

Abstract
Cerebral edema, a common and often fatal companion to most forms of acute central nervous system disease, has been recognized since the time of ancient Egypt. Unfortunately, our therapeutic armamentarium remains limited, in part due to historic limitations in our understanding of cerebral edema pathophysiology. Recent advancements have led to a number of clinical trials for novel therapeutics that could fundamentally alter the treatment of cerebral edema. In this review, we discuss these agents, their targets, and the data supporting their use, with a focus on agents that have progressed to clinical trials.
Keywords: cerebral edema, clinical trial, antiedema drug
INTRODUCTION
Cerebral edema, defined as a net increase in brain water mass, is present in most types of acute central nervous system (CNS) injury or insult. In ischemic stroke, severe cerebral edema can increase mortality to nearly 80% (1) and is an independent risk factor for poor outcomes (2). In traumatic brain injury (TBI), brain swelling accounts for nearly 50% of all mortalities (3). In glioblastoma, peritumoral edema is a strong independent predictor for reduced survival (4–6). And in intracerebral hemorrhage (ICH), perihematoma edema volume is associated with increased morbidity and mortality (7).
All current therapies for cerebral edema are nonspecific to the underlying pathophysiology. Instead, they primarily seek to minimize critical downstream consequences of edema: mass effect and increased intracranial pressure (ICP). For example, decompressive craniectomy does not inhibit the formation of edema fluid but rather enables the brain to expand further, thereby reducing pressure. While hyperosmotic therapies such as mannitol do reduce edema fluid volume, they simply compete with, rather than inhibit, the driving forces that promote edema formation. As a consequence, these therapies are typically given only after the ICP reaches a critical level and when brain perfusion is threatened. Ideally, antiedema therapy would be given prophylactically, thereby avoiding any risk to tissue perfusion. However, this paradigm requires new antiedema drugs that block edema formation itself.
Several pharmacological agents have shown promise in preclinical models and are currently being tested in clinical trials. In this review, we briefly present the pathophysiology of cerebral edema. We then discuss several potential antiedema drugs (Table 1), but only those that have progressed to clinical trials, focusing on their mechanisms of action and the data that support their efficacy.
Table 1.
Prior and ongoing clinical trials of antiedema drugs
| Trial | Trial details | Drug | Studied population | Salient results (references) |
|---|---|---|---|---|
| NCT03000283 | Ongoing pilot, goal of 7 participants | Conivaptan | Patients with ICH greater than 20 cc not due to infection, thrombolysis, SAH, trauma or tumor who are expected to survive more than 48 hours | Currently recruiting |
| NCT02002390 | Phase 2 completed in 2014, 22 participants | Fingolimod | Patients with either supratentorial ICH of 5–30 cc or ischemic stroke Excluded if taken for surgical evacuation or if ICH due to coagulopathy, trauma, or thrombocytopenia |
In ICH, fingolimod improved neurological function and reduced edema (41) In stroke, fingolimod reduced lesion volume and lesion growth and improved neurological function at 90 days (160) |
| NCT00526214 (ACE-ICH) | Pilot completed in 2009,44 participants | Celecoxib | Patients presenting with supratentorial ICH not due to trauma, aneurysm rupture, or anticoagulation Excluded if planned surgical evacuation within 24 h |
Celecoxib reduced hematoma expansion and perihematoma edema expansion (40) |
| NCT01268683 (GAMES-PILOT) | Phase 2a completed in 2013,10 participants | Glyburide | Patients with large (82–210 cc) acute MCA or MCA/ACA ischemic stroke Excluded if patients had prior commitment to DC, treatment with IA rtPA or mechanical thrombectomy, herniation signs, or hemorrhage |
Glyburide was feasible and well tolerated with no symptomatic hypoglycemia (119) Glyburide reduced radiographic markers ofvasogenic edema (120) and improved clinical outcomes (161) |
| NCT01794182 (GAMES-RP) | Phase 2 completed in 2016, 83 participants | Patients with large (82–300 cc) acute MCA ischemic stroke Excluded if patients had prior commitment to DC, treatment with IA rtPA or mechanical thrombectomy, contralateral infarction, signs of herniation, or hemorrhage |
Primary and secondary outcomes not met In adjudicated posthoc analysis, glyburide reduced midline shift, serum MMP9, NIHSS, edema-related deaths, and 30-day all-cause mortality (43,44,121) |
|
| NCT02864953 (CHARM) | Ongoing phase 3, goal of 680 participants | Patients with large (80–300 cc) acute MCA ischemic stroke or large hemispheric infarction with NIHSS ≥10 Excluded if patients likely to have withdrawal of care on day 1, have prior commitment to DC, or have contralateral infarction |
Currently recruiting | |
| No identifier | Phase 1 completed in 1998, 17 participants | Xerecept | Patients with primary or secondary brain tumor with evidence of edema on CT; included patients had stable steroid dose and were not submitted to concomitant chemotherapy or radiation | Xerecept was well tolerated and improved neurological symptoms (137) |
| NCT00088166 | Phase 3 completed in 2008, 200 participants | Patients with histologically malignant brain tumor and ≥1 steroid side effects; included patients had stable steroid use and were not treated with surgery, radiosurgery, or radiation within 5 weeks of enrollment | Primary outcome not met Secondary outcomes met significance Xerecept reduced dexamethasone requirements, improved myopathy, and reduced risk of Cushing syndrome (138) |
|
| No identifier | Phase 2 completed in 2007, 32 participants | Bevacizumab | Patients with histologically confirmed progressive or recurrent grade III-IV glioma, post radiation therapy; patients not concomitantly treated with surgery, radiation, or chemotherapy | Bevacizumab reduced tumor cross-sectional area, radiographic markers of edema, and glucocorticoid requirements and resulted in neurological improvement (153) |
| NCT00943826 (AVAglio) | Phase 3 completed in 2015, 921 participants | Patients with newly diagnosed, histologically confirmed glioblastoma, and stable or decreasing glucocorticoid use Excluded if patients had hemorrhage or prior treatment for glioblastoma |
Bevacizumab reduced glucocorticoid use and increased the time-to-initiation of glucocorticoid treatment (146) | |
| NCT00305656 (NCT00254943) | Phase 2 completed in 2012, 31 participants | Cediranib | Patients with histologically confirmed glioblastoma and stable dose of corticosteroids | Cediranib reduced glucocorticoid use (151) Cediranib induced vessel normalization and reduced radiographic edema (152) |
Abbreviations: ACA, anterior cerebral artery; CT, computed tomography; DC, decompressive craniectomy; IA, intra-arterial; ICH, intracerebral hemorrhage; MCA, middle cerebral artery; MMP9, matrix metallopeptidase 9; NIHSS, National Institutes of Health stroke scale; rtPA, recombinant tissue plasminogen activator; SAH, subarachnoid hemorrhage.
CEREBRAL EDEMA PATHOPHYSIOLOGY
General Concepts: Ischemia and Trauma
For a detailed description of cerebral edema pathophysiology, please refer to Stokum et al. (8). Briefly, cerebral edema develops in several stages following CNS injury, although the precise details depend on the type and severity of the insult. Within minutes after injury, the neuroparenchyma exhibits cytotoxic edema (used here to refer only to cellular swelling), which consists mainly of astrocyte swelling (9, 10). Multiple ion transporters contribute to cytotoxic edema formation by mediating astrocyte osmolyte uptake, which in turn drives astrocyte water uptake. Examples of ion transporters include the sulfonylurea receptor 1–transient receptor potential melastatin 4 (SUR1-TRPM4) channel (11, 12), the sodium-potassium-chloride transporter subtype 1 transporter (13, 14), the sodium-hydrogen exchanger (15), and the excitatory amino acid transporters (16).
As an isolated rearrangement of parenchymal water, cytotoxic (cellular) edema by itself does not directly produce brain swelling. However, cytotoxic edema does generate the major driving force for downstream edema formation. By depleting extracellular sodium ions (Na+), cytotoxic edema generates a new Na+ gradient across the blood–brain barrier (BBB) that favors the influx of vascular Na+ (17). Various Na+ transporters expressed by brain endothelial cells then enable Na+ osmolytes to follow this new electrochemical gradient inward across the BBB (18–20). Water follows, resulting in the formation of a subtype of cerebral edema called ionic edema, which does result in brain swelling. In reality, cytotoxic edema is not thought to occur in isolation from ionic edema, but conceptually separating the two processes aids in understanding the role of distinct molecular mechanisms that contribute separately to the two processes.
During ionic edema formation, the BBB remains impermeable to circulating plasma proteins and erythrocytes. However, as the brain injury matures, plasma proteins appear in edema fluid due to the formation of permeability pores in the BBB. A variety of mechanisms, including vascular endothelial growth factor (VEGF) upregulation (21), matrix metalloproteinase activation (22), and changes in endothelial morphology, mediate the formation of permeability pores (23). Edema fluid that contains plasma proteins but still excludes erythrocytes is called vasogenic edema.
Peritumoral Edema
Peritumoral edema, which mostly consists of vasogenic edema, is formed by the disordered and proangiogenic tumor vasculature. The relatively unique mechanisms that underlie its formation are described below.
Relative to normal tissue, tumor vessels are serpiginous, irregular, and disorganized, resulting in large avascular areas and patchy necrosis (24). Up to 15% of tumor vessels may be mosaic, wherein the luminal wall comprises both endothelial and tumor cells (25). Strangely, some tumors contain isolated networks of vessel-like channels formed directly by tumor cells (26). The cells that form the tumor vasculature are also abnormal. Glioblastoma endothelial cells are proliferative and hypertrophic (27). Furthermore, many tumor endothelial cells and pericytes are derived from tumor stem cells rather than from stromal tissue (28–30).
Despite the increased vascularity present in many tumors, tumor perfusion is generally poor (31), in part because only 50–70% of newly formed vessels are capable of carrying erythrocytes (32). The poor perfusion provided by the abnormal tumor vasculature, combined with the heightened metabolic demand of the growing tumor, results in a hypoxic tumor microenvironment that promotes angiogenesis. In newly formed vessels, the BBB is not fully developed (33) and permits the passage of molecules up to ~550 nm (33, 34). In tumor vessels, interendothelial junctional proteins are often downregulated or undetectable (30). The increased permeability of tumor vessels encourages the extravasation of plasma, i.e., formation of vasogenic edema.
Peritumoral edema is an important barrier to tumor treatment (35). The combined mass effect of the tumor plus the peritumoral edema can drive local hydrostatic pressure in excess of 12 mm Hg (36). The increased tumor interstitial pressure reduces the hydrostatic pressure gradient between blood and tumor, inhibiting the delivery of chemotherapeutics. Furthermore, increased tumor interstitial pressure promotes bulk flow of fluid away from the tumor, which limits the efficacy of convection-enhanced delivery. Unsurprisingly, the magnitude of peritumoral edema is highly predictive of reduced patient survival (37).
CLINICAL TRIAL DESIGN
A number of antiedema agents have progressed to human clinical trial. However, the design of antiedema drug trials is still evolving as lessons are learned from the recent trials focused on this target. Here, we present several aspects of trial design that are salient to antiedema drugs.
Adequate preclinical animal experiments are critical prior to any clinical trial. Experiments should be performed using multiple animal models and should be conducted in multiple laboratories to ascertain drug dosing, time window, efficacy, and drug accumulation in the target organ. Several previous trials have suffered from inadequate preclinical data. In the head injury trials (HITs), nimodipine, an L-type voltage-gated calcium channel blocker, was tested in TBI without any preclinical data (38). The HITs failed to show any therapeutic benefit, and in the case of HIT-2, nimodipine was associated with worsened outcome. In the Selfotel (CGS-19755) TBI trial where a glutamate N-methyl-d-aspartate receptor antagonist was tested, the primary outcome failed. However, since it was unknown whether peripherally administered Selfotel accumulated in the CNS, the negative result was difficult to interpret (38). Exploration of intermediate or pharmacodynamic end points that may be relevant to human translation is often helpful. For example, imaging markers of water content or plasma biomarkers may help identify potential candidate biomarkers in humans.
Well-designed inclusion and exclusion criteria can strongly influence the ability of a study to detect a therapeutic effect. It is important to identify patients that are likely to have the biological target of interest. Framing the study design so that the population is enriched with patients with a high likelihood of developing the problem of interest can maximize both the chance of drug effect and the magnitude of that effect, assuming the target problem has a causal relationship with clinical outcome. In addition, patients with relatively mild or relatively severe CNS insults are very likely to either improve or deteriorate, regardless of therapeutic intervention. Thus, if patient outcome is the desired end point, the ideal cohort may consist of patients that lie on an inflection point of disease severity (38). Even with stringent inclusion/exclusion criteria, patient matching may be difficult due to disease-related heterogeneity. For example, the heterogeneity of TBI patients has complicated data interpretation in prior TBI clinical trials (39). To address this problem, some have suggested stratification by clinically important, disease-specific variables, such as hemorrhagic shock or the presence of intraventricular hemorrhage (39).
Appropriate end point selection is a major issue in antiedema drug trial design. A number of previous trials have utilized radiographic end points, such as the quantification of edema using computed tomography ( NCT03000283) (40) or using edema-sensitive MRI sequences such as gradient echo and fluid-attenuated inversion recovery (FLAIR) (41). Although several radiographic measurements are broadly accepted for cerebral edema, few have been validated as a quantitative end point for clinical trials. Ipsilateral swelling, lesional swelling, and midline shift are valuable radiographic markers (42, 43), but the relationship between these parameters and the magnitude of edema has yet to be established. Conversely, a clear association between midline shift and patient survival was established in a clinical trial of intravenous (IV) glyburide in large hemispheric infarction (43, 44) (Figure 1).
Figure 1.
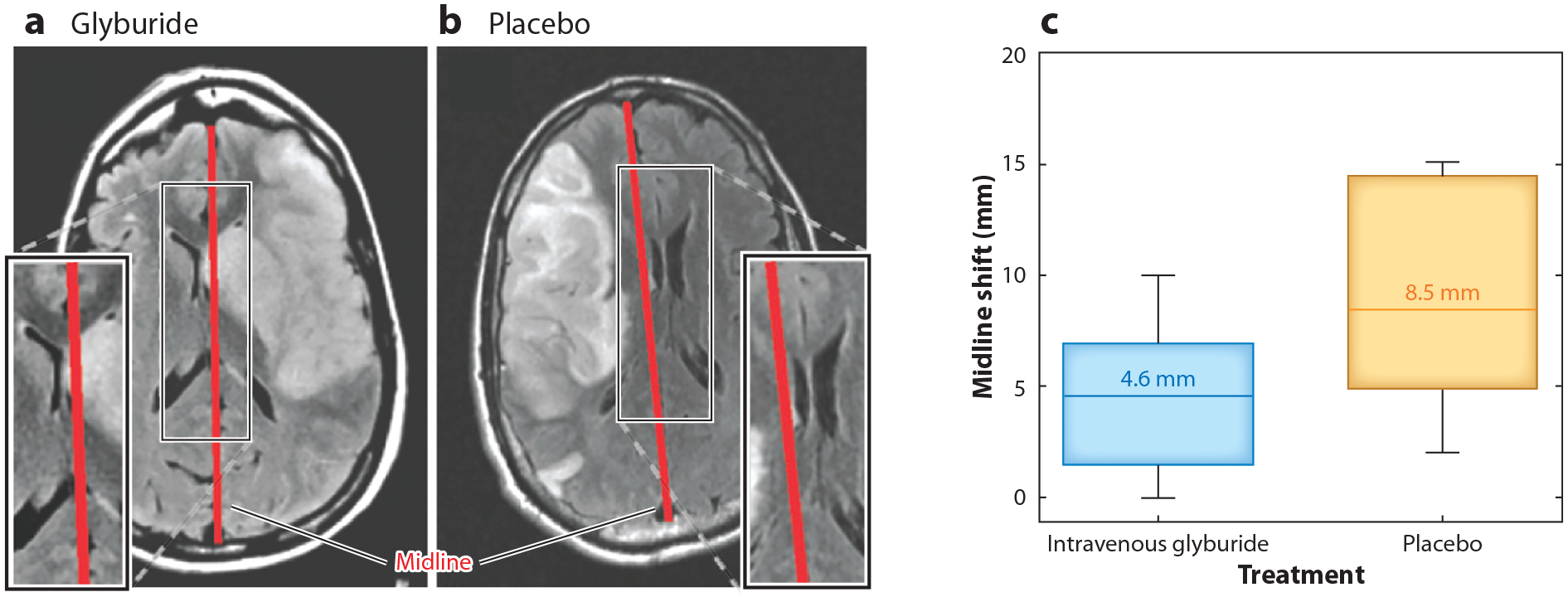
(a,b) Axial brain T2 fluid-attenuated inversion recovery MRI images from the Glyburide Advantage in Malignant Edema and Stroke study ( NCT01794182) showing midline (red bar) shift in patients with large middle cerebral artery territory ischemic stroke who were given intravenous (IV) (a) glyburide or (b) placebo. (c) Graph showing the reduced median midline shift in patients given IV glyburide, with boxes depicting interquartile range, whiskers representing 10th to 90th percentiles, and bars showing 95% confidence intervals. Figure adapted with permission from Reference 43.
Several antiedema drug trials have used patient outcome end points, such as in-hospital mortality ( NCT03000283) and clinical improvement as measured by the modified Rankin Scale, Glasgow Coma Scale, or National Institutes of Health stroke scale (40, 43). While patient outcomes are essential in determining the ultimate efficacy of an intervention, these end points are vulnerable to confounding. For example, in the Glyburide Advantage in Malignant Edema and Stroke (GAMES-RP) trial, where patients with large hemispheric infarction were treated with IV glyburide versus placebo to reduce edema, the primary end point was the proportion of patients with a modified Rankin Scale (mRS) score of ≤4 at 90 days without decompressive craniectomy (43). The study was complicated by significant intercenter variability in the application of decompressive craniectomy, with over 90% of the surgeries performed by approximately 50% of the study sites. Intercenter variability has complicated other trials, such as the TBI tirilazad trial in which intercenter variability accounted for over 40% of the study variance (38). This is especially true for nonrandom variability that occurs postrandomization.
ARGININE VASOPRESSIN AND THE VAPTANS
Arginine Vasopressin Is a Central Mediator of Brain Edema
Arginine vasopressin (AVP), a nine–amino acid peptide primarily produced by the posterior pituitary, was detected in mammalian cerebrospinal fluid (CSF) in 1978 (45, 46). In early experiments, CSF AVP increased after CSF harvest, which indicated a possible role for AVP in the regulation of brain water content (45, 46).
In the mammalian CNS, vasopressin exerts its effects primarily via the V1 receptor (47, 48), which is widely expressed throughout the adult brain (49, 50). AVP reaches the neuroparenchyma through multiple routes: Circulating AVP can be transported across the BBB via a carrier-mediated system (51); AVP can be centrally secreted by neurons and the choroid plexus epithelium (52, 53); and hypothalmo-extrahypophyseal vasopressin pathways innervate the ventricular walls, which may release AVP into ventricular CSF (54).
In the healthy brain, exogenous AVP delivered into the ventricles modestly increases brain water content by ~1.3% (55). AVP mediates changes in brain water content by regulating capillary permeability (56), astrocyte volume (57, 58), CSF production and absorption (59, 60), and cerebral blood flow (61). In contrast to its modest role in the healthy brain, central AVP signaling is a potent regulator of edema in the injured brain. In models of cerebral ischemia and TBI, AVP and its V1 receptor are upregulated (53, 62, 63). Experiments have consistently shown that AVP worsens cerebral edema in models of ischemic stroke (47, 48, 62, 64), after brain cryoinjury (65), and in models of TBI (66).
Circulating AVP also worsens brain edema, albeit indirectly. Among all hospitalized patients, hyponatremia, even when asymptomatic, is associated with increased brain edema and worsened mortality (67, 68). Hyponatremia is present in ~10% of patients with TBI and in ~20% of patients with subarachnoid hemorrhage (69). The syndrome of inappropriate antidiuretic hormone secretion (SIADH) is the underlying etiology of hyponatremia in ~62% of neurosurgical patients (69). SIADH occurs mainly through AVP stimulation of renal V2 receptors, resulting in antidiuresis and euvolemic hyponatremia (70).
Vaptans: Arginine Vasopressin Receptor Antagonists
Vaptans are nonpeptide small-molecule inhibitors of vasopressin receptors, with varying receptor subtype specificity. Two vaptans—conivaptan, a V1a and V2 receptor antagonist, and tolvaptan, a V2 specific antagonist—are currently approved to treat hyponatremia (71).
Preclinical experiments have shown that vasopressin receptor inhibition effectively reduces cerebral edema formation after CNS injury. Vasopressin antagonism reduces brain edema formation after cardiac arrest (72) and in models of ischemic stroke (47), subarachnoid hemorrhage (73), and TBI (66, 74, 75). Additional studies have found that vaptans exert their antiedema effects mainly via the inhibition of V1 receptors (47, 48) and through the regulation of aquaporin-4 (48), an aquaporin expressed in the CNS that plays a major role in edema dynamics (8).
Currently, there are limited clinical data supporting the use of vaptans for the treatment of brain edema. One case report demonstrated reduced ICP in a patient with occlusive carotid dissection treated with a vaptan (76). Another case report showed reduced edema in a patient with a midbrain and thalamic hemorrhage who was treated with a vaptan (77). There is an ongoing clinical trial ( NCT03000283) to test the efficacy of conivaptan in the treatment of cerebral edema in patients with nontraumatic ICH.
SPHINGOSINE-1-PHOSPHATE AND FINGOLIMOD
Sphingosine-1-Phosphate Signaling and Cerebral Edema
Sphingosine-1-phosphate (S1P) is a sphingolipid derivative that signals through S1P receptor subtypes 1–5 (S1P1–5) (78). Circulating S1P is derived from multiple sources, including platelets (79), erythrocytes (80), and endothelial cells (81). In the healthy CNS, S1P receptors are widely expressed by all cell types (82). Following CNS injury and during neuroinflammation, S1P and S1P receptors are upregulated in the CNS (83, 84).
S1P signaling is complex and has different roles in different cell types. Classically, S1P was considered critical in the regulation of lymphocyte trafficking. Via S1P1, S1P signaling is necessary for the egress of lymphocytes from peripheral lymphoid tissues (85). Consequently, S1P1 knockout lymphocytes become sequestered in lymphoid tissues (86).
There is growing recognition of the nonimmunological roles of S1P signaling. In endothelial cells, the major S1P receptor subtypes are S1P1, S1P2, and S1P3 (87), which regulate vascular and BBB permeability. S1P1 receptor signaling is particularly important in the development and maintenance of the vascular barrier via its effects upon the actin cytoskeleton and endothelial morphology (88). S1P1 knockout mice die around embryonic day 12–14 from hemorrhage due to inhibited vessel maturation (89). In conditions of anaphylaxis, histamine stimulation, or inflammation, erythrocyte-derived S1P stimulates endothelial S1P1 to help maintain vascular integrity (90).
Interestingly, in contrast to S1P1, endothelial S1P2 disrupts intercellular adherens junctions and promotes increased vascular permeability (91). During inflammation, S1P2 is upregulated, which could reflect a mechanism that enables context-specific tuning of endothelial permeability (87). Vascular S1P3 regulates vascular tone and perfusion by mediating cytoskeletal changes and activation of endothelial nitric oxide synthetase (92, 93).
Fingolimod: S1P Receptor Modulator
Fingolimod (FTY720) is an S1P receptor modulator that was approved in 2010 to treat multiple sclerosis (94–98). In vivo, FTY720 is activated to fingolimod phosphate by sphingosine kinase 2. Upon ligation of S1P1 and S1P3–5 receptors, fingolimod briefly acts as an agonist (99). However, upon fingolimod stimulation, S1P receptors are internalized, thereby quenching their biological activity (86). Thus, fingolimod ultimately acts as a functional S1P receptor antagonist.
In preclinical experiments, fingolimod reduced cerebral edema in models of ICH (100, 101) and ischemic stroke (102). Unfortunately, due to the pleiotropic roles of S1P in the cerebral vasculature and circulating immune cells, the precise mechanism of its antiedema effects is unclear.
There is growing clinical evidence in support of fingolimod as an antiedema drug, particularly following ICH. In one study that included 23 patients with ICH, fingolimod was shown to improve neurological status, perihematomal edema, and three-month mRS scores (41, 103). In a second study that included 22 patients with acute ischemic stroke, fingolimod improved neurological status and reduced microvascular permeability (104). Fingolimod is relatively well tolerated in patients, although it is linked with some instances of minor infections, bradycardia, and decreased pulmonary function (99).
CYCLOOXYGENASE AND NONSTEROIDAL ANTI-INFLAMMATORY DRUGS
Cyclooxygenase (COX) enzymes process arachidonic acid to generate proinflammatory prostaglandins and thromboxanes and come in three isoforms: COX1–3. Inflammation plays a key role in the pathophysiology of many CNS injuries and is particularly important in ICH (105). After ICH, COX2 is upregulated in the endothelium and invading leukocytes (106). In animal models of ICH, COX2 worsens neuronal death, neurological outcome, infarct volume, and brain edema (107, 108).
Two studies have examined the efficacy of the COX2 inhibitor celecoxib in reducing hematoma volume and cerebral edema after ICH. In a retrospective study of patients with ICH given celecoxib versus no celecoxib, celecoxib was found to reduce edema volume and hematoma expansion (109). In a randomized prospective study where patients with ICH were treated with celecoxib (n = 20) versus standard therapy (n = 24), celecoxib reduced perihematomal edema and hematoma expansion (40).
SUR1-TRPM4 AND GLYBURIDE
SUR1-TRPM4 is a monovalent cation channel that is de novo upregulated after CNS injury. The pore-forming subunit of the SUR1-TRPM4 channel is composed of TRPM4, a constitutively expressed monovalent cation channel that opens in response to increased intracellular calcium (11, 110, 111). After injury, SUR1, an adenosine triphosphate (ATP)-binding cassette, is de novo upregulated and coassociates with TRPM4, which doubles TRPM4 calcium sensitivity and sensitizes TRPM4 to intracellular ATP depletion (11, 110, 112).
In conditions of ATP depletion, such as acute CNS injury, SUR1-TRPM4 mediates the influx of Na+ osmolytes, resulting in oncotic cell swelling and cell death (11, 12, 113). This ionic redistribution promotes transcapillary ion and water influx, driving brain edema and brain swelling (8). Furthermore, SUR1-TRPM4 also mediates the oncotic cell death of the capillary endothelium, resulting in capillary fragmentation, secondary hemorrhage, and worsened edema (114).
Glyburide is a sulfonylurea drug that inhibits SUR1-containing channel complexes. When given after cerebral ischemia, glyburide inhibits newly expressed SUR1-TRPM4 channels in the BBB (20). Glyburide reduces brain edema in animal models of ischemic stroke (20, 115), TBI (116), and subarachnoid hemorrhage (117). SUR1 inhibitors were also found to decrease peritumoral edema in animal models of cerebral metastases (118).
Several clinical trials have sought to assess the efficacy of glyburide for the treatment of malignant cerebral edema after large hemispheric infarction. In the first trial—the GAMES pilot—10 patients with large anterior circulation stroke were treated with IV glyburide, demonstrating treatment feasibility (119). A follow-up analysis of the GAMES pilot data showed reduced T2 FLAIR ratio and reduced water diffusivity in the ischemic tissue, indicating that glyburide reduced vasogenic edema (120). In the phase 2 GAMES-RP trial (43), patients 18–80 years old with large (80–300 cm3) anterior circulation infarctions were randomized to glyburide (n = 41) versus placebo (n = 36). The primary outcome was the proportion of patients with mRS scores of 0–4 at 90 days without decompressive craniectomy. Secondary outcomes included the proportion of patients that underwent decompressive craniectomy or were dead within 14 days and the change from baseline in ipsilateral hemispheric or lesional swelling within 72–96 h measured by MRI. The primary end point was not met, possibly due to high intercenter variability in the application of surgical decompression (90% of the surgeries in the trial occurred in half of the trial sites). However, glyburide was shown to improve mortality at 30 days, reduce median midline shift from 8.5 to 4.6 mm (Figure 1), and lower total plasma matrix metallopeptidase 9 levels. Furthermore, posthoc analyses showed significantly reduced adjudicated neurological and edema-related deaths as well as favorable long-term outcomes in patients <70 years old (44, 121). The phase 3 Study to Evaluate the Efficacy and Safety of Intravenous BIIB093 (IV glyburide) for Severe Cerebral Edema Following Large Hemispheric Infarction (CHARM) is currently recruiting patients ( NCT02864953). The prespecified outcome in the CHARM trial does not include surgical decompression and instead includes the mRS score at 90 days and the reduction of midline shift at 72 h.
CORTICOSTEROIDS AND XERECEPT FOR PERITUMORAL EDEMA
Dexamethasone
The first documented use of corticosteroids to treat edema was in 1957 when they were used in patients with cerebral breast cancer metastases (122). However, their use did not become widespread until the work of Joseph Galicich. In 1958, Dr. Galicich noted that BBB permeability varied diurnally with plasma cortisol levels, an observation that prompted him to treat peritumoral edema with corticosteroids (123). In 1961, his seminal work demonstrated the efficacy of dexamethasone for the treatment of peritumoral edema (124). Importantly, dexamethasone was the first drug taken to US Food and Drug Administration (FDA) approval by a neurosurgeon. While all subsequent randomized trials have tested dexamethasone for the treatment of peritumoral edema surrounding brain metastases (125–127), corticosteroids are now used in a variety of brain neoplasms.
Dexamethasone, which diffuses freely across the BBB (128), exerts pluripotent effects on the cerebral vasculature. Corticosteroids downregulate proinflammatory cytokines (129), reduce endothelial VEGF production (130), increase vascular differentiation (131), and induce expression of tight junction proteins (132). Together, these changes reduce the permeability of tumor microvessels (133).
Unfortunately, the side effect profile of corticosteroids is a major limiting factor to their use. Peripheral edema, hyperglycemia, and Cushing’s syndrome occur in up to 15%, 72.3%, and 15% of patients, respectively (134). Thromboembolism, infections, delayed wound healing, gastrointestinal ulcers, and psychiatric issues are other common side effects (134).
Corticotrophin-Releasing Factor (Xerecept)
The side effect profile of corticosteroids prompted the development of the so-called steroid-sparing therapies. Human corticotrophin-releasing factor (hCRF), alternatively called corticorelin acetate and Xerecept, is a synthesized form of the endogenous 41–amino acid hypothalamus-derived peptide. When given peripherally, hCRF stabilizes the brain endothelium and reduces vasogenic edema in cold injury (135), and it reduces vascular permeability in rat models of glioma (136).
hCRF has been tested in human patients with brain tumors. A phase 1 trial showed improved neurological examination in 10 out of 17 patients following hCRF treatment. Hypotension was reported as a side effect in 2 out of 4 patients treated with high-dose hCRF (137). In a second study of 200 patients given hCRF versus placebo, hCRF significantly reduced dexamethasone requirements, improved myopathy symptoms, and reduced the rate of Cushing’s syndrome (138). With further study, hCRF could help to control peritumoral edema in patients with severe steroid side effects.
VEGF, BEVACIZUMAB, AND RECEPTOR TYROSINE KINASE INHIBITORS
Antiangiogenic Therapies for Glioblastoma
Tumor angiogenesis and peritumoral edema formation is primarily driven by the overexpression of VEGFs (139), which include VEGF-A–D (140). VEGFs bind to the receptor tyrosine kinase VEGF receptor (VEGFR) 1–3 and can also activate a number of alternative coreceptors (140). VEGF is a potent mediator of angiogenesis. Only 30 min after the intraparenchymal infusion of VEGF, 90% of neighboring brain vessels develop interendothelial gaps, lose basement membrane integrity, and become permeable to albumin (141). VEGF is highly upregulated in brain tumors, and its expression is strongly correlated with tumor grade (142).
Anti-VEGF Therapy Does Not Improve Survival
Various VEGF inhibitors have been developed due to its strong role in tumor angiogenesis. Bevacizumab is a monoclonal immunoglobulin G humanized antibody targeted against VEGF-A. Cediranib and enzastaurin are notable examples of small-molecule inhibitors of the tyrosine kinase VEGFR.
There have been several clinical trials assessing VEGF inhibitors for the treatment of glioblastoma. Initial findings were encouraging. In the BRAIN study, there was improved, progression-free survival (PFS) in patients with recurrent glioblastoma who were treated with bevacizumab plus irinotecan versus irinotecan alone at 6 months (42.6% versus 50.3%) (143). A follow-up trial showed 29% PFS at 6 months with bevacizumab plus irinotecan (144). These two trials led the FDA to approve bevacizumab for the treatment of recurrent glioblastoma in 2009.
Unfortunately, no follow-up study has shown any improvement in overall survival with anti-VEGF therapy (145). Both the AVAglio and RTOG 0825 studies failed to show improved survival in patients with newly diagnosed glioblastoma who were treated with anti-VEGF therapy (146, 147). Studies of receptor tyrosine kinase inhibitors have been equally disappointing. In phase 3 trials, both cediranib and enzastaurin failed to improve overall survival in patients with recurrent glioblastoma (148, 149). A recent meta-analysis of 14 clinical trials of VEGF inhibitors confirmed these disappointing findings (150).
Antiangiogenic Therapy Improves Peritumoral Edema
While anti-VEGF therapy does not improve overall survival, clinical trials have consistently shown that the inhibition of VEGF signaling reduces peritumoral edema. A number of studies reported that anti-VEGF therapy reduces corticosteroid requirements (146, 148, 151–153). Radiographically, both bevacizumab and cediranib reduced peritumoral T2 and FLAIR signals (151, 153). Cediranib also reduced tumor mass effect (152). Interestingly, the antiedema effects of anti-VEGF therapy are reversible upon discontinuation of therapy, which can result in a significant rebound of edema (152).
VEGF inhibition is thought to reduce edema by inducing normalization of brain tumor vasculature. In tumor vessel normalization, the dysregulated balance between pro- versus antiangiogenesis is shifted towards antiangiogenesis (154). In support of this hypothesis, bevacizumab treatment reduced the expression of VEGF and reduced tumor vascularity (155).
There are several important caveats to the widespread use of anti-VEGF drugs as antiedema therapies. Firstly, their effects are reversible upon discontinuation and can cause rebound edema. Secondly, tumor progression inevitably occurs with anti-VEGF therapy, and overall survival is unchanged. Progression may be due to nonangiogenic mechanisms of neovascularization, compensatory increases in non-VEGF angiogenesis (156), or heterogeneity in tumor responsiveness (157). Lastly, and most disturbingly, VEGF inhibition has been associated with greatly increased satellite tumor formation, perhaps because VEGF inhibitors worsen hypoxia and thereby promote tumor cell migration (158, 159).
VEGF inhibition has garnered great disappointment due to its poor performance in increasing patient survival. However, given the impressive effects of VEGF inhibition on peritumoral edema and the important role of peritumoral edema in patient morbidity and mortality, these drugs may deserve reconsideration. VEGF inhibitors may be useful as antiedema agents that might best be used in conjunction with other therapies.
CONCLUSION
Cerebral edema is a major cause of morbidity and mortality in patients with neurological and neurosurgical diseases. While our current therapeutic options are limited, we are presently in a period of great potential, with several new antiedema agents being tested in the clinic (Table 1). With these new agents, brain edema could be treated prophylactically to prevent the formation of edema, thereby making the terrible consequences of mass effect and increased ICP less common events.
Glyburide:
a sulfonylurea drug that inhibits SUR1-containing channel complexes, including the SUR1-TRPM4 channel
Vaptans:
a class of small-molecule vasopressin receptor inhibitors that include conivaptan and tolvaptan
Fingolimod:
a sphingosine-1-phosphate (S1P) receptor modulator that acts as a functional S1P receptor antagonist
Celecoxib:
a nonsteroidal anti-inflammatory drug inhibitor of cyclooxygenase-2
Dexamethasone:
a corticosteroid commonly used to treat peritumoral edema
Xerecept:
a synthesized form of the endogenous hypothalamic human corticotrophin-releasing factor developed to treat peritumoral edema in patients with severe steroid side effects
Bevacizumab:
a monoclonal immunoglobulin G humanized antibody targeted against vascular endothelial growth factor A
Cediranib and enzastaurin:
small-molecule inhibitors of the vascular endothelial growth factor receptor
ACKNOWLEDGMENTS
V.G. is supported by a grant from the National Institute of Neurological Disorders and Stroke (NINDS) (5R01NS061934). W.T.K. is supported by grants from NINDS (R01NS099209), Bio-gen (research grant), the American Heart Association (AHA) (17CSA33550004), and the National Aeronautics and Space Administration (17-BPBA_2-0160). K.N.S. is supported by grants from NINDS (U24NS107136, U24NS107215, U01NS106513, and RO1NR018335) and the AHA (17CSA33550004). J.M.S. is supported by grants from the National Heart, Lung, and Blood Institute (R01HL082517) and NINDS (R01NS060801, R01NS102589, and R01NS105633).
Footnotes
DISCLOSURE STATEMENT
J.M.S. holds a US patent (7,285,574) and is a member of the scientific advisory board of and holds shares in Remedy Pharmaceuticals. W.T.K. and K.N.S. have received grants from Remedy Pharmaceuticals.
LITERATURE CITED
- 1.Kochanek KD, Xu J, Murphy SL, Minino AM, Kung HC. 2011. Deaths: final data for 2009. Natl. Vital Stat. Rep 60:1–116 [PubMed] [Google Scholar]
- 2.Battey TW, Karki M, Singhal AB, Wu O, Sadaghiani S, et al. 2014. Brain edema predicts outcome after nonlacunar ischemic stroke. Stroke 45:3643–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Donkin JJ, Vink R. 2010. Mechanisms of cerebral edema in traumatic brain injury: therapeutic developments. Curr. Opin. Neurol 23:293–99 [DOI] [PubMed] [Google Scholar]
- 4.Wu CX, Lin GS, Lin ZX, Zhang JD, Liu SY, Zhou CF. 2015. Peritumoral edema shown by MRI predicts poor clinical outcome in glioblastoma. World J. Surg. Oncol 13:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hammoud MA, Sawaya R, Shi W, Thall PF, Leeds NE. 1996. Prognostic significance of preoperative MRI scans in glioblastoma multiforme. J. Neurooncol 27:65–73 [DOI] [PubMed] [Google Scholar]
- 6.Pope WB, Sayre J, Perlina A, Villablanca JP, Mischel PS, Cloughesy TF. 2005. MR imaging correlates of survival in patients with high-grade gliomas. Am. J. Neuroradiol 26:2466–74 [PMC free article] [PubMed] [Google Scholar]
- 7.Arima H, Wang JG, Huang Y, Heeley E, Skulina C, et al. 2009. Significance of perihematomal edema in acute intracerebral hemorrhage: the INTERACT trial. Neurology 73:1963–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stokum JA, Gerzanich V, Simard JM. 2016. Molecular pathophysiology of cerebral edema. J. Cereb. Blood Flow Metab 36:513–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Norenberg MD. 1994. Astrocyte responses to CNS injury. J. Neuropathol. Exp. Neurol 53:213–20 [DOI] [PubMed] [Google Scholar]
- 10.Risher WC, Andrew RD, Kirov SA. 2009. Real-time passive volume responses of astrocytes to acute osmotic and ischemic stress in cortical slices and in vivo revealed by two-photon microscopy. Glia 57:207–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen M, Dong Y, Simard JM. 2003. Functional coupling between sulfonylurea receptor type 1 and a nonselective cation channel in reactive astrocytes from adult rat brain. J. Neurosci 23:8568–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen M, Simard JM. 2001. Cell swelling and a nonselective cation channel regulated by internal Ca2+ and ATP in native reactive astrocytes from adult rat brain. J. Neurosci 21:6512–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Su G, Kintner DB, Flagella M, Shull GE, Sun D. 2002. Astrocytes from Na+-K+-Cl− cotransporter-null mice exhibit absence of swelling and decrease in EAA release. Am. J. Physiol. Cell Physiol 282:C1147–60 [DOI] [PubMed] [Google Scholar]
- 14.Su G, Kintner DB, Sun D. 2002. Contribution of Na+-K+-Cl− cotransporter to high-[K+]o- induced swelling and EAA release in astrocytes. Am. J. Physiol. Cell Physiol 282:C1136–46 [DOI] [PubMed] [Google Scholar]
- 15.Jakubovicz DE, Klip A. 1989. Lactic acid-induced swelling in C6 glial cells via Na+/H+ exchange. Brain Res. 485:215–24 [DOI] [PubMed] [Google Scholar]
- 16.Hansson E, Muyderman H, Leonova J, Allansson L, Sinclair J, et al. 2000. Astroglia and glutamate in physiology and pathology: aspects on glutamate transport, glutamate-induced cell swelling and gap-junction communication. Neurochem. Int 37:317–29 [DOI] [PubMed] [Google Scholar]
- 17.Mori K, Miyazaki M, Iwase H, Maeda M. 2002. Temporal profile of changes in brain tissue extracellular space and extracellular ion (Na+, K+) concentrations after cerebral ischemia and the effects of mild cerebral hypothermia. J. Neurotrauma 19:1261–70 [DOI] [PubMed] [Google Scholar]
- 18.Kitayama J, Kitazono T, Yao H, Ooboshi H, Takaba H, et al. 2001. Inhibition of Na+/H+ exchanger reduces infarct volume of focal cerebral ischemia in rats. Brain Res. 922:223–28 [DOI] [PubMed] [Google Scholar]
- 19.Yan Y, Dempsey RJ, Flemmer A, Forbush B, Sun D. 2003. Inhibition of Na+-K+-Cl− cotransporter during focal cerebral ischemia decreases edema and neuronal damage. Brain Res. 961:22–31 [DOI] [PubMed] [Google Scholar]
- 20.Simard JM, Chen M, Tarasov KV, Bhatta S, Ivanova S, et al. 2006. Newly expressed SUR1-regulated NCCa-ATP channel mediates cerebral edema after ischemic stroke. Nat. Med 12:433–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kovacs Z, Ikezaki K, Samoto K, Inamura T, Fukui M. 1996. VEGF and flt: expression time kinetics in rat brain infarct. Stroke 27:1865–72 [DOI] [PubMed] [Google Scholar]
- 22.Mun-Bryce S, Rosenberg GA. 1998. Matrix metalloproteinases in cerebrovascular disease. J. Cereb. Blood Flow Metab 18:1163–72 [DOI] [PubMed] [Google Scholar]
- 23.Garcia JG, Siflinger-Birnboim A, Bizios R, Del Vecchio PJ, Fenton JW 2nd, Malik AB. 1986. Thrombin-induced increase in albumin permeability across the endothelium. J. Cell Physiol 128:96–104 [DOI] [PubMed] [Google Scholar]
- 24.Vajkoczy P, Menger MD. 2000. Vascular microenvironment in gliomas. J. Neurooncol 50:99–108 [DOI] [PubMed] [Google Scholar]
- 25.Chang YS, di Tomaso E, McDonald DM, Jones R, Jain RK, Munn LL. 2000. Mosaic blood vessels in tumors: frequency of cancer cells in contact with flowing blood. PNAS 97:14608–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mao JM, Liu J, Guo G, Mao XG, Li CX. 2015. Glioblastoma vasculogenic mimicry: signaling pathways progression and potential anti-angiogenesis targets. Biomark. Res 3:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nyström SHM. 1959. Electron microscopical structure of the wall of small blood vessels in human multiform glioblastoma. Nature 184:65. [PubMed] [Google Scholar]
- 28.Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, et al. 2013. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 153:139–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, et al. 2010. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 468:829–33 [DOI] [PubMed] [Google Scholar]
- 30.Liebner S, Fischmann A, Rascher G, Duffner F, Grote EH, et al. 2000. Claudin-1 and claudin-5 expression and tight junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropathol. 100:323–31 [DOI] [PubMed] [Google Scholar]
- 31.Groothuis DR, Pasternak JF, Fischer JM, Blasberg RG, Bigner DD, Vick NA. 1983. Regional measurements of blood flow in experimental RG-2 rat gliomas. Cancer Res. 43:3362–67 [PubMed] [Google Scholar]
- 32.Bernsen HJ, Rijken PF, Oostendorp T, van der Kogel AJ. 1995. Vascularity and perfusion of human gliomas xenografted in the athymic nude mouse. Br. J. Cancer 71:721–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, et al. 1998. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. PNAS 95:4607–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vajkoczy P, Schilling L, Ullrich A, Schmiedek P, Menger MD. 1998. Characterization of angiogenesis and microcirculation of high-grade glioma: an intravital multifluorescence microscopic approach in the athymic nude mouse. J. Cereb. Blood Flow Metab 18:510–20 [DOI] [PubMed] [Google Scholar]
- 35.Gerstner ER, Duda DG, di Tomaso E, Ryg PA, Loeffler JS, et al. 2009. VEGF inhibitors in the treatment of cerebral edema in patients with brain cancer. Nat. Rev. Clin. Oncol 6:229–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee CG, Heijn M, di Tomaso E, Griffon-Etienne G, Ancukiewicz M, et al. 2000. Anti-vascular endothelial growth factor treatment augments tumor radiation response under normoxic or hypoxic conditions. Cancer Res. 60:5565–70 [PubMed] [Google Scholar]
- 37.Carrillo JA, Lai A, Nghiemphu PL, Kim HJ, Phillips HS, et al. 2012. Relationship between tumor enhancement, edema, IDH1 mutational status, MGMT promoter methylation, and survival in glioblastoma. Am. J. Neuroradiol 33:1349–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Narayan RK, Michel ME, Ansell B, Baethmann A, Biegon A, et al. 2002. Clinical trials in head injury. J. Neurotrauma 19:503–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marshall LF, Maas AI, Marshall SB, Bricolo A, Fearnside M, et al. 1998. A multicenter trial on the efficacy of using tirilazad mesylate in cases of head injury. J. Neurosurg 89:519–25 [DOI] [PubMed] [Google Scholar]
- 40.Lee SH, Park HK, Ryu WS, Lee JS, Bae HJ, et al. 2013. Effects of celecoxib on hematoma and edema volumes in primary intracerebral hemorrhage: a multicenter randomized controlled trial. Eur. J. Neurol 20:1161–69 [DOI] [PubMed] [Google Scholar]
- 41.Fu Y, Hao J, Zhang N, Ren L, Sun N, et al. 2014. Fingolimod for the treatment of intracerebral hemorrhage: a 2-arm proof-of-concept study. JAMA Neurol. 71:1092–101 [DOI] [PubMed] [Google Scholar]
- 42.Yoo AJ, Sheth KN, Kimberly WT, Chaudhry ZA, Elm JJ, et al. 2013. Validating imaging biomarkers of cerebral edema in patients with severe ischemic stroke. J. Stroke Cerebrovasc. Dis 22:742–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sheth KN, Elm JJ, Molyneaux BJ, Hinson H, Beslow LA, et al. 2016. Safety and efficacy of intravenous glyburide on brain swelling after large hemispheric infarction (GAMES-RP): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 15:1160–69 [DOI] [PubMed] [Google Scholar]
- 44.Kimberly WT, Bevers MB, von Kummer R, Demchuk AM, Romero JM, et al. 2018. Effect of IV glyburide on adjudicated edema endpoints in the GAMES-RP trial. Neurology 91:e2163–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dogterom J, Van Wimersma Greidanus TB, Swabb DF. 1977. Evidence for the release of vasopressin and oxytocin into cerebrospinal fluid: measurements in plasma and CSF of intact and hypophysectomized rats. Neuroendocrinology 24:108–18 [DOI] [PubMed] [Google Scholar]
- 46.Dogterom J, van Wimersma Greidanus TB, De Wied D. 1978. Vasopressin in cerebrospinal fluid and plasma of man, dog, and rat. Am. J. Physiol 234:E463–67 [DOI] [PubMed] [Google Scholar]
- 47.Vakili A, Kataoka H, Plesnila N. 2005. Role of arginine vasopressin V1 and V2 receptors for brain damage after transient focal cerebral ischemia. J. Cereb. Blood Flow Metab 25:1012–19 [DOI] [PubMed] [Google Scholar]
- 48.Liu X, Nakayama S, Amiry-Moghaddam M, Ottersen OP, Bhardwaj A. 2010. Arginine-vasopressin V1 but not V2 receptor antagonism modulates infarct volume, brain water content, and aquaporin-4 expression following experimental stroke. Neurocrit. Care 12:124–31 [DOI] [PubMed] [Google Scholar]
- 49.Corbani M, Marir R, Trueba M, Chafai M, Vincent A, et al. 2018. Neuroanatomical distribution and function of the vasopressin V1B receptor in the rat brain deciphered using specific fluorescent ligands. Gen. Comp. Endocrinol 258:15–32 [DOI] [PubMed] [Google Scholar]
- 50.Szot P, Bale TL, Dorsa DM. 1994. Distribution of messenger RNA for the vasopressin V1a receptor in the CNS of male and female rats. Mol. Brain Res 24:1–10 [DOI] [PubMed] [Google Scholar]
- 51.Zlokovic BV, Hyman S, McComb JG, Lipovac MN, Tang G, Davson H. 1990. Kinetics of arginine-vasopressin uptake at the blood-brain barrier. Biochim. Biophys. Acta 1025:191–98 [DOI] [PubMed] [Google Scholar]
- 52.Chodobski A, Loh YP, Corsetti S, Szmydynger-Chodobska J, Johanson CE, et al. 1997. The presence of arginine vasopressin and its mRNA in rat choroid plexus epithelium. Brain Res. Mol. Brain Res 48:67–72 [DOI] [PubMed] [Google Scholar]
- 53.Szmydynger-Chodobska J, Zink BJ, Chodobski A. 2011. Multiple sites of vasopressin synthesis in the injured brain. J. Cereb. Blood Flow Metab 31:47–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buijs RM, Swaab DF, Dogterom J, van Leeuwen FW. 1978. Intra- and extrahypothalamic vasopressin and oxytocin pathways in the rat. Cell Tissue Res. 186:423–33 [DOI] [PubMed] [Google Scholar]
- 55.Doczi T, Szerdahelyi P, Gulya K, Kiss J. 1982. Brain water accumulation after the central administration of vasopressin. Neurosurgery 11:402–7 [DOI] [PubMed] [Google Scholar]
- 56.Raichle ME, Grubb RL Jr. 1978. Regulation of brain water permeability by centrally-released vasopressin. Brain Res. 143:191–94 [DOI] [PubMed] [Google Scholar]
- 57.Sarfaraz D, Fraser CL. 1999. Effects of arginine vasopressin on cell volume regulation in brain astrocyte in culture. Am. J. Physiol 276:E596–601 [DOI] [PubMed] [Google Scholar]
- 58.Latzkovits L, Cserr HF, Park JT, Patlak CS, Pettigrew KD, Rimanoczy A. 1993. Effects of arginine vasopressin and atriopeptin on glial cell volume measured as 3-MG space. Am. J. Physiol 264:C603–8 [DOI] [PubMed] [Google Scholar]
- 59.Faraci FM, Mayhan WG, Heistad DD. 1990. Effect of vasopressin on production of cerebrospinal fluid: possible role of vasopressin (V1)-receptors. Am. J. Physiol 258:R94–98 [DOI] [PubMed] [Google Scholar]
- 60.Seckl JR, Lightman SL. 1991. Intracerebroventricular vasopressin reduces CSF absorption rate in the conscious goat. Exp. Brain Res 84:173–76 [DOI] [PubMed] [Google Scholar]
- 61.Fernandez N, Martinez MA, Garcia-Villalon AL, Monge L, Dieguez G. 2001. Cerebral vasoconstriction produced by vasopressin in conscious goats: role of vasopressin V1 and V2 receptors and nitric oxide. Br. J. Pharmacol 132:1837–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu X, Jin Y, Zheng H, Chen G, Tan B, Wu B. 2000. Arginine vasopressin gene expression in supraoptic nucleus and paraventricular nucleus of hypothalamus following cerebral ischemia and reperfusion. Chin. Med. Sci. J 15:157–61 [PubMed] [Google Scholar]
- 63.Szmydynger-Chodobska J, Chung I, Kozniewska E, Tran B, Harrington FJ, et al. 2004. Increased expression of vasopressin V1a receptors after traumatic brain injury. J. Neurotrauma 21:1090–102 [DOI] [PubMed] [Google Scholar]
- 64.Zeynalov E, Jones SM, Seo JW, Snell LD, Elliott JP. 2015. Arginine-vasopressin receptor blocker conivaptan reduces brain edema and blood-brain barrier disruption after experimental stroke in mice. PLOS ONE 10:e0136121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reeder RF, Nattie EE, North WG. 1986. Effect of vasopressin on cold-induced brain edema in cats. J. Neurosurg 64:941–50 [DOI] [PubMed] [Google Scholar]
- 66.Krieg SM, Trabold R, Plesnila N. 2017. Time-dependent effects of arginine-vasopressin V1 receptor inhibition on secondary brain damage after traumatic brain injury. J. Neurotrauma 34:1329–36 [DOI] [PubMed] [Google Scholar]
- 67.Arieff AI, Llach F, Massry SG. 1976. Neurological manifestations and morbidity of hyponatremia: correlation with brain water and electrolytes. Medicine 55:121–29 [DOI] [PubMed] [Google Scholar]
- 68.Gill G, Huda B, Boyd A, Skagen K, Wile D, et al. 2006. Characteristics and mortality of severe hyponatraemia—a hospital-based study. Clin. Endocrinol 65:246–49 [DOI] [PubMed] [Google Scholar]
- 69.Sherlock M, O’Sullivan E, Agha A, Behan LA, Owens D, et al. 2009. Incidence and pathophysiology of severe hyponatraemia in neurosurgical patients. Postgrad Med. J 85:171–75 [DOI] [PubMed] [Google Scholar]
- 70.Knepper MA. 1997. Molecular physiology of urinary concentrating mechanism: regulation of aquaporin water channels by vasopressin. Am. J. Physiol 272:F3–12 [DOI] [PubMed] [Google Scholar]
- 71.Kleindienst A, Hannon MJ, Buchfelder M, Verbalis JG. 2016. Hyponatremia in neurotrauma: the role of vasopressin. J. Neurotrauma 33:615–24 [DOI] [PubMed] [Google Scholar]
- 72.Nakayama S, Amiry-Moghaddam M, Ottersen OP, Bhardwaj A. 2016. Conivaptan, a selective arginine vasopressin V1a and V2 receptor antagonist attenuates global cerebral edema following experimental cardiac arrest via perivascular pool of aquaporin-4. Neurocrit. Care 24:273–82 [DOI] [PubMed] [Google Scholar]
- 73.Hockel K, Scholler K, Trabold R, Nussberger J, Plesnila N. 2012. Vasopressin V1a receptors mediate posthemorrhagic systemic hypertension thereby determining rebleeding rate and outcome after experimental subarachnoid hemorrhage. Stroke 43:227–32 [DOI] [PubMed] [Google Scholar]
- 74.Marmarou CR, Liang X, Abidi NH, Parveen S, Taya K, et al. 2014. Selective vasopressin-1a receptor antagonist prevents brain edema, reduces astrocytic cell swelling and GFAP, V1aR and AQP4 expression after focal traumatic brain injury. Brain Res. 1581:89–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Krieg SM, Sonanini S, Plesnila N, Trabold R. 2015. Effect of small molecule vasopressin V1a and V2 receptor antagonists on brain edema formation and secondary brain damage following traumatic brain injury in mice. J. Neurotrauma 32:221–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dhar R, Murphy-Human T. 2011. A bolus of conivaptan lowers intracranial pressure in a patient with hyponatremia after traumatic brain injury. Neurocrit. Care 14:97–102 [DOI] [PubMed] [Google Scholar]
- 77.Hedna VS, Bidari S, Gubernick D, Ansari S, Satriotomo I, et al. 2014. Treatment of stroke related refractory brain edema using mixed vasopressin antagonism: a case report and review of the literature. BMC Neurol. 14:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Groves A, Kihara Y, Chun J. 2013. Fingolimod: direct CNS effects of sphingosine 1-phosphate (S1P) receptor modulation and implications in multiple sclerosis therapy. J. Neurol. Sci 328:9–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yatomi Y, Yamamura S, Ruan F, Igarashi Y. 1997. Sphingosine 1-phosphate induces platelet activation through an extracellular action and shares a platelet surface receptor with lysophosphatidic acid. J. Biol. Chem 272:5291–97 [DOI] [PubMed] [Google Scholar]
- 80.Pappu R, Schwab SR, Cornelissen I, Pereira JP, Regard JB, et al. 2007. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 316:295–98 [DOI] [PubMed] [Google Scholar]
- 81.Venkataraman K, Lee YM, Michaud J, Thangada S, Ai Y, et al. 2008. Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ. Res 102:669–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Prager B, Spampinato SF, Ransohoff RM. 2015. Sphingosine 1-phosphate signaling at the blood-brain barrier. Trends Mol. Med 21:354–63 [DOI] [PubMed] [Google Scholar]
- 83.Fischer I, Alliod C, Martinier N, Newcombe J, Brana C, Pouly S. 2011. Sphingosine kinase 1 and sphingosine 1-phosphate receptor 3 are functionally upregulated on astrocytes under pro-inflammatory conditions. PLOS ONE 6:e23905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kimura A, Ohmori T, Ohkawa R, Madoiwa S, Mimuro J, et al. 2007. Essential roles of sphingosine 1-phosphate/S1P1 receptor axis in the migration of neural stem cells toward a site of spinal cord injury. Stem Cells 25:115–24 [DOI] [PubMed] [Google Scholar]
- 85.Pham THM, Okada T, Matloubian M, Lo CG, Cyster JG. 2008. S1P1 receptor signaling overrides retention mediated by Gαi-coupled receptors to promote T cell egress. Immunity 28:122–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, et al. 2004. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 427:355–60 [DOI] [PubMed] [Google Scholar]
- 87.Du J, Zeng C, Li Q, Chen B, Liu H, et al. 2012. LPS and TNF-α induce expression of sphingosine-1-phosphate receptor-2 in human microvascular endothelial cells. Pathol. Res. Pract 208:82–88 [DOI] [PubMed] [Google Scholar]
- 88.Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, et al. 2001. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J. Clin. Investig 108:689–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liu Y, Wada R, Yamashita T, Mi Y, Deng CX, et al. 2000. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J. Clin. Investig 106:951–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Camerer E, Regard JB, Cornelissen I, Srinivasan Y, Duong DN, et al. 2009. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J. Clin. Investig 119:1871–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sanchez T, Skoura A, Wu MT, Casserly B, Harrington EO, Hla T. 2007. Induction of vascular permeability by the sphingosine-1-phosphate receptor-2 (S1P2R) and its downstream effectors ROCK and PTEN. Arterioscler. Thromb. Vasc. Biol 27:1312–18 [DOI] [PubMed] [Google Scholar]
- 92.Nofer JR, van der Giet M, Tolle M, Wolinska I, von Wnuck Lipinski K, et al. 2004. HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. J. Clin. Investig 113:569–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Salomone S, Potts EM, Tyndall S, Ip PC, Chun J, et al. 2008. Analysis of sphingosine 1-phosphate receptors involved in constriction of isolated cerebral arteries with receptor null mice and pharmacological tools. Br. J. Pharmacol 153:140–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cohen JA, Chun J. 2011. Mechanisms of fingolimod’s efficacy and adverse effects in multiple sclerosis. Ann. Neurol 69:759–77 [DOI] [PubMed] [Google Scholar]
- 95.Kappos L, Radue E-W, O’Connor P, Polman C, Hohlfeld R, et al. 2010. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N. Engl. J. Med 362:387–401 [DOI] [PubMed] [Google Scholar]
- 96.Kappos L, O’Connor P, Radue E-W, Polman C, Hohlfeld R, et al. 2015. Long-term effects of fingolimod in multiple sclerosis: the randomized FREEDOMS extension trial. Neurology 84:1582–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cohen JA, Barkhof F, Comi G, Hartung H-P, Khatri BO, et al. 2010. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N. Engl. J. Med 362:402–15 [DOI] [PubMed] [Google Scholar]
- 98.Cohen JA, Khatri B, Barkhof F, Comi G, Hartung H-P, et al. 2016. Long-term (up to 4.5 years) treatment with fingolimod in multiple sclerosis: results from the extension of the randomised TRANSFORMS study. J. Neurol. Neurosurg. Psychiatry 87:468–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chun J, Hartung HP. 2010. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin. Neuropharmacol 33:91–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rolland WB, Lekic T, Krafft PR, Hasegawa Y, Altay O, et al. 2013. Fingolimod reduces cerebral lymphocyte infiltration in experimental models of rodent intracerebral hemorrhage. Exp. Neurol 241:45–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wei Y, Yemisci M, Kim HH, Yung LM, Shin HK, et al. 2011. Fingolimod provides long-term protection in rodent models of cerebral ischemia. Ann. Neurol 69:119–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lu L, Barfejani AH, Qin T, Dong Q, Ayata C, Waeber C. 2014. Fingolimod exerts neuroprotective effects in a mouse model of intracerebral hemorrhage. Brain Res. 1555:89–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li YJ, Chang GQ, Liu Y, Gong Y, Yang C, et al. 2015. Fingolimod alters inflammatory mediators and vascular permeability in intracerebral hemorrhage. Neurosci. Bull 31:755–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fu Y, Zhang N, Ren L, Yan Y, Sun N, et al. 2014. Impact of an immune modulator fingolimod on acute ischemic stroke. PNAS 111:18315–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang J, Dore S. 2007. Inflammation after intracerebral hemorrhage. J. Cereb. Blood Flow Metab 27:894–908 [DOI] [PubMed] [Google Scholar]
- 106.Gong C, Ennis SR, Hoff JT, Keep RF. 2001. Inducible cyclooxygenase-2 expression after experimental intracerebral hemorrhage. Brain Res. 901:38–46 [DOI] [PubMed] [Google Scholar]
- 107.Nagayama M, Niwa K, Nagayama T, Ross ME, Iadecola C. 1999. The cyclooxygenase-2 inhibitor NS-398 ameliorates ischemic brain injury in wild-type mice but not in mice with deletion of the inducible nitric oxide synthase gene. J. Cereb. Blood Flow Metab 19:1213–19 [DOI] [PubMed] [Google Scholar]
- 108.Chu K, Jeong SW, Jung KH, Han SY, Lee ST, et al. 2004. Celecoxib induces functional recovery after intracerebral hemorrhage with reduction of brain edema and perihematomal cell death. J. Cereb. Blood Flow Metab 24:926–33 [DOI] [PubMed] [Google Scholar]
- 109.Park HK, Lee SH, Chu K, Roh JK. 2009. Effects of celecoxib on volumes of hematoma and edema in patients with primary intracerebral hemorrhage. J. Neurol. Sci 279:43–46 [DOI] [PubMed] [Google Scholar]
- 110.Woo SK, Kwon MS, Ivanov A, Gerzanich V, Simard JM. 2013. The sulfonylurea receptor 1 (Sur1)-transient receptor potential melastatin 4 (Trpm4) channel. J. Biol. Chem 288:3655–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nilius B, Prenen J, Tang J, Wang C, Owsianik G, et al. 2005. Regulation of the Ca2+ sensitivity of the nonselective cation channel TRPM4. J. Biol. Chem 280:6423–33 [DOI] [PubMed] [Google Scholar]
- 112.Mehta RI, Ivanova S, Tosun C, Castellani RJ, Gerzanich V, Simard JM. 2013. Sulfonylurea receptor 1 expression in human cerebral infarcts. J. Neuropathol. Exp. Neurol 72:871–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Stokum JA, Kwon MS, Woo SK, Tsymbalyuk O, Vennekens R, et al. 2018. SUR1-TRPM4 and AQP4 form a heteromultimeric complex that amplifies ion/water osmotic coupling and drives astrocyte swelling. Glia 66:108–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gerzanich V, Woo SK, Vennekens R, Tsymbalyuk O, Ivanova S, et al. 2009. De novo expression of Trpm4 initiates secondary hemorrhage in spinal cord injury. Nat. Med 15:185–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Simard JM, Yurovsky V, Tsymbalyuk N, Melnichenko L, Ivanova S, Gerzanich V. 2009. Protective effect of delayed treatment with low-dose glibenclamide in three models of ischemic stroke. Stroke 40:604–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zweckberger K, Hackenberg K, Jung CS, Hertle DN, Kiening KL, et al. 2014. Glibenclamide reduces secondary brain damage after experimental traumatic brain injury. Neuroscience 272:199–206 [DOI] [PubMed] [Google Scholar]
- 117.Simard JM, Geng Z, Woo SK, Ivanova S, Tosun C, et al. 2009. Glibenclamide reduces inflammation, vasogenic edema, and caspase-3 activation after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab 29:317–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Thompson EM, Pishko GL, Muldoon LL, Neuwelt EA. 2013. Inhibition of SUR1 decreases the vascular permeability of cerebral metastases. Neoplasia 15:535–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sheth KN, Kimberly WT, Elm JJ, Kent TA, Mandava P, et al. 2014. Pilot study of intravenous glyburide in patients with a large ischemic stroke. Stroke 45:281–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kimberly WT, Battey TW, Pham L, Wu O, Yoo AJ, et al. 2014. Glyburide is associated with attenuated vasogenic edema in stroke patients. Neurocrit. Care 20:193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sheth KN, Petersen NH, Cheung K, Elm JJ, Hinson HE, et al. 2018. Long-term outcomes in patients aged ≤70 years with intravenous glyburide from the phase II GAMES-RP study of large hemispheric infarction: an exploratory analysis. Stroke 49:1457–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kofman S, Garvin JS, Nagamani D, Taylor SG 3rd. 1957. Treatment of cerebral metastases from breast carcinoma with prednisolone. J. Am. Med. Assoc 163:1473–76 [DOI] [PubMed] [Google Scholar]
- 123.McClelland S 3rd, Long DM. 2008. Genesis of the use of corticosteroids in the treatment and prevention of brain edema. Neurosurgery 62:965–67 [DOI] [PubMed] [Google Scholar]
- 124.Galicich JH, French LA. 1961. Use of dexamethasone in the treatment of cerebral edema resulting from brain tumors and brain surgery. Am. Pract. Dig. Treat 12:169–74 [PubMed] [Google Scholar]
- 125.Horton J, Baxter DH, Olson KB. 1971. The management of metastases to the brain by irradiation and corticosteroids. Am. J. Roentgenol. Radium. Ther. Nucl. Med 111:334–36 [DOI] [PubMed] [Google Scholar]
- 126.Vecht CJ, Hovestadt A, Verbiest HB, van Vliet JJ, van Putten WL. 1994. Dose-effect relationship of dexamethasone on Karnofsky performance in metastatic brain tumors: a randomized study of doses of 4, 8, and 16 mg per day. Neurology 44:675–80 [DOI] [PubMed] [Google Scholar]
- 127.Wolfson AH, Snodgrass SM, Schwade JG, Markoe AM, Landy H, et al. 1994. The role of steroids in the management of metastatic carcinoma to the brain. A pilot prospective trial. Am. J. Clin. Oncol 17:234–38 [DOI] [PubMed] [Google Scholar]
- 128.Piette C, Munaut C, Foidart JM, Deprez M. 2006. Treating gliomas with glucocorticoids: from bedside to bench. Acta Neuropathol. 112:651–64 [DOI] [PubMed] [Google Scholar]
- 129.Stellato C 2004. Post-transcriptional and nongenomic effects of glucocorticoids. Proc. Am. Thorac. Soc 1:255–63 [DOI] [PubMed] [Google Scholar]
- 130.Nauck M, Karakiulakis G, Perruchoud AP, Papakonstantinou E, Roth M. 1998. Corticosteroids inhibit the expression of the vascular endothelial growth factor gene in human vascular smooth muscle cells. Eur. J. Pharmacol 341:309–15 [DOI] [PubMed] [Google Scholar]
- 131.Guerin C, Wolff JE, Laterra J, Drewes LR, Brem H, Goldstein GW. 1992. Vascular differentiation and glucose transporter expression in rat gliomas: effects of steroids. Ann. Neurol 31:481–87 [DOI] [PubMed] [Google Scholar]
- 132.Gu YT, Qin LJ, Qin X, Xu F. 2009. The molecular mechanism of dexamethasone-mediated effect on the blood-brain tumor barrier permeability in a rat brain tumor model. Neurosci. Lett 452:114–18 [DOI] [PubMed] [Google Scholar]
- 133.Siegal T, Soti F, Biegon A, Pop E, Brewster ME. 1997. Effect of a chemical delivery system for dexamethasone (Dex-CDS) on peritumoral edema in an experimental brain tumor model. Pharm. Res 14:672–75 [DOI] [PubMed] [Google Scholar]
- 134.Hempen C, Weiss E, Hess CF. 2002. Dexamethasone treatment in patients with brain metastases and primary brain tumors: do the benefits outweigh the side-effects? Support. Care Cancer 10:322–28 [DOI] [PubMed] [Google Scholar]
- 135.Wei ET, Gao GC. 1991. Corticotropin-releasing factor: an inhibitor of vascular leakage in rat skeletal muscle and brain cortex after injury. Regul. Pept 33:93–104 [DOI] [PubMed] [Google Scholar]
- 136.Tjuvajev J, Uehara H, Desai R, Beattie B, Matei C, et al. 1996. Corticotropin-releasing factor decreases vasogenic brain edema. Cancer Res. 56:1352–60 [PubMed] [Google Scholar]
- 137.Villalona-Calero MA, Eckardt J, Burris H, Kraynak M, Fields-Jones S, et al. 1998. A phase I trial of human corticotropin-releasing factor (hCRF) in patients with peritumoral brain edema. Ann. Oncol 9:71–77 [DOI] [PubMed] [Google Scholar]
- 138.Recht L, Mechtler LL, Wong ET, O’Connor PC, Rodda BE. 2013. Steroid-sparing effect of corticorelin acetate in peritumoral cerebral edema is associated with improvement in steroid-induced myopathy. J. Clin. Oncol 31:1182–87 [DOI] [PubMed] [Google Scholar]
- 139.Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. 2007. Angiogenesis in brain tumours. Nat. Rev. Neurosci 8:610–22 [DOI] [PubMed] [Google Scholar]
- 140.Simons M, Gordon E, Claesson-Welsh L. 2016. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol 17:611–25 [DOI] [PubMed] [Google Scholar]
- 141.Dobrogowska DH, Lossinsky AS, Tarnawski M, Vorbrodt AW. 1998. Increased blood-brain barrier permeability and endothelial abnormalities induced by vascular endothelial growth factor. J. Neurocytol 27:163–73 [DOI] [PubMed] [Google Scholar]
- 142.Schmidt NO, Westphal M, Hagel C, Ergun S, Stavrou D, et al. 1999. Levels of vascular endothelial growth factor, hepatocyte growth factor/scatter factor and basic fibroblast growth factor in human gliomas and their relation to angiogenesis. Int. J. Cancer 84:10–18 [DOI] [PubMed] [Google Scholar]
- 143.Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, et al. 2009. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J. Clin. Oncol 27:4733–40 [DOI] [PubMed] [Google Scholar]
- 144.Kreisl TN, Kim L, Moore K, Duic P, Royce C, et al. 2009. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J. Clin. Oncol 27:740–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.van den Bent M, Gorlia T, Bendszus M, Sahm F, Domont J, et al. 2016. EH1.3 EORTC 26101 phase III trial exploring the combination of bevacizumab and lomustine versus lomustine in patients with first progression of a glioblastoma. Neuro-Oncology 18(Suppl. 4):iv1–2 [Google Scholar]
- 146.Chinot OL, Wick W, Mason W, Henriksson R, Saran F, et al. 2014. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med 370:709–22 [DOI] [PubMed] [Google Scholar]
- 147.Gilbert MR, Dignam J, Won M, Blumenthal DT, Vogelbaum MA, et al. 2013. RTOG 0825: phase III double-blind placebo-controlled trial evaluating bevacizumab (Bev) in patients (Pts) with newly diagnosed glioblastoma (GBM). J. Clin. Oncol 31(Suppl. 18):123129739 [Google Scholar]
- 148.Batchelor TT, Mulholland P, Neyns B, Nabors LB, Campone M, et al. 2013. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J. Clin. Oncol 31:3212–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wick W, Puduvalli VK, Chamberlain MC, van den Bent MJ, Carpentier AF, et al. 2010. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J. Clin. Oncol 28:1168–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lombardi G, Pambuku A, Bellu L, Farina M, Della Puppa A, et al. 2017. Effectiveness of antiangiogenic drugs in glioblastoma patients: a systematic review and meta-analysis of randomized clinical trials. Crit. Rev. Oncol. Hematol 111:94–102 [DOI] [PubMed] [Google Scholar]
- 151.Batchelor TT, Duda DG, di Tomaso E, Ancukiewicz M, Plotkin SR, et al. 2010. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J. Clin. Oncol 28:2817–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, et al. 2007. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 11:83–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Vredenburgh JJ, Desjardins A, Herndon JE 2nd, Dowell JM, Reardon DA, et al. 2007. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin. Cancer Res 13:1253–59 [DOI] [PubMed] [Google Scholar]
- 154.Jain RK. 2001. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat. Med 7:987–89 [DOI] [PubMed] [Google Scholar]
- 155.Tamura R, Tanaka T, Miyake K, Tabei Y, Ohara K, et al. 2016. Histopathological investigation of glioblastomas resected under bevacizumab treatment. Oncotarget 7:52423–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Peterson TE, Kirkpatrick ND, Huang Y, Farrar CT, Marijt KA, et al. 2016. Dual inhibition of Ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. PNAS 113:4470–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Batchelor TT, Gerstner ER, Emblem KE, Duda DG, Kalpathy-Cramer J, et al. 2013. Improved tumor oxygenation and survival in glioblastoma patients who show increased blood perfusion after cediranib and chemoradiation. PNAS 110:19059–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, et al. 2009. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 15:220–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rubenstein JL, Kim J, Ozawa T, Zhang M, Westphal M, et al. 2000. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia 2:306–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhu Z, Fu Y, Tian D, Sun N, Han W, et al. 2015. Combination of the immune modulator fingolimod with alteplase in acute ischemic stroke: a pilot trial. Circulation 132:1104–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sheth KN, Kimberly WT, Elm JJ, Kent TA, Yoo AJ, et al. 2014. Exploratory analysis of glyburide as a novel therapy for preventing brain swelling. Neurocrit. Care 21:43–51 [DOI] [PMC free article] [PubMed] [Google Scholar]