

We revise the interpretation of coherence signals in photosynthetic systems and clarify what they tell us about light harvesting.
Abstract
Photosynthesis is a highly optimized process from which valuable lessons can be learned about the operating principles in nature. Its primary steps involve energy transport operating near theoretical quantum limits in efficiency. Recently, extensive research was motivated by the hypothesis that nature used quantum coherences to direct energy transfer. This body of work, a cornerstone for the field of quantum biology, rests on the interpretation of small-amplitude oscillations in two-dimensional electronic spectra of photosynthetic complexes. This Review discusses recent work reexamining these claims and demonstrates that interexciton coherences are too short lived to have any functional significance in photosynthetic energy transfer. Instead, the observed long-lived coherences originate from impulsively excited vibrations, generally observed in femtosecond spectroscopy. These efforts, collectively, lead to a more detailed understanding of the quantum aspects of dissipation. Nature, rather than trying to avoid dissipation, exploits it via engineering of exciton-bath interaction to create efficient energy flow.
INTRODUCTION
Over the past decade, the field of quantum biology has seen an enormous increase in activity, with detailed studies of phenomena ranging from the primary processes in vision and photosynthesis to avian navigation (1, 2). In principle, the study of quantum effects in complex biological systems has a history stretching back to the early years of quantum mechanics (3); however, only recently has it truly taken center stage as a scientifically testable concept. While the overall discussion has wide-ranging ramifications, for the purposes of this Review, we will focus on the subfield where the debate is most amenable to direct experimental tests of purported quantum effects—photosynthetic light harvesting.
In femtosecond multidimensional spectroscopy of several pigment-protein complexes (PPCs), we find what has been widely considered the experimental signature of nontrivial quantum effects in light harvesting: oscillatory signals—the spectroscopic characteristic of “quantum coherence.” These signals, or rather their interpretation with the associated claims of a direct link to the system’s “quantumness” (4), have drawn enormous attention, much of it from scientists outside the immediate community of photosynthetic light harvesting (5). While significant efforts have been spent on interpreting these weak signals, the overall debate has raised important questions of a general nature (6). What is uniquely “quantum” in biology? What “nontrivial quantum effects” can be considered as the origin of observable biological phenomena?
While addressing these questions has been extremely productive in terms of stimulating experimental and theoretical work, it has seemingly moved the discussion in photosynthesis away from actual biological function. The strong focus on coherence, specifically, has then led to a distorted view of natural photosynthesis. We identify two underlying assumptions in recent discussions: First, it occurs in both specialist and nonspecialist literature that coherence and quantumness are taken as equivalent terms and crucial to photosynthetic function (see the “Theoretical considerations: Coherence and quantumness” section for further discussion). Second, the narrow focus on the initial femtosecond dynamics draws attention away from the fact that light harvesting is, to a large extent, ruled by processes on time scales of tens of picoseconds (7–9). Thus, the efficiency bottlenecks are not found in the subpicosecond intraprotein relaxation, but rather in the orders-of-magnitude slower processes, such as intercomplex energy transfer and subsequent energy transduction steps in the form of electron transfer at the reaction center as discussed in the “Collective excitations and energy migration in light-harvesting systems” section below (10, 11).
While it is crucial that rate-limiting processes are kept in mind, the main goal of this Review is to critically assess the persistence and role of quantum coherence in photosynthetic light harvesting. In more general terms, we believe that there is a deep understanding to be gained in tackling the emergence of the essentially classical world of biology from its quantized molecular origins. To collectively make progress in this interdisciplinary field, however, we find that having well-defined terminology and transparent definitions of fundamental concepts is of great importance. In this regard, we outline here what we consider the most useful picture of photoexcitation and energy migration in multipigment systems such as PPCs. We will then clarify the terms “coherence” and quantumness in the context of ultrafast spectroscopy of molecular systems, meanwhile posing suggestions for a transparent use of these terms. Following these definitions [see also (6)], we analyze recent work on coherence in PPCs. Although we believe that our observations here generalize to a wide range of PPCs, we pay special attention to the Fenna-Matthews-Olson (FMO) protein, a light-harvesting complex from green sulfur bacteria, which has taken on an exemplary role in quantum biology.
Collective excitations and energy migration in light-harvesting systems
In essence, photosynthetic antennae are collections of pigments, such as (bacterio)chlorophylls and carotenoids, usually held in close proximity by a protein scaffold. The coupling between the pigments results in redistribution of transition energies and oscillator strengths, and when interacting with light, the pigments can no longer act as independent units. Because of this correlation between pigments, it is customary to describe transport within PPCs in terms of collective excitations—called “excitons” when vibrational-electronic mixing is weak—whose wave functions depend on the specifics of the coupling but generally extend over more than one pigment (12).
While the spectral observables of PPCs can be calculated in any basis of quantum mechanical states—for example, using the individual pigments (site basis) or otherwise—an excitonic description is desirable, because excitons represent the stationary eigenstates of the system. It is the signals from these states that are observed, e.g., in an optical absorption spectrum, and they are distinctly different from those associated with the isolated pigments.
In Fig. 1, we depict how the tuning of pigment energies and their coupling result in the formation of delocalized excitons, whose spatial structure is used to direct energy transfer in the FMO complex. This excitonic level-to-level transfer has recently been fully mapped out (13) with the help of two-dimensional electronic spectroscopy (2DES) (14). More specifically, electrostatic interactions with the protein and solvent environment tune local pigment excitation energies (termed site energies) (15–18), and interaction between these energetically varying local states results in a ladder of excitonic states, where the higher energy states are localized toward the peripheral antenna complexes, while lower energy excitons are close to the photosynthetic reaction center (19–21). The protein and solvent environment not only act to tune the energy of the collective excitations but also play an essential function as the thermal bath into which excess energy can be dissipated. This efficient dissipation of excess energy, enabled by coupling between the excitons and vibrations (21–25), is crucial for fast and efficient energy transfer among the excitonic states.
Fig. 1. Excitation energy transfer and decay of coherences in the FMO protein.
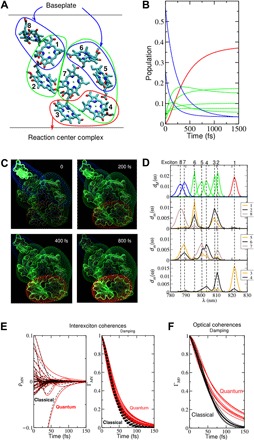
(A) Illustration of the excitation energy transfer in the FMO protein of green sulfur bacteria. The eight BChl a pigments of the monomeric subunit of the trimeric FMO protein are oriented as depicted. The excitation energy enters from the baseplate at the top and is transferred to the reaction center complex at the bottom. The blue, green, and red surroundings of the pigments indicate high-, intermediate-, and low-energy exciton states, respectively, to which the respective pigments contribute, as analyzed in detail in (D). (B) Time-dependent population of the exciton states [same color code as in (A)], assuming that the initial state is created by incoherent exciton transfer from the baseplate (section S5). (C) Time-dependent populations of local excited states, illustrated at four different times by illuminating the pigments accordingly. In addition, the exciton states are included as surroundings of pigments that appear and fade away according to the populations of these states in (B). (D) Analysis of the spatial extent of the different exciton states, using the density of exciton states dM(ω), eq. S20, shown in the top part, where the same color code is used for the different exciton states as in (A) to (D) and the exciton states pigment distribution functions dm(ω), eq. S21, shown in the lower three parts. (E) Interexciton coherences (left part) and their damping function (right part). (F) Damping functions of the optical coherences. In both (E) and (F), the coherences are initiated by assuming a δ-pulse excitation at time zero, and the quantum mechanical treatment of nuclear motion (red lines; eqs. S26, S27, S30, and S31) is compared with a classical treatment; see eqs. S33, S34, and S37, black lines. These calculations, as well as the calculations of the population transfer (B and C) were carried out for room temperature (300 K). The lower parts of figures S4, S6, and S8 show the population transfer obtained for a classical treatment of the nuclear motion, which fails to thermalize correctly. Two movies illustrating the spatial energy transfer as in (C) are available in the Supplementary Materials.
We must emphasize here that the warm, wet, and disordered environment of pigments in biological systems is far from the situation found in strongly coupled highly ordered solid-state systems, where excitations can be delocalized over the whole crystal. The interpigment coupling strength is often on the same order of magnitude as the interaction with the environment (bath), which, in combination with static disorder, results in a tendency to localize the excitation over a small number of pigments even in strongly coupled antenna complexes (e.g., order three to five pigments for LH1 and LH2) (26, 27).
Theoretical considerations: Coherence and quantumness
In the recent literature, there are extended discussions of quantumness of energy transfer and its importance, e.g., for the robustness or efficiency of photosynthetic processes. It is increasingly common to see an equivalence being made between coherence and “nontrivial” quantum effects. However, coherence is not at all uniquely quantum but is also a well-known property of classical systems, e.g., for the motion of a pendulum or the propagation of electromagnetic waves (28, 29), in which a well-defined phase relationship is maintained. As the existence of coherence by itself does not imply quantumness, its use as a descriptive term in discussions of “quantum coherent energy transport” calls for specification (6). However, the precise meaning of the term coherence is often left ambiguous, resulting in difficulty in discerning exactly what underlying physical phenomena are being discussed. In the interest of clarity, we provide a functional definition of coherence in the context of the observables in ultrafast spectroscopy as described in the following (6). It is interesting to realize that the issue of the relationships and subtle differences between coherences, “correlations,” and “intermolecular couplings,” as well as fundamental issues about how to treat the thermal equilibration of a quantum subsystem coupled to an environment (see below) also arose in great detail in the development of nuclear magnetic resonance a few decades ago, while considering coherent superpositions of spins on molecules separated by micrometers or millimeters in solution (30, 31). Moreover, it had been clarified that a classical bath does not lead to a proper thermalization at low temperature (30, 31).
Technically, the term coherence is used to denote off-diagonal elements in any density matrix. As the physical meaning of these off-diagonal matrix elements is completely dependent on the choice of basis (e.g., site or excitonic basis), however, we find this general definition too broad to be useful. Here, we thus prefer a more restrictive terminology, which essentially corresponds to the common use in recent ultrafast spectroscopy literature.
This problem of the definition of terms is well illustrated by the difficulties in communication between the experimental and theoretical communities. Specifically, we mean the difficulties arising when coherence is introduced as off-diagonal elements of the density matrix in the basis of localized pigment states. These coherences “appear” as oscillations in the site basis in theoretical simulations (fig. S5 shows an example of strongly damped oscillations in site basis coherences); however, the physical interpretation is simply that there is some degree of spatial delocalization of the excitations in the system (6, 32). This information is useful when thinking about spatial relationships and degrees of localization of the excitons. However, there are only time-dependent (oscillatory) spectroscopic signals associated with these coherences whenever it is possible to selectively photoexcite isolated pigments in a coupled system. While careful tailoring of the laser pulse amplitude and phase may allow one (for a small number of systems) to create the necessary linear superposition of system eigenstates to achieve this, this approach is obviously confined to highly specialized laboratory settings.
We find a more useful definition of coherence, and the one most closely associated with the meaning of the term in recent experimental literature, to be the off-diagonal elements in the density matrix in the basis of system eigenstates (i.e., corresponding to the excitonic basis in the absence of vibronic mixing). The physical interpretation of these elements is a measure of the degree to which the (light-induced) state of the molecular system corresponds to a linear superposition of different eigenstates (e.g., excitons) of the system. These superpositions, when excited by short laser pulses, are nonstationary, evolving in time as damped oscillations with a frequency corresponding to the energy difference between the involved eigenstates.
In the context of dynamics, it is useful to make a further distinction: We refer to the specific superposition of ground and excited states as optical coherences. The evolution of optical coherence determines the transition frequencies and homogeneous linewidth of the absorption spectrum. In optical 2D experiments, it appears during the coherence times t1 and t3 (see Figs. 2 and 3 for details). Optical coherence provides information on the system-bath interactions relevant to electronic decoherence but has no simple relationship to energy transfer, which is a meaningful concept in the site basis. However, the two are not completely unrelated either, as our analytical estimate below will show.
Fig. 2. What is a 2D spectrum?
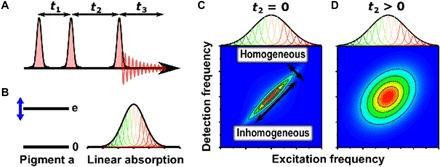
(A) In 2DES (14), a sequence of three laser pulses interacts with the sample, causing emission of a signal that is recorded as a function of the three time delays. For a given “population time” t2, the Fourier transform of the signal with respect to the t1 and t3 delays provides the respective excitation and detection frequency axes of the 2D spectrum. (B) Simple quantum two-level model for the energy levels of a photosynthetic pigment, giving rise to an inhomogeneously broadened absorption spectrum. In photosynthetic complexes, the protein environment tunes the electronic energy gap, and small conformational differences among proteins probed in an ensemble measurement cause shifts of energy gaps, broadening the absorption from its inherent homogeneous width. (C) A 2D spectrum recorded at t2 = 0 separates homogeneous and inhomogeneous broadening, which are manifest as the antidiagonal and diagonal widths, respectively. (D) At later times (t2 > 0), dynamical interactions between the pigment and protein environment lead to energy gap fluctuations that broaden the antidiagonal width in a process termed spectral diffusion. The 2D spectrum contains both absorptive and refractive responses of the system; however, usually only the real part of the 2D spectrum is presented, corresponding to the absorptive part.
Fig. 3. What can we learn from 2D spectra?
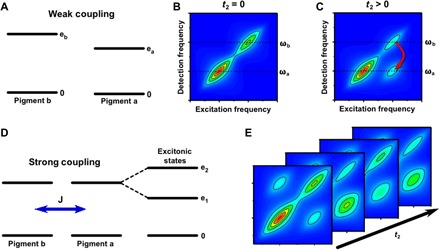
2D spectra have rich information about electronic structure and dynamics (14). Photosynthetic complexes contain light-absorbing pigments that are held in place by a protein scaffold that controls their relative distance and orientation, determining their coupling. In (A), we consider a common case of two weakly coupled pigments a and b. (B) At t2 = 0, the 2D spectrum displays peaks along the diagonal that reveal the inhomogeneously broadened peaks at ωa and ωb, corresponding to absorption by pigments a and b, respectively. (C) At later times, if pigments a and b are sufficiently close in space and favorably oriented, then energy transfer may occur between them, with higher probability that energy flows “downhill” from the higher energy state of pigment b to pigment a. The energy transfer process leads to the formation of a cross-peak in the 2D spectrum. Recording 2D spectra as a function of population time t2 enables the mapping of energy transfer pathways and time scales. In (D), we consider the case of two strongly coupled photosynthetic pigments. The strong coupling mixes the energy levels of the individual pigments, leading to excitons in which excitations are delocalized across the coupled pigments. Excitonic coupling between transitions is revealed by cross-peaks in the t2 = 0, so-called correlation spectrum. Besides the population relaxation between two excitonic states, observed as growing of the lower cross peak, coherence is manifest as t2-dependent oscillations as shown in (E). The distribution of the oscillating signals on 2D maps can provide important insight into the physical origin of the coherence as discussed in Fig. 4.
The coherences interpreted to be directly related to energy transfer correspond to superpositions of different excited states. These superpositions are nonstationary and evolve during the population time t2, e.g., between the pump and probe pulses in a transient absorption experiment. While superpositions between any set of system eigenstates (e.g., excitonic and vibronic) can be generated with the appropriate optical pulses, in particular, superpositions of excitonic states have been considered as important in the context of energy transfer. In the literature, these have been referred to as electronic or excitonic coherences. Here, we will refer to these specific coherences as interexciton coherences. Just as optical coherences, they evolve as damped oscillations in accordance with the energy differences between the involved system eigenstates. A number of factors, including the strength of the interaction with the bath, the exciton wave function overlap, and the lifetime of the involved states, determine the dephasing time of these interexciton coherences. As the strength of coupling to the bath influences the dephasing of both optical and interexcitonic coherences, fast-decaying optical coherence (observed as broad homogeneous line shapes) also indicates fast interexcitonic coherence decay. The specific relationship to make this connection depends on the degree of exciton localization and the bath dynamics and is detailed below.
The brief summary above provides what we consider a practical and useful definition of coherence as used in ultrafast spectroscopy studies. Note that these observations are valid both for classical and quantum representations, and the association of coherence in PPCs with classical oscillators turns out to be remarkably accurate (vide infra). The question remains, however: Where do we find quantumness in biology? Obviously, at the length scale of atoms, the world is governed by the wave properties of matter. The classical observables of biology are only concepts derived from this “true” quantum reality (33). But what truly quantum effects remain prominent in the classical, macroscopic world of biological systems, where much of the quantum mechanical “strangeness” is erased?
This question can be addressed by simplifying complex calculations and developing analytical theory to provide physical insight into quantum and classical aspects of the problem. The approach requires making controlled approximations to quantum complexity by introducing classical or semiclassical treatments in such a way as to preserve the fundamental bedrock of reality. These approximation methods can then be used to address the question of what quantum characteristics are essential to explain a particular phenomenon. A useful approach partitions the degrees of freedom of the full problem into those of the system of interest, and the rest involved in describing its surroundings, or the bath. The latter is typically formed by the protein scaffold and the solvent, which hosts the entire complex. There is a long history of mixing different types of models for these subsystems together with classical (34), quasiclassical (35), semiclassical (36–39), and fully quantum mechanical (40–45) descriptions of their dynamics, particularly in the context of how the spectrum of the system, and its relaxation and dissipation after excitation, can be influenced by the environment (46, 47). Such a mapping can even be made exact (37, 48). In the context of light harvesting, it has become apparent that electronic excitation may be described entirely by classical models (49) and that the coherent and incoherent regimes are common for both quantum and classical descriptions of their dynamics (50). Furthermore, it has been shown (29, 47, 51) that the excitons in PPCs can be treated as a set of classical oscillators, so even here, the strictly quantum nature of biology appears hidden. With the above qualification about mixing quantum and classical descriptions of the dynamics, it may be counterintuitive that a quantum description is most useful for the description of the bath dynamics—and in the coupling between the system and this bath (29, 52).
To capture the relevant dynamics, we built the simplest approach that yields an analytical theory (outlined in the Supplementary Materials) on an approximate quantum dynamical description of the system interacting with a classical dynamical model for the bath. This approach gives an analytic result, and we shall use it for illustrative purposes. The critical observation in this quantum system/classical bath description of the dynamics is that it fails to capture thermalization of the system (29, 52–54). In simple words, in a world where the behavior of electrons is governed by the fundamental equations of quantum mechanics and that of the nuclei by classical physics, there would be no preferential “downhill” energy flow, strongly impairing the macroscopic function of these complexes (figs. S4, S6, and S8). This relaxation process by coupling to bath modes is ultimately responsible for the directed transport of excitation energy (Fig. 1, A and C), the central function of these light-harvesting complexes. In contrast to the population of exciton states, which fail to correctly thermalize, the interexciton coherences can be well described by using a classical description of nuclear motion (Fig. 1E). Both the mixed quantum-classical and fully quantum descriptions agree that dephasing of interexciton coherences typically results in sub–100-fs decay times at room temperature. This is substantially shorter than intercomplex energy transfer times and hence cannot play any functional role.
Since the short lifetime of interexciton coherences is in the focus of this Review, in the following, we provide its theoretical foundation and possible functional implication with the above considerations by properly treating the bath interaction. The good agreement between the fully quantum mechanical and the mixed quantum-classical damping of interexciton coherences, discussed above, allows us to use the simple mixed quantum-classical expression, derived in the Supplementary Materials (sections S4 and S6), to reliably capture the general behavior. In this classical environment limit and within the Frenkel exciton model, the decay of interexciton coherence ρMN between the Mth and Nth exciton states is given as , where the first factor contains the dephasing constants τM and τN that equal twice the lifetimes of the exciton states ∣M〉 and ∣N〉, which are determined by exciton relaxation (eq. S18). In the second factor, termed pure dephasing, Eλ is the reorganization energy of the pigment’s local transition energy fluctuations, and contains the probabilities and of the mth pigment being excited in exciton states ∣M〉 and ∣N〉. The inverse of κMN is a measure for the intrinsic correlation in the fluctuations of the Mth and Nth exciton energies. For any two excited states, which delocalize exactly the same way over the same pigments, κMN becomes zero, and the correlation is perfect. For the optical coherence ρM0 (0 denotes the global ground state), the damping function reduces to , where the inverse participation ratio quantifies the delocalization of the Mth exciton state. In the limit of localized electronic states, we have ΛM = 1 and κMN = 2, there is no lifetime dephasing (; see eq. S18, because of zero spatial overlap between excited states). In this case, the interexciton coherences decay faster than the optical coherences. For completely delocalized excited states, κMN becomes zero, and we have ΛM = 1/NP, with the number of pigments Np. Thus, the pure dephasing does not affect interexciton coherences, whereas its effect on optical coherences is suppressed. In this case, it is likely that the lifetime dephasing, which is enhanced for strong spatial overlap of exciton states (eq. S19), will dominate the dephasing of both types of coherences. If so, there is a general trend that the interexciton coherences for delocalized states (and within the Frenkel exciton model) will also decay faster than the optical coherences. We note that these arguments rely on several approximations, most notably, a secular approximation (justified for the FMO complex in fig. S2), which we use here to illustrate the central issue of coherence lifetimes. To achieve a more accurate description of reality, more refined and numerically exact tools might have to be used (42, 55–58). In addition, alternative quantifiers of quantumness on the basis of energy current operators can be applied (42).
For the FMO protein, the calculated dephasing times of interexciton and optical coherences are in the range of 50 and 75 fs, respectively (Fig. 1, E and F), significantly shorter than the lifetimes of the exciton states (Fig. 1B), showing the dominance of pure dephasing processes. This result reflects the partial localization of excited states and their modest spatial overlap in this system (Fig. 1D).
With the expected extremely fast decoherence, it is appropriate to consider an alternative mechanism (beyond the spatial overlap of excitons) that could lead to long-lived interexciton coherences: correlations in site energy fluctuations of different pigments. This correlation has not been included in the theoretical considerations above. A number of studies have advocated such “environmental protection of excitonic coherence” as the source of long-lived oscillations in 2D spectra (59–61). However, no quantum mechanics/molecular mechanics based dynamic studies of the FMO protein could identify correlations in site energy fluctuations (22, 25). In a normal mode analysis of the FMO spectral density, correlations were found but only at very low vibrational frequencies (23). These correlations were calculated to have practically no influence on the populations of exciton states. Moreover, while artificially introducing these correlations for higher-frequency components of the spectral density can lead to protection of interexciton coherences, they will, at the same time, markedly hamper exciton relaxation and, thereby, the spatial transfer of excitation energy. Hence, it has to be concluded that correlations in site energy fluctuations, which would allow for long-lived interexciton coherences are detrimental for the light-harvesting function (23). Whether coherence is actually generated under natural excitation conditions (i.e., by sunlight) is still a heavily debated topic (62–64), with some works dismissing the idea of coherence under sunlight (62, 63), while others follow an early suggestion of representing sunlight by a series of ultrashort bursts (65). We would like to point out that, in a secular approximation (justified for the FMO protein; see fig. S2), the evolution of interexciton coherences is independent of the evolution of populations, obviously excluding a direct functional influence of these coherences.
From a structural point of view, it is the inhomogeneous charge distribution in the FMO protein that, on one hand, leads to varying site energies of the pigments and, on the other hand, gives rise to different local exciton-vibrational coupling constants, suppressing correlations in site energy fluctuations. The first effect is used to direct the excitation energy toward the reaction center, the second effect leads to an efficient dissipation of the excess energy of excitons. Both lead to the observation of a fast decay of interexciton coherences in femtosecond spectroscopy experiments. As the electrostatic tuning of site energies by the protein environment is used by many photosynthetic PPCs, e.g., those of higher plants (9, 66), we think that the mechanisms analyzed above for the FMO protein are quite general.
In the following, we will investigate how coherence manifests itself in experimental spectra—oscillatory features in specific spectral regions—and critically evaluate the interpretation of these experimental observables for the representative FMO case. We will point out that the interpretation of these oscillations as originating from superpositions of exciton states (rather than from vibrations) is incorrect and needs to be revised on the basis of several experimental and theoretical studies (39, 67–69).
Experimental considerations: Coherence in ultrafast spectroscopy
When experimentally addressing nonstationary coherences, it is important to understand how these coherences, appearing as oscillating signals and referred to here as “quantum beats” (QBs), are excited. Fundamentally, observation of QBs requires a laser spectrum broad enough to cover transitions of all the states involved in the coherence, in other words, the laser must contain the resonance frequencies of all involved oscillators. In addition, the laser pulses have to be equal or shorter than the period of the QBs to provide the required time resolution. As laser excitation creates superpositions of any states with allowed transition dipole moments, care has to be exercised to distinguish coherences associated with excitons. For example, if a laser spectrum covers two states in a vibrational progression, either in the ground or in the excited electronic state, the induced signal is due to concerted motion of a nuclear mode in the molecules, i.e., the observation of vibrational coherence. Conversely, in the case of two electronic (or exciton) states, a well-defined phase relation will be initiated—electronic (or interexcitonic) coherence (70). In between these limiting cases arises the general situation of superpositions of states with mixed vibrational-electronic character, which defines vibronic coherence—a field of significant current interest (71). As both electronic (72) and purely vibrational coherences (73–75) modulate ultrafast spectra in the form of periodic oscillations, the need to distinguish between them is obvious. As discussed previously (76, 77), the assignment of long-lived small-amplitude QBs in several photosynthetic systems to interexciton coherences based solely on their frequencies and sometimes phase (78–80) is insufficient.
Further complications in assignment arise because of disorder and spectral congestion. 2DES (see Figs. 2 and 3) was initially introduced to remove inhomogeneous broadening with the hope to directly observe interexciton couplings and fully resolve the energy transfer pathways (81, 82). However, this hope has not been generally realized. Severe spectral congestion for multipigment systems often leads to overlap of oscillatory signals, with strongly distorted features due to interference effects. Given the number of possible spectral features in PPCs, their assignment is far from trivial even in the well-resolved 2DES experiment.
Studies of photosynthetic excitons
We reiterate that the core of the initial argument for significant involvement of interexciton coherence—or any coherence—in photosynthetic systems was the observation of long-lived oscillations in electronic 2D spectra (4). These were interpreted to originate from linear superpositions of excitonic states, exhibiting dephasing times of several hundred femtoseconds or more. This was taken to imply a connection to energy transfer dynamics. While these experiments were fundamental to the development of quantum biology as a field, it is important to recognize that coherence dynamics has a much longer history than this recent explosion of interest might suggest. The first such observation was made already in 1991 by Vos et al. (83) in a low-temperature study of the purple bacteria reaction center. Here, however, the authors assigned the QBs to vibrational wave packet motion on the excited state (84). In another early study, Chachisvilis et al. (85) came to a similar conclusion in their study of the core light-harvesting complexes (LH1 and LH2) of purple bacteria: The observed QBs were caused by vibrations. The first observation of interexciton coherences contributing to the signals of PPCs was made by Savikhin et al. (86), who, in 1997, observed oscillations in the pump-probe anisotropy of the FMO complex at 19 K. The fast dephasing of ~200 fs agreed well with naïve expectations for dephasing of interexciton coherence in a biological system at low temperatures and suggested that this dephasing of coherences in biological conditions should be too fast to contribute significantly to the light-harvesting function. It thus came as a substantial surprise when, a decade later, oscillatory signals persisting for >600 fs (4) in the same complex at 77 K were reported and assigned to long-lived interexciton coherences. Not only did this proposal imply that decoherence was much slower than allowed by a realistic physical model at the time but also that the extremely successful paradigm of energy transfer in light-harvesting systems, based on incoherent transport of energy between partially-localized exciton states, would have to be revised. Similar spectral signatures—small-amplitude oscillations—were reported in other organisms (79, 80, 87, 88), leading to speculations that long-lived interexciton coherence was ubiquitous in natural photosynthesis, i.e., nature had discovered a design principle to exploit quantum coherences to direct biological functions.
While these studies have led to enormous interest in coherent phenomena, realistically, the experimental basis for the excitonic interpretation suffered from a selective view of the 2D spectra. In particular, for practical signal-to-noise reasons, these studies largely relied on kinetic traces extracted from only a very limited—typically one or two—areas of the 2D spectra. As we show in Fig. 4, coherence signals are often complex and difficult to reliably identify (67), calling for a more holistic interpretation of the entire 2D dataset. In the presence of vibronic mixing of electronic and vibrational states—which seems to be the case in many photosynthetic complexes—coherence signals are very much entangled, and an oscillation map analysis (Fig. 4) is indispensable (89). To outline a basic framework for the interpretation of QBs in 2D spectra and to provide readers with some basic tools to qualitatively assess 2D experiments on QBs, we schematically show their analysis in two relevant model systems in Fig. 4.
Fig. 4. Assigning QBs to physical processes.
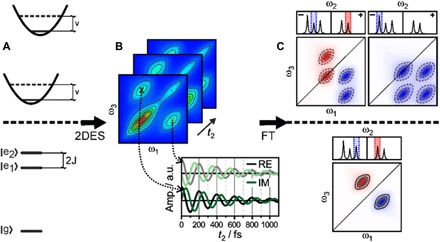
Coherences of different physical origin—vibrational or excitonic—lead to different characteristic patterns in the so-called oscillation maps. The characteristics of these signals—such as frequency, pump dependence, and detection dependence—provide unique identifiers, at least in idealized systems. Comparing the complicated signals from these systems to model systems proves very helpful. In (A), simple and useful model systems are the displaced oscillator (top) featuring ground- and excited-state vibrations and the excitonic dimer (bottom), where excited-state splitting is induced by coupling between pigments. (B) Even these simple models may yield virtually indistinguishable 2D spectra, with coherence manifested as QBs in the signal amplitude at specific spectral coordinates. When following these beats along the population time t2, one can observe periodic modulations of the real (RE; absorption) and imaginary (IM; dispersion) parts of the signal (black/gray and green/light green, respectively). a.u., arbitrary units. (C) Successful assignment of oscillatory signals to physical phenomena requires simultaneous analysis of beats in the entire 2D map, yielding oscillation maps after complex Fourier transformation (FT). These oscillation maps are most insightful when retrieved separately for rephasing (photon-echo) and nonrephasing responses. We here sketch rephasing data of a system with three closely spaced, but distinguishable, coherences: an excited state vibrational wave packet (top left), a Raman-active ground-state vibrational mode (top right), and an excitonic coherence (bottom). The oscillation maps show unique patterns, allowing unambiguous identification of coherences.
Present status of experiments: Revisiting FMO
In the debate on the importance of quantum coherences in photosynthesis, the FMO complex has again taken center stage because of its exemplar status in quantum biology. Here, we outline several recent studies of this complex, each reaching the same conclusion: The observed long-lived QBs are inconsistent with interexciton coherence. Instead, these oscillations mostly show characteristics of Raman-active vibrational modes on the electronic ground-state surface, unrelated to the energy transfer process.
The collaborative work of the Miller, Thorwart, and Cogdell groups, Duan et al. (90), addressed the question of whether the oscillatory signals were observable on functionally relevant time scales at biologically relevant temperatures. In contrast to the earlier studies, mostly performed at cryogenic temperatures [with a notable study at ~4°C (277 K) (88)], the authors found no low-frequency QBs with dephasing times beyond 60 fs. On the basis of the observed time scales for energy transfer within the exciton manifold, this work demonstrated that interexciton coherence cannot contribute meaningfully to energy transfer dynamics at physiological temperatures. The correct picture for energy transport involves incoherent relaxation between exciton states, downhill in energy. Since these exciton states are partially localized, this relaxation corresponds to a hopping between different spatial regions of the complex, giving the transport a direction (Fig. 1).
The Scholes and Blankenship groups, Maiuri et al. (91), came to a similar conclusion in a study at cryogenic temperatures. They took an approach relying on the direct correspondence between the energy gap between excitonic levels and the frequency of the interexciton QB signal. By altering the energy gaps in the excitonic structure using genetic engineering, one would expect the frequency of the observed oscillation should change. In contrast, the authors’ observations for a series of transient absorption experiments on FMO mutants with radically different excitonic splittings were that the QB frequencies were essentially unchanged. While this mutation-based approach is difficult to implement as a general analysis strategy for all light harvesters, for FMO, it provided unambiguous evidence for vibrational rather than interexciton coherence.
Last, the Zigmantas, Knoester, and Jansen groups, Thyrhaug et al. (92), investigated the wild-type FMO complex at cryogenic temperatures using polarization-controlled 2DES. They relied on pattern analysis (Fig. 4) to identify the QBs corresponding to specific types of coherences. This approach has been validated in studies on isolated bacteriochlorophyll a pigments (93). Again, the authors concluded that the long-lived QBs in the FMO protein were predominantly originating from Raman-active ground-state vibrations, with some contribution from excited state vibrational coherences. While electronic coherences were also identified, these were found (92) to be fully damped within 240 fs—in agreement with the earlier work of Savikhin et al. (86). Note that, through the use of polarization-controlled spectroscopy, it was possible to identify vibronically mixed excited states in the complex.
Individually and collectively, these studies demonstrate that the long-lived QBs in the FMO protein—one of the most heavily studied PPCs—are vibrational in origin. Thus, the interpretation of long-lived QBs in the FMO protein, as characteristics of interexciton coherence posed in 2007 (4) and in several subsequent studies (79, 80, 87, 88), has been replaced by a well-founded vibrational picture.
The working paradigm of an energy gradient and spatial proximity guided incoherent exciton transport, rather than the “wave-like” dynamics implied by the interexciton coherence picture, has been reestablished as the framework of choice in photosynthetic light harvesting. Is this conclusion reached for the FMO complex generic? We believe, yes, as the general mechanism of variable site energies directing the energy automatically leads to partial localization of the exciton states, with spatially uncorrelated bath fluctuations over these sites. Correspondingly, the loss of interexciton coherence is rapid under ambient conditions. Other ubiquitous natural photoactive complexes such as the Light-Harvesting Complex II (94) or the reaction center of the Photosystem II (95) show similarly fast electronic decoherence rates.
We must note that the world of photosynthetic light harvesting still remains interesting, as recent work has shown that much more complicated dynamics may appear when vibrations and electronic energy gaps are in resonance. These resonances lead to inseparability of nuclear and electronic degrees of freedom (96, 97), which some theoretical work has suggested may affect energy transfer (98). These resonances have also been reported in photosynthetic reaction centers (89, 99, 100). Yet, the character of a quantum state, which is initially strongly vibronic in nature, changes over time in a nontrivial way when the electronic and vibrational sectors are exposed to different dephasing and relaxation channels on very different time scales. The amplitude of the antiresonant vibrational mode of two coupled monomers can be enhanced by a strong coupling to a long-lived coherent electronic state when the latter evolves without (96) or under weak (101) electronic dephasing. However, the fast electronic dephasing, which appears to be general for light-harvesting systems, destroys coherence in the electronic sector faster than the vibrational period. Consequently, the vibrational coherence in the sector of the antiresonant vibrational mode, under realistic conditions, remains unaffected in this limit of short-lived electronic coherences (102). In this respect, we believe that considerable care must be taken in interpreting the resulting spectral signatures of vibronic coherences—and, in particular, when attempting to place this physics in the context of biological function. The rich spectroscopic information obtainable by 2DES should provide essential input for models of these complex situations, and these models will be an important next step for testing the putative functional significance of electronic-vibrational resonances for photosynthetic function. We hope that forthcoming studies will ultimately improve both our understanding of nature’s remarkable photosynthetic processes and our ability to mimic nature’s best “ideas” in artificial light-harvesting materials.
CONCLUSIONS
In summary, we have revisited the quantum aspects of photosynthetic light harvesting. It has become clear from basic considerations that there is no equivalence between quantumness of the processes and coherences observed in femtosecond spectroscopy experiments. Even the very fundamental question if nonstationary coherences in photosynthetic systems can be excited by sunlight still awaits full clarification (62–64, 103). Whatever the state preparation is, the dynamics will be governed by the associated couplings of the system and its interaction with the bath. Furthermore, the claims of the persistence of these coherences in femtosecond experiments have been critically reevaluated. In particular, detailed analysis of the exemplar system in quantum biology—the FMO complex—shows unambiguously the absence of long-lived interexciton coherence on relevant time scales in this system, both at cryogenic and physiological temperatures. Instead, it has become clear that the long-lived oscillating signals originate from vibrational modes predominantly on the electronic ground state. More advanced data analysis and theoretical treatments using realistic parametrization of the bath are needed for clear identification of coherence signals. The extensive discussion of earlier assignment of these spectral signatures, propagating in the community for a decade, underlines this need.
The major positive outcome is the improvement of theoretical and experimental methods that have led to a deeper understanding of the system-bath interactions responsible for decoherence and dissipation in biological systems. Nature does not engineer the bath to avoid decoherence to direct functional processes; such an approach almost certainly would not be robust. Nature, rather than trying to avoid dissipation, specifically exploits it together with the engineering of site energies and excitonic coupling to direct energy transport. The role of thermodynamic parameters in driving biological functions is well appreciated on other levels. Here, we see that this principle applies even to the energy transfer processes involved in photosynthesis that occur on the fastest possible time scales. The basic physics behind thermalization is used to impose direction. This simple concept, mastered by nature over all relevant time and spatial dimensions, is truly a marvel of biology.
METHODS
Time-local density matrix theory in the representation of exciton states (see the Supplementary Materials for details) is applied using a Markov and a secular approximation for the off-diagonal elements of the exciton-vibrational coupling (104) for the description of optical line shapes (section S3), the decay of optical and interexciton coherences (section S4), and exciton relaxation (sections S3 to S5). The parameters of the Frenkel exciton Hamiltonian of the FMO protein (section S2) are taken from a quantum chemical/electrostatic study (17) (site energies and excitonic couplings; see table S1) and from an analysis of fluorescence line-narrowing and temperature-dependent absorption spectra (spectral density of exciton-vibrational coupling) (105). These parameters, which are tested against linear optical spectra (fig. S3), are used in calculations of exciton relaxation initiated by incoherent transfer from the baseplate (Fig. 1, B and C, section S5, and figs. S6 to S8) and in the calculations of the decay of interexciton (Fig. 1E) and optical (Fig. 1F) coherences initiated by a δ-pulse excitation (section S4). The classical limit of the nuclear motion (black lines in Fig. 1, E and F; and figs. S4, S6, and S8) is obtained by solving Hamilton’s equations of motion for the nuclei and performing an average over Boltzmann-distributed initial coordinates and momenta in the calculation of the energy gap correlation function of the pigments, which enters the damping function of coherences and the rate constant (section S6).
Supplementary Material
Acknowledgments
Funding: D.F.C. acknowledges the support of U.S. National Science Foundation (NSF) grant CHE-1665367. D.Z. acknowledges support from the Swedish Research Council. H.-G.D. acknowledges financial support by the Joachim-Herz-Stiftung Hamburg within a PIER fellowship. The work of H.-G.D. and R.J.D.M. was supported by the Max Planck Society. Moreover, H.-G.D., M.T., and R.J.D.M. were supported by the Cluster of Excellence “CUI: Advanced Imaging of Matter” of the Deutsche Forschungsgemeinschaft (DFG) - EXC 2056 - project ID 390715994. H.-S.T. acknowledges support from the Singapore Ministry of Education Academic Research Fund (Tier 2 MOE2015-T2-1-039). U.K. is grateful for a Tan Chin Tuan Exchange Fellowship for a research stay at Nanyang Technological University, Singapore. J.C. acknowledges funding through NSF CHE 1836913 and NSF CHE 1800301. J.H. acknowledges funding by the DFG (German Research Foundation) under Germany’s Excellence Strategy EXC 2089/1390776260. J.P.O. acknowledges support from the Office of Basic Energy Sciences, the U.S. Department of Energy under grant number DE-SC0016384, and the NSF under grant number PHY-1607570. R.J.C. gratefully acknowledges support from the Photosynthetic Antenna Research Center, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under award number DE-SC 0001035. S.W. and D.Z. acknowledge support from the Knut and Alice Wallenberg Foundation. T.M. is supported by the Czech Science Foundation (GACR) grant 17-22160S. Author contributions: The authors met many times over the past 2 1/2 years, both in person at meetings and by videoconference to thoroughly discuss and debate the reviewed topics in the field to arrive at the consensus view presented in this paper. T.R. carried out the calculations presented in Fig. 1 and in the Supplementary Materials. All authors contributed to the planning, writing, and all stages of review of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/14/eaaz4888/DC1
REFERENCES AND NOTES
- 1.Lambert N., Chen Y.-N., Cheng Y.-C., Li C.-M., Chen G.-Y., Nori F., Quantum biology. Nat. Phys. 9, 10–18 (2013). [Google Scholar]
- 2.Brookes J. C., Quantum effects in biology: Golden rule in enzymes, olfaction, photosynthesis and magnetodetection. Proc. Math Phys. Eng. Sci. 473, 20160822 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.E. Schrödinger, What is Life? (Cambridge Univ. Press, Cambridge, 1944). [Google Scholar]
- 4.Engel G. S., Calhoun T. R., Read E. L., Ahn T.-K., Mančal T., Cheng Y.-C., Blankenship R. E., Fleming G. R., Evidence for wavelike energy transfer through quantum coherence in photosynthetic systems. Nature 446, 782–786 (2007). [DOI] [PubMed] [Google Scholar]
- 5.J. McFadden, J. Al-Khalili, Life on the Edge: The Coming of Age of Quantum Biology. (Broadway Books, 2014). [Google Scholar]
- 6.Mukamel S., Comment on “Coherence and uncertainty in nanostructured organic photovoltaics”. J. Phys. Chem. A 117, 10563–10564 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Sundström V., Pullerits T., van Grondelle R., Photosynthetic light-harvesting: Reconciling dynamics and structure of purple bacterial LH2 reveals function of photosynthetic unit. J. Phys. Chem. B 103, 2327–2346 (1999). [Google Scholar]
- 8.Dostál J., Pšenčík J., Zigmantas D., In situ mapping of the energy flow through the entire photosynthetic apparatus. Nat. Chem. 8, 705–710 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Raszewski G., Renger T., Light harvesting in photosystem II core complexes is limited by the transfer to the trap: Can the core complex turn into a photoprotective mode? J. Am. Chem. Soc. 130, 4431–4446 (2008). [DOI] [PubMed] [Google Scholar]
- 10.R. E. Blankenship, Molecular Mechanisms of Photosynthesis (Wiley-Blackwell, ed. 2, 2014). [Google Scholar]
- 11.R. Croce, R. van Grondelle, H. van Amerogen, I. Van Stokkum, Light-Harvesting in Photosynthesis (CRC Press, 2018). [Google Scholar]
- 12.H. van Amerongen, R. van Grondelle, L. Valkunas, Photosynthetic Excitons (World Scientific, 2000). [Google Scholar]
- 13.Thyrhaug E., Židek K., Dostál J., Bína D., Zigmantas D., Exciton structure and energy transfer in the Fenna-Matthews-Olson complex. J. Phys. Chem. Lett. 7, 1653–1660 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Abramavicius D., Palmieri B., Voronine D. V., Šanda F., Mukamel S., Coherent multidimensional optical spectroscopy of excitons in molecular aggregates; quasiparticle versus Supermolecule Perspectives. Chem. Rev. 109, 2350–2408 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Müh F., Madjet M. E.-A., Adolphs J., Abdurahman A., Rabenstein B., Ishikita H., Knapp E.-W., Renger T., α-helices direct excitation energy flow in the Fenna–Matthews–Olson protein. Proc. Natl. Acad. Sci. U.S.A. 104, 16862–16867 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adolphs J., Müh F., Madjet M. E.-A., Renger T., Calculation of pigment transition energies in the FMO protein: From simplicity to complexity and back. Photosynth. Res. 95, 197–209 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Schmidt am Busch M., Müh F., Madjet M. E.-A., Renger T., The eighth bacteriochlorophyll completes the excitation energy funnel in the FMO protein. J. Phys. Chem. Lett. 2, 93–98 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Saer R. G., Stadnytskyi V., Magdaong N. C., Goodson C., Savikhin S., Blankenship R. E., Probing the excitonic landscape of the chlorobaculum tepidum Fenna-Matthews-Olson (FMO) complex: A mutagenesis approach. Biochim. Biophys. Acta Bioenerg. 1858, 288–296 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Adolphs J., Renger T., How proteins trigger excitation energy transfer in the FMO complex of green sulfur bacteria. Biophys. J. 91, 2778–2797 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wen J., Zhang H., Gross M. L., Blankenship R. E., Membrane orientation of the FMO antenna protein from chlorobaculum tepidum as determined by mass spectrometry-based footprinting. Proc. Natl. Acad. Sci. U.S.A. 106, 6134–6139 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moix J., Wu J., Huo P., Coker D., Cao J., Efficient energy transfer in light-harvesting systems, III: The influence of the eighth bacteriochlorophyll on the dynamics and efficiency in FMO. J. Phys. Chem. Lett. 2, 3045–3052 (2011). [Google Scholar]
- 22.Olbrich C., Strümpfer J., Schulten K., Kleinekathöfer U., Theory and simulation of the environmental effects on FMO electronic transitions. J. Phys. Chem. Lett. 2, 1771–1776 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Renger T., Klinger A., Steinecker F., Schmidt am Busch M., Numata J., Müh F., Normal mode analysis of the spectral density of the Fenna-Matthews-Olson Light-Harvesting protein: How the protein dissipates the excess energy of excitons. J. Phys. Chem. B 116, 14565–14580 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chandrasekan S., Aghtar M., Valleau S., Aspuru-Guzik A., Kleinekathöfer U., Influence of force fields and quantum chemistry approach on spectral density of Bchl a in solution and in FMO proteins. J. Phys. Chem. B 119, 9995–10004 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Rivera E., Montemayor D., Masia M., Coker D. F., Influence of site-dependent pigment–Protein interactions on excitation energy transfer in photosynthetic light harvesting. J. Phys. Chem. B 117, 5510–5521 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Pullerits T., Chachisvilis M., Sundström V., Exciton delocalization length in the B850 antenna of Rhodobacter sphaeroides. J. Phys. Chem. 100, 10787–10792 (1996). [Google Scholar]
- 27.Monshouwer R., Abrahamsson M., van Mourik F., van Grondelle R., Superradiance and exciton delocalization in bacterial photosynthetic light-harvesting systems. J. Phys. Chem. B 37, 7241–7248 (1997). [Google Scholar]
- 28.C. Shane, An Introduction to Electromagnetic Wave Propagation and Antennas (Springer Science and Business Media, 1996). [Google Scholar]
- 29.Mančal T., Excitation energy transfer in a classical analogue of photosynthetic antennae. J. Phys. Chem. B 117, 11282–11291 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Lee S., Richter W., Vathyam S., Warren W. S., Quantum treatment of the effects of dipole-dipole interactions in liquid nuclear magnetic resonance. J. Chem. Phys. 105, 874–900 (1996). [Google Scholar]
- 31.Jeener J., Vlassenbroek A., Broekaert P., Unified derivation of the dipolar field and relaxation terms in the Bloch-Redfield equations of liquid NMR. J. Chem. Phys. 103, 1309–1332 (1995). [Google Scholar]
- 32.Sumi H., Theory on rates of excitation-energy transfer between molecular aggregates through distributed transition dipoles with application to the antenna system in bacterial photosynthesis. J. Phys. Chem. B 103, 252–260 (1999). [Google Scholar]
- 33.Bohr N., Über die serienspektra der elemente. Zeitschrift für Physik 2, 423–478 (1920). [Google Scholar]
- 34.A. Nitzan, Chemical Dynamics in the Condensed Phase (Oxford Univ. Press, 2006). [Google Scholar]
- 35.Cotton S. J., Miller W. H., The symmetrical quasi-classical model for electronically non-adiabatic processes applied to energy transfer dynamics in site-exciton models of light-harvesting complexes. J. Chem. Theory Comput. 12, 983–991 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Meyera H.-D., Miller W. H., A classical analog for electronic degrees of freedom in nonadiabatic collision processes. J. Chem. Phys. 70, 3214–3223 (1979). [Google Scholar]
- 37.Stock G., Thoss M., Semiclassical description of nonadiabatic quantum dynamics. Phys. Rev. Lett. 78, 578–581 (1997). [DOI] [PubMed] [Google Scholar]
- 38.Thoss M., Stock G., Mapping approach to the semiclassical description of nonadiabatic quantum dynamics. Phys. Rev. A 59, 64–79 (1999). [Google Scholar]
- 39.Tempelaar R., Jansen T. L. C., Knoester J., Vibrational beatings conceal evidence of electronic coherence in the FMO light-harvesting complex. J. Phys. Chem. B 118, 12865–12872 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Makri N., Makarov D. E., Tensor propagator for iterative quantum time evolution of reduced density matrices. I. Theory. J. Chem. Phys. 102, 4600–4610 (1995). [Google Scholar]
- 41.Makri N., Makarov D. E., Tensor propagator for iterative quantum time evolution of reduced density matrices. II. Numerical methodology. J. Chem. Phys. 102, 4611–4618 (1995). [Google Scholar]
- 42.Nalbach P., Braun D., Thorwart M., Exciton transfer dynamics and quantumness of energy transfer in the Fenna-Matthews-Olson complex. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 84, 041926 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Mujica-Martinez C. A., Nalbach P., Thorwart M., Quantification of non-Markovian effects in the Fenna-Matthews-Olson complex. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 88, 062719 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Nalbach P., Mujica-Martinez C. A., Thorwart M., Vibronically coherent speed-up of the excitation energy transfer in the Fenna–Matthews–Olson complex. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 91, 022706 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Tanimura Y., Kubo R., Time evolution of quantum system in contact with a nearly Gaussian-Markovian noise bath. J. Phys. Soc. Jpn. 58, 101–114 (1989). [Google Scholar]
- 46.Bader J. S., Berne B. J., Quantum and classical relaxation rates from classical simulations. J. Chem. Phys. 100, 8359–8366 (1994). [Google Scholar]
- 47.Egorov S. A., Rabani E., Berne B. J., Vibronic spectra in condensed matter: A comparison of exact quantum mechanical and various semiclassical treatments for harmonic baths. J. Chem. Phys. 108, 1407–1422 (1998). [Google Scholar]
- 48.Miller W. H., Perspective: Quantum or classical coherence? J. Chem. Phys. 136, 210901 (2012). [DOI] [PubMed] [Google Scholar]
- 49.Briggs J. S., Eisfeld A., Equivalence of quantum and classical coherence in electronic energy transfer. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 83, 051911 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Zimanyi E. N., Silbey R. J., Unified treatment of coherent and incoherent electronic energy transfer dynamics using classical electrodynamics. J. Chem. Phys. 133, 144107 (2010). [DOI] [PubMed] [Google Scholar]
- 51.Mukamel S., Communications: Signature of quasiparticle entanglement in multidimensional nonlinear optical spectroscopy of aggregates. J. Chem. Phys. 132, 241105 (2010). [DOI] [PubMed] [Google Scholar]
- 52.Reppert M., Brumer P., Quantumness in light harvesting is determined by vibrational dynamics. J. Chem. Phys. 149, 234102 (2018). [DOI] [PubMed] [Google Scholar]
- 53.U. Weiss, Quantum Dissipative Systems (World Scientific, Singapore, ed. 3, 2008). [Google Scholar]
- 54.Feynman R. P., Vernon F. L. Jr., The theory of a general quantum system interacting with a linear dissipative system. Ann. Phys-New York 24, 118–173 (1963). [Google Scholar]
- 55.Chin A. W., Rivas A., Huelga S. F., Plenio M. B., Exact mapping between system-reservoir quantum models and semi-infinite discrete chains using orthogonal polynomials. J. Math. Phys. 51, 092109 (2010). [Google Scholar]
- 56.Huo P., Coker D. F., Iterative linearized density matrix propagation for modeling coherent excitation energy transfer in photosynthetic light harvesting. J. Chem. Phys. 133, 184108 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Moix J. M., Ma J., Cao J., Förster reson0061nce energy transfer, absorption and emission spectra in multichromophoric systems. III. Exact stochastic path integral evaluation. J. Chem. Phys. 142, 094108 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Tanimura Y., Reduced hierarchy equation of motion approach with Drude plus Brownian spectral distribution: Probing electron transfer processes by means of two-dimensional correlation spectroscopy. J. Chem. Phys. 137, 22A550 (2012). [DOI] [PubMed] [Google Scholar]
- 59.Lee H., Cheng Y.-C., Fleming G. R., Coherence dynamics in photosynthesis: protein protection of excitonic coherence. Science 316, 1462–1465 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Chin A. W., Huelga S. F., Plenio M. B., Coherence and decoherence in biological systems: Principles of noise-assisted transport and the origin of long-lived coherences. Philos. Trans. A Math Phys. Eng. Sci. 370, 3638–3657 (2012). [DOI] [PubMed] [Google Scholar]
- 61.Rolczynski B. S., Zheng H., Singh V. P., Navotnaya P., Ginzburg A. R., Caram J. R., Ashraf K., Gardiner A. T., Yeh S.-H., Kais S., Cogdell R. J., Engel G. S., Correlated protein environments drive quantum coherence lifetimes in photosynthetic pigment-protein complexes. Chem 4, 138–149 (2018). [Google Scholar]
- 62.Brumer P., Shapiro M., Molecular response in one-photon absorption via natural thermal light vs. pulsed laser excitation. Proc. Natl. Acad. Sci. U.S.A. 109, 19575–19578 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mančal T., Valkunas L., Exciton dynamics in photosynthetic complexes: Excitation by coherent and incoherent light. New J. Phys. 12, 065044 (2010). [Google Scholar]
- 64.J. Olšina, A. G. Dijkstra, C. Wang, J. Cao, Can natural sunlight induce coherent exciton dynamics? arXiv:1408.5385 (2014).
- 65.Chan H. C. H., Gamel O. E., Fleming G. R., Whaley K. B., Single-photon absorption by single photosynthetic light-harvesting complexes. J. Phys. B At. Mol. Opt. Phys. 51, 054002 (2018). [Google Scholar]
- 66.Müh F., Plöckinger M., Renger T., Electrostatic asymmetry in the Reaction Center of Photosystem II. J. Phys. Chem. Lett. 8, 850–858 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Butkus V., Zigmantas D., Valkunas L., Abramavicius D., Vibrational vs. electronic coherences in 2D spectrum of molecular systems. Chem. Phys. Lett. 545, 40–43 (2012). [Google Scholar]
- 68.Duan H.-G., Nalbach P., Prokhorenko V. I., Mukamel S., Thorwart M., On the origin of oscillations in two-dimensional spectra of excitonically-coupled molecular systems. New J. Phys. 17, 072002 (2015). [Google Scholar]
- 69.Halpin A., Johnson P. J. M., Tempelaar R., Murphy R. S., Knoester J., Jansen T. L. C., Miller R. J. D., Two-dimensional spectroscopy of a molecular dimer unveils the effects of vibronic coupling on exciton coherences. Nat. Chem. 6, 196–201 (2014). [DOI] [PubMed] [Google Scholar]
- 70.Aslangul C., Kottis P., Density operator description of excitons in molecular aggregates: Optical absorption and motion. I. The dimer problem. Phys. Rev. B 10, 4364–4382 (1974). [Google Scholar]
- 71.Wang L., Allodi M. A., Engel G. S., Quantum coherences reveal excited-state dynamics in biophysical systems. Nat. Rev. Chem. 3, 477–490 (2019). [Google Scholar]
- 72.Cheng Y.-C., Fleming G. R., Coherence quantum beats in two-dimensional electronic spectroscopy. J. Phys. Chem. A 112, 4254–4260 (2008). [DOI] [PubMed] [Google Scholar]
- 73.Nemeth A., Milota F., Mančal T., Lukeš V., Hauer J., Kauffmann H. F., Sperling J., Vibrational wave packet induced oscillations in two-dimensional electronic spectra. I. Experiments. J. Chem. Phys. 132, 184514 (2010). [Google Scholar]
- 74.Mančal T., Nemeth A., Milota F., Lukeš V., Kauffmann H. F., Sperling J., Vibrational wave packet induced oscillations in two-dimensional electronic spectra. II. Theory. J. Chem. Phys. 132, 184515 (2010). [Google Scholar]
- 75.Dostál J., Mančal T., Vácha F., Pšenčík J., Zigmantas D., Unraveling the nature of coherent beatings in chlorosomes. J. Chem. Phys. 140, 115103 (2014). [DOI] [PubMed] [Google Scholar]
- 76.Butkus V., Zigmantas D., Abramavicius D., Valkunas L., Distinctive character of electronic and vibrational coherences in disordered molecular aggregates. Chem. Phys. Lett. 587, 93–98 (2013). [Google Scholar]
- 77.Mančal T., Christensson N., Lukeš V., Milota F., Bixner O., Kauffmann H. F., Hauer J., System-dependent signatures of electronic and vibrational coherences in electronic two-dimensional spectra. J. Phys. Chem. Lett. 3, 1497–1502 (2012). [DOI] [PubMed] [Google Scholar]
- 78.Yue S., Wang Z., Leng X., Zhu R.-D., Chen H.-L., Weng Y.-X., Coupling of multi-vibrational modes in bacteriochlorophyll a in solution observed with 2D electronic spectroscopy. Chem. Phys. Lett. 683, 591–597 (2017). [Google Scholar]
- 79.Collini E., Wong C. Y., Wilk K. E., Curmi P. M. G., Brumer P., Scholes G. D., Coherently wired light-harvesting in photosynthetic marine algae at ambient temperature. Nature 463, 644–647 (2010). [DOI] [PubMed] [Google Scholar]
- 80.Harel E., Engel G. S., Quantum coherence spectroscopy reveals complex dynamics in bacterial light-harvesting complex 2 (LH2). Proc. Natl. Acad. Sci. U.S.A. 109, 706–711 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brixner T., Stenger J., Vaswani H. M., Cho M., Blankenship R. E., Fleming G. R., Two-dimensional spectroscopy of electronic couplings in photosynthesis. Nature 434, 625–628 (2005). [DOI] [PubMed] [Google Scholar]
- 82.Zigmantas D., Read E. L., Mančal T., Brixner T., Gardiner A. T., Cogdell R. J., Fleming G. R., Two-dimensional electronic spectroscopy of the B800-B820 light-harvesting complex. Proc. Natl. Acad. Sci. U.S.A. 103, 12672–12677 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vos M. H., Lambry J. C., Robles S. J., Youvan D. C., Breton J., Martin J. L., Direct observation of vibrational coherence in bacterial reaction centers using femtosecond absorption-spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 88, 8885–8889 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vos M. H., Rappaport F., Lambry J.-C., Breton J., Martin J.-L., Visualization of coherent nuclear motion in a membrane-protein by femtosecond spectroscopy. Nature 363, 320–325 (1993). [Google Scholar]
- 85.Chachisvilis M., Pullerits T., Jones M. R., Hunter C. N., Sundström V., Vibrational dynamics in the light-harvesting complexes of the photosynthetic bacterium Rhodobacter sphaeroides. Chem. Phys. Lett. 224, 345–354 (1994). [Google Scholar]
- 86.Savikhin S., Buck D. R., Struve W. S., Oscillating anisotropies in a bacteriochlorophyll protein: Evidence for quantum beating between exciton levels. Chem. Phys. 223, 303–312 (1997). [Google Scholar]
- 87.Schlau-Cohen G. S., Ishizaki A., Calhoun T. R., Ginsberg N. S., Ballottari M., Bassi R., Fleming G. R., Elucidation of the timescales and origin of quantum electronic coherence in LHCII. Nat. Chem. 4, 389–395 (2012). [DOI] [PubMed] [Google Scholar]
- 88.Panitchayangkoon G., Hayes D., Fransted K. A., Caram J. R., Harel E., Wen J., Blankenship R. E., Engel G. S., Long-lived quantum coherence in photosynthetic complexes at physiological temperature. Proc. Natl. Acad. Sci. U.S.A. 107, 12766–12770 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Paleček D., Edlund P., Westenhoff S., Zigmantas D., Quantum coherence as a witness of vibronically hot energy transfer in bacterial reaction center. Sci. Adv. 3, e1603141 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Duan H.-G., Prokhorenko V. I., Cogdell R. J., Ashraf K., Stevens A. L., Thorwart M., Miller R. J. D., Nature does not rely on long-lived electronic quantum coherence for photosynthetic energy transfer. Proc. Natl. Acad. Sci. U.S.A. 114, 8493–8498 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Maiuri M., Ostroumov E. E., Saer R. G., Blankenship R. E., Scholes G. D., Coherent wavepackets in the Fenna-Matthews-Olson complex are robust to excitonic-structure perturbations caused by mutagenesis. Nat. Chem. 10, 177–183 (2018). [DOI] [PubMed] [Google Scholar]
- 92.Thyrhaug E., Tempelaar R., Alcocer M. J. P., Žídek K., Bína D., Knoester J., Jansen T. L. C., Zigmantas D., Identification and characterization of diverse coherences in the Fenna-Matthews-Olson complex. Nat. Chem. 10, 780–786 (2018). [DOI] [PubMed] [Google Scholar]
- 93.Policht V. R., Niedringhaus A., Ogilvie J. P., Characterization of vibrational coherence in monomeric bacteriochlorophyll a by two-dimensional electronic spectroscopy. J. Phys. Chem. Lett. 9, 6631–6637 (2018). [DOI] [PubMed] [Google Scholar]
- 94.Duan H.-G., Stevens A. L., Nalbach P., Thorwart M., Prokhorenko V. I., Miller R. J. D., Two-dimensional electronic spectroscopy of light-harvesting complex II at ambient temperature: A joint experimental and theoretical study. J. Phys. Chem. B 119, 12017–12027 (2015). [DOI] [PubMed] [Google Scholar]
- 95.Duan H.-G., Prokhorenko V. I., Wientjes E., Croce R., Thorwart M., Miller R. J. D., Primary charge separation in the Photosystem II reaction center revealed by a Global Analysis of the two-dimensional electronic spectra. Sci. Rep. 7, 12347 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tiwari V., Peters W. K., Jonas D. M., Electronic resonance with anticorrelated pigment vibrations drives photosynthetic energy transfer outside the adiabatic framework. Proc. Natl. Acad. Sci. U.S.A. 110, 1203–1208 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Christensson N., Kauffmann H. F., Pullerits T., Mančal T., Origin of long-lived coherences in light-harvesting complexes. J. Phys. Chem. B 116, 7449–7454 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Womick J. M., Moran A. M., Vibronic enhancement of exciton sizes and energy transport in photosynthetic complexes. J. Phys. Chem. B 115, 1347–1356 (2011). [DOI] [PubMed] [Google Scholar]
- 99.Fuller F. D., Pan J., Gelzinis A., Butkus V., Senlik S. S., Wilcox D. E., Yocum C. F., Valkunas L., Abramavicius D., Ogilvie J. P., Vibronic coherence in oxygenic photosynthesis. Nat. Chem. 6, 706–711 (2014). [DOI] [PubMed] [Google Scholar]
- 100.Romero E., Augulis R., Novoderezhkin V. I., Ferretti M., Thieme J., Zigmantas D., van Grondelle R., Quantum coherence in photosynthesis for efficient solar-energy conversion. Nat. Phys. 10, 676–682 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yeh S.-H., Hoehn R. D., Allodi M. A., Engel G. S., Kais S., Elucidation of near-resonance vibronic coherence lifetimes by nonadiabatic electronic-vibrational state character mixing. Proc. Natl. Acad. Sci. U.S.A. 116, 18263–18268 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Duan H.-G., Thorwart M., Miller R. J. D., Does electronic coherence enhance anticorrelated pigment vibrations under realistic conditions? J. Chem. Phys. 151, 114115 (2019). [DOI] [PubMed] [Google Scholar]
- 103.Jiang X.-P., Brumer P., Creation and dynamics of molecular states prepared with coherent vs partially coherent pulsed light. J. Chem. Phys. 94, 5833–5843 (1991). [Google Scholar]
- 104.Renger T., Marcus R. A., On the relation of protein dynamics and exciton relaxation in pigment-protein complexes: An estimation of the spectral density and a theory for the calculation of optical spectra. J. Chem. Phys. 116, 9997–10019 (2002). [Google Scholar]
- 105.Wendling M., Pullerits T., Przyjalgowski M. A., Vulto S. I. E., Aartsma T. J., van Grondelle R., van Amerongen H., Electron-vibrational coupling in the Fenna-Matthews-Olson complex of Prosthecochloris aestuarii determined by temperature-dependent absorption and fluorescence line-narrowing measurements. J. Phys. Chem. B 104, 5825–5831 (2000). [Google Scholar]
- 106.Tronrud D. E., Wen J., Gay L., Blankenship R. E., The structural basis for the difference in absorbance spectra for the FMO antenna protein from various green sulfur bacteria. Photosynth. Res. 100, 79–87 (2009). [DOI] [PubMed] [Google Scholar]
- 107.Adolphs J., Maier F., Renger T., Wavelength-dependent exciton-vibrational coupling in the water-soluble chlorophyll binding protein revealed by multilevel theory of difference fluorescence line-narrowing. J. Phys. Chem. B 122, 8891–8899 (2018). [DOI] [PubMed] [Google Scholar]
- 108.Hein B., Kreisbeck C., Kramer T., Rodríguez M., Modelling of oscillation in two-dimensional echo-spectra of the Fenna–Matthews–Olson complex. New J. Phys. 14, 023018 (2012). [Google Scholar]
- 109.Dinh T.-C., Renger T., Towards an exact theory of linear absorbance and circular dichroism of pigment-protein complexes: Importance of non-secular contribution. J. Chem. Phys. 142, 034104 (2015). [DOI] [PubMed] [Google Scholar]
- 110.Ma J., Cao J., Förster resonance energy transfer, absorption and emission spectra in multichromophoric systems. I. Full cumulant expansions and system-bath entanglement. J. Chem. Phys. 142, 094106 (2015). [DOI] [PubMed] [Google Scholar]
- 111.Wendling M., Przyjalgowski M. A., Gülen D., Vulto S. I. E., Aartsma T. J., van Grondelle R., van Amerongen H., The quantitative relationship between structure and polarized spectroscopy in the FMO complex of Prosthecochloris aestuarii: Refining experiments and simulations. Photosynth. Res. 71, 99–123 (2002). [DOI] [PubMed] [Google Scholar]
- 112.Raszewski G., Saenger W., Renger T., Theory of optical spectra of photosystem II reaction centers: Location of the triplet state and the identity of the primary electron donor. Biophys. J. 88, 986–998 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Parendekar P. V., Tully J. C., Mixed quantum-classical equilibrium. J. Chem. Phys. 122, 094102 (2005). [DOI] [PubMed] [Google Scholar]
- 114.Miller W. H., Cotton S. J., Communication: Note on detailed balance in symmetrical quasi-classical models for electronically non-adiabatic dynamics. J. Chem. Phys. 142, 131103 (2015). [DOI] [PubMed] [Google Scholar]
- 115.Lindorfer D., Renger T., Theory of anisotropic circular dichroism of excitonically coupled systems: Application to the baseplate of green sulfur bacteria. J. Phys. Chem. B 122, 2747–2756 (2018). [DOI] [PubMed] [Google Scholar]
- 116.Lax M., The Franck-Condon principle and its application to crystals. J. Chem. Phys. 20, 1752–1760 (1952). [Google Scholar]
- 117.Dostál J., Vácha F., Pšenčík J., Zigmantas D., 2D electronic spectroscopy reveals excitonic structure in the baseplate of a chlorosome. J. Phys. Chem. Lett. 5, 1743–1747 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/14/eaaz4888/DC1