responses, Nuclear factor erythroid 2 (NF-E2)-related factor 2 (Nrf2) (Hashimoto et al. 2016). Selenoproteins, Gpx2 and Txnrd1, are targets of Nrf2 in duodenum (Muller M et al. 2010). Phosphorylated IKKs phosphorylate IκB subunits leading to their proteasomal degradation following dissociation, making the NF-κB dimers translocate to nucleus to activate transcription of a plethora of genes, including COX-2, reviewed in (Tanabe and Tohnai 2002; Gilmore 2006) (Fig. 3).
Figure 3. Regulation of COX-2 expression by Se/selenoproteins summarized from different cells.
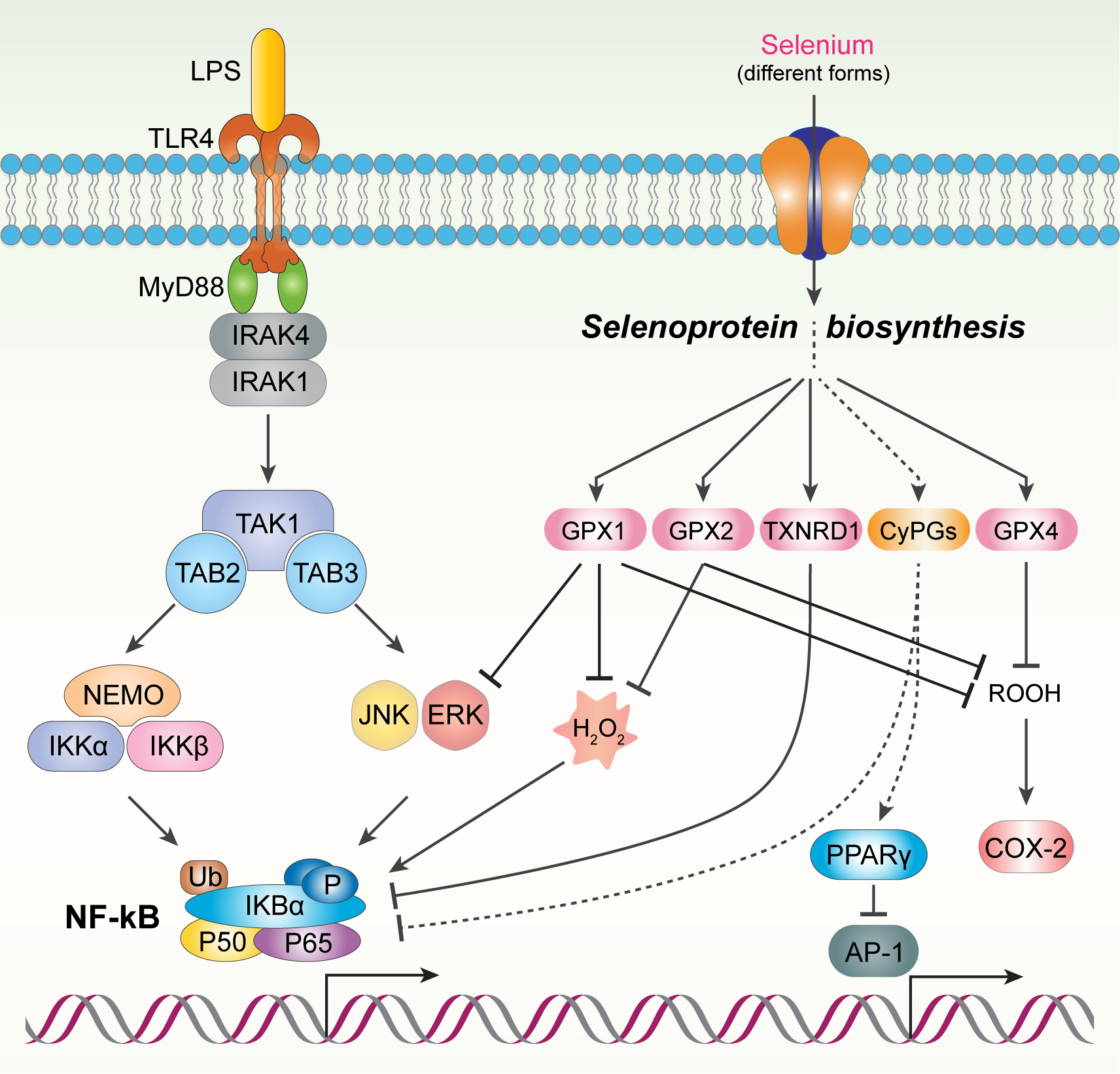
In LPS-stimulated macrophages, LPS binds TLR4 and recruits MyD88, IRAK4, IRAK1, TRAF6. TRAF6 induces the activation of TAK1–3 which phosphorylates IKK leading to the degradation of IĸB. As a result, NF-ĸB binds to the promoter region of COX-2 and induces the transcription of COX-2. In the presence of Se, GPX1 inhibits two MAPK (JNK and Erk) which can mediate the dissociation of IĸB from NF-ĸB. In human breat T47D cell line, overexpression of GPX2 inhibits oxidative stress and IĸB transactivation and NF-ĸB activation. In Hela cells, Txnrd1 inhibits NF-ĸB via mechanisms involving the phosphorylation of p65 in NF-ĸB. In CaSki cells, 15d-PGJ2 inhibits the binding affinity of AP-1 to CRE in the promoter region of COX-2 through PPARγ which downregulates the expression of COX-2. Additionally, 15d-PGJ2 can directly bind NF-ĸB and inactivate it which leads to the downregulation of COX-2.
Se supplementation downregulated the expression of COX-2 by targeting NF-κB through various mechanisms. Se supplementation (0.05 – 1.5 μM, Na2SeO3) induced the enhanced production and activity of GPX1 in Se-deficient RAW264.7 macrophages and deceased the expression of COX-2 (≥0.2 μM, Na2SeO3) (Vunta et al. 2008). GPX1 was able to protect against oxidative stress-induced by LPS in the Se-deficient macrophages, possibly resulting in the inhibition of mitogen-activated protein kinases (MAPKs), JNK, and extracellular-signal-regulated kinase (Erk) (Kretz-Remy et al. 1996; Vunta et al. 2008). As a result, the dissociation of IκB from NF-κB complex was inhibited leading to its attenuated nuclear translocation and decreased expression of COX-2. Besides, selenite was reported to directly inhibit DNA binding activity of NF-κB by the oxidizing cysteine residue suggesting short-term effects of high dose selenite by acting on NF-κB per se (Kim and Stadtman 1997). Overexpression of GPX2 in human breast T47D cell line inhibited H2O2-induced oxidative stress and IκB transactivation as well as NF-κB activation (Kretz-Remy et al. 1996) (Fig. 3). A recent study using mice and human colorectal adenocarcinoma (HT-29) cells with stable knockdown of Gpx1 and Gpx2 showed that the inhibitory effect of Gpx2 on NF-κB signaling and its target gene expression was more pronounced than that of Gpx1, while eicosanoid products of COX- and LOX-mediated metabolism increased more strongly in cells that lacked Gpx1 than in those without Gpx2 expression (Koeberle et al. 2020). Other studies have suggested thioredoxin reductase-1 (Txnrd1) to control the transactivation potential of NF-κB in Hela cells via mechanisms that involves phosphorylation of Ser536 within the transactivation domain of p65 in addition to activation of enzymes involved in the pathway of NF-κB activation as mentioned above (Heilman et al. 2011). An alternative mechanism involves the increased production of cyclopentenone PGs (CyPGs) by Se supplementation that inhibits the kinase activity of IKK1/2 via a direct covalent Michael addition of reactive Cys residue in the kinase domain (Rossi et al. 2000). CyPG, 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), inhibited the binding activity of AP-1 [to 3’, 5’-cyclic adenosine monophosphate (cAMP) response element (CRE) binding site] and NF-κB, resulting in the attenuated expression of COX-2 through peroxisome proliferator-activated receptor gamma (PPARγ) signaling in CaSki human cervical cancer cells (Han S et al. 2003) (Fig. 3). In addition, epigenetic mechanisms were also shown to regulate COX-2 expression by Se. Treatment of WT BMDMs with Se (500 nM Na2SeO3) decreased acetylation of H4K12 at the promoter of COX-2. However, a complete lack of selenoproteins, as in Trsp knockout BMDMs, failed to modulate histone acetylation at the site of COX-2 promoter (Narayan et al. 2015). These studies clearly indicated that incorporation of Se into selenoproteins was essential for the regulation of COX-2 expression by Se at multiple levels in activated macrophages.
Studies by Gandhi, Kaushal et al (Gandhi et al. 2011) and Hema et al (2007) together indicated that Se status affected the function of COX-2, but not COX-1, leading to the production of downstream metabolites via the eicosanoid class switching paradigm. As shown in Fig. 4, Se status also regulated the expression of specific PG synthases, such as hematopoietic PGD synthase (H-PGDS), microsomal PGE synthase-1 (mPGES-1), PGI synthase (PGIS), and TX synthase (TXS), which are downstream enzymes that catalyze the conversion of PGH2, a highly labile product of COX, into specific effector PGs (Fig. 2). The role of some of these products in immunity and their regulation by Se will be described in the following sections.
Figure 4. Schematic representation of “eicosanoid class switching” mediated by selenoproteins generated from macrophages.
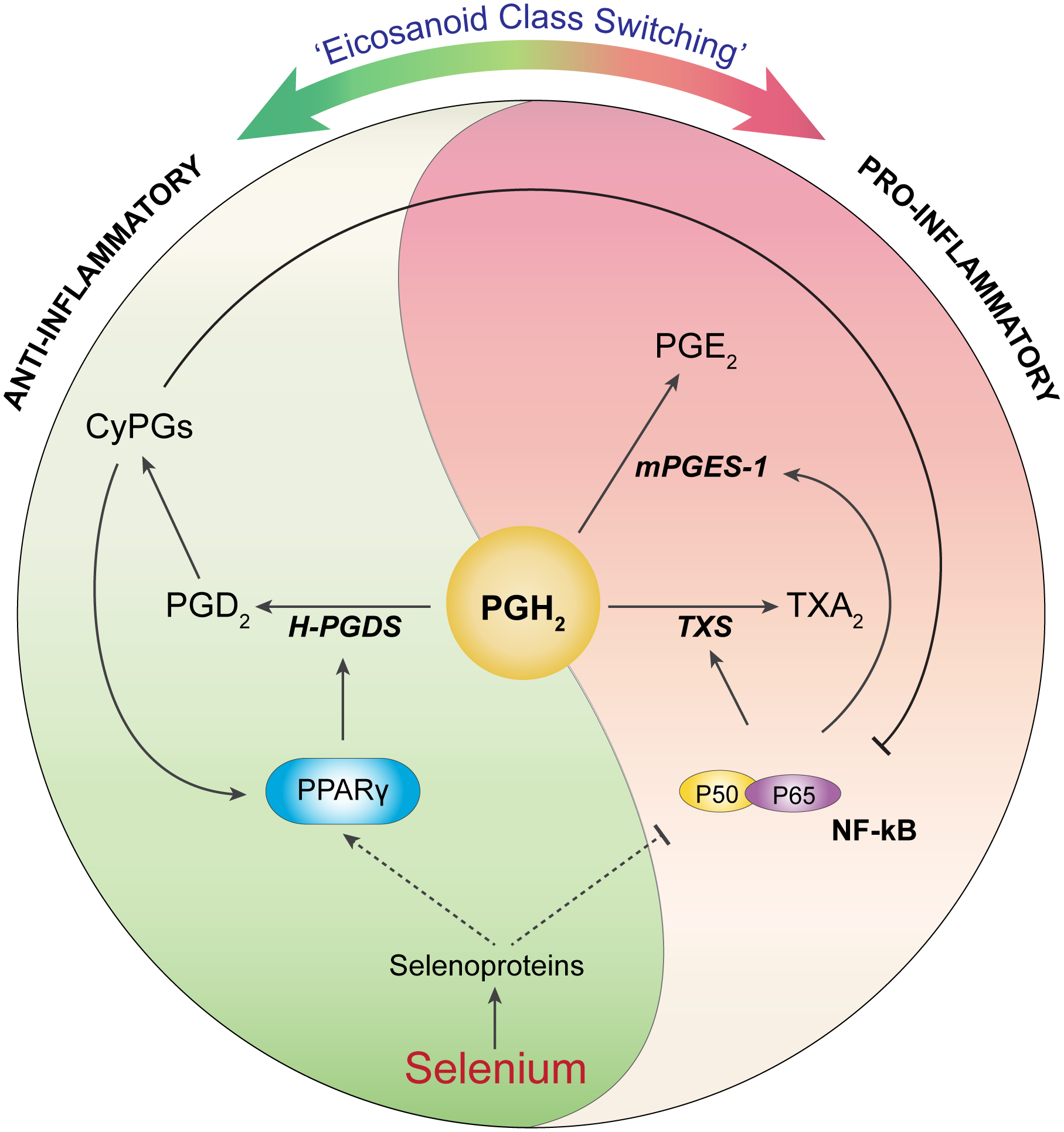
Supplementation of Se upregulates the expression of selenoproteins, which mediate the shift of the ARA pathway to produce more anti-inflammatory PGD2 and its downstream CyPG metabolites, while decreasing the levels of pro-inflammatory PGE2 and TXA2 via the regulation of PG synthases (mPGES-1, TXS, and H-PGDS) as well as two transcription factors, i.e. NF-ĸB and PPARγ.
B. Role of PGD2 and CyPGs in immunity
As described in the preceding section, PGH2 is metabolized by enzymes, PGDS to form PGD2, which is the major PG in the central neuron system (CNS) (Yermakova and O’banion 2000) and is also of great importance as a mediator in allergy and asthma suggesting its role in extra-neuronal systems, including respiratory system and immune system (O’Sullivan 1999; Matsuoka et al. 2000; Mitsumori 2004). H-PGDS (Kanaoka and Urade 2003) and lipocalin-type PGDS (L-PGDS) (Tanaka T et al. 1997) are two enzymes that catalyze the conversion of PGH2 to PGD2 in a glutathione (GSH)-dependent or GSH-independent manner, respectively.
H-PGDS, a sigma-class glutathione S-transferase (GST), is mainly expressed in the hematopoietic and inflammatory cells including mast cells, dendritic cells (DCs), Th2 cells, and other antigen-presenting cells (APCs), such as monocytes and macrophages. This unique expression pattern implies that H-PGDS may contribute to a type-2 immune response through the production of PGD2 and its downstream CyPGs. But the role of H-PGDS in Th2 immunity may suggest divergent effects depending on the nature of inflammation, type of participating cells, and cytokines produced in addition to the Se status. For instance, H-PGDS acts in a pro-inflammatory manner in airway hyperresponsiveness (AHR) or asthma by producing localized PGD2, which is characterized by bronchoconstriction and secretion of allergic cytokines, as well as immune cell infiltration (O’Sullivan 1999). PGD2 can bind to a G-protein coupled receptor (GPCR), Gpr44 [also termed as chemoattractant receptor-homologous molecule expressed on Th2 cells (Crth2) or PGD2 receptor 2 (DP2)] on type 2 innate lymphoid cells (ILC2) to upregulate the production of type 2 cytokines, such as IL-4, IL-5, and IL-13 by activating ILC2. Moreover, Gpr44 activation by PGD2 also enhances the production of two ILC2 activators, IL-25 and IL-33, to mediate ILC2-dependent immune responses (Xue et al. 2014). Mast cells, as another major contributor of type 2 immune response, also produce PGD2 to activate Gpr44 on ILC2. Furthermore, activated ILC2 can in turn stimulate mast cell activation by secreting IL-3, IL-4, and IL-13 (Xue et al. 2014). However, CyPGs were shown to act preferentially on Gpr44. Interestingly, Se treatment (> 200 nM) led to increased activation of Gpr44 through these non-enzymatic metabolites of PGD2 (as described below) that decreased COX-2 expression via increased Ca2+ mobilization and expression of miR155, which attenuates the activation of NF-κB via downregulating the autocrine loop involving TNF-α in macrophages (Diwakar et al. 2019). Given that oxidative stress is involved in asthma, the interplay of two GPCRs (Gpr44 and another PGD2 receptor, DP1) and their ligand-specific and temporal regulation may dictate the outcome of whether Se supplementation would be beneficial or not.
In the context of inflammatory bowel disease (IBD), studies showed the pro-resolution effect of H-PGDS where activation of Gpr44 may be involved, while DP1 exerts the opposite function (Sturm et al. 2014). Thus, it appears that differential expression of PGD2 receptors may play an important role in dictating the outcome. As mentioned earlier, the regulatory role of Gpr44 in IBD and LPS-induced inflammation by a subgroup of PGD2 metabolites generated via multiple non-enzymatic reactions from PGD2 (Straus and Glass 2001) cannot be ignored. These metabolites have an alkylidene cyclopentenone structure, and therefore are commonly referred to as CyPGs, which comprise Δ13-PGJ2, Δ12-PGJ2, and 15d-PGJ2 (Fig. 2). Although these small molecules are short-lived bioactive lipids, they are implicated in various physiological and pathological activities (Willoughby et al. 2000). Interestingly, Gpr44 expression was found in the mucosa of patients with IBD and ulcerative colitis patients in remission that also had higher levels of PGD2 (Vong et al. 2010). Even though the levels of CyPGs were not measured in this study, given that CyPGs can preferentially activate Gpr44 with a high affinity, in addition to the DP1 receptor, Gpr44-dependent activation by these CyPGs may likely mediate the resolution in colitis.
1. Regulation of CyPGs by Se/selenoprotein(s)
Previous studies in our laboratory have indicated a dose-dependent effect of Se (as selenite, Na2SeO3; 50–500 nM) on the production of PGD2 and CyPGs upon LPS stimulation of murine macrophages, while decreasing other PGs (PGE2, TXA2, and PGF2α) as a result of differential regulation of PG synthases leading to the “eicosanoid class switching” paradigm (Fig. 4) (Gandhi et al. 2011). Intriguingly, Se-supplementation led to increased activation of PPARγ, while inhibition of enzymatic activity of H-PGDS abrogated this increased PPARγ activation, thereby suggesting an interaction of Se and PPARγ where H-PGDS activity was indispensable (Fig. 4). These studies are in agreement with the role of CyPGs as endogenous ligands of PPARγ (Forman et al. 1995; Kliewer et al. 1995) and the trans-repression of NF-κB by ligand-dependent activation of PPARγ (Pascual et al. 2005).
In support, studies in our laboratory have provided evidence of the endogenous production of high levels (high nM) of CyPGs in macrophages treated with Se (>200 nM Se as selenite). A feedback loop involving the PPARγ-dependent mechanism to sustain such an increase in endogenous CyPGs came from our studies showing two PPAR-response elements (PPREs) in the proximal promoter of H-PGDS that were critical in the upregulation of H-PGDS by Se (Gandhi et al. 2011). The effect of Se was abrogated in the absence of selenoprotein synthesis suggesting that the “eicosanoid class switching” mechanism involved selenoproteins.
To be able to exert physiological effects, CyPGs produced upon Se supplementation need to be protected from subsequent metabolic inactivation. Notably, the degradation of CyPGs is mediated by a key enzyme, 15-hydroxyprostaglandin dehydrogenase (15-PGDH) that catalyzes the oxidation of the hydroxyl group in many bioactive lipid mediators, with some preference to PGE2 (Ensor and Tai 1995). Recent studies in our laboratory focused on leukemia have found an interesting link between Se, CyPGs, and leukemia stem cells (LSCs) in a chronic myeloid leukemia (CML) model, where supraphysiological levels (dose levels above the requirement where key markers of Se status, such as SELENOP and GPX1 are not elevated with any further increase in dose) of Se were protective. Interestingly, the decreased expression of 15-PGDH was accompanied by enhanced production of CyPGs upon dietary Se supplementation in these mice (Finch et al. 2017). Thus, Se may affect the production of CyPGs through multiple mechanisms involving the upstream and downstream signals of CyPGs, which in turn modulate immune function through diverse mechanisms as described earlier.
C. PGE2 and its role in immunity
PGES catalyzes the conversion of PGH2 to form PGE2, which exerts its biological functions by binding to one of the GPCRs, PGE2 receptors1–4 (EP1–4). Activation of some isoforms (EP2 or EP3) leads to the elevation of intracellular cAMP, which may in turn result in immune dampening events (Lycke et al. 1990; Phipps et al. 1990; Roper et al. 1990). Contrary to PGD2 as well as CyPGs, PGE2 is regarded as a pro-inflammatory mediator in most conditions; however, PGE2 is also a vital signal that induces the production of other potent pro-resolution mediators such as lipoxins via the modulation of LOX (Serhan and Savill 2005). PGE2 also plays a role in the regulation of vascular permeability, body temperature, and aging. In particular, the protective role of PGE2 in gastric mucosa against acid exposure was identified by the administration of NSAIDs. Gastrointestinal bleeding associated with non-selective NSAID usage was ameliorated with PGE1 analogue, misoprostol (Hunt et al. 1983).
Interestingly, PGE2 reportedly suppressed the immune functions by inhibiting mitogenesis and thus inducing apoptosis of T cells (Goodwin et al. 1977). In addition, PGE2 also downregulated the synthesis of Th1 cytokines (e.g., IL-2 and IFNγ), while stimulating Th2 cytokines (e.g., IL-4 and IL-10) (Fedyk et al. 1997), which may also be a part of the pro-resolution program. With regard to the effect of PGE2 on B cells, studies have shown that PGE2 may affect the outcome of B cells due to its impact on B cell development. In other words, PGE2 can induce the apoptosis or proliferation of immature or mature B cells, respectively (Goodwin et al. 1977). In addition to the proliferating activity on mature B cells, PGE2 can induce an immunoglobulin (Ig)-class switch favoring the production of IgG1 and IgE, while diminishing the synthesis of IgM and IgG3 (Phipps et al. 1991). The profoundly enhanced production of IgG1 and IgE by PGE2 in mature B cells may result from the response to LPS synergizing with IL-4 (Morawetz et al. 1996). Intriguingly, the activation of EP2 or EP2 and EP4 mediated Ig-class switching in mature B cells has been reported to require intracellular cAMP (Fedyk and Phipps 1996). While the above data were generated in in vitro studies, in vivo studies also indicated high levels of PGE2 to be associated with elevated secretion of IgE (Gao et al. 2016). In an EP2 knockout mouse, IgE production was markedly suppressed in response to ovalbumin (OVA) challenge. Mechanistically, interaction between EP2 and PGE2 enhanced the activation of Signal Transducer And Activator Of Transcription 6 (STAT6) that is stimulated by IL-4, thus promoting the Ig-class switching favoring the production of IgE (Gao et al. 2016). Accordingly, antagonists of EP2 or EP4 or inhibitors of PGES are thought to be therapeutic candidates for type I hypersensitivity reaction, which is an immediate allergic reaction mediated by IgE. In contrast to this notion, exogenous PGE2 as well as EP2-selective agonists significantly caused attenuated AHR induced by house dust mite (HDM) or other allergens (Melillo et al. 1994; Sestini et al. 1996; Gauvreau et al. 1999; Herrerias et al. 2009; Serra-Pages et al. 2015). However, other studies showed that inhibition of COXs worsened airway inflammation in mice exposed to OVA (Gavett et al. 1999; Peebles et al. 2002; Hashimoto et al. 2005; Nakata et al. 2005; Peebles et al. 2005; Torres et al. 2009). Much like the literature related to Se in allergy and asthma, a similar inconsistency exists in the role of PGE2 in allergy and asthma that may be ascribed to experimental models (Maunsell et al. 1968; Van Hove et al. 2007).
1. Regulation by Se/Selenoprotein(s)
Given that the function of COX-2 is regulated by Se levels, it is speculated that Se may also modulate the allergic disorders by changing the metabolism of PGE2 by regulating the production of PGH2, the precursor of PGE2. Given that signaling mechanisms involving STAT activation by IL-4 can be increased by Se supplementation (Nelson et al. 2011), it is very likely that some of the downstream pathways activated by PGE2-EP receptor could be also impacted by Se; however, further studies are necessary to tease out the underlying intricate mechanisms. In a clinical study of ulcerative colitis, increased plasma or intestinal mucosal PGE2 was reported to be associated with disease activity (Wiercinska-Drapalo et al. 1999). Previously, studies in our laboratory demonstrated that Se status was inversely correlated with IBD-induced inflammation in a murine model, where mice treated with DSS-induced acute colitis maintained on either Se-deficient (<0.01 ppm; Na2SeO3) or Se-adequate (0.08 ppm; Na2SeO3) diets showed exacerbated inflammation and poor survival compared to mice on Se-supplemented (0.4 ppm; Na2SeO3) or Se-high (1.0 ppm; Na2SeO3) diets (Kaushal et al. 2014). Similarly, Trspfl/flLysMCre mice lacking selenoprotein expression in monocyte/macrophage cells showed comparable disease pattern, as seen in Se-deficient WT mice, despite being on a Se-supplemented diet (Kaushal et al. 2014). Furthermore, increased production of PGE2 were seen in DSS-treated WT mice on Se-deficient or Se-adequate diets for eight weeks compared to those on Se-supplemented diet suggesting Se status clearly had an impact on PGE2 levels. Koeberle et al (Koeberle et al. 2020) demonstrated that unstimulated HT-29 cells lacking Gpx1 and Gpx2 showed higher baseline levels of PGE2 and PGD2 compared to the control cells. Interestingly, IL-1β stimulation of Gpx1 knockdown cells increased levels of PGE2 more than PGD2 suggesting distinct and overlapping roles of Gpx1 and Gpx2 in controlling PGE2 during colonic inflammation.
High levels of PGE2 induced by Se deficiency was a combined effect of increased mPGES-1 expression in addition to low expression and activity of 15-PGDH, which was differentially regulated by Se supplementation (Kaushal et al. 2014). 15-PGDH, a nicotinamide adenine dinucleotide (NAD)+-dependent enzyme, can oxidize many prostanoid metabolites. Even though 15-PGDH is reported to preferentially mediate the oxidative reaction from PGE2 to 15-keto PGE2, 15-PGDH could potentially inactivate CyPGs. Besides, specialized proresolving mediators (SPMs) metabolized from EPA and docosahexaenoic acid (DHA), such as resolvin E1 (RvE1) and protectins, are also substrates for 15-PGDH-catalyzed reaction (Ensor and Tai 1995; Fredman et al. 2012). These results suggest that 15-PGDH may possess a dual role in inflammation and resolution that need to be further elucidated. More recently, studies in our laboratory have found that the colonic expression and activity of 15-PGDH was increased by Se supplementation to inactivate PGE2 in DSS-induced colitis, which was corroborated by increased levels of 15-keto PGE2 (Kaushal et al. 2014). A specific inhibitor for 15-PGDH, CAY10397, abrogated the anti-inflammatory effect of Se supplementation on colitis. Similar results were observed in a second model of IBD induced by Citrobacter rodentium infection (unpublished results).
Furthermore, elevated expression of GPX2 in Se supplemented mice with colitis may elucidate how selenoproteins regulate the activity of 15-PGDH to further impact the production and activity of PGE2. In addition, other selenoproteins [selenoprotein P (SEPP1) (Andoh et al. 2005), selenoprotein S (SelenoS) (Seiderer et al. 2007), selenoprotein K (SelenoK) (Marciel and Hoffmann 2019)] are also implicated in inflammation associated with IBD. With respect to the production of PGE2, selenoproteins may also regulate the synthases, particularly PGES enzymes. For example, GPX2 knockdown in HT-29 colon cancer cells led to increased expression of microsomal PGES-1 (mPGES-1), one of the inducible isoforms of PGES, accompanied by increased production of PGE2 (Banning et al. 2008). It is well-known that mPGES-1 expression can be induced by pro-inflammatory cytokines, IL-1β (Leclerc et al. 2013) and TNF-α (Bage et al. 2010) that are both downregulated by Se (Zhang F et al. 2002). On the other hand, pediatric patients with Kashin–Beck disease (KBD), a Se deficiency-induced cardiomyopathy, had increased serum levels of IL-6, IL-1β, and TNF-α (Zhou et al. 2014). An increased serum level of TNF-α is interesting in the context as it is known to destabilize GPX4. Under severe Se deficiency as in KBD, an elevated level of TNF-α could have profound effects on GPX4 with possible implications on ferroptotic cell death of cardiomyocytes. In fact, it was shown that whole blood level of GPX4 was about 10 times lower in the disease group when compared to the control group (Du et al. 2012).
It has been reported that TNF-α and IL-1β have a common downstream target molecule, NF-κB, which is also identified as a regulator for PGE2 production. Thus, based on the data, we speculate that Se supplementation may orchestrate the production of PGE2 through GPXs/IL-1β and/or TNF-α/NF-κB/mPGES-1 signaling axis. Moreover, another key transcription factor, PPARγ, may also participate to mediate the anti-inflammatory effects of Se supplementation by counteracting the activity of NF-κB, while increasing the expression of H-PGDS to dampen the expression of other PG synthases (Gandhi et al. 2011).
Unlike mPGES-1, there is not much evidence available with regard to the role of mPGES-2, which is highly expressed in intestine, heart, and brain, where it is thought to maintain the physiological biosynthesis of PGE2 (Tanikawa et al. 2002). While mPGES-1 is inducible upon LPS treatment or pathological triggers such as ischemia, hypoxia, and physical stress, mPGES-2 is more likely to play a constitutive role in physiological and pathological conditions (Murakami et al. 2003). Interestingly, mPGES-2 knockout mice had no measurable changes of PGE2 production in any tissues (Jania et al. 2009). Further observations did not reveal any lymphoid tumors in mice lacking mPGES-2 expression. Taken together, it appears that mPGES-2 may be dispensable in mice and humans (Nakanishi et al. 2010). In addition to the two microsomal forms, a third isoform, cytosolic PGES (cPGES), is reported to be important for the development of pulmonary system. cPGES knockout mice succumbed to pulmonary immaturity in the perinatal period, likely due to the defective stabilization of the glucocorticoid receptor complex (Lovgren et al. 2007). Preliminary studies in our laboratory suggest that dietary Se does not impact the regulation of mPGES-2 and cPGES in activated macrophages (Kaushal et al. 2014).
D. The role of PGI2 in immunity
Prostacyclin (PGI2), first isolated from swine and rabbit aortas, is formed from its precursor PGH2 through the activity of PGIS (CYP8A1) predominantly expressed within the cardiovascular and pulmonary endothelium (Moncada et al. 1976; Mubarak 2010; Chu et al. 2015). Many studies identified PGI2 to antagonize the activity of TXA2 in the vasculature by inhibiting platelet aggregation and promoting vasodilation (Rustin et al. 1987; Kinsella 2001; Gleim et al. 2009; Smyth et al. 2009; Tonelli et al. 2015). Interestingly, such a counter regulation is also observed between PGI2 and PGE2, specifically during the development of atherogenesis. In this case, augmented production of PGI2 mediated a cardioprotective effect in mPGES-1 knockout mice where the production of PGE2 was attenuated. Surprisingly, suppression of thrombogenesis by the interaction of PGI2 and its receptor, IP, limits atherogenesis (Tang et al. 2016). In addition, in human lung carcinoma, elevated PGE2 was implicated in tumor progression and inhibition of apoptosis, whereas PGI2 as well as its analog, iloprost, inhibited initiation of lung cancer (Keith et al. 2002; Keith et al. 2004; Nemenoff et al. 2008).
From an immune context, both PGI2 and PGE2 are considered pro-inflammatory mediators in the initial phase of inflammation by enhancing vascular permeability and subsequently facilitating leukocyte infiltration (Hata and Breyer 2004). However, on its own, the immunomodulatory role of PGI2 includes both anti-inflammatory [e.g., atherosclerosis, pulmonary arterial hypertension (PAH)] and pro-inflammatory [e.g., rheumatoid arthritis (RA), osteoarthritis (OA)] roles depending on the downstream signaling pathways following IP activation (Stitham et al. 2011). An IP receptor-independent mechanism involving PPARβ/δ activation can be induced by PGI2 analogs and IP agonists that subsequently results in vasodilation by inducing phosphoinositide 3-kinase (PI3K)–Akt–nitric oxide synthase (NOS) pathway (Jimenez et al. 2010) to favor the proresolving effects of PGI2 in cardio-pulmonary vascular diseases.
Apart from the vascular endothelium, PGIS is also expressed by macrophages. Previous studies in our laboratory demonstrated a biphasic dose response to increasing Se levels in LPS-treated macrophages, where doses up to 100 nM (as selenite) increased the transcript levels of PGIS. However, further addition to Se (above 100 nM) led to drastic transcriptional downregulation (Gandhi et al. 2011). The role of such a biphasic regulation of PGIS by Se on macrophage physiology and function is currently unknown.
1. Regulation of PGI2 by Se/selenoprotein(s)
Although the regulation of PGI2 by Se is relatively less well studied, literature is replete with studies demonstrating the role of PGI2 in the context of atherosclerosis with reference to endothelial cells. Evidence from bovine mammary endothelial cells (BMECs) has shown that Se deficient (0 μg/L, Na2SeO3) culture conditions resulted in significantly decreased production of PGI2 and PGE2, whereas increased hydroperoxyeicosatetraenoic acid (15-HPETE) was produced compared to Se-adequate (10 μg/L, Na2SeO3) treatment (Cao et al. 2000). In another study related to the vascular homeostasis in rats, experimental Se deficiency (0.022 mg/kg diet versus adequate 0.159 mg/kg diet) led to markedly decreased concentration of plasma 6-keto-PGF1α, a stable metabolite of PGI2 (Qu et al. 2000). Thus, it appears that Se status plays a role in the maintenance of vascular system homeostasis by regulating the production of PGI2 in the endothelium. Although exact mechanisms of regulation of PGI2 by Se remain unknown, accumulating evidence has provided the following ideas. In atherosclerosis, observational studies revealed an inverse relationship between selenoproteins (GPX1, Txnrd1, SelenoP, and SelenoS) and the development of atherosclerosis. Specifically, Gpx1 knockout mice on an ApoE knockout background (Gpx1−/−ApoE−/− double knockout) accelerated the progression of atherosclerosis compared to control mice (ApoE single-knockout) (Torzewski et al. 2007). In another study, ebselen, a synthetic organoselenium GPX1 mimetic and a substrate for Txnrd1, significantly improved the atherosclerotic lesions in the aorta of diabetic ApoE knockout mice (Chew et al. 2009). In vitro studies also found that GPX1 activity and PGI2 production were concurrently enhanced in human endothelial cells cultured in Se-containing medium (Na2SeO3, 30 or 90 μg/L) (Ricetti et al. 1994). Furthermore, Se deficient endothelial cells stimulated with TNF-α or H2O2 produced a much less PGI2; while Se adequate endothelial cells treated with exogenous lipid hydroperoxide, 15-HPETE, led to decreased expression of PGIS (Weaver et al. 2001). In summary, based on the available data, it appears that Se supplementation-induced expression of GPX1 attenuates the synthesis of 15-HPETE as well as other ROS to derepress PGIS expression leading to substantial increase in PGI2 and its related anti-atherosclerotic functions within the vascular system.
E. The role of TXA2 in immunity
TXA2 and its stable metabolite, TXB2, are generated from PGH2 by the enzyme TXS (Neri Serneri et al. 1983). Apart from platelets, macrophages also express TXS and produce TXA2 (Wetzka et al. 1997). Although it has a very short half-life of 30 seconds at 37 ˚C, TXA2 is a potent vascular constrictor and platelet activator, which antagonizes the effect of PGI2. Upon induction by stimulation such as ischemia-reperfusion in the lungs, TXA2 can contribute to pulmonary hypertension and bronchoconstriction (Zamora et al. 1993). In IBD (both Crohn’s disease and ulcerative colitis) patients, high levels of TXA2 accumulated in the intestinal epithelia (Zifroni et al. 1983), which might be a therapeutic target for IBD treatment (Taniguchi et al. 1997; Howes et al. 2007). Regarding its involvement in immune disorders, previous studies also identified the contribution of TXA2 in asthma (Dogne et al. 2002), atopic dermatitis (Tanaka K et al. 2002), and inflammatory tachycardia (Takayama et al. 2005). In the thymus, CD4+CD8+ immature thymocytes exhibited elevated expression of TXA2 receptor (TP) as in platelets (Ushikubi et al. 1993). Activation of TP by an agonist (STA2) induced apoptosis in these cells (Ushikubi et al. 1993). Furthermore, the ability of TXA2 to impair the interaction between DCs and T cells as well as DC-dependent T cell proliferation was reported (Kabashima et al. 2003). Collectively, TXA2-TP signaling may affect the adaptive immunity by interfering the maturation and activity of T cells.
1. Regulation of TXA2 by Se/selenoprotein(s)
Studies in pregnancy-induced hypertension (PIH) have suggested a potential link between TXA2 and Se (Fitzgerald et al. 1990; Han L and Zhou 1994). Clinical studies found that PIH patients had increased TP expression on platelets (Liel et al. 1993). In a TXS knockout mouse study, pregnant mice showed decreased hypertension upon TXS deletion (Pai et al. 2016). Interestingly, a decreased incidence of PIH upon Se supplementation was seen in pregnant women (Han L and Zhou 1994). Moreover, other research findings correlated Se supplementation with diminished production of TXA2 (Meydani 1992; Haberland et al. 2001; Arnaud et al. 2007).
Previous studies in our laboratory provided a mechanism mediated by Se and/or selenoproteins on the modulation of pro-inflammatory or anti-inflammatory prostanoids that we termed as “eicosanoid class switching” (Gandhi et al. 2011; Nelson et al. 2011; Diwakar B.T. 2016) (Fig. 4). In both RAW 264.7 macrophage-like cells and primary BMDMs, supplementation with selenite significantly reduced the markers for pro-inflammatory macrophage phenotype (M1) in the presence of LPS stimulation. In IL-4 treated macrophages, Se supplementation upregulated the expression of markers for anti-inflammatory macrophage phenotype (M2) (Nelson et al. 2011). As a result, “eicosanoid class switching” favored the production of D-series PGs (PGD2 and CyPGs), while inhibiting the production of E-series (PGE2) and TXA2 (Gandhi et al. 2011; Kudva et al. 2015). Se supplementation downregulated the expression of TXS-1 and TXA2 in macrophages (Gandhi et al. 2011) and endothelial cells (Cao et al. 2000). Use of macrophages in which selenoprotein machinery was disrupted led to the conclusion that Sec incorporation in selenoproteins was key in the downregulation of TXS (Gandhi et al. 2011). Much like mPGES-1, NF-κB also serves as a key transcription factor that drives the expression of TXS (Stachowska et al. 2009; He et al. 2013). In LPS-treated macrophages, downregulation of NF-κB by Se supplementation was associated with an increased activation of PPARγ, leading to the attenuation of TXA2 production (Prabhu et al. 2002; Zamamiri-Davis et al. 2002; Vunta et al. 2008). It is thought that signaling by Se/selenoprotein/NF-κB/PPARγ axis impacts the TXS-mediated regulation of TXA2 synthesis by Se.
In summary, Se and selenoproteins regulate the production of various PGs through “eicosanoid class switching” where proresolving PGD2 and downstream CyPGs are increased, while others like PGE2, PGI2, and TXA2 are inhibited (Fig. 4). Previous clinical studies have emphasized the importance of measurement of baseline Se levels in patients, which is a useful guideline for Se intake (Pazirandeh et al. 1999; Hadjibabaie et al. 2008; Khanna et al. 2010; Stevens et al. 2011). However, none of the clinical studies, including the SELECT (Selenium and Vitamin E Cancer Prevention Trial) for prostate cancer, have considered the NSAIDs as a confounding factor to explain if the health benefits of Se supplementation are associated with prostanoids. Further studies are required to associate modulation of PGs as a function of Se in diet, with better biomarkers of Se status (such as SELENOP), to clearly exclude the confounding role of NSAIDs.
Along with prostanoids, hydroperoxy- and hydroxy- metabolites of ARA derived from LOXs and their involvement in inflammation and resolution of inflammation are equally important. Early studies confirmed that Se/selenoproteins also regulated biosynthesis of leukotrienes by inhibiting different LOXs. 5-LOX from rat basophilic leukemia cells was inhibited by GPX1 (Haurand and Flohe 1988) and GPX4 (Weitzel and Wendel 1993); and Se deficiency-induced overproduction of leukotrienes was normalized by restoration of GPX4 (Hatzelmann et al. 1989). Similarly, 5-LOX in B cells was reported to be inhibited by Se/GPX4 (Werz and Steinhilber 1996). Moreover, other LOXs, such as 12-LOX in human platelets (Hill TD et al. 1989) and 15-LOX from rabbit reticulocytes (Schnurr et al. 1996), were also negatively regulated by GPX3 (Jin et al. 2011) and GPX4, respectively. Using a GPX4 knockout mouse model, the regulation of this selenoprotein on the inhibition of 12,15-LOX was confirmed (Seiler et al. 2008). These findings have expanded our understanding of the role of Se/selenoproteins beyond prostanoid metabolism, to include other eicosanoids, such as leukotrienes; for more information, see (Samuelsson et al. 1987; Back et al. 2011; Back et al. 2014).
IV. Se/selenoproteins in Immunity
A. Immune cells
As the first line of defense, innate immune cells possesses immediate protective mechanisms including antimicrobial enzymes and peptides, as well as the complement system, which can target various pathogens for lysis or phagocytosis by phagocytic cells (Murphy and Weaver 2016). Furthermore, additional mechanisms are activated by pathogen-associated molecular patterns (PAMPs) to subsequently induce an inflammatory response by producing chemical mediators, enzymes, chemokines, and cytokines (Murphy and Weaver 2016). Adequate consumption of Se has been demonstrated to be important in the functions of various innate immune cells including granulocytes, monocytes and macrophages, DCs, and natural killer (NK) cells and their functions (McKenzie et al. 1998).
1. Polymorphonuclear (PMN) leukocytes
Granulocytes, also called PMNs, are comprised of neutrophils, eosinophils, basophils, and mast cells. Although PMN leukocytes are all short-lived in circulating blood, they mediate the innate immunity through the production of antimicrobial factors and the participation in the development of inflammation and the phagocytosis (Havemann and Gramse 1984). PMN leukocytes therefore build up a powerful barrier to defend against pathogens that involves ROS as part of their arsenal. Interestingly, Se and/or selenoproteins have been indicated to play a role in modulating PMN leukocyte functions.
Neutrophils isolated from Se adequate cattle showed a greater ability to kill the ingested intracellular fungi (e.g., Candida albicans (Boyne and Arthur 1979)) and bacteria (Staphylococcus aureus and Escherichia coli (Grasso et al. 1990; Hogan et al. 1990)). However, the phagocytic capacity of neutrophils was reportedly not dependent on Se status (Grasso et al. 1990; Hogan et al. 1990). During phagocytosis, neutrophils generate large amounts of ROS including superoxide and H2O2 through NADPH oxidases (NOXs, mainly NOX2), which together induce oxidative burst and have a strong antimicrobial effect (Babior et al. 2002). Antioxidant selenoproteins reduced the respiratory burst cascade by lowering the activity of NOXs (Venardos et al. 2007). On the other hand, excessive levels of ROS can also be detrimental to the neutrophils (Arruda and Barja-Fidalgo 2009). Therefore, to overcome the detrimental effect of ROS during the early (or initiation) phase of inflammation, self-protective mechanisms within the neutrophils are of paramount importance. Human PMNs treated with TNF-α led to increased expression of GPX4 via CCAAT-enhancer-binding protein ε (C/EBPε) through ROS signaling, suggesting that selenoproteins also mediate the self-protection of neutrophils against oxidative damage during activation (Hattori et al. 2005). These results support the notion that Se/selenoproteins may play a key role in neutrophil physiology.
Eosinophils participate in the defense against parasitic and fungal infections in addition to their role in allergy and asthma. GPX activity in eosinophils from asthmatic patients was enhanced indicating that the antioxidant protection against the large amount of ROS may help eosinophils survive (Misso et al. 1998). Given that eosinophils can also produce PGD2, it will be interesting to examine the exact role of selenoproteins and the impact on eicosanoid class switching mechanisms (Fig. 4) and their role in eosinophil functions, which are currently unknown.
2. Macrophages
Macrophages are not only phagocytes that play a role as the first-line of defense in innate immunity, but also as APCs to present antigen in combination with major histocompatibility complex (MHC) II proteins to their cognate T cell receptors (TCRs) on CD4+ T cells to mediate adaptive immune responses. In addition, macrophages play a pivotal role in supporting efficient erythroid differentiation (Sadahira and Mori 1999; Chow et al. 2013; Liao, Carlson, et al. 2018).
An in vitro study on J774.1murine macrophages indicated that Se supplementation increased phagocytosis of macrophages stimulated with LPS, where SelenoK was required for phagocytosis (Safir et al. 2003). Macrophages from SelenoK knockout mice were less effective in FcγR-mediated phagocytosis (Norton et al. 2017). On the other hand, overexpression of SelenoK in BV2 microglial cells, which are CNS resident macrophages, increased the phagocytosis through upregulation of inositol trisphosphate (IP3)-Ca2+ signaling (Meng et al. 2019). In addition, selenoproteins were shown to regulate macrophage migration as in the case of mice lacking Trsp in macrophages, which exhibited a diminished migration of macrophages (Carlson et al. 2009; Norton et al. 2017; Meng et al. 2019). Recent studies in our laboratory have indicated macrophages lacking selenoproteins poorly support stress-erythroid differentiation during short-term irradiation of the bone marrow or treatment with hemolytic agents (phenylhydrazine) suggesting a pivotal role for selenoproteins (Liao, Hardison, et al. 2018). Monocyte-derived macrophages form a rosette structure, termed as erythroblastic islands, in physical association with erythroid cells in an effort to provide nutrients, including key signaling molecules such as heme, supporting the notion that macrophages act as nursing cells. Our studies indicated that during Se deficiency in murine models, these rosette structures were poorly organized that, in part, led to poor recovery during stress-erythropoiesis (Liao, Hardison, et al. 2018). Current studies are focused on elucidating the underlying mechanisms and the role of specific selenoproteins in regulating these functions.
Macrophages partake important roles in both the inflammatory and resolution phases of an infection by differentiating into two contrasting phenotypes, the pro-inflammatory M1 macrophage (also termed as classically-activated macrophage) and anti-inflammatory M2 macrophage (or alternatively-activated macrophage) that represent two ends of the spectrum with several intermediate stages that are poorly defined with regard to their pathophysiological roles. M1 macrophages are formed upon the stimulation by IFN-γ secreted from type 1 innate lymphoid cells (ILC1) or NK cells and are involved in type 1 immune response (Barros et al. 2013). On the other hand, M2 macrophages are induced by IL-4, IL-13, or IL-10 produced by mast cells, eosinophils, neutrophils, and other Th2 cells and is linked to a type 2 immune response (Barros et al. 2013). Currently, this dichotomy is challenged by two paradigms: First, in vitro studies overlook the complexity and combined effects of various stimuli, which are important in vivo, making it difficult to apply the in vitro results to in vivo situations. Second, the disparities within macrophage activation and disease progression in humans and mice pose a problem when translating the results from murine experiments to humans since the markers used to define M1 and M2 polarization vary between mouse and human (Martinez and Gordon 2014). Instead the commonly used mouse proteins Ym1, Fizz1, and arginase-1 that have no homologs in human, use of conserved markers such as transglutaminase-2 (TGM2), mannose receptor C type 1 (MRC1/CD206), and CD68 antibodies may help in translation. However, this classification is still valuable for providing basic understanding of the mechanisms in the context of inflammation and resolution.
Previously, we discussed that Se mediated the phenotypic switching from M1 to M2 during inflammation associated with sterile or parasitic infection. Se supplementation significantly attenuated the phenotypic markers for M1 in the presence of LPS stimulation, while upregulating M2 markers, which increased further following IL-4 stimulation or during helminth infection (Nelson et al. 2016; Shay et al. 2017). Studies with various pharmacological inhibitors to inhibit CyPGs in Se-supplemented mice or administration of CyPGs to Se-deficient mice suggested that such an effect was a result of “eicosanoid class switching” as described earlier (Fig. 4). The key question that remains to be answered is the identity of specific selenoproteins that aid in macrophage polarization. Based on the literature, it is believed that selenoproteins GPXs (Nelson et al. 2011), SelenoP (Bosschaerts et al. 2008), and methionine sulfoxide reductase B1 (MsrB1) (Lee et al. 2017), may represent a growing list of candidate selenoproteins that display a potential in regulating the polarization of macrophages.
3. T cells and B cells
Mounting evidence has shown that in vivo Se levels may modulate the proliferation and differentiation of T cells. In a study using porcine splenocytes, Se was reported to promote TCR activation-induced T cell proliferation in association with increased mRNA expression of GPX1 and Txnrd1 (Ren et al. 2012). T cells from mice fed high Se diet (1.0 mg/kg) had a much higher proliferation rate than those from Se low (0.08 mg/kg) and medium (0.25 mg/kg) diets (Hoffmann FW et al. 2010). An in vivo study using athymic, immune-deficient NU/J nude mice showed that CD4+ T cells were increased in Se-sufficient mice (1.0 mg/kg) compared to mice on Se-deficient (0 mg/kg) or Se-adequate (0.15 mg/kg) diets (Cheng et al. 2012). Furthermore, higher Se levels increased free thiols in activated CD4+ T cells, which likely played an important role in the clonal expansion of these cells by favoring the TCR-induced oxidative burst in cells (Hoffmann FW et al. 2010). Interestingly, some selenoproteins including Gpx1, Txnrd1, and other antioxidant proteins in the same cells showed high enzymatic activity implying a potential protective mechanism to counter the high demand for ROS and the need to mitigate possible damage by ROS (Hoffmann FW et al. 2010).
In addition to the effect on T cell proliferation, Se was also reported to modulate T cell differentiation. Studies by the Hoffmann laboratory showed that high Se diet (1.0 mg/kg) induced a biased differentiation of CD4+ T cells toward Th1 immunity with a high level of IFN-γ (Hoffmann FW et al. 2010). Similarly, mice fed Se-nanoparticles exhibited an enhanced Th1 response after Hepatitis B virus (HBV) vaccination (Mahdavi et al. 2017). Trsp deletion in T cells showed atrophy of thymus and exhibited decreased numbers of mature T cells along with hyperproduction of ROS (Shrimali et al. 2007; Shrimali et al. 2008) and attenuated TCR signaling (Shrimali et al. 2007). This suggests that selenoproteins regulate T cell immunity, in part, by scavenging excess ROS.
B cell-mediated humoral immunity is dependent on the formation of various antibodies. Naïve B cells can be activated by antigens in two ways: thymus-independent or thymus-dependent, where the latter derives help from TFH cells, which are already activated by the same antigens (Murphy and Weaver 2016). These antibodies can exert their functions by binding to pathogens and prevent their entry into cells or indirectly activate complement system to effect killing by phagocytes.
Several reports have suggested that Se levels might regulate the function of B cells. Mice fed Se-deficient (0 mg/kg) or Se-high (1.0 mg/kg) diets showed decreased numbers of peripheral B cells (Cheng et al. 2012). Similar trends were seen in SeMet-treated mice where mice on low (0.02 ppm) and above-adequate (2 ppm) diets had reduced B cell numbers in the spleen (Vega et al. 2007). On the contrary, a small human study where dietary Se intake showed increased B lymphocyte numbers in a time-dependent manner. B cells in the Se supplemented group were only increased at day 45, but returned to levels as in Se low group at day 100 (Hawkes et al. 2001). A clinical trial showed that subjects consuming Se at 50 or 100 μg/day for 15 weeks had an improved anti-viral immunity; however, titration of antibodies against poliovirus was unchanged by Se intake (Broome et al. 2004). In contrast, other research also identified enhanced antibody response against diphtheria vaccine upon Se supplementation (Hawkes et al. 2001). It is unclear whether Se regulates antibody formation depending on the type of infection. More recently, researchers found that splenic B cells from Sep15−/− mice synthesized increased IgM in vitro upon LPS stimulation, although in vivo and in vitro clearance of bacterial infection was not changed (Yim et al. 2018). Still, limited information regarding the role of Se in B cells warrants further studies.
B. Immune responses
1. Bacterial infection
Se deficiency is often associated with bacterial infections. For instance, Se status is implicated in the progression of tuberculosis in patients infected with Mycobacterium tuberculosis (Grobler et al. 2016). Particularly, patients with pulmonary tuberculosis showed decreased Se levels compared to normal subjects (Ramakrishnan et al. 2012). Dietary supplementation with Se was reported to reduce the mortality in tuberculosis patients concomitantly infected with human immunodeficiency virus (HIV) (Range et al. 2006). Another study also found that Se supplementation could attenuate oxidative stress in tuberculosis patients receiving chemotherapy, which might be helpful for protecting host cells from excess ROS (Seyedrezazadeh et al. 2008). Other than Mycobacterium tuberculosis, Se may also play a role in the defense against other bacterial infections such as Helicobacter pylori (Ustundag et al. 2001), Dichelobacter nodosus (Hall et al. 2013), and Listeria monocytogenes (Wang C et al. 2009).
Interestingly, in a mouse model of Se and vitamin E deficiency, mice infected with Citrobacter rodentium showed significantly higher bacterial burden in mice maintained on diets devoid of Se and vitamin E than mice on nutritionally adequate diets. Se- and vitamin E-deficient mice also had upregulated expression of pro-inflammatory cytokines such as Il-6, Il-17, Il-22, Tnf-α, and Ifn-γ in addition to decreased expression of Gpx1 (Smith AD et al. 2011). Although it is possible that the anti-bacterial effect is exerted by a combination of Se and vitamin E, the alteration of Gpx1 suggests the beneficial role of Se, which is currently being investigated in our laboratory and will be reported in the near future.
2. Viral infection
Se levels are closely related to viral infections, as exemplified by Keshan disease, which is caused by the deficiency of dietary Se (Chen et al. 1980). Coxsackie B viruses (CVB), as a preeminent etiology of human acute myocarditis and a common cause of dilated cardiomyopathy are present in cardiomyocytes in patients with Keshan disease (Levander and Beck 1997). Supplementation of dietary Se eliminated the infection of CVB and effectively treated Keshan disease (Cermelli et al. 2002). Gpx1 knockout mice infected with CVB3 developed a disease resembling Keshan disease, whereas mice expressing normal Gpx1= could not be induced to develop any myocarditis by CVB3 infection (Beck et al. 1998). The protective effect of Gpx1 was speculated to modulate the viral genome via the incorporation of Se into viral GPX and that the viral GPX could lead to the transformation of a pathogenic phenotype to a mutant nonpathogenic phenotype (Zhang W et al. 1999). Another mitochondrial selenoprotein, Txnrd2, was suggested to play an important role in the development of Keshan disease. Cardiac specific ablation of Txnrd2 expression led to a dysfunction in the heart that was reminiscent of the cardiac abnormality in Keshan disease (Hara et al. 2001). As an antioxidant factor expressed in mitochondria, Txnrd2 is responsible for scavenging excess ROS (Chen C et al. 2017). Thus, disturbance of mitochondrial function due to the lack of Txnrd2 led to exacerbated oxidative stress, which may cause damage to heart (Rohrbach et al. 2006; Stanley et al. 2011). Whether Txnrd2 protects myocardium from CVB infection and if CVB infection affects the expression of Txnrd2 are important questions that should be further studied. In addition to CVB, HIV/AIDS is another disease where a strong inverse association with Se has been established in preclinical and clinical studies.
AIDS is caused by the infection of HIV and characterized by a progressive disease latency accompanied by gradual loss of CD4+ T cells. Previous findings have associated Se deficiency with accelerated disease progression (Bologna et al. 1994; Baum and Shor-Posner 1997; Campa et al. 1999). For example, HIV-1-seropositive subjects were reported to present low levels of zinc, Se, vitamin A, and vitamin B12. However, Se was the only predictor significantly associated with mortality. Se-deficient and Se-adequate participants had an overall survival of 31.4 months and 57.4 months, respectively (Baum 2000). As in Keshan disease, manifestations of cardiomyopathy were also reported in HIV/AIDS patients (Kavanaugh-McHugh et al. 1991). Interestingly, Se supplementation improved the general health of these patients, including cardiac function (Kavanaugh-McHugh et al. 1991). These findings suggest that Se supplementation may be a candidate for adjunct therapy in HIV/AIDS.
Clinical trials were carried out to identify the potential effects of Se supplementation on HIV disease progression. An early study showed that patients taking 100 μg/day of Se for one year had better outcomes in terms of decreased hospitalizations (Burbano et al. 2002). In another cohort that analyzed 450 HIV-1+ patients taking Se yeast 200 μg/day for 9 months, Se was found to reduce the viral burden and improve CD4+ cell counts (Hurwitz et al. 2007). However, in a randomized double-blinded trial of supplementation with different nutrient combinations (multivitamin, Se, multivitamin + Se, placebo), a significant improvement of CD4+ cell counts was only observed in multivitamin + Se group (Baum et al. 2013). Still, these data support a beneficial role of Se supplementation in HIV spectrum disease with a caveat of its coadministration in combination with other anti-viral drugs. In-vitro studies by Kalantari et al (2008) showed Se supplementation in the form of selenite (100 nM) reduced pro-viral transcription in human macrophages. This effect was dependent, in part, on the redox modulation of HIV-1 Tat (transactivating protein) by Txnrd1. Furthermore, the reduction of disulfides in Tat by Txnrd1 appeared to increase the covalent interaction of electrophiles, such as 15d-PGJ2, with Cys-SH group in Tat thus limiting the reoxidation of Cys-SH as well as ensuring sustained inhibition of Tat-dependent transcription activity (Kalantari et al. 2008; Kalantari et al. 2009). These encouraging preclinical studies provide a strong rationale to revisit the role of Se in HIV/AIDS with an emphasis on new mechanisms in patient derived cells harboring the virus and if NSAIDs interfere in the biological activity of Se.
1. Parasitic infection
The role of Se and/or selenoproteins in parasitic infections have also been examined. Compelling data have shown that antioxidant-dependent properties of Se may induce the resistance to parasitic infection by Trypanosoma (Smith A et al. 2005). In a mouse model of Trypanosoma cruzi infection, 3 ppm of dietary Se alleviated intestinal megasyndromes (de Souza et al. 2010). In addition, Se deficiency was also linked to megasyndromes in clinical patients (Jelicks et al. 2011). Trypanosoma cruzi infection causes Chagas disease, a progressive cardiomyopathy. Literature is replete with studies demonstrating low Se levels are associated with the increased severity of Chagas disease (de Souza et al. 2002; Gomez et al. 2002; Rivera et al. 2002). Furthermore, supplementation of Se from 2 ppm to 16 ppm was shown to play a beneficial role in the resolution of Chagas disease in mice (Davis et al. 1998; de Souza et al. 2003; de Souza et al. 2010; Souza et al. 2010). It is well-established that oxidative burst contributes to disease progression in Chagasic patients (Wen et al. 2006; de Oliveira et al. 2007) and such an excess of oxidative stress is associated with decline in mitochondrial functional (Wen et al. 2006). High doses of Se exhibited a preventive effect of mitochondrial dysfunction in Chagas disease (Tirosh and Reifen 2007; Pineyro et al. 2008).
Previously, our laboratory reported the role of Se and selenoproteins in a helminthic parasitic infection caused by Nippostrongylus brasiliensis (Nelson et al. 2016). Mice on Se-adequate diet (0.08 ppm) showed a significantly enhanced adult worm clearance compared to those on a Se-deficient diet (<0.01 ppm). This was associated with a phenotypic switching of macrophages from a pro-inflammatory M1 towards an anti-inflammatory M2 phenotype in the jejunum. Furthermore, use of Trspfl/flCreLysM mice that lack selenoproteins in monocytes and macrophages indicated that despite the presence of a higher level of Se in the diet (Se-supplemented: 0.4 ppm), there were more adult worms in Trspfl/flCreLysM mice compared to the infected WT control mice (Nelson et al. 2016). This implies that incorporation of Se into selenoproteins is indispensable for the parasite clearance activities of macrophages. As mentioned above, phenotypic switching of macrophages was accompanied by “eicosanoid class switching” favoring the production of CyPGs (Fig. 4). Use of indomethacin, a COX inhibitor, clearly showed the interaction between Se and eicosanoids during infection. Interestingly, inhibition of COX activity worsened the outcome of infection in these mice. However, treatment of mice with 15d-PGJ2 protected the mice that lacked endogenous eicosanoids following indomethacin treatment. Thus, Se-dependent CyPG production and the consequent induction of M2 macrophages was critical in the mediation of the anti-parasitic function of Se (Nelson et al. 2016). In addition to innate immune responses, it appears that adaptive immune mechanisms mediated by T effector cells are also involved in the elimination of Nippostrongylus brasiliensis mediated by Se. Enhanced production of Th2 signature cytokines (e.g., IL-13) and increased amounts of IL-4 producing T cells indicated that Th2 immune response might also play a role here (Nelson et al. 2016). However, further studies are required to explain how selenoproteins regulate the Th2 immune response during such infections.
2. Cancer immunity
The interaction between cancer cells and the immune system is well recognized. The origin of this interaction stems from the “cancer immunoediting” theory, which divides the process of tumor growth into three phases: elimination, equilibrium, and escape (Mittal et al. 2014). Immune cells recognize and try to destroy initial tumor cells in the elimination phase; however, if such an anti-tumor activity fails, tumor cells and immune system tend to establish an equilibrium where tumor cells gain more mutations to aid in their survival. In the escape phase, tumor cells escape the immune surveillance and avoid being targeted by the immune system.
CD8+ cytotoxic T cells are thus far considered as a key element of anti-tumor immunity. These cells attack tumor cells presenting peptide:MHC I complex and induce their apoptosis. Studies showed that Se supplementation might enhance the proliferation and differentiation of cytotoxic T cells (Petrie et al. 1989). This effect was likely mediated by the increased levels of IL-2 and IFN-γ in tumor microenvironment (TME) (Farrar et al. 1981; Kern et al. 1981; Knuth et al. 1984; Roy et al. 1993). Besides, the presence of Th1 cells was also correlated with a better prognosis in tumors (Haabeth et al. 2011; Fridman et al. 2012). As mentioned above, Se supplementation causes skewing of CD4+ T cells differentiation towards Th1 effector cells (Hoffmann FW et al. 2010; Mahdavi et al. 2017). Furthermore, under certain circumstances, Se showed positive anti-tumor effects through the induction of a Th1 immune response (Yazdi et al. 2012; Yazdi et al. 2015).
However, tumor cells also succeed in circumventing the killing by cytotoxic T cells by downregulating MHC I molecules (Leone et al. 2013). Such an adaptive change of MHC I can trigger the targeting by NK cells through “missing-self” recognition (Karre 2002). Interestingly, Se was reported to facilitate anti-tumor function of NK cells by enhancing IL-2/IL-2R interaction (Kiremidjian-Schumacher L. et al. 1996; Enqvist et al. 2011). Unfortunately, it is still unclear which selenoprotein(s) in NK cells regulate this function or whether cellular niche that responds to Se activates the NK cells to cause such an effect.
Abundant experimental evidence has identified tumor-associated macrophages (TAMs) as M2-like phenotype that supports tumor progression and metastasis (Sica and Mantovani 2012). Macrophage reprogramming from M2 phenotype to M1 phenotype contributes to enhancing the efficacy of cancer drugs and suppressing tumor growth (Larionova et al. 2019). Interestingly, numerous studies confirmed that Se could shift M2 to M1 in various cancers to effectively retard tumor progression (Kiremidjian-Schumacher L et al. 1991; Mao et al. 2016; Gautam et al. 2017). It is also believed that enhanced differentiation of Th1 cells is responsible to secrete polarizing cytokines for M1 macrophages (Barros et al. 2013). Compared to the macrophage phenotypic switching induced by Se supplementation mentioned above, these studies suggest that Se might affect macrophages in a tumor context differently from those in inflammatory scenarios leading to a broader question regarding the diversity of signals and/or cells within inflammatory networks.
Studies in our laboratory on the role of Se in myeloid leukemias have suggested the interaction of the immune system with cancer stem cells. Acute myeloid leukemia (AML) represents a malignant disorder that is characterized by infiltration of abnormally proliferated hematopoietic stem and progenitor cells in bone marrow, peripheral blood, and occasionally other tissues (Sabattini et al. 2010). As in many other hematologic malignancies, AML has a high relapse rate following current induction therapies, where initial remission rates after induction therapy decline from 90%, 70%, 60%, to 40% in pediatric, young, middle-aged, and older patients, respectively (Prchal and Levi 2010). Such a poor prognosis is due to the ability of proliferation and differentiation of leukemia stem cells (LSCs), where patients tend to relapse leading to minimal residual disease (MRD) (van Rhenen et al. 2005). Despite the poor prognosis, we have achieved exciting developments in the insight into the cytogenetic and molecular heterogeneity of AML, even though most of the novel therapies have failed to effectively bring about overall and long-term survival benefit.
In 1956, Se was reported to be effective to treat leukemia where leukemic patients (two AML and two CML) had improved leukocytosis and a decreased spleen size after administration of Se cystine (Weisberger and Suhrland 1956). Since then, various studies have demonstrated anti-leukemic effect of different forms of Se using in vitro and in vivo model systems (Batist et al. 1986; Li J et al. 2003; Philchenkov et al. 2007). Taking cues from our previous research that has indicated the ability of Se at supraphysiological levels to inhibit the viability of CML LSCs (Gandhi et al. 2014) involving the endogenous CyPGs as well as the eicosanoid class switching paradigms (Finch et al. 2017), we have embarked on targeting the self-renewing and drug resistant LSCs that are responsible for the post-remission relapse of AML to achieve the eradication of LSCs as our goal to treat AML using new paradigms. In support of the role of Se therapy in AML, several studies have reported significantly decreased serum levels of Se were observed in patients with AML, CML, acute lymphoblastic leukemia (ALL), and acute nonlymphocytic leukemia (ANLL) (Pazirandeh et al. 1999; Hadjibabaie et al. 2008; Khanna et al. 2010; Stevens et al. 2011). These observations imply an inverse causality between serum levels of Se and the progression of hematopoietic malignancies.
Despite this, further research is required to understand if Se supplementation is beneficial in leukemia patients given the role of selenoprotein, GPX4-induced ferroptosis in the context of leukemias. Ferroptosis is a unique type of iron-dependent programmed cell death driven by the accumulated lipid peroxides (Yang and Stockwell 2016). Blocking the activity of GPX4 or inhibiting GSH can induce ferroptosis (Yang et al. 2014). Besides, Se utilization by GPX4 is critical for the inactivation of GPX4 by ferroptosis inducers (Ingold et al. 2018). Ferroptosis inducers inactivate GPX4 by covalently modifying the active site selenocysteine (Yang et al. 2016). This distinct cell death involving the inhibition of GPX4 has been demonstrated to be a candidate mechanism for leukemia treatment. In ALL, inhibition of GPX4 by RSL3 triggered the production of ROS and led to ferroptosis. GPX4 inhibited LOXs, while inhibitors of LOXs reversed the cell death induced by GPX4 in ALL cells further lending credence to the idea that GPX4-regulation of LOXs prevents ferroptosis as a result to protect ALL cells from cell death (Probst et al. 2017). In AML, Erastin, a ferroptosis inducer, also induced ferroptosis in AML cell line, HL-60 cells and enhanced the sensitivity of these cells to chemotherapy (Yu et al. 2015). However, it will be interesting to see if a similar mechanism is functional in a primary cell system involving AML LSCs.
Compared to the anti-tumor effect of Th1 cells, Th2 cells are generally regarded as tumor-promoting cells (Fridman 2012; Fridman et al. 2012). However, in the context of hematologic cancers, this could challenge the existing paradigm. In high-risk ALL, IL-4, a signature cytokine for Th2 immunity, was able to induce apoptosis of leukemia cells (Manabe et al. 1994). Similar results were corroborated from an in vitro study on B cell ALL (Manabe et al. 1994). In children with M5-AML, IL-4 was reported to inhibit the proliferation of leukemia cells (McKenna et al. 1996). To gain the insight of the anti-tumor role of Th2 immune response in leukemias, further studies are essential. AML leukemia blasts-induced T cell anergy may be a potential explanation where the effect of IL-4 treatment may be less severe than a full-blown Th2-mediated immune response (Narita et al. 2001). The profound effect of Se on IL-4-induced M2 macrophages has been reported in our laboratory which may also reflect Se’s role in Th2-M2 immune response (Shay et al. 2017). More studies are required to address the role of Se and Th2 immunity in cancers, particularly hematologic malignancies.
3. Allergic asthma
Asthma is a common inflammatory disease affecting the respiratory system where both innate and adaptive immune mechanisms are involved. Its pathobiological mechanisms include AHR, excess mucus secretion, and reversible narrowing and obstruction of airways (Maddox and Schwartz 2002). Compared to nonallergic (idiopathic) asthma, allergic asthma is more often discussed in the context of immunology as the concomitant presence of humoral immune (high serum IgE) and cell-mediated immune (increased Th2 cells and related cytokines) responses (Grove et al. 1975).
Interestingly, oxidative stress has been identified as an important contributor to the pathogenesis of allergic asthma (Riedl and Nel 2008). Several epidemiological analyses provided the association between asthma and Se status. Literature is replete with studies that have shown low Se levels to be negatively associated with lung function and positively correlated with the severity of asthma (Omland et al. 2002; de Luis et al. 2003; Qujeq et al. 2003; Guo et al. 2011). Furthermore, serum Se levels as well as GPX expression were decreased in asthmatic adults (Guo et al. 2011) and children (Kocyigit et al. 2004). However, other studies have indicated no association between Se and asthma (Ford et al. 2004). In several studies, high Se levels and GPX activity were thought to exacerbate the condition of asthma leading to the notion of a “U” shaped relationship that remains to be clearly established (Dunstan et al. 2006; Garcia-Larsen et al. 2007). In addition, a few interventional studies were carried out by supplementing asthmatics with dietary Se; however, due to the differences of formulation, duration, dosage of Se used, it is impossible to derive any conclusions regarding the benefits of Se intake in these settings (Dunstan et al. 2007; Shaheen et al. 2007).
Studies with an allergic asthma model (with OVA challenge in OT-1 and OT-2 mice) suggested that mice fed 0.25 ppm Se diet had the more severe asthma than mice on diet with 2.7 ppm Se (Hoffmann PR et al. 2007). High Se (2.7 ppm) level was reported to mediate the skewing of Th0 cells towards Th1 and Treg differentiation away from Th2 differentiation (Hoffmann PR et al. 2007). Given that Th2 phenotype is a hallmark feature of asthma, this differentiational switching of Th cells is recognized as a major mechanism that provides some rationale to suggest the benefit of high Se in asthma. Furthermore, Gpx1 knockout mice failed to develop allergic asthma when challenged with OVA (Won et al. 2010); while disruption of Gpx2 exhibited a protective role in a murine asthma model (Dittrich et al. 2010). It is still unclear why these two GPXs have opposing effects in asthma. Future studies are needed in order to understand the functions of selenoproteins in asthma.
V. Summary
Optimal Se status is critically important for health/physiological homeostasis. It is very clear that perturbation of Se levels may result in severe consequences on human health. But the concept of dietary supplementation needs to be optimized taking into account the baseline levels of Se as defined by better biomarkers that are more informative than plasma or serum Se. Studies from many laboratories, including ours, have suggested a strong-link between Se, selenoproteins, and novel bioactive lipid mediators, in the form of CyPGs, which clearly impacts inflammation and pro-resolution pathways at various levels in a multicellular network consisting of various immune cells. However, time seems propitious to make a translational leap where this mechanism is put to its final test in well controlled studies. This is particularly true since many of the clinical trials with Se supplementation performed to date, including the SELECT for prostate cancer (Lippman et al. 2009; Klein et al. 2011), did not exclude the use of NSAIDs suggesting a possible interference in the ability of bioactive and proresolving lipid mediators to effect resolution of inflammation. Such studies will ultimately give us a better appreciation of the role of this unique trace element in pathology and therapy.
Acknowledgements
This work was supported, in part, by grants from the National Institutes of Health R01 DK077152, Office of Dietary Supplements, R01 CA162665, USDA-NIFA Hatch project number 4605; Accession # 1010021, American Institute for Cancer Research (K.S.P). We thank Shaneice Nettleford and Tai-Jung Lee for their timely help and suggestions.
Abbreviations used:
- AHR
airway hyperresponsiveness
- AIDS
acquired immunodeficiency syndrome
- AKR1B1
Aldo-keto reductase family 1, member B1
- ALL
acute lymphoblastic leukemia
- AML
acute myeloid leukemia
- ANLL
acute nonlymphocytic leukemia
- APC
antigen-presenting cell
- ARA
arachidonic acid
- BMDM
bone marrow-derived macrophage
- BMEC
bovine mammary endothelial cell
- cAMP
3’, 5’-cyclic adenosine monophosphate
- CAR
chimeric antigen receptor
- C/EBP
CCAAT-enhancer-binding protein
- CML
chronic myeloid leukemia
- CNS
central neuron system
- COX
cyclooxygenase
- cPGES
cytosolic PGES
- CRE
cAMP response element
- Crth2
chemoattractant receptor-homologous molecule expressed on Th2 cells
- CVB
coxsackie B virus
- CyPG
cyclopentenone PG
- Cys
cysteine
- DC
dendritic cell
- DHA
docosahexaenoic acid
- DP2
PGD2 receptor 2
- 15d-PGJ2
15-deoxy-Δ12,14-prostaglandin J2
- DSS
dextran sodium sulfate
- EFSec
Sec-specific elongation factor
- EPA
eicosapentaenoic acid
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- Erk
extracellular-signal-regulated kinase
- GI
gastrointestinal
- GPCR
G-protein coupled receptor
- GPX
glutathione peroxidase
- GSH
glutathione
- GST
glutathione S-transferase
- HBV
Hepatitis B virus
- HDM
house dust mite
- HIV
human immunodeficiency virus
- H2O2
hydrogen peroxide
- 15-HPETE
hydroperoxyeicosatetraenoic acid
- H-PGDS
hematopoietic PGDS
- HSe−
selenide
- H2SePO3−
selenophosphate
- IBD
inflammatory bowel disease
- IFN
interferon
- Ig
immunoglobulin
- IKK
IκB kinase
- IL
interleukin
- ILC
innate lymphoid cell
- IP3
inositol trisphosphate
- IRAK
IL-1 receptor-activated kinase
- JNK
c-Jun N-terminal kinase
- KBD
Kashin–Beck disease
- LOX
lipoxygenase
- L-PGDS
lipocalin-type PGDS
- LPS
lipopolysaccharide
- LSC
leukemia stem cell
- mAb
monoclonal antibody
- MAIT
mucosal associated invariant T cell
- MAPK
mitogen-activated protein kinase
- MDSC
myeloid derived suppressor cell
- MHC
major histocompatibility complex
- mPGES
microsomal PGES
- MRD
minimal residual disease
- MsrB1
methionine sulfoxide reductase B1
- MyD88
myeloid differentiation factor 88
- NAD
nicotinamide adenine dinucleotide
- Na2SeO3
sodium selenite
- NF
nuclear factor
- NK
Natural killer
- NOS
nitric oxide synthase
- NOX
NADPH oxidase
- NF-E2
nuclear factor erythroid 2
- Nrf2
NF-E2-related factor 2
- NSAIDs
non-steroidal anti-inflammatory drugs
- O
oxygen
- OA
osteoarthritis
- OVA
ovalbumin
- PAH
pulmonary arterial hypertension
- PAMP
pathogen-associated molecular pattern
- PBMC
peripheral blood mononuclear cell
- PD-1
programmed death 1
- PG
prostaglandin
- 15-PGDH
15-hydroxyprostaglandin dehydrogenase
- PG
prostaglandin
- PGDS
PGD synthase
- PGES
PGE synthase
- PGI2
prostacyclin
- PGIS
PGI synthase
- PHGPX
phospholipid hydroperoxide GPX
- PIH
pregnancy-induced hypertension
- PI3K
phosphoinositide 3-kinase
- PLA2
phospholipase A2
- PMN
polymorphonuclear
- Po
polonium
- PPARγ
peroxisome proliferator-activated receptor gamma
- PPRE
PPAR-response element
- PSTK
phosphoseryl-tRNA kinase
- Ptgs
prostaglandin-endoperoxide synthase
- PUFA
polyunsaturated fatty acid
- RA
rheumatoid arthritis
- ROS
reactive oxygen species
- RvE1
resolvin E1
- S
sulfur
- SBP2
SECIS binding protein 2
- Se
selenium
- Sec
selenocysteine
- SECIS
Sec insertion sequence
- SELECT
Selenium and Vitamin E Cancer Prevention
- SelenoK
selenoprotein K
- SelenoP
selenoprotein P
- SelenoS
selenoprotein S
- SeMet
selenomethionine
- SEPP1
selenoprotein P
- SerS
seryl-tRNA synthase
- SPM
specialized proresolving mediator
- SPS2
selenophosphate sythetase-2
- STAT6
Signal Transducer And Activator Of Transcription 6
- TAB
TAK1-binding protein
- TAK1
TGFβ-activated kinase 1
- TCR
T cell receptor
- Te
tellurium
- TFH
follicular T helper
- TGFβ
transforming growth factor β
- Th
T helper
- TLR4
toll-like-receptor 4
- TME
tumor microenvironment
- TNF
tumor necrosis factor
- TRAF6
TNF receptor-associated factor 6
- Treg
regulatory T cell
- Trsp
Sec-tRNA[Ser]Sec gene
- TX
thromboxane
- Txnrd1
thioredoxin reductase-1
- TXS
TX synthase
- 3’-UTR
3’-untranslated region
- WT
wild type
References
- Ajibade AA, Wang HY, Wang RF. 2013. Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol. 34(7):307–316. [DOI] [PubMed] [Google Scholar]
- Andoh A, Hirashima M, Maeda H, Hata K, Inatomi O, Tsujikawa T, Sasaki M, Takahashi K, Fujiyama Y. 2005. Serum selenoprotein-P levels in patients with inflammatory bowel disease. Nutrition. 21(5):574–579. [DOI] [PubMed] [Google Scholar]
- Arnaud J, Bost M, Vitoux D, Labarere J, Galan P, Faure H, Hercberg S, Bordet JC, Roussel AM, Chappuis P et al. 2007. Effect of low dose antioxidant vitamin and trace element supplementation on the urinary concentrations of thromboxane and prostacyclin metabolites. J Am Coll Nutr. 26(5):405–411. [DOI] [PubMed] [Google Scholar]
- Arner ES. 2010. Selenoproteins-What unique properties can arise with selenocysteine in place of cysteine? Exp Cell Res. 316(8):1296–1303. [DOI] [PubMed] [Google Scholar]
- Arruda MA, Barja-Fidalgo C. 2009. NADPH oxidase activity: In the crossroad of neutrophil life and death. Front Biosci (Landmark Ed). 14:4546–4556. [DOI] [PubMed] [Google Scholar]
- Babior BM, Lambeth JD, Nauseef W. 2002. The neutrophil NADPH oxidase. Arch Biochem Biophys. 397(2):342–344. [DOI] [PubMed] [Google Scholar]
- Back M, Dahlen SE, Drazen JM, Evans JF, Serhan CN, Shimizu T, Yokomizo T, Rovati GE. 2011. International Union of Basic and Clinical Pharmacology. LXXXIV: leukotriene receptor nomenclature, distribution, and pathophysiological functions. Pharmacol Rev. 63(3):539–584. [DOI] [PubMed] [Google Scholar]
- Back M, Powell WS, Dahlen SE, Drazen JM, Evans JF, Serhan CN, Shimizu T, Yokomizo T, Rovati GE. 2014. Update on leukotriene, lipoxin and oxoeicosanoid receptors: IUPHAR Review 7. Br J Pharmacol. 171(15):3551–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bage T, Lindberg J, Lundeberg J, Modeer T, Yucel-Lindberg T. 2010. Signal pathways JNK and NF-kappaB, identified by global gene expression profiling, are involved in regulation of TNFalpha-induced mPGES-1 and COX-2 expression in gingival fibroblasts. BMC Genomics. 11:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banning A, Florian S, Deubel S, Thalmann S, Muller-Schmehl K, Jacobasch G, Brigelius-Flohe R. 2008. GPx2 counteracts PGE2 production by dampening COX-2 and mPGES-1 expression in human colon cancer cells. Antioxid Redox Signal. 10(9):1491–1500. [DOI] [PubMed] [Google Scholar]
- Barrett CW, Ning W, Chen X, Smith JJ, Washington MK, Hill KE, Coburn LA, Peek RM, Chaturvedi R, Wilson KT et al. 2013. Tumor suppressor function of the plasma glutathione peroxidase gpx3 in colitis-associated carcinoma. Cancer Res. 73(3):1245–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros MH, Hauck F, Dreyer JH, Kempkes B, Niedobitek G. 2013. Macrophage polarisation: an immunohistochemical approach for identifying M1 and M2 macrophages. PLoS One. 8(11):e80908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batist G, Katki AG, Klecker RW Jr., Myers CE. 1986. Selenium-induced cytotoxicity of human leukemia cells: interaction with reduced glutathione. Cancer Res. 46(11):5482–5485. [PubMed] [Google Scholar]
- Baum MK. 2000. Role of micronutrients in HIV-infected intravenous drug users. J Acquir Immune Defic Syndr. 25 Suppl 1:S49–52. [DOI] [PubMed] [Google Scholar]
- Baum MK, Campa A, Lai S, Sales Martinez S, Tsalaile L, Burns P, Farahani M, Li Y, van Widenfelt E, Page JB et al. 2013. Effect of micronutrient supplementation on disease progression in asymptomatic, antiretroviral-naive, HIV-infected adults in Botswana: a randomized clinical trial. JAMA. 310(20):2154–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum MK, Shor-Posner G. 1997. Nutritional status and survival in HIV-1 disease. AIDS. 11(5):689–690. [PubMed] [Google Scholar]
- Beck MA, Esworthy RS, Ho YS, Chu FF. 1998. Glutathione peroxidase protects mice from viral-induced myocarditis. FASEB J. 12(12):1143–1149. [DOI] [PubMed] [Google Scholar]
- Berry MJ, Banu L, Chen YY, Mandel SJ, Kieffer JD, Harney JW, Larsen PR. 1991. Recognition of UGA as a selenocysteine codon in type I deiodinase requires sequences in the 3’ untranslated region. Nature. 353(6341):273–276. [DOI] [PubMed] [Google Scholar]
- Bilski J, Mazur-Bialy A, Wojcik D, Magierowski M, Surmiak M, Kwiecien S, Magierowska K, Hubalewska-Mazgaj M, Sliwowski Z, Brzozowski T. 2019. Effect of Forced Physical Activity on the Severity of Experimental Colitis in Normal Weight and Obese Mice. Involvement of Oxidative Stress and Proinflammatory Biomarkers. Nutrients. 11(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bologna R, Indacochea F, Shor-Posner G, Mantero-Atienza E, Grazziutti M, Sotomayor M-C, Fletcher M, Cabrejos C, Scott GB, Baum MK. 1994. Selenium and immunity in HIV-1 infected pediatric patients. Journal of Nutritional immunology. 3(1):41–49. [Google Scholar]
- Bosl MR, Seldin MF, Nishimura S, Taketo M. 1995. Cloning, structural analysis and mapping of the mouse selenocysteine tRNA([Ser]Sec) gene (Trsp). Mol Gen Genet. 248(3):247–252. [DOI] [PubMed] [Google Scholar]
- Bosschaerts T, Guilliams M, Noel W, Herin M, Burk RF, Hill KE, Brys L, Raes G, Ghassabeh GH, De Baetselier P et al. 2008. Alternatively activated myeloid cells limit pathogenicity associated with African trypanosomiasis through the IL-10 inducible gene selenoprotein P. J Immunol. 180(9):6168–6175. [DOI] [PubMed] [Google Scholar]
- Boyne R, Arthur JR. 1979. Alterations of neutrophil function in selenium-deficient cattle. J Comp Pathol. 89(1):151–158. [DOI] [PubMed] [Google Scholar]
- Brigelius-Flohe R, Kipp AP. 2012. Physiological functions of GPx2 and its role in inflammation-triggered carcinogenesis. Ann N Y Acad Sci. 1259:19–25. [DOI] [PubMed] [Google Scholar]
- Brock TG, Peters-Golden M. 2007. Activation and regulation of cellular eicosanoid biosynthesis. ScientificWorldJournal. 7:1273–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broome CS, McArdle F, Kyle JA, Andrews F, Lowe NM, Hart CA, Arthur JR, Jackson MJ. 2004. An increase in selenium intake improves immune function and poliovirus handling in adults with marginal selenium status. Am J Clin Nutr. 80(1):154–162. [DOI] [PubMed] [Google Scholar]
- Brown KM, Pickard K, Nicol F, Beckett GJ, Duthie GG, Arthur JR. 2000. Effects of organic and inorganic selenium supplementation on selenoenzyme activity in blood lymphocytes, granulocytes, platelets and erythrocytes. Clin Sci (Lond). 98(5):593–599. [PubMed] [Google Scholar]
- Burbano X, Miguez-Burbano MJ, McCollister K, Zhang G, Rodriguez A, Ruiz P, Lecusay R, Shor-Posner G. 2002. Impact of a selenium chemoprevention clinical trial on hospital admissions of HIV-infected participants. HIV Clin Trials. 3(6):483–491. [DOI] [PubMed] [Google Scholar]
- Campa A, Shor-Posner G, Indacochea F, Zhang G, Lai H, Asthana D, Scott GB, Baum MK. 1999. Mortality risk in selenium-deficient HIV-positive children. J Acquir Immune Defic Syndr Hum Retrovirol. 20(5):508–513. [DOI] [PubMed] [Google Scholar]
- Cao YZ, Reddy CC, Sordillo LM. 2000. Altered eicosanoid biosynthesis in selenium-deficient endothelial cells. Free Radic Biol Med. 28(3):381–389. [DOI] [PubMed] [Google Scholar]
- Carlson BA, Yoo MH, Sano Y, Sengupta A, Kim JY, Irons R, Gladyshev VN, Hatfield DL, Park JM. 2009. Selenoproteins regulate macrophage invasiveness and extracellular matrix-related gene expression. BMC Immunol. 10:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cermelli C, Vinceti M, Scaltriti E, Bazzani E, Beretti F, Vivoli G, Portolani M. 2002. Selenite inhibition of Coxsackie virus B5 replication: implications on the etiology of Keshan disease. J Trace Elem Med Biol. 16(1):41–46. [DOI] [PubMed] [Google Scholar]
- Chambers I, Frampton J, Goldfarb P, Affara N, McBain W, Harrison PR. 1986. The structure of the mouse glutathione peroxidase gene: the selenocysteine in the active site is encoded by the ‘termination’ codon, TGA. EMBO J. 5(6):1221–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Chen H, Zhou HJ, Ji W, Min W. 2017. Mechanistic Role of Thioredoxin 2 in Heart Failure. Adv Exp Med Biol. 982:265–276. [DOI] [PubMed] [Google Scholar]
- Chen J, Pan T, Wan N, Sun Z, Zhang Z, Li S. 2017. Cadmium-induced endoplasmic reticulum stress in chicken neutrophils is alleviated by selenium. J Inorg Biochem. 170:169–177. [DOI] [PubMed] [Google Scholar]
- Chen X, Yang G, Chen J, Chen X, Wen Z, Ge K. 1980. Studies on the relations of selenium and Keshan disease. Biol Trace Elem Res. 2(2):91–107. [DOI] [PubMed] [Google Scholar]
- Cheng WH, Holmstrom A, Li X, Wu RT, Zeng H, Xiao Z. 2012. Effect of dietary selenium and cancer cell xenograft on peripheral T and B lymphocytes in adult nude mice. Biol Trace Elem Res. 146(2):230–235. [DOI] [PubMed] [Google Scholar]
- Chew P, Yuen DY, Koh P, Stefanovic N, Febbraio MA, Kola I, Cooper ME, de Haan JB. 2009. Site-specific antiatherogenic effect of the antioxidant ebselen in the diabetic apolipoprotein E-deficient mouse. Arterioscler Thromb Vasc Biol. 29(6):823–830. [DOI] [PubMed] [Google Scholar]
- Chow A, Huggins M, Ahmed J, Hashimoto D, Lucas D, Kunisaki Y, Pinho S, Leboeuf M, Noizat C, van Rooijen N et al. 2013. CD169(+) macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat Med. 19(4):429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu LY, Liou JY, Wu KK. 2015. Prostacyclin protects vascular integrity via PPAR/14–3-3 pathway. Prostaglandins Other Lipid Mediat. 118–119:19–27. [DOI] [PubMed] [Google Scholar]
- Conrad M, Jakupoglu C, Moreno SG, Lippl S, Banjac A, Schneider M, Beck H, Hatzopoulos AK, Just U, Sinowatz F et al. 2004. Essential role for mitochondrial thioredoxin reductase in hematopoiesis, heart development, and heart function. Mol Cell Biol. 24(21):9414–9423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland PR, Fletcher JE, Carlson BA, Hatfield DL, Driscoll DM. 2000. A novel RNA binding protein, SBP2, is required for the translation of mammalian selenoprotein mRNAs. EMBO J. 19(2):306–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CD, Brooks L, Calisi C, Bennett BJ, McElroy DM. 1998. Beneficial effect of selenium supplementation during murine infection with Trypanosoma cruzi. J Parasitol. 84(6):1274–1277. [PubMed] [Google Scholar]
- de Luis DA, Izaola O, Aller R, Armentia A, Cuellar L. 2003. [Antioxidant and fat intake in patients with polinic asthma]. Med Clin (Barc). 121(17):653–654. [PubMed] [Google Scholar]
- de Oliveira TB, Pedrosa RC, Filho DW. 2007. Oxidative stress in chronic cardiopathy associated with Chagas disease. Int J Cardiol. 116(3):357–363. [DOI] [PubMed] [Google Scholar]
- de Souza AP, de Oliveira GM, Vanderpas J, de Castro SL, Rivera MT, Araujo-Jorge TC. 2003. Selenium supplementation at low doses contributes to the decrease in heart damage in experimental Trypanosoma cruzi infection. Parasitol Res. 91(1):51–54. [DOI] [PubMed] [Google Scholar]
- de Souza AP, Melo de Oliveira G, Neve J, Vanderpas J, Pirmez C, de Castro SL, Araujo-Jorge TC, Rivera MT. 2002. Trypanosoma cruzi: host selenium deficiency leads to higher mortality but similar parasitemia in mice. Exp Parasitol. 101(4):193–199. [DOI] [PubMed] [Google Scholar]
- de Souza AP, Sieberg R, Li H, Cahill HR, Zhao D, Araujo-Jorge TC, Tanowitz HB, Jelicks LA. 2010. The role of selenium in intestinal motility and morphology in a murine model of Typanosoma cruzi infection. Parasitol Res. 106(6):1293–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis EA, Cao J, Hsu YH, Magrioti V, Kokotos G. 2011. Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem Rev. 111(10):6130–6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond A, Dudock B, Hatfield D. 1981. Structure and properties of a bovine liver UGA suppressor serine tRNA with a tryptophan anticodon. Cell. 25(2):497–506. [DOI] [PubMed] [Google Scholar]
- Dittrich AM, Meyer HA, Krokowski M, Quarcoo D, Ahrens B, Kube SM, Witzenrath M, Esworthy RS, Chu FF, Hamelmann E. 2010. Glutathione peroxidase-2 protects from allergen-induced airway inflammation in mice. Eur Respir J. 35(5):1148–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diwakar BTFER, Liao C, Shay AE, Prabhu KS . 2016. The Role of Selenoproteins in Resolution of Inflammation. . 4th ed. Cham: Springer; (Selenium. [Google Scholar]
- Diwakar BT, Yoast R, Nettleford S, Qian F, Lee TJ, Berry S, Huffnagle I, Rossi RM, Trebak M, Paulson RF et al. 2019. Crth2 receptor signaling down-regulates lipopolysaccharide-induced NF-kappaB activation in murine macrophages via changes in intracellular calcium. FASEB J. 33(11):12838–12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS et al. 2012. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 149(5):1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogne JM, de Leval X, Benoit P, Rolin S, Pirotte B, Masereel B. 2002. Therapeutic potential of thromboxane inhibitors in asthma. Expert Opin Investig Drugs. 11(2):275–281. [DOI] [PubMed] [Google Scholar]
- Dogne JM, Hanson J, Supuran C, Pratico D. 2006. Coxibs and cardiovascular side-effects: from light to shadow. Curr Pharm Des. 12(8):971–975. [DOI] [PubMed] [Google Scholar]
- Doroudi R, Gan LM, Selin Sjogren L, Jern S. 2000. Effects of shear stress on eicosanoid gene expression and metabolite production in vascular endothelium as studied in a novel biomechanical perfusion model. Biochem Biophys Res Commun. 269(1):257–264. [DOI] [PubMed] [Google Scholar]
- Du XH, Dai XX, Xia Song R, Zou XZ, Yan Sun W, Mo XY, Lu Bai G, Xiong YM. 2012. SNP and mRNA expression for glutathione peroxidase 4 in Kashin-Beck disease. Br J Nutr. 107(2):164–169. [DOI] [PubMed] [Google Scholar]
- Dunstan JA, Breckler L, Hale J, Lehmann H, Franklin P, Lyons G, Ching SY, Mori TA, Barden A, Prescott SL. 2006. Associations between antioxidant status, markers of oxidative stress and immune responses in allergic adults. Clin Exp Allergy. 36(8):993–1000. [DOI] [PubMed] [Google Scholar]
- Dunstan JA, Breckler L, Hale J, Lehmann H, Franklin P, Lyons G, Ching SY, Mori TA, Barden A, Prescott SL. 2007. Supplementation with vitamins C, E, beta-carotene and selenium has no effect on anti-oxidant status and immune responses in allergic adults: a randomized controlled trial. Clin Exp Allergy. 37(2):180–187. [DOI] [PubMed] [Google Scholar]
- Emmink BL, Laoukili J, Kipp AP, Koster J, Govaert KM, Fatrai S, Verheem A, Steller EJ, Brigelius-Flohe R, Jimenez CR et al. 2014. GPx2 suppression of H2O2 stress links the formation of differentiated tumor mass to metastatic capacity in colorectal cancer. Cancer Res. 74(22):6717–6730. [DOI] [PubMed] [Google Scholar]
- Enqvist M, Nilsonne G, Hammarfjord O, Wallin RP, Bjorkstrom NK, Bjornstedt M, Hjerpe A, Ljunggren HG, Dobra K, Malmberg KJ et al. 2011. Selenite induces posttranscriptional blockade of HLA-E expression and sensitizes tumor cells to CD94/NKG2A-positive NK cells. J Immunol. 187(7):3546–3554. [DOI] [PubMed] [Google Scholar]
- Ensor CM, Tai HH. 1995. 15-Hydroxyprostaglandin dehydrogenase. J Lipid Mediat Cell Signal. 12(2–3):313–319. [DOI] [PubMed] [Google Scholar]
- Farrar WL, Johnson HM, Farrar JJ. 1981. Regulation of the production of immune interferon and cytotoxic T lymphocytes by interleukin 2. J Immunol. 126(3):1120–1125. [PubMed] [Google Scholar]
- Fedyk ER, Brown DM, Phipps RP. 1997. PGE2 regulation of B lymphocytes and T helper 1 and T helper 2 cells: induction of inflammatory versus allergic responses. Adv Exp Med Biol. 407:237–242. [DOI] [PubMed] [Google Scholar]
- Fedyk ER, Phipps RP. 1996. Prostaglandin E2 receptors of the EP2 and EP4 subtypes regulate activation and differentiation of mouse B lymphocytes to IgE-secreting cells. Proc Natl Acad Sci U S A. 93(20):10978–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Xia Y, Garcia GE, Hwang D, Wilson CB. 1995. Involvement of reactive oxygen intermediates in cyclooxygenase-2 expression induced by interleukin-1, tumor necrosis factor-alpha, and lipopolysaccharide. J Clin Invest. 95(4):1669–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson AD, Labunskyy VM, Fomenko DE, Arac D, Chelliah Y, Amezcua CA, Rizo J, Gladyshev VN, Deisenhofer J. 2006. NMR structures of the selenoproteins Sep15 and SelM reveal redox activity of a new thioredoxin-like family. J Biol Chem. 281(6):3536–3543. [DOI] [PubMed] [Google Scholar]
- Finch ER, Tukaramrao DB, Goodfield LL, Quickel MD, Paulson RF, Prabhu KS. 2017. Activation of PPARgamma by endogenous prostaglandin J2 mediates the antileukemic effect of selenium in murine leukemia. Blood. 129(13):1802–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald DJ, Rocki W, Murray R, Mayo G, FitzGerald GA. 1990. Thromboxane A2 synthesis in pregnancy-induced hypertension. Lancet. 335(8692):751–754. [DOI] [PubMed] [Google Scholar]
- Ford ES, Mannino DM, Redd SC. 2004. Serum antioxidant concentrations among U.S. adults with self-reported asthma. J Asthma. 41(2):179–187. [DOI] [PubMed] [Google Scholar]
- Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 1995. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 83(5):803–812. [DOI] [PubMed] [Google Scholar]
- Fredericks GJ, Hoffmann FW, Rose AH, Osterheld HJ, Hess FM, Mercier F, Hoffmann PR. 2014. Stable expression and function of the inositol 1,4,5-triphosphate receptor requires palmitoylation by a DHHC6/selenoprotein K complex. Proc Natl Acad Sci U S A. 111(46):16478–16483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredman G, Li Y, Dalli J, Chiang N, Serhan CN. 2012. Self-limited versus delayed resolution of acute inflammation: temporal regulation of pro-resolving mediators and microRNA. Sci Rep. 2:639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman WH. 2012. The immune microenvironment as a guide for cancer therapies. Oncoimmunology. 1(3):261–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman WH, Pages F, Sautes-Fridman C, Galon J. 2012. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 12(4):298–306. [DOI] [PubMed] [Google Scholar]
- Galasso G, Schiekofer S, Sato K, Shibata R, Handy DE, Ouchi N, Leopold JA, Loscalzo J, Walsh K. 2006. Impaired angiogenesis in glutathione peroxidase-1-deficient mice is associated with endothelial progenitor cell dysfunction. Circ Res. 98(2):254–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi UH, Kaushal N, Hegde S, Finch ER, Kudva AK, Kennett MJ, Jordan CT, Paulson RF, Prabhu KS. 2014. Selenium suppresses leukemia through the action of endogenous eicosanoids. Cancer Res. 74(14):3890–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi UH, Kaushal N, Ravindra KC, Hegde S, Nelson SM, Narayan V, Vunta H, Paulson RF, Prabhu KS. 2011. Selenoprotein-dependent up-regulation of hematopoietic prostaglandin D2 synthase in macrophages is mediated through the activation of peroxisome proliferator-activated receptor (PPAR) gamma. J Biol Chem. 286(31):27471–27482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Zhao C, Wang W, Jin R, Li Q, Ge Q, Guan Y, Zhang Y. 2016. Prostaglandins E2 signal mediated by receptor subtype EP2 promotes IgE production in vivo and contributes to asthma development. Sci Rep. 6:20505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Larsen V, Chinn S, Arts IC, Amigo H, Rona RJ. 2007. Atopy, wheeze and bronchial responsiveness in young Chilean adults. Do dietary antioxidants matter? Allergy. 62(6):714–715. [DOI] [PubMed] [Google Scholar]
- Gautam PK, Kumar S, Tomar MS, Singh RK, Acharya A, Kumar S, Ram B. 2017. Selenium nanoparticles induce suppressed function of tumor associated macrophages and inhibit Dalton’s lymphoma proliferation. Biochem Biophys Rep. 12:172–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauvreau GM, Watson RM, O’Byrne PM. 1999. Protective effects of inhaled PGE2 on allergen-induced airway responses and airway inflammation. Am J Respir Crit Care Med. 159(1):31–36. [DOI] [PubMed] [Google Scholar]
- Gavett SH, Madison SL, Chulada PC, Scarborough PE, Qu W, Boyle JE, Tiano HF, Lee CA, Langenbach R, Roggli VL et al. 1999. Allergic lung responses are increased in prostaglandin H synthase-deficient mice. J Clin Invest. 104(6):721–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore TD. 2006. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 25(51):6680–6684. [DOI] [PubMed] [Google Scholar]
- Gladyshev VN, Hatfield DL. 1999. Selenocysteine-containing proteins in mammals. J Biomed Sci. 6(3):151–160. [DOI] [PubMed] [Google Scholar]
- Gleim S, Kasza Z, Martin K, Hwa J. 2009. Prostacyclin receptor/thromboxane receptor interactions and cellular responses in human atherothrombotic disease. Curr Atheroscler Rep. 11(3):227–235. [DOI] [PubMed] [Google Scholar]
- Gomez RM, Solana ME, Levander OA. 2002. Host selenium deficiency increases the severity of chronic inflammatory myopathy in Trypanosoma cruzi-inoculated mice. J Parasitol. 88(3):541–547. [DOI] [PubMed] [Google Scholar]
- Goodwin JS, Bankhurst AD, Messner RP. 1977. Suppression of human T-cell mitogenesis by prostaglandin. Existence of a prostaglandin-producing suppressor cell. J Exp Med. 146(6):1719–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso PJ, Scholz RW, Erskine RJ, Eberhart RJ. 1990. Phagocytosis, bactericidal activity, and oxidative metabolism of milk neutrophils from dairy cows fed selenium-supplemented and selenium-deficient diets. Am J Vet Res. 51(2):269–274. [PubMed] [Google Scholar]
- Green GA. 2001. Understanding NSAIDs: from aspirin to COX-2. Clin Cornerstone. 3(5):50–60. [DOI] [PubMed] [Google Scholar]
- Grobler L, Nagpal S, Sudarsanam TD, Sinclair D. 2016. Nutritional supplements for people being treated for active tuberculosis. Cochrane Database Syst Rev.(6):CD006086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove DI, Burston TO, Wellby ML, Ford RM, Forbes IJ. 1975. Humoral and cellular immunity in asthma. J Allergy Clin Immunol. 55(3):152–163. [DOI] [PubMed] [Google Scholar]
- Guimaraes MJ, Peterson D, Vicari A, Cocks BG, Copeland NG, Gilbert DJ, Jenkins NA, Ferrick DA, Kastelein RA, Bazan JF et al. 1996. Identification of a novel selD homolog from eukaryotes, bacteria, and archaea: is there an autoregulatory mechanism in selenocysteine metabolism? Proc Natl Acad Sci U S A. 93(26):15086–15091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo CH, Liu PJ, Hsia S, Chuang CJ, Chen PC. 2011. Role of certain trace minerals in oxidative stress, inflammation, CD4/CD8 lymphocyte ratios and lung function in asthmatic patients. Ann Clin Biochem. 48(Pt 4):344–351. [DOI] [PubMed] [Google Scholar]
- Haabeth OA, Lorvik KB, Hammarstrom C, Donaldson IM, Haraldsen G, Bogen B, Corthay A. 2011. Inflammation driven by tumour-specific Th1 cells protects against B-cell cancer. Nat Commun. 2:240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland A, Neubert K, Kruse I, Behne D, Schimk I. 2001. Consequences of long-term selenium-deficient diet on the prostacyclin and thromboxane release from rat aorta. Biol Trace Elem Res. 81(1):71–78. [DOI] [PubMed] [Google Scholar]
- Hadjibabaie M, Iravani M, Shamshiri AR, Zaker Z, Mousavi A, Alimoghaddam K, Bahar B, Kalantar E, Ghavamzadeh A. 2008. The prevalence of low selenium levels in adult patients undergoing bone marrow transplantation: a brief communication. Nutr Cancer. 60(6):837–839. [DOI] [PubMed] [Google Scholar]
- Hall JA, Vorachek WR, Stewart WC, Gorman ME, Mosher WD, Pirelli GJ, Bobe G. 2013. Selenium supplementation restores innate and humoral immune responses in footrot-affected sheep. PLoS One. 8(12):e82572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Zhou SM. 1994. Selenium supplement in the prevention of pregnancy induced hypertension. Chin Med J (Engl). 107(11):870–871. [PubMed] [Google Scholar]
- Han S, Inoue H, Flowers LC, Sidell N. 2003. Control of COX-2 gene expression through peroxisome proliferator-activated receptor gamma in human cervical cancer cells. Clin Cancer Res. 9(12):4627–4635. [PubMed] [Google Scholar]
- Hara S, Shoji Y, Sakurai A, Yuasa K, Himeno S, Imura N. 2001. Effects of selenium deficiency on expression of selenoproteins in bovine arterial endothelial cells. Biol Pharm Bull. 24(7):754–759. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Sheller JR, Morrow JD, Collins RD, Goleniewska K, O’Neal J, Zhou W, Ji S, Mitchell DB, Graham BS et al. 2005. Cyclooxygenase inhibition augments allergic inflammation through CD4-dependent, STAT6-independent mechanisms. J Immunol. 174(1):525–532. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Simmons AN, Kajino-Sakamoto R, Tsuji Y, Ninomiya-Tsuji J. 2016. TAK1 Regulates the Nrf2 Antioxidant System Through Modulating p62/SQSTM1. Antioxid Redox Signal. 25(17):953–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata AN, Breyer RM. 2004. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther. 103(2):147–166. [DOI] [PubMed] [Google Scholar]
- Hattori H, Imai H, Furuhama K, Sato O, Nakagawa Y. 2005. Induction of phospholipid hydroperoxide glutathione peroxidase in human polymorphonuclear neutrophils and HL60 cells stimulated with TNF-alpha. Biochem Biophys Res Commun. 337(2):464–473. [DOI] [PubMed] [Google Scholar]
- Hatzelmann A, Schatz M, Ullrich V. 1989. Involvement of glutathione peroxidase activity in the stimulation of 5-lipoxygenase activity by glutathione-depleting agents in human polymorphonuclear leukocytes. Eur J Biochem. 180(3):527–533. [DOI] [PubMed] [Google Scholar]
- Haurand M, Flohe L. 1988. Kinetic studies on arachidonate 5-lipoxygenase from rat basophilic leukemia cells. Biol Chem Hoppe Seyler. 369(2):133–142. [DOI] [PubMed] [Google Scholar]
- Havemann K, Gramse M. 1984. Physiology and pathophysiology of neutral proteinases of human granulocytes. Adv Exp Med Biol. 167:1–20. [DOI] [PubMed] [Google Scholar]
- Hawkes WC, Kelley DS, Taylor PC. 2001. The effects of dietary selenium on the immune system in healthy men. Biol Trace Elem Res. 81(3):189–213. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Ueno Y, Okamoto T. 1993. Oxidoreductive regulation of nuclear factor kappa B. Involvement of a cellular reducing catalyst thioredoxin. J Biol Chem. 268(15):11380–11388. [PubMed] [Google Scholar]
- He J, Zhou Y, Xing J, Wang Q, Zhu H, Zhu Y, Zou MH. 2013. Liver kinase B1 is required for thromboxane receptor-dependent nuclear factor-kappaB activation and inflammatory responses. Arterioscler Thromb Vasc Biol. 33(6):1297–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilman JM, Burke TJ, McClain CJ, Watson WH. 2011. Transactivation of gene expression by NF-kappaB is dependent on thioredoxin reductase activity. Free Radic Biol Med. 51(8):1533–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrerias A, Torres R, Serra M, Marco A, Roca-Ferrer J, Picado C, de Mora F. 2009. Subcutaneous prostaglandin E(2) restrains airway mast cell activity in vivo and reduces lung eosinophilia and Th(2) cytokine overproduction in house dust mite-sensitive mice. Int Arch Allergy Immunol. 149(4):323–332. [DOI] [PubMed] [Google Scholar]
- Hill KE, Zhou J, McMahan WJ, Motley AK, Atkins JF, Gesteland RF, Burk RF. 2003. Deletion of selenoprotein P alters distribution of selenium in the mouse. J Biol Chem. 278(16):13640–13646. [DOI] [PubMed] [Google Scholar]
- Hill TD, White JG, Rao GH. 1989. Role of glutathione and glutathione peroxidase in human platelet arachidonic acid metabolism. Prostaglandins. 38(1):21–32. [DOI] [PubMed] [Google Scholar]
- Hoffmann FW, Hashimoto AC, Shafer LA, Dow S, Berry MJ, Hoffmann PR. 2010. Dietary selenium modulates activation and differentiation of CD4+ T cells in mice through a mechanism involving cellular free thiols. J Nutr. 140(6):1155–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann PR, Jourdan-Le Saux C, Hoffmann FW, Chang PS, Bollt O, He Q, Tam EK, Berry MJ. 2007. A role for dietary selenium and selenoproteins in allergic airway inflammation. J Immunol. 179(5):3258–3267. [DOI] [PubMed] [Google Scholar]
- Hogan JS, Smith KL, Weiss WP, Todhunter DA, Schockey WL. 1990. Relationships among vitamin E, selenium, and bovine blood neutrophils. J Dairy Sci. 73(9):2372–2378. [DOI] [PubMed] [Google Scholar]
- Hondal RJ, Nilsson BL, Raines RT. 2001. Selenocysteine in native chemical ligation and expressed protein ligation. J Am Chem Soc. 123(21):5140–5141. [DOI] [PubMed] [Google Scholar]
- Hong Y, Li CH, Burgess JR, Chang M, Salem A, Srikumar K, Reddy CC. 1989. The role of selenium-dependent and selenium-independent glutathione peroxidases in the formation of prostaglandin F2 alpha. J Biol Chem. 264(23):13793–13800. [PubMed] [Google Scholar]
- Howes LG, James MJ, Florin T, Walker C. 2007. Nv-52: a novel thromboxane synthase inhibitor for the treatment of inflammatory bowel disease. Expert Opin Investig Drugs. 16(8):1255–1266. [DOI] [PubMed] [Google Scholar]
- Hunt JN, Smith JL, Jiang CL, Kessler L. 1983. Effect of synthetic prostaglandin E1 analog on aspirin-induced gastric bleeding and secretion. Dig Dis Sci. 28(10):897–902. [DOI] [PubMed] [Google Scholar]
- Hurwitz BE, Klaus JR, Llabre MM, Gonzalez A, Lawrence PJ, Maher KJ, Greeson JM, Baum MK, Shor-Posner G, Skyler JS et al. 2007. Suppression of human immunodeficiency virus type 1 viral load with selenium supplementation: a randomized controlled trial. Arch Intern Med. 167(2):148–154. [DOI] [PubMed] [Google Scholar]
- Hwang JT, Kim YM, Surh YJ, Baik HW, Lee SK, Ha J, Park OJ. 2006. Selenium regulates cyclooxygenase-2 and extracellular signal-regulated kinase signaling pathways by activating AMP-activated protein kinase in colon cancer cells. Cancer Res. 66(20):10057–10063. [DOI] [PubMed] [Google Scholar]
- Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, Roveri A, Peng X, Porto Freitas F, Seibt T et al. 2018. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell. 172(3):409–422 e421. [DOI] [PubMed] [Google Scholar]
- Jakupoglu C, Przemeck GK, Schneider M, Moreno SG, Mayr N, Hatzopoulos AK, de Angelis MH, Wurst W, Bornkamm GW, Brielmeier M et al. 2005. Cytoplasmic thioredoxin reductase is essential for embryogenesis but dispensable for cardiac development. Mol Cell Biol. 25(5):1980–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jania LA, Chandrasekharan S, Backlund MG, Foley NA, Snouwaert J, Wang IM, Clark P, Audoly LP, Koller BH. 2009. Microsomal prostaglandin E synthase-2 is not essential for in vivo prostaglandin E2 biosynthesis. Prostaglandins Other Lipid Mediat. 88(3–4):73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelicks LA, de Souza AP, Araujo-Jorge TC, Tanowitz HB. 2011. Would selenium supplementation aid in therapy for Chagas disease? Trends Parasitol. 27(3):102–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez R, Sanchez M, Zarzuelo MJ, Romero M, Quintela AM, Lopez-Sepulveda R, Galindo P, Gomez-Guzman M, Haro JM, Zarzuelo A et al. 2010. Endothelium-dependent vasodilator effects of peroxisome proliferator-activated receptor beta agonists via the phosphatidyl-inositol-3 kinase-Akt pathway. J Pharmacol Exp Ther. 332(2):554–561. [DOI] [PubMed] [Google Scholar]
- Jin RC, Mahoney CE, Coleman Anderson L, Ottaviano F, Croce K, Leopold JA, Zhang YY, Tang SS, Handy DE, Loscalzo J. 2011. Glutathione peroxidase-3 deficiency promotes platelet-dependent thrombosis in vivo. Circulation. 123(18):1963–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashima K, Murata T, Tanaka H, Matsuoka T, Sakata D, Yoshida N, Katagiri K, Kinashi T, Tanaka T, Miyasaka M et al. 2003. Thromboxane A2 modulates interaction of dendritic cells and T cells and regulates acquired immunity. Nat Immunol. 4(7):694–701. [DOI] [PubMed] [Google Scholar]
- Kalantari P, Narayan V, Henderson AJ, Prabhu KS. 2009. 15-Deoxy-Δ12, 14-prostaglandin J2 inhibits HIV-1 transactivating protein, Tat, through covalent modification. The FASEB Journal. 23(8):2366–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalantari P, Narayan V, Natarajan SK, Muralidhar K, Gandhi UH, Vunta H, Henderson AJ, Prabhu KS. 2008. Thioredoxin reductase-1 negatively regulates HIV-1 transactivating protein Tat-dependent transcription in human macrophages. Journal of Biological Chemistry. 283(48):33183–33190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaoka Y, Urade Y. 2003. Hematopoietic prostaglandin D synthase. Prostaglandins, leukotrienes and essential fatty acids. 69(2–3):163–167. [DOI] [PubMed] [Google Scholar]
- Karre K 2002. NK cells, MHC class I molecules and the missing self. Scand J Immunol. 55(3):221–228. [DOI] [PubMed] [Google Scholar]
- Kaushal N, Kudva AK, Patterson AD, Chiaro C, Kennett MJ, Desai D, Amin S, Carlson BA, Cantorna MT, Prabhu KS. 2014. Crucial role of macrophage selenoproteins in experimental colitis. J Immunol. 193(7):3683–3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanaugh-McHugh AL, Ruff A, Perlman E, Hutton N, Modlin J, Rowe S. 1991. Selenium deficiency and cardiomyopathy in acquired immunodeficiency syndrome. JPEN J Parenter Enteral Nutr. 15(3):347–349. [DOI] [PubMed] [Google Scholar]
- Keith RL, Miller YE, Hoshikawa Y, Moore MD, Gesell TL, Gao B, Malkinson AM, Golpon HA, Nemenoff RA, Geraci MW. 2002. Manipulation of pulmonary prostacyclin synthase expression prevents murine lung cancer. Cancer Res. 62(3):734–740. [PubMed] [Google Scholar]
- Keith RL, Miller YE, Hudish TM, Girod CE, Sotto-Santiago S, Franklin WA, Nemenoff RA, March TH, Nana-Sinkam SP, Geraci MW. 2004. Pulmonary prostacyclin synthase overexpression chemoprevents tobacco smoke lung carcinogenesis in mice. Cancer Res. 64(16):5897–5904. [DOI] [PubMed] [Google Scholar]
- Kern DE, Gillis S, Okada M, Henney CS. 1981. The role of interleukin-2 (IL-2) in the differentiatin of cytotoxic T cells: the effect of monoclonal anti-IL-2 antibody and absorption with IL-2 dependent T cell lines. J Immunol. 127(4):1323–1328. [PubMed] [Google Scholar]
- Khanna RS, Negi R, Pande D, Khanna HD. 2010. Clinical significance and analytical determination of trace amounts of selenium in human blood–spectrophotometeric technique. Open Med Devices J. 2:69–72. [Google Scholar]
- Kim IY, Stadtman TC. 1997. Inhibition of NF-kappaB DNA binding and nitric oxide induction in human T cells and lung adenocarcinoma cells by selenite treatment. Proc Natl Acad Sci U S A. 94(24):12904–12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsella BT. 2001. Thromboxane A2 signalling in humans: a ‘Tail’ of two receptors. Biochem Soc Trans. 29(Pt 6):641–654. [DOI] [PubMed] [Google Scholar]
- Kiremidjian-Schumacher L, Roy M, Wishe H, Cohen M, Stotzky G. 1991. Effects of selenium supplementation on macrophage-mediated tumor cytodestruction. Journal of Nutritional Immunology. 1(1):65–79. [Google Scholar]
- Kiremidjian-Schumacher L, Roy M, Wishe HI, Cohen MW, Stotzky G. 1996. Supplementation with selenium augments the functions of natural killer and lymphokine-activated killer cells. Biol Trace Elem Res. 52(3):227–239. [DOI] [PubMed] [Google Scholar]
- Klein EA, Thompson IM Jr., Tangen CM, Crowley JJ, Lucia MS, Goodman PJ, Minasian LM, Ford LG, Parnes HL, Gaziano JM et al. 2011. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 306(14):1549–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. 1995. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell. 83(5):813–819. [DOI] [PubMed] [Google Scholar]
- Knuth A, Danowski B, Oettgen HF, Old LJ. 1984. T-cell-mediated cytotoxicity against autologous malignant melanoma: analysis with interleukin 2-dependent T-cell cultures. Proc Natl Acad Sci U S A. 81(11):3511–3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocyigit A, Armutcu F, Gurel A, Ermis B. 2004. Alterations in plasma essential trace elements selenium, manganese, zinc, copper, and iron concentrations and the possible role of these elements on oxidative status in patients with childhood asthma. Biol Trace Elem Res. 97(1):31–41. [DOI] [PubMed] [Google Scholar]
- Koeberle SC, Gollowitzer A, Laoukili J, Kranenburg O, Werz O, Koeberle A, Kipp AP. 2020. Distinct and overlapping functions of glutathione peroxidases 1 and 2 in limiting NF-kappaB-driven inflammation through redox-active mechanisms. Redox Biol. 28:101388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkov KV, Novoselov SV, Hatfield DL, Gladyshev VN. 2002. Mammalian selenoprotein in which selenocysteine (Sec) incorporation is supported by a new form of Sec insertion sequence element. Mol Cell Biol. 22(5):1402–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krehl S, Loewinger M, Florian S, Kipp AP, Banning A, Wessjohann LA, Brauer MN, Iori R, Esworthy RS, Chu FF et al. 2012. Glutathione peroxidase-2 and selenium decreased inflammation and tumors in a mouse model of inflammation-associated carcinogenesis whereas sulforaphane effects differed with selenium supply. Carcinogenesis. 33(3):620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretz-Remy C, Mehlen P, Mirault ME, Arrigo AP. 1996. Inhibition of I kappa B-alpha phosphorylation and degradation and subsequent NF-kappa B activation by glutathione peroxidase overexpression. J Cell Biol. 133(5):1083–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigo R, Gladyshev VN. 2003. Characterization of mammalian selenoproteomes. Science. 300(5624):1439–1443. [DOI] [PubMed] [Google Scholar]
- Kudva AK, Shay AE, Prabhu KS. 2015. Selenium and inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. 309(2):G71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulmacz RJ LWEM. 1997. Peroxide Tone in Eicosanoid Signaling. Boston, MA: Springer; (Oxidative Stress and Signal Transduction. [Google Scholar]
- Kulmacz RJ. 1986. Prostaglandin H synthase and hydroperoxides: peroxidase reaction and inactivation kinetics. Arch Biochem Biophys. 249(2):273–285. [DOI] [PubMed] [Google Scholar]
- Kulmacz RJ, Lands WE. 1983. Requirements for hydroperoxide by the cyclooxygenase and peroxidase activities of prostaglandin H synthase. Prostaglandins. 25(4):531–540. [DOI] [PubMed] [Google Scholar]
- Kumaraswamy E, Malykh A, Korotkov KV, Kozyavkin S, Hu Y, Kwon SY, Moustafa ME, Carlson BA, Berry MJ, Lee BJ et al. 2000. Structure-expression relationships of the 15-kDa selenoprotein gene. Possible role of the protein in cancer etiology. J Biol Chem. 275(45):35540–35547. [DOI] [PubMed] [Google Scholar]
- Larionova I, Cherdyntseva N, Liu T, Patysheva M, Rakina M, Kzhyshkowska J. 2019. Interaction of tumor-associated macrophages and cancer chemotherapy. Oncoimmunology. 8(7):1596004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclerc P, Wahamaa H, Idborg H, Jakobsson PJ, Harris HE, Korotkova M. 2013. IL-1beta/HMGB1 complexes promote The PGE2 biosynthesis pathway in synovial fibroblasts. Scand J Immunol. 77(5):350–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BC, Lee SG, Choo MK, Kim JH, Lee HM, Kim S, Fomenko DE, Kim HY, Park JM, Gladyshev VN. 2017. Selenoprotein MsrB1 promotes anti-inflammatory cytokine gene expression in macrophages and controls immune response in vivo. Sci Rep. 7(1):5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BC, Peterfi Z, Hoffmann FW, Moore RE, Kaya A, Avanesov A, Tarrago L, Zhou Y, Weerapana E, Fomenko DE et al. 2013. MsrB1 and MICALs regulate actin assembly and macrophage function via reversible stereoselective methionine oxidation. Mol Cell. 51(3):397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinfelder W, Zehelein E, Mandrand-Berthelot MA, Bock A. 1988. Gene for a novel tRNA species that accepts L-serine and cotranslationally inserts selenocysteine. Nature. 331(6158):723–725. [DOI] [PubMed] [Google Scholar]
- Leone P, Shin EC, Perosa F, Vacca A, Dammacco F, Racanelli V. 2013. MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J Natl Cancer Inst. 105(16):1172–1187. [DOI] [PubMed] [Google Scholar]
- Leslie CC. 2015. Cytosolic phospholipase A(2): physiological function and role in disease. J Lipid Res. 56(8):1386–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levander OA, Beck MA. 1997. Interacting nutritional and infectious etiologies of Keshan disease. Insights from coxsackie virus B-induced myocarditis in mice deficient in selenium or vitamin E. Biol Trace Elem Res. 56(1):5–21. [DOI] [PubMed] [Google Scholar]
- Li C, Deng X, Xie X, Liu Y, Friedmann Angeli JP, Lai L. 2018. Activation of Glutathione Peroxidase 4 as a Novel Anti-inflammatory Strategy. Front Pharmacol. 9:1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zuo L, Shen T, Xu CM, Zhang ZN. 2003. Induction of apoptosis by sodium selenite in human acute promyelocytic leukemia NB4 cells: involvement of oxidative stress and mitochondria. J Trace Elem Med Biol. 17(1):19–26. [DOI] [PubMed] [Google Scholar]
- Li Y, Fang M, Zhang J, Wang J, Song Y, Shi J, Li W, Wu G, Ren J, Wang Z et al. 2016. Hydrogel dual delivered celecoxib and anti-PD-1 synergistically improve antitumor immunity. Oncoimmunology. 5(2):e1074374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YB, Han JY, Jiang W, Wang J. 2011. Selenium inhibits high glucose-induced cyclooxygenase-2 and P-selectin expression in vascular endothelial cells. Mol Biol Rep. 38(4):2301–2306. [DOI] [PubMed] [Google Scholar]
- Liao C, Carlson BA, Paulson RF, Prabhu KS. 2018. The intricate role of selenium and selenoproteins in erythropoiesis. Free Radic Biol Med. 127:165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao C, Hardison RC, Kennett MJ, Carlson BA, Paulson RF, Prabhu KS. 2018. Selenoproteins regulate stress erythroid progenitors and spleen microenvironment during stress erythropoiesis. Blood. 131(23):2568–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liel N, Nathan I, Yermiyahu T, Zolotov Z, Lieberman JR, Dvilansky A, Halushka PV. 1993. Increased platelet thromboxane A2/prostaglandin H2 receptors in patients with pregnancy induced hypertension. Thromb Res. 70(3):205–210. [DOI] [PubMed] [Google Scholar]
- Lippman SM, Klein EA, Goodman PJ, Lucia MS, Thompson IM, Ford LG, Parnes HL, Minasian LM, Gaziano JM, Hartline JA et al. 2009. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 301(1):39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovgren AK, Kovarova M, Koller BH. 2007. cPGES/p23 is required for glucocorticoid receptor function and embryonic growth but not prostaglandin E2 synthesis. Mol Cell Biol. 27(12):4416–4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low SC, Harney JW, Berry MJ. 1995. Cloning and functional characterization of human selenophosphate synthetase, an essential component of selenoprotein synthesis. J Biol Chem. 270(37):21659–21664. [DOI] [PubMed] [Google Scholar]
- Lu YC, Yeh WC, Ohashi PS. 2008. LPS/TLR4 signal transduction pathway. Cytokine. 42(2):145–151. [DOI] [PubMed] [Google Scholar]
- Lycke N, Severinson E, Strober W. 1990. Cholera toxin acts synergistically with IL-4 to promote IgG1 switch differentiation. J Immunol. 145(10):3316–3324. [PubMed] [Google Scholar]
- Maddipati KR, Marnett LJ. 1987. Characterization of the major hydroperoxide-reducing activity of human plasma. Purification and properties of a selenium-dependent glutathione peroxidase. J Biol Chem. 262(36):17398–17403. [PubMed] [Google Scholar]
- Maddox L, Schwartz DA. 2002. The pathophysiology of asthma. Annu Rev Med. 53:477–498. [DOI] [PubMed] [Google Scholar]
- Mahdavi M, Mavandadnejad F, Yazdi MH, Faghfuri E, Hashemi H, Homayouni-Oreh S, Farhoudi R, Shahverdi AR. 2017. Oral administration of synthetic selenium nanoparticles induced robust Th1 cytokine pattern after HBs antigen vaccination in mouse model. J Infect Public Health. 10(1):102–109. [DOI] [PubMed] [Google Scholar]
- Manabe A, Coustan-Smith E, Kumagai M, Behm FG, Raimondi SC, Pui CH, Campana D. 1994. Interleukin-4 induces programmed cell death (apoptosis) in cases of high-risk acute lymphoblastic leukemia. Blood. 83(7):1731–1737. [PubMed] [Google Scholar]
- Mao GH, Ren Y, Li Q, Wu HY, Jin D, Zhao T, Xu CQ, Zhang DH, Jia QD, Bai YP et al. 2016. Anti-tumor and immunomodulatory activity of selenium (Se)-polysaccharide from Se-enriched Grifola frondosa. Int J Biol Macromol. 82:607–613. [DOI] [PubMed] [Google Scholar]
- Marciel MP, Hoffmann PR. 2019. Molecular Mechanisms by Which Selenoprotein K Regulates Immunity and Cancer. Biol Trace Elem Res. 192(1):60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Romero FJ, Kryukov GV, Lobanov AV, Carlson BA, Lee BJ, Gladyshev VN, Hatfield DL. 2001. Selenium metabolism in Drosophila: selenoproteins, selenoprotein mRNA expression, fertility, and mortality. J Biol Chem. 276(32):29798–29804. [DOI] [PubMed] [Google Scholar]
- Martinez FO, Gordon S. 2014. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 6:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka T, Hirata M, Tanaka H, Takahashi Y, Murata T, Kabashima K, Sugimoto Y, Kobayashi T, Ushikubi F, Aze Y. 2000. Prostaglandin D2 as a mediator of allergic asthma. science. 287(5460):2013–2017. [DOI] [PubMed] [Google Scholar]
- Maunsell K, Wraith DG, Cunnington AM. 1968. Mites and house-dust allergy in bronchial asthma. Lancet. 1(7555):1267–1270. [DOI] [PubMed] [Google Scholar]
- McBride OW, Rajagopalan M, Hatfield D. 1987. Opal suppressor phosphoserine tRNA gene and pseudogene are located on human chromosomes 19 and 22, respectively. J Biol Chem. 262(23):11163–11166. [PubMed] [Google Scholar]
- McKenna HJ, Smith FO, Brasel K, Hirschstein D, Bernstein ID, Williams DE, Lyman SD. 1996. Effects of flt3 ligand on acute myeloid and lymphocytic leukemic blast cells from children. Exp Hematol. 24(2):378–385. [PubMed] [Google Scholar]
- McKenzie RC, Rafferty TS, Beckett GJ. 1998. Selenium: an essential element for immune function. Immunol Today. 19(8):342–345. [DOI] [PubMed] [Google Scholar]
- Melillo E, Woolley KL, Manning PJ, Watson RM, O’Byrne PM. 1994. Effect of inhaled PGE2 on exercise-induced bronchoconstriction in asthmatic subjects. Am J Respir Crit Care Med. 149(5):1138–1141. [DOI] [PubMed] [Google Scholar]
- Meng XL, Chen CL, Liu YY, Su SJ, Gou JM, Huan FN, Wang D, Liu HS, Ben SB, Lu J. 2019. Selenoprotein SELENOK Enhances the Migration and Phagocytosis of Microglial Cells by Increasing the Cytosolic Free Ca(2+) Level Resulted from the Up-Regulation of IP3R. Neuroscience. 406:38–49. [DOI] [PubMed] [Google Scholar]
- Meydani M 1992. Modulation of the platelet thromboxane A2 and aortic prostacyclin synthesis by dietary selenium and vitamin E. Biol Trace Elem Res. 33:79–86. [DOI] [PubMed] [Google Scholar]
- Misso NL, Peroni DJ, Watkins DN, Stewart GA, Thompson PJ. 1998. Glutathione peroxidase activity and mRNA expression in eosinophils and neutrophils of asthmatic and non-asthmatic subjects. J Leukoc Biol. 63(1):124–130. [DOI] [PubMed] [Google Scholar]
- Mitsumori S 2004. Recent progress in work on PGD2 antagonists for drugs targeting allergic diseases. Current pharmaceutical design. 10(28):3533–3538. [DOI] [PubMed] [Google Scholar]
- Mittal D, Gubin MM, Schreiber RD, Smyth MJ. 2014. New insights into cancer immunoediting and its three component phases—elimination, equilibrium and escape. Current opinion in immunology. 27:16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghadaszadeh B, Petit N, Jaillard C, Brockington M, Quijano Roy S, Merlini L, Romero N, Estournet B, Desguerre I, Chaigne D et al. 2001. Mutations in SEPN1 cause congenital muscular dystrophy with spinal rigidity and restrictive respiratory syndrome. Nat Genet. 29(1):17–18. [DOI] [PubMed] [Google Scholar]
- Moncada S, Gryglewski R, Bunting S, Vane JR. 1976. An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature. 263(5579):663–665. [DOI] [PubMed] [Google Scholar]
- Morawetz RA, Gabriele L, Rizzo LV, Noben-Trauth N, Kuhn R, Rajewsky K, Muller W, Doherty TM, Finkelman F, Coffman RL et al. 1996. Interleukin (IL)-4-independent immunoglobulin class switch to immunoglobulin (Ig)E in the mouse. J Exp Med. 184(5):1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen R, Clemmensen HS, Woodworth JS, Therkelsen ML, Mustafa T, Tonby K, Jenum S, Agger EM, Dyrhol-Riise AM, Andersen P. 2019. Cyclooxygenase inhibitors impair CD4 T cell immunity and exacerbate Mycobacterium tuberculosis infection in aerosol-challenged mice. Commun Biol. 2:288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mubarak KK. 2010. A review of prostaglandin analogs in the management of patients with pulmonary arterial hypertension. Respir Med. 104(1):9–21. [DOI] [PubMed] [Google Scholar]
- Muller M, Banning A, Brigelius-Flohe R, Kipp A. 2010. Nrf2 target genes are induced under marginal selenium-deficiency. Genes Nutr. 5(4):297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller MF, Florian S, Pommer S, Osterhoff M, Esworthy RS, Chu FF, Brigelius-Flohe R, Kipp AP. 2013. Deletion of glutathione peroxidase-2 inhibits azoxymethane-induced colon cancer development. PLoS One. 8(8):e72055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Nakashima K, Kamei D, Masuda S, Ishikawa Y, Ishii T, Ohmiya Y, Watanabe K, Kudo I. 2003. Cellular prostaglandin E2 production by membrane-bound prostaglandin E synthase-2 via both cyclooxygenases-1 and −2. J Biol Chem. 278(39):37937–37947. [DOI] [PubMed] [Google Scholar]
- Murphy K, Weaver C. 2016. Janeway’s immunobiology. 9th edition ed. New York, NY: Garland Science/Taylor & Francis Group, LLC. [Google Scholar]
- Nakanishi M, Gokhale V, Meuillet EJ, Rosenberg DW. 2010. mPGES-1 as a target for cancer suppression: A comprehensive invited review “Phospholipase A2 and lipid mediators”. Biochimie. 92(6):660–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata J, Kondo M, Tamaoki J, Takemiya T, Nohara M, Yamagata K, Nagai A. 2005. Augmentation of allergic inflammation in the airways of cyclooxygenase-2-deficient mice. Respirology. 10(2):149–156. [DOI] [PubMed] [Google Scholar]
- Narayan V, Ravindra KC, Liao C, Kaushal N, Carlson BA, Prabhu KS. 2015. Epigenetic regulation of inflammatory gene expression in macrophages by selenium. J Nutr Biochem. 26(2):138–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Takahashi M, Liu A, Nikkuni K, Furukawa T, Toba K, Koyama S, Takai K, Sanada M, Aizawa Y. 2001. Leukemia blast-induced T-cell anergy demonstrated by leukemia-derived dendritic cells in acute myelogenous leukemia. Exp Hematol. 29(6):709–719. [DOI] [PubMed] [Google Scholar]
- Nelson SM, Lei X, Prabhu KS. 2011. Selenium levels affect the IL-4-induced expression of alternative activation markers in murine macrophages. J Nutr. 141(9):1754–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson SM, Shay AE, James JL, Carlson BA, Urban JF Jr., Prabhu KS. 2016. Selenoprotein Expression in Macrophages Is Critical for Optimal Clearance of Parasitic Helminth Nippostrongylus brasiliensis. J Biol Chem. 291(6):2787–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemenoff R, Meyer AM, Hudish TM, Mozer AB, Snee A, Narumiya S, Stearman RS, Winn RA, Weiser-Evans M, Geraci MW et al. 2008. Prostacyclin prevents murine lung cancer independent of the membrane receptor by activation of peroxisomal proliferator--activated receptor gamma. Cancer Prev Res (Phila). 1(5):349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri Serneri GG, Abbate R, Gensini GF, Panetta A, Casolo GC, Carini M. 1983. TxA2 production by human arteries and veins. Prostaglandins. 25(6):753–766. [DOI] [PubMed] [Google Scholar]
- Norton RL, Fredericks GJ, Huang Z, Fay JD, Hoffmann FW, Hoffmann PR. 2017. Selenoprotein K regulation of palmitoylation and calpain cleavage of ASAP2 is required for efficient FcgammaR-mediated phagocytosis. J Leukoc Biol. 101(2):439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan S 1999. On the role of PGD2 metabolites as markers of mast cell activation in asthma. Acta Physiologica Scandinavica Supplementum. 644:1–74. [PubMed] [Google Scholar]
- Omland O, Deguchi Y, Sigsgaard T, Hansen JC. 2002. Selenium serum and urine is associated to mild asthma and atopy. The SUS study. J Trace Elem Med Biol. 16(2):123–127. [DOI] [PubMed] [Google Scholar]
- Ouchi N, Walsh K. 2007. Adiponectin as an anti-inflammatory factor. Clinica chimica acta. 380(1–2):24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai CH, Yen CT, Chen CP, Yu IS, Lin SW, Lin SR. 2016. Lack of Thromboxane Synthase Prevents Hypertension and Fetal Growth Restriction after High Salt Treatment during Pregnancy. PLoS One. 11(3):e0151617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. 2005. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 437(7059):759–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazirandeh A, Assadi Nejad M, Vossogh P. 1999. Determination of selenium in blood serum of children with acute leukemia and effect of chemotherapy on serum selenium level. J Trace Elem Med Biol. 13(4):242–246. [DOI] [PubMed] [Google Scholar]
- Peebles RS Jr., Hashimoto K, Morrow JD, Dworski R, Collins RD, Hashimoto Y, Christman JW, Kang KH, Jarzecka K, Furlong J et al. 2002. Selective cyclooxygenase-1 and −2 inhibitors each increase allergic inflammation and airway hyperresponsiveness in mice. Am J Respir Crit Care Med. 165(8):1154–1160. [DOI] [PubMed] [Google Scholar]
- Peebles RS Jr., Hashimoto K, Sheller JR, Moore ML, Morrow JD, Ji S, Elias JA, Goleniewska K, O’Neal J, Mitchell DB et al. 2005. Allergen-induced airway hyperresponsiveness mediated by cyclooxygenase inhibition is not dependent on 5-lipoxygenase or IL-5, but is IL-13 dependent. J Immunol. 175(12):8253–8259. [DOI] [PubMed] [Google Scholar]
- Petrie HT, Klassen LW, Klassen PS, O’Dell JR, Kay HD. 1989. Selenium and the Immune Response: 2. Enhancement of Murine Cytotoxic T‐Lymphocyte and Natural Killer Cell Cytotoxicity In Vivo. Journal of leukocyte biology. 45(3):215–220. [DOI] [PubMed] [Google Scholar]
- Philchenkov A, Zavelevich M, Khranovskaya N, Surai P. 2007. Comparative analysis of apoptosis induction by selenium compounds in human lymphoblastic leukemia MT-4 cells. Exp Oncol. 29(4):257–261. [PubMed] [Google Scholar]
- Phipps RP, Roper RL, Stein SH. 1990. Regulation of B-cell tolerance and triggering by macrophages and lymphoid dendritic cells. Immunol Rev. 117:135–158. [DOI] [PubMed] [Google Scholar]
- Phipps RP, Stein SH, Roper RL. 1991. A new view of prostaglandin E regulation of the immune response. Immunol Today. 12(10):349–352. [DOI] [PubMed] [Google Scholar]
- Pineyro MD, Parodi-Talice A, Arcari T, Robello C. 2008. Peroxiredoxins from Trypanosoma cruzi: virulence factors and drug targets for treatment of Chagas disease? Gene. 408(1–2):45–50. [DOI] [PubMed] [Google Scholar]
- Prabhu KS, Zamamiri-Davis F, Stewart JB, Thompson JT, Sordillo LM, Reddy CC. 2002. Selenium deficiency increases the expression of inducible nitric oxide synthase in RAW 264.7 macrophages: role of nuclear factor-kappaB in up-regulation. Biochem J. 366(Pt 1):203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prchal J, Levi MM. 2010. Williams hematology. New York, NY: The McGraw-Hill Companies. [Google Scholar]
- Probst L, Dachert J, Schenk B, Fulda S. 2017. Lipoxygenase inhibitors protect acute lymphoblastic leukemia cells from ferroptotic cell death. Biochem Pharmacol. 140:41–52. [DOI] [PubMed] [Google Scholar]
- Qu X, Huang K, Deng L, Xu H. 2000. Selenium deficiency-induced alterations in the vascular system of the rat. Biol Trace Elem Res. 75(1–3):119–128. [DOI] [PubMed] [Google Scholar]
- Qujeq D, Hidari B, Bijani K, Shirdel H. 2003. Glutathione peroxidase activity and serum selenium concentration in intrinsic asthmatic patients. Clin Chem Lab Med. 41(2):200–202. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan K, Shenbagarathai R, Kavitha K, Thirumalaikolundusubramanian P, Rathinasabapati R. 2012. Selenium levels in persons with HIV/tuberculosis in India, Madurai City. Clin Lab. 58(1–2):165–168. [PubMed] [Google Scholar]
- Range N, Changalucha J, Krarup H, Magnussen P, Andersen AB, Friis H. 2006. The effect of multi-vitamin/mineral supplementation on mortality during treatment of pulmonary tuberculosis: a randomised two-by-two factorial trial in Mwanza, Tanzania. Br J Nutr. 95(4):762–770. [DOI] [PubMed] [Google Scholar]
- Ren F, Chen X, Hesketh J, Gan F, Huang K. 2012. Selenium promotes T-cell response to TCR-stimulation and ConA, but not PHA in primary porcine splenocytes. PLoS One. 7(4):e35375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricetti MM, Guidi GC, Bellisola G, Marrocchella R, Rigo A, Perona G. 1994. Selenium enhances glutathione peroxidase activity and prostacyclin release in cultured human endothelial cells. Concurrent effects on mRNA levels. Biol Trace Elem Res. 46(1–2):113–123. [DOI] [PubMed] [Google Scholar]
- Riedl MA, Nel AE. 2008. Importance of oxidative stress in the pathogenesis and treatment of asthma. Curr Opin Allergy Clin Immunol. 8(1):49–56. [DOI] [PubMed] [Google Scholar]
- Rivera MT, de Souza AP, Moreno AH, Xavier SS, Gomes JA, Rocha MO, Correa-Oliveira R, Neve J, Vanderpas J, Araujo-Jorge TC. 2002. Progressive Chagas’ cardiomyopathy is associated with low selenium levels. Am J Trop Med Hyg. 66(6):706–712. [DOI] [PubMed] [Google Scholar]
- Rohrbach S, Gruenler S, Teschner M, Holtz J. 2006. The thioredoxin system in aging muscle: key role of mitochondrial thioredoxin reductase in the protective effects of caloric restriction? Am J Physiol Regul Integr Comp Physiol. 291(4):R927–935. [DOI] [PubMed] [Google Scholar]
- Rollins TE, Smith WL. 1980. Subcellular localization of prostaglandin-forming cyclooxygenase in Swiss mouse 3T3 fibroblasts by electron microscopic immunocytochemistry. J Biol Chem. 255(10):4872–4875. [PubMed] [Google Scholar]
- Roper RL, Conrad DH, Brown DM, Warner GL, Phipps RP. 1990. Prostaglandin E2 promotes IL-4-induced IgE and IgG1 synthesis. J Immunol. 145(8):2644–2651. [PubMed] [Google Scholar]
- Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG. 2000. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature. 403(6765):103–108. [DOI] [PubMed] [Google Scholar]
- Rotruck JT, Pope AL, Ganther HE, Swanson A, Hafeman DG, Hoekstra W. 1973. Selenium: biochemical role as a component of glutathione peroxidase. Science. 179(4073):588–590. [DOI] [PubMed] [Google Scholar]
- Roy M, Kiremidjian-Schumacher L, Wishe HI, Cohen MW, Stotzky G. 1993. Selenium supplementation enhances the expression of interleukin 2 receptor subunits and internalization of interleukin 2. Proc Soc Exp Biol Med. 202(3):295–301. [DOI] [PubMed] [Google Scholar]
- Rustin MH, Bull HA, Machin SJ, Koro O, Dowd PM. 1987. Serum from patients with Raynaud’s phenomenon inhibits prostacyclin production. J Invest Dermatol. 89(6):555–559. [DOI] [PubMed] [Google Scholar]
- Sabattini E, Bacci F, Sagramoso C, Pileri SA. 2010. WHO classification of tumours of haematopoietic and lymphoid tissues in 2008: an overview. Pathologica. 102(3):83–87. [PubMed] [Google Scholar]
- Sadahira Y, Mori M. 1999. Role of the macrophage in erythropoiesis. Pathol Int. 49(10):841–848. [DOI] [PubMed] [Google Scholar]
- Safir N, Wendel A, Saile R, Chabraoui L. 2003. The effect of selenium on immune functions of J774.1 cells. Clin Chem Lab Med. 41(8):1005–1011. [DOI] [PubMed] [Google Scholar]
- Sakamoto H, Imai H, Nakagawa Y. 2000. Involvement of phospholipid hydroperoxide glutathione peroxidase in the modulation of prostaglandin D2 synthesis. J Biol Chem. 275(51):40028–40035. [DOI] [PubMed] [Google Scholar]
- Salinas GRH, Xu XM, Carlson BA, Hatfield DL, Gladyshev VN 2006. Evolution of selenocysteine decoding and the key role of selenophosphate synthetase in the pathway of selenium utilization. Boston, MA: Springer. [Google Scholar]
- Samuelsson B, Dahlen SE, Lindgren JA, Rouzer CA, Serhan CN. 1987. Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science. 237(4819):1171–1176. [DOI] [PubMed] [Google Scholar]
- Santesmasses D, Mariotti M, Gladyshev VN. 2019. Tolerance to selenoprotein loss differs between human and mouse. Mol Biol Evol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnurr K, Belkner J, Ursini F, Schewe T, Kuhn H. 1996. The selenoenzyme phospholipid hydroperoxide glutathione peroxidase controls the activity of the 15-lipoxygenase with complex substrates and preserves the specificity of the oxygenation products. J Biol Chem. 271(9):4653–4658. [DOI] [PubMed] [Google Scholar]
- Schomburg L, Schweizer U. 2009. Hierarchical regulation of selenoprotein expression and sex-specific effects of selenium. Biochim Biophys Acta. 1790(11):1453–1462. [DOI] [PubMed] [Google Scholar]
- Schomburg L, Schweizer U, Holtmann B, Flohe L, Sendtner M, Kohrle J. 2003. Gene disruption discloses role of selenoprotein P in selenium delivery to target tissues. Biochem J. 370(Pt 2):397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiderer J, Dambacher J, Kuhnlein B, Pfennig S, Konrad A, Torok HP, Haller D, Goke B, Ochsenkuhn T, Lohse P et al. 2007. The role of the selenoprotein S (SELS) gene −105G>A promoter polymorphism in inflammatory bowel disease and regulation of SELS gene expression in intestinal inflammation. Tissue Antigens. 70(3):238–246. [DOI] [PubMed] [Google Scholar]
- Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Radmark O, Wurst W et al. 2008. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 8(3):237–248. [DOI] [PubMed] [Google Scholar]
- Sengupta A, Lichti UF, Carlson BA, Cataisson C, Ryscavage AO, Mikulec C, Conrad M, Fischer SM, Hatfield DL, Yuspa SH. 2013. Targeted disruption of glutathione peroxidase 4 in mouse skin epithelial cells impairs postnatal hair follicle morphogenesis that is partially rescued through inhibition of COX-2. J Invest Dermatol. 133(7):1731–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Savill J. 2005. Resolution of inflammation: the beginning programs the end. Nat Immunol. 6(12):1191–1197. [DOI] [PubMed] [Google Scholar]
- Serra-Pages M, Torres R, Plaza J, Herrerias A, Costa-Farre C, Marco A, Jimenez M, Maurer M, Picado C, de Mora F. 2015. Activation of the Prostaglandin E2 receptor EP2 prevents house dust mite-induced airway hyperresponsiveness and inflammation by restraining mast cells’ activity. Clin Exp Allergy. 45(10):1590–1600. [DOI] [PubMed] [Google Scholar]
- Sestini P, Armetti L, Gambaro G, Pieroni MG, Refini RM, Sala A, Vaghi A, Folco GC, Bianco S, Robuschi M. 1996. Inhaled PGE2 prevents aspirin-induced bronchoconstriction and urinary LTE4 excretion in aspirin-sensitive asthma. Am J Respir Crit Care Med. 153(2):572–575. [DOI] [PubMed] [Google Scholar]
- Seyedrezazadeh E, Ostadrahimi A, Mahboob S, Assadi Y, Ghaemmagami J, Pourmogaddam M. 2008. Effect of vitamin E and selenium supplementation on oxidative stress status in pulmonary tuberculosis patients. Respirology. 13(2):294–298. [DOI] [PubMed] [Google Scholar]
- Shaheen SO, Newson RB, Rayman MP, Wong AP, Tumilty MK, Phillips JM, Potts JF, Kelly FJ, White PT, Burney PG. 2007. Randomised, double blind, placebo-controlled trial of selenium supplementation in adult asthma. Thorax. 62(6):483–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay AE, Diwakar BT, Guan BJ, Narayan V, Urban JF Jr., Prabhu KS 2017. IL-4 up-regulates cyclooxygenase-1 expression in macrophages. J Biol Chem. 292(35):14544–14555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shchedrina VA, Everley RA, Zhang Y, Gygi SP, Hatfield DL, Gladyshev VN. 2011. Selenoprotein K binds multiprotein complexes and is involved in the regulation of endoplasmic reticulum homeostasis. J Biol Chem. 286(50):42937–42948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- mShrimali RK, Irons RD, Carlson BA, Park JM, Hatfield DL. 2007. Selenoprotein deficiency adversely affects T cell development, proliferation, TCR signaling and cell mediated immunity. Federation of American Societies for Experimental Biology. [Google Scholar]
- Shrimali RK, Irons RD, Carlson BA, Sano Y, Gladyshev VN, Park JM, Hatfield DL. 2008. Selenoproteins mediate T cell immunity through an antioxidant mechanism. J Biol Chem. 283(29):20181–20185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica A, Mantovani A. 2012. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 122(3):787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A, Madden KB, Yeung KJ, Zhao A, Elfrey J, Finkelman F, Levander O, Shea-Donohue T, Urban JF Jr., 2005. Deficiencies in selenium and/or vitamin E lower the resistance of mice to Heligmosomoides polygyrus infections. J Nutr. 135(4):830–836. [DOI] [PubMed] [Google Scholar]
- Smith AD, Botero S, Shea-Donohue T, Urban JF Jr., 2011. The pathogenicity of an enteric Citrobacter rodentium Infection is enhanced by deficiencies in the antioxidants selenium and vitamin E. Infect Immun. 79(4):1471–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WL, Lands WE. 1972. Oxygenation of polyunsaturated fatty acids during prostaglandin biosynthesis by sheep vesicular gland. Biochemistry. 11(17):3276–3285. [DOI] [PubMed] [Google Scholar]
- Smyth EM, Grosser T, Wang M, Yu Y, FitzGerald GA. 2009. Prostanoids in health and disease. J Lipid Res. 50 Suppl:S423–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza AP, Jelicks LA, Tanowitz HB, Olivieri BP, Medeiros MM, Oliveira GM, Pires AR, Santos AM, Araujo-Jorge TC. 2010. The benefits of using selenium in the treatment of Chagas disease: prevention of right ventricle chamber dilatation and reversion of Trypanosoma cruzi-induced acute and chronic cardiomyopathy in mice. Mem Inst Oswaldo Cruz. 105(6):746–751. [DOI] [PubMed] [Google Scholar]
- St Germain DL, Galton VA. 1997. The deiodinase family of selenoproteins. Thyroid. 7(4):655–668. [DOI] [PubMed] [Google Scholar]
- Stachowska E, Dolegowska B, Dziedziejko V, Rybicka M, Kaczmarczyk M, Bober J, Rac M, Machalinski B, Chlubek D. 2009. Prostaglandin E2 (PGE2) and thromboxane A2 (TXA2) synthesis is regulated by conjugated linoleic acids (CLA) in human macrophages. J Physiol Pharmacol. 60(1):77–85. [PubMed] [Google Scholar]
- Stanley BA, Sivakumaran V, Shi S, McDonald I, Lloyd D, Watson WH, Aon MA, Paolocci N. 2011. Thioredoxin reductase-2 is essential for keeping low levels of H(2)O(2) emission from isolated heart mitochondria. J Biol Chem. 286(38):33669–33677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens J, Waters R, Sieniawska C, Kassam S, Montoto S, Fitzgibbon J, Rohatiner A, Lister A, Joel S. 2011. Serum selenium concentration at diagnosis and outcome in patients with haematological malignancies. Br J Haematol. 154(4):448–456. [DOI] [PubMed] [Google Scholar]
- Stitham J, Midgett C, Martin KA, Hwa J. 2011. Prostacyclin: an inflammatory paradox. Front Pharmacol. 2:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straus DS, Glass CK. 2001. Cyclopentenone prostaglandins: new insights on biological activities and cellular targets. Med Res Rev. 21(3):185–210. [DOI] [PubMed] [Google Scholar]
- Sturm EM, Radnai B, Jandl K, Stancic A, Parzmair GP, Hogenauer C, Kump P, Wenzl H, Petritsch W, Pieber TR et al. 2014. Opposing roles of prostaglandin D2 receptors in ulcerative colitis. J Immunol. 193(2):827–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su D, Novoselov SV, Sun QA, Moustafa ME, Zhou Y, Oko R, Hatfield DL, Gladyshev VN. 2005. Mammalian selenoprotein thioredoxin-glutathione reductase. Roles in disulfide bond formation and sperm maturation. J Biol Chem. 280(28):26491–26498. [DOI] [PubMed] [Google Scholar]
- Takayama K, Yuhki K, Ono K, Fujino T, Hara A, Yamada T, Kuriyama S, Karibe H, Okada Y, Takahata O et al. 2005. Thromboxane A2 and prostaglandin F2alpha mediate inflammatory tachycardia. Nat Med. 11(5):562–566. [DOI] [PubMed] [Google Scholar]
- Tanabe T, Tohnai N. 2002. Cyclooxygenase isozymes and their gene structures and expression. Prostaglandins Other Lipid Mediat. 68–69:95–114. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Roberts MH, Yamamoto N, Sugiura H, Uehara M, Mao XQ, Shirakawa T, Hopkin JM. 2002. Genetic variants of the receptors for thromboxane A2 and IL-4 in atopic dermatitis. Biochem Biophys Res Commun. 292(3):776–780. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Urade Y, Kimura H, Eguchi N, Nishikawa A, Hayaishi O. 1997. Lipocalin-type prostaglandin D synthase (β-trace) is a newly recognized type of retinoid transporter. Journal of Biological Chemistry. 272(25):15789–15795. [DOI] [PubMed] [Google Scholar]
- Tang SY, Monslow J, G RG, Todd L, Pawelzik SC, Chen L, Lawson J, Pure E, FitzGerald GA. 2016. Cardiovascular Consequences of Prostanoid I Receptor Deletion in Microsomal Prostaglandin E Synthase-1-Deficient Hyperlipidemic Mice. Circulation. 134(4):328–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi T, Tsukada H, Nakamura H, Kodama M, Fukuda K, Tominaga M, Seino Y. 1997. Effects of a thromboxane A2 receptor antagonist in an animal model of inflammatory bowel disease. Digestion. 58(5):476–478. [DOI] [PubMed] [Google Scholar]
- Tanikawa N, Ohmiya Y, Ohkubo H, Hashimoto K, Kangawa K, Kojima M, Ito S, Watanabe K. 2002. Identification and characterization of a novel type of membrane-associated prostaglandin E synthase. Biochem Biophys Res Commun. 291(4):884–889. [DOI] [PubMed] [Google Scholar]
- Thu VT, Kim HK, Ha SH, Yoo JY, Park WS, Kim N, Oh GT, Han J. 2010. Glutathione peroxidase 1 protects mitochondria against hypoxia/reoxygenation damage in mouse hearts. Pflugers Arch. 460(1):55–68. [DOI] [PubMed] [Google Scholar]
- mTirosh O, Reifen R. 2007. Treatment and prevention of inflammatory disease and mitochondrial dysfunction with high dose selenium. Google Patents. [Google Scholar]
- Tonelli AR, Ahmed MK, Alkukhun L, Cikach F, Aulak K, Dweik RA. 2015. Treprostinil Iontophoresis in Idiopathic Pulmonary Arterial Hypertension. Am J Respir Crit Care Med. 192(8):1014–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres R, Perez M, Marco A, Picado C, de Mora F. 2009. [A cyclooxygenase-2 selective inhibitor worsens respiratory function and enhances mast cell activity in ovalbumin-sensitized mice]. Arch Bronconeumol. 45(4):162–167. [DOI] [PubMed] [Google Scholar]
- Torzewski M, Ochsenhirt V, Kleschyov AL, Oelze M, Daiber A, Li H, Rossmann H, Tsimikas S, Reifenberg K, Cheng F et al. 2007. Deficiency of glutathione peroxidase-1 accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 27(4):850–857. [DOI] [PubMed] [Google Scholar]
- Tujebajeva RM, Copeland PR, Xu XM, Carlson BA, Harney JW, Driscoll DM, Hatfield DL, Berry MJ. 2000. Decoding apparatus for eukaryotic selenocysteine insertion. EMBO Rep. 1(2):158–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turanov AA, Shchedrina VA, Everley RA, Lobanov AV, Yim SH, Marino SM, Gygi SP, Hatfield DL, Gladyshev VN. 2014. Selenoprotein S is involved in maintenance and transport of multiprotein complexes. Biochem J. 462(3):555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursini F, Heim S, Kiess M, Maiorino M, Roveri A, Wissing J, Flohe L. 1999. Dual function of the selenoprotein PHGPx during sperm maturation. Science. 285(5432):1393–1396. [DOI] [PubMed] [Google Scholar]
- Ursini F, Maiorino M, Valente M, Ferri L, Gregolin C. 1982. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim Biophys Acta. 710(2):197–211. [DOI] [PubMed] [Google Scholar]
- Ushikubi F, Aiba Y, Nakamura K, Namba T, Hirata M, Mazda O, Katsura Y, Narumiya S. 1993. Thromboxane A2 receptor is highly expressed in mouse immature thymocytes and mediates DNA fragmentation and apoptosis. J Exp Med. 178(5):1825–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ustundag Y, Boyacioglu S, Haberal A, Demirhan B, Bilezikci B. 2001. Plasma and gastric tissue selenium levels in patients with Helicobacter pylori infection. J Clin Gastroenterol. 32(5):405–408. [DOI] [PubMed] [Google Scholar]
- Van Hove CL, Maes T, Joos GF, Tournoy KG. 2007. Prolonged inhaled allergen exposure can induce persistent tolerance. Am J Respir Cell Mol Biol. 36(5):573–584. [DOI] [PubMed] [Google Scholar]
- van Rhenen A, Feller N, Kelder A, Westra AH, Rombouts E, Zweegman S, van der Pol MA, Waisfisz Q, Ossenkoppele GJ, Schuurhuis GJ. 2005. High stem cell frequency in acute myeloid leukemia at diagnosis predicts high minimal residual disease and poor survival. Clin Cancer Res. 11(18):6520–6527. [DOI] [PubMed] [Google Scholar]
- Vane JR, Bakhle YS, Botting RM. 1998. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 38:97–120. [DOI] [PubMed] [Google Scholar]
- Vega L, Rodriguez-Sosa M, Garcia-Montalvo EA, Del Razo LM, Elizondo G. 2007. Non-optimal levels of dietary selenomethionine alter splenocyte response and modify oxidative stress markers in female mice. Food Chem Toxicol. 45(7):1147–1153. [DOI] [PubMed] [Google Scholar]
- Venardos KM, Perkins A, Headrick J, Kaye DM. 2007. Myocardial ischemia-reperfusion injury, antioxidant enzyme systems, and selenium: a review. Curr Med Chem. 14(14):1539–1549. [DOI] [PubMed] [Google Scholar]
- Vendeland SC, Beilstein MA, Yeh JY, Ream W, Whanger PD. 1995. Rat skeletal muscle selenoprotein W: cDNA clone and mRNA modulation by dietary selenium. Proc Natl Acad Sci U S A. 92(19):8749–8753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vong L, Ferraz JG, Panaccione R, Beck PL, Wallace JL. 2010. A pro-resolution mediator, prostaglandin D(2), is specifically up-regulated in individuals in long-term remission from ulcerative colitis. Proc Natl Acad Sci U S A. 107(26):12023–12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vunta H, Belda BJ, Arner RJ, Channa Reddy C, Vanden Heuvel JP, Sandeep Prabhu K. 2008. Selenium attenuates pro-inflammatory gene expression in macrophages. Mol Nutr Food Res. 52(11):1316–1323. [DOI] [PubMed] [Google Scholar]
- Wang C, Wang H, Luo J, Hu Y, Wei L, Duan M, He H. 2009. Selenium deficiency impairs host innate immune response and induces susceptibility to Listeria monocytogenes infection. BMC Immunol. 10:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhang L, O’Neill A, Bahamon B, Alsop DC, Mier JW, Goldberg SN, Signoretti S, Atkins MB, Wood CG et al. 2013. Cox-2 inhibition enhances the activity of sunitinib in human renal cell carcinoma xenografts. Br J Cancer. 108(2):319–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver JA, Maddox JF, Cao YZ, Mullarky IK, Sordillo LM. 2001. Increased 15-HPETE production decreases prostacyclin synthase activity during oxidant stress in aortic endothelial cells. Free Radic Biol Med. 30(3):299–308. [DOI] [PubMed] [Google Scholar]
- Weisberger AS, Suhrland LG. 1956. Studies on analogues of L-cysteine and L-cystine. III. The effect of selenium cystine on leukemia. Blood. 11(1):19–30. [PubMed] [Google Scholar]
- Weitzel F, Wendel A. 1993. Selenoenzymes regulate the activity of leukocyte 5-lipoxygenase via the peroxide tone. J Biol Chem. 268(9):6288–6292. [PubMed] [Google Scholar]
- Wen JJ, Yachelini PC, Sembaj A, Manzur RE, Garg NJ. 2006. Increased oxidative stress is correlated with mitochondrial dysfunction in chagasic patients. Free Radic Biol Med. 41(2):270–276. [DOI] [PubMed] [Google Scholar]
- Werz O, Steinhilber D. 1996. Selenium-dependent peroxidases suppress 5-lipoxygenase activity in B-lymphocytes and immature myeloid cells. The presence of peroxidase-insensitive 5-lipoxygenase activity in differentiated myeloid cells. Eur J Biochem. 242(1):90–97. [DOI] [PubMed] [Google Scholar]
- Wetzka B, Clark DE, Charnock-Jones DS, Zahradnik HP, Smith SK. 1997. Isolation of macrophages (Hofbauer cells) from human term placenta and their prostaglandin E2 and thromboxane production. Hum Reprod. 12(4):847–852. [DOI] [PubMed] [Google Scholar]
- White WB, West CR, Borer JS, Gorelick PB, Lavange L, Pan SX, Weiner E, Verburg KM. 2007. Risk of cardiovascular events in patients receiving celecoxib: a meta-analysis of randomized clinical trials. Am J Cardiol. 99(1):91–98. [DOI] [PubMed] [Google Scholar]
- Wiercinska-Drapalo A, Flisiak R, Prokopowicz D. 1999. Effects of ulcerative colitis activity on plasma and mucosal prostaglandin E2 concentration. Prostaglandins Other Lipid Mediat. 58(2–4):159–165. [DOI] [PubMed] [Google Scholar]
- Willoughby DA, Moore AR, Colville-Nash PR. 2000. Cyclopentenone prostaglandins-new allies in the war on inflammation. Nat Med. 6(2):137–138. [DOI] [PubMed] [Google Scholar]
- Won HY, Sohn JH, Min HJ, Lee K, Woo HA, Ho YS, Park JW, Rhee SG, Hwang ES. 2010. Glutathione peroxidase 1 deficiency attenuates allergen-induced airway inflammation by suppressing Th2 and Th17 cell development. Antioxid Redox Signal. 13(5):575–587. [DOI] [PubMed] [Google Scholar]
- Xue L, Salimi M, Panse I, Mjosberg JM, McKenzie AN, Spits H, Klenerman P, Ogg G. 2014. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J Allergy Clin Immunol. 133(4):1184–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. 2016. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 113(34):E4966–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB et al. 2014. Regulation of ferroptotic cancer cell death by GPX4. Cell. 156(1–2):317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Stockwell BR. 2016. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 26(3):165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdi MH, Mahdavi M, Faghfuri E, Faramarzi MA, Sepehrizadeh Z, Hassan ZM, Gholami M, Shahverdi AR. 2015. Th1 Immune Response Induction by Biogenic Selenium Nanoparticles in Mice with Breast Cancer: Preliminary Vaccine Model. Iran J Biotechnol. 13(2):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdi MH, Mahdavi M, Varastehmoradi B, Faramarzi MA, Shahverdi AR. 2012. The immunostimulatory effect of biogenic selenium nanoparticles on the 4T1 breast cancer model: an in vivo study. Biol Trace Elem Res. 149(1):22–28. [DOI] [PubMed] [Google Scholar]
- Yermakova A, O’banion M. 2000. Cyclooxygenases in the central nervous system: implications for treatment of neurological disorders. Current pharmaceutical design. 6(17):1755–1776. [DOI] [PubMed] [Google Scholar]
- Yim SH, Everley RA, Schildberg FA, Lee SG, Orsi A, Barbati ZR, Karatepe K, Fomenko DE, Tsuji PA, Luo HR et al. 2018. Role of Selenof as a Gatekeeper of Secreted Disulfide-Rich Glycoproteins. Cell Rep. 23(5):1387–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze MT, Zeh HJ, Kang R, Tang D. 2015. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol. 2(4):e1054549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamamiri-Davis F, Lu Y, Thompson JT, Prabhu KS, Reddy PV, Sordillo LM, Reddy CC. 2002. Nuclear factor-kappaB mediates over-expression of cyclooxygenase-2 during activation of RAW 264.7 macrophages in selenium deficiency. Free Radic Biol Med. 32(9):890–897. [DOI] [PubMed] [Google Scholar]
- Zamora CA, Baron DA, Heffner JE. 1993. Thromboxane contributes to pulmonary hypertension in ischemia-reperfusion lung injury. J Appl Physiol (1985). 74(1):224–229. [DOI] [PubMed] [Google Scholar]
- Zhang F, Yu W, Hargrove JL, Greenspan P, Dean RG, Taylor EW, Hartle DK. 2002. Inhibition of TNF-alpha induced ICAM-1, VCAM-1 and E-selectin expression by selenium. Atherosclerosis. 161(2):381–386. [DOI] [PubMed] [Google Scholar]
- Zhang JL, Xu B, Huang XD, Gao YH, Chen Y, Shan AS. 2016. Selenium Deficiency Affects the mRNA Expression of Inflammatory Factors and Selenoprotein Genes in the Kidneys of Broiler Chicks. Biol Trace Elem Res. 171(1):201–207. [DOI] [PubMed] [Google Scholar]
- Zhang W, Ramanathan CS, Nadimpalli RG, Bhat AA, Cox AG, Taylor EW. 1999. Selenium-dependent glutathione peroxidase modules encoded by RNA viruses. Biol Trace Elem Res. 70(2):97–116. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Guo J, Wang G, Chu Y, Hu X. 2017. Suppression of immune regulatory cells with combined therapy of celecoxib and sunitinib in renal cell carcinoma. Oncotarget. 8(1):1668–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Wang Z, Chen J, Wang W, Song D, Li S, Yang H, Xue S, Chen C. 2014. Increased levels of IL-6, IL-1beta, and TNF-alpha in Kashin-Beck disease and rats induced by T-2 toxin and selenium deficiency. Rheumatol Int. 34(7):995–1004. [DOI] [PubMed] [Google Scholar]
- Zifroni A, Treves AJ, Sachar DB, Rachmilewitz D. 1983. Prostanoid synthesis by cultured intestinal epithelial and mononuclear cells in inflammatory bowel disease. Gut. 24(7):659–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinoni F, Birkmann A, Stadtman TC, Bock A. 1986. Nucleotide sequence and expression of the selenocysteine-containing polypeptide of formate dehydrogenase (formate-hydrogen-lyase-linked) from Escherichia coli. Proc Natl Acad Sci U S A. 83(13):4650–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]