

Abstract
Following the discovery of a new class of compounds that inhibit the mucosa-associated lymphoid tissue lymphoma translocation 1 (MALT1) protease in a prior study, further chemical investigation of the Dictyosporium digitatum fungus resulted in the identification of 16 additional metabolites, including 12 undescribed compounds (1-12). The constitution and relative configuration of these new molecules were established by comprehensive NMR and HRMS analyses. Their absolute configurations were determined by employing Mosher’s ester analysis and TDDFT ECD calculations. Two sesquiterpenes, dictyosporins A (1) and B (2), possess an undescribed eudesmen-type of structural scaffold. The ability of the isolated compounds to inhibit MALT1 proteolytic activity was evaluated, but none of them exhibited significant inhibition.
Keywords: Dictyosporium digitatum, Dictyosporiaceae, structure elucidation, Mosher’s ester analysis, DFT calculations, dictyosporin
A comprehensive suite of contemporary spectroscopic and computational methodologies for small molecule structure determination were employed to define the structural motifs of twelve new fungal metabolites. These studies illustrate the broad utility and power of NMR experiments combined with computational methods to verify the constitution and configuration of new molecular scaffolds.
Graphical Abstract
The structure and absolute configuration of twelve metabolites from the fungus Dictyosporium digitatum were established by NMR and HRMS, in conjunction with Mosher’s ester analysis and quantum mechanical ECD calculations.
1. Introduction
Fungi are widely distributed from terrestrial environments to freshwater and marine habitats (Rédou et al., 2015), and it is estimated that about 5.1 million unique fungal species exist, but only approximately 100,000 of these have been taxonomically described (Blackwell, 2011). These eukaryotic microbes produce specialized metabolites involved in a variety of ecological functions such as quorum sensing, chemical defense, allelopathy, and maintenance of symbiotic interactions (Spiteller, 2015). A recent chemoinformatic study revealed that fungal metabolites often contain unique structural scaffolds and functional group arrays that are not represented in most commercial chemical libraries which contain both natural and semi-synthetic products (Gonzalez-Medina et al., 2017). Historically, most of the focus was on terrestrial saprophytic fungi as sources of new metabolites, but more recently attention has broadened to include fungi from marine habitats and endophytic strains living in plants. Therefore, fungi remain a vast and promising resource for bioactive natural products screening and discovery efforts.
Fungi in the genus Dictyosporium belong to the family Dictyosporiaceae (Tanaka et al., 2015) and they are commonly found on submerged, decaying wood (Goh et al., 1999). This genus of fungi has only been sparsely evaluated in prior chemical investigations (Prasher and Verma, 2015). Our previous chemical study on a soil isolate of Dictyosporum sp. (now identified as Dictyosporum digitatum) resulted in identification of the first non-quinone natural products which can inhibit the mucosa-associated lymphoid tissue lymphoma translocation 1 (MALT1) protease (Tran et al., 2019). Inspired by the previous findings, we searched for other minor components of the fungal extract and isolated 12 undescribed and four known compounds. Herein their isolation, structure elucidation, and biological evaluation are reported.
2. Results and Discussion
The Dictyosporum digitatum extract was chromatographed on a Diol MPLC column and subsequently separated by repeated C18 HPLC to yield 12 new metabolites (1-12), along with four known compounds (13-16) (Fig. 1). Dictyosporin A (1) was obtained as a white powder. The (+)-HRESIMS spectrum displayed a sodium adduct ion [M + Na]+ at m/z 273.1464 corresponding to a molecular formula of C15H22O3 with a hydrogen deficiency index of five. NMR data (Table 1) confirmed that compound 1 contained 15 carbons including one sp2 quaternary carbon (δC 146.8), two sp2 methines (δC 129.7 and 128.9), one sp2 methylene (δC 111.2), two oxygenated sp3 quaternary carbons (δC 86.4 and 81.0), one sp3 quaternary carbon (δC 31.4), one oxygenated sp3 methine (δC 69.9), one sp3 methine (δC 45.4), one oxygenated sp3 methylene (δC 66.6), three sp3 methylenes (δC 40.0, 39.0, and 27.4), and two methyls (δC 28.1 and 19.4). COSY data identified four isolated proton-proton spin systems of –C1–C2=C3–, –C5–C6–, –C8(OH)–C9–, and –C15(OH) (Fig. 2A). HMBC data revealed the presence of an iso-propenyl group C13=C11(C12)–. HMBC correlations of H-1/C-10, C-5, and C-9; H-5/C-10 and C-9; H-9/C-10, C-1, and C-5; and H-14/C-1, C-5, C-9, and C-10 established the connections of C-1 to both C-5 and C-9 via C-10, and the location of the isolated methyl C-14 (δC 28.1) at C-10. The quaternary carbon C-4 was linked to C-3, C-5, and C-15 by HMBC correlations of H-15/C-3, C-4, and C-5; and H-3/C-4 and C-5. The connections of C-6, C-8, and the iso-propenyl group via C-7 were determined by HMBC correlations from H-6, H-8, 8-OH, H-12, and H-13 to C-7. C-4 and C-7 were connected via an oxygen atom to provide a fused tetrahydrofuran-type of ring which fulfilled the remaining unsaturation equivalent and molecular formula requirements. Therefore, the planar structure of dictyosporin A (1) was established as shown in Fig. 2A.
Fig. 1.
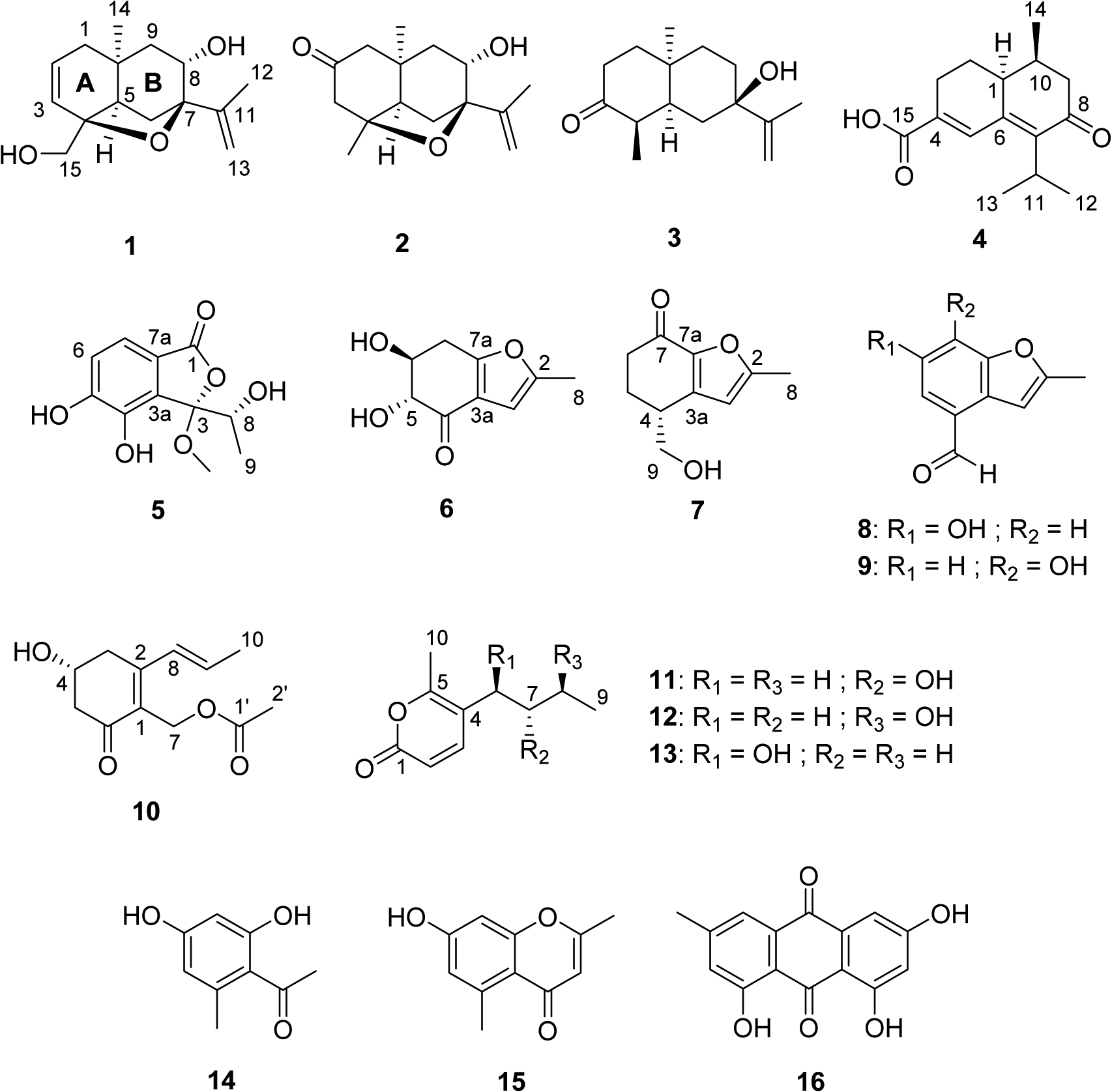
Structures of compounds 1-16.
Table 1.
1H NMR (600 MHz) and 13C NMR (150 MHz) data for compounds 1-4 in DMSO-d6.
| Pos. | 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|---|
| δC | δH | δC | δH | δC | δH | δC | δH | |
| 1 | 40.0 | Hα: 1.73, d (17.4) | 53.9 | Hα: 2.29, d (14.4) | 31.0 | Hα: 1.30 (m) | 38.7 | 2.69, dt (4.4, 13.2) |
| Hβ: 1.67, dd (5.4, 17.4) | Hβ: 1.99a | Hβ: 2.24, dt (5.4, 13.8) | ||||||
| 2 | 128.9 | 5.75, dd (5.1, 10.2) | 209.5 | 37.4 | Hα: 2.57, dt (7.1, 13.8) | 26.7 | Hα 1.74 (m) | |
| Hβ: 2.04, ddd (1.8, 5.4, 13.8) | Hβ: 1.48, dq (4.7, 13.2) | |||||||
| 3 | 129.7 | 5.68, d (10.2) | 49.8 | Hα: 2.44, d (17.4) | 212.3 | 25.0 | Hα: 2.31, m | |
| Hp: 2.55, d (17.4) | Hβ: 2.59, m | |||||||
| 4 | 81.0 | 83.7 | 42.4 | 2.92, pent (6.6) | 138.0 | |||
| 5 | 45.4 | 1.92, d (4.8) | 50.9 | 2.03, d (4.2) | 48.1 | 1.55, m | 131.5 | 7.63, s |
| 6 | 27.4 | Hα: 2.45, d (12.0) | 28.5 | Hα: 2.59, d (12.0) | 33.1 | Hα: 1.83, m | 144.5 | |
| Hβ: 1.74, dd (4.8, 12.0) | Hβ: 1.99a | Hβ: 0.81, t (13.8) | ||||||
| 7 | 86.4 | 86.4 | 72.2 | 140.6 | ||||
| 8 | 69.9 | 3.61, dd (4.2, 4.6) | 69.6 | 3.60, dd (4.3, 5.6) | 31.4 | Hα: 1.82, m | 198.2 | |
| Hβ: 1.50, dt (4.8, 13.8) | ||||||||
| 9 | 39.0 | Hα: 1.12, d (14.8) | 40.0 | Hα: 1.26, d (14.4) | 36.8 | Hα: 1.29, m | 47.0 | Hα: 2.24a |
| Hβ: 2.07, dd (5.3, 14.4) | Hβ: 1.53, dd (5.6, 14.4) | Hβ: 1.33, m | Hβ: 2.64, dd (5.4, 16.2) | |||||
| 10 | 31.4 | 35.3 | 33.0 | 31.6 | 2.24a | |||
| 11 | 146.8 | 146.9 | 146.4 | 26.0 | 3.16, heptet (6.6) | |||
| 12 | 19.4 | 1.71, s | 19.4 | 1.72, s | 18.5 | 1.66, s | 20.9 | 1.13, d (6.6) |
| 13 | 111.2 | Hα: 4.84, s | 111.1 | Hα: 4.87, s | 112.1 | Hα: 4.92, s | 21.4 | 1.17, d (6.6) |
| Hβ: 4.76, s | Hβ: 4.78, s | Hβ: 4.85, s | ||||||
| 14 | 28.1 | 1.15, s | 28.8 | 1.17, s | 26.9 | 1.19, s | 12.5 | 0.81, d (6.6) |
| 15 | 66.6 | Hα: 3.23, dd (5.4, 10.2) | 30.0 | 1.18, s | 11.4 | 0.83, d (6.6) | 167.7 | |
| Hβ: 3.13, dd (4.8, 10.2) | ||||||||
| 7-OH | 4.47, s | |||||||
| 8-OH | 4.52, d (3.8) | 4.75, d (3.6) | ||||||
| 15-OH | 4.47, t (5.9) | 12.76, br s | ||||||
Overlapping signals
Fig. 2.
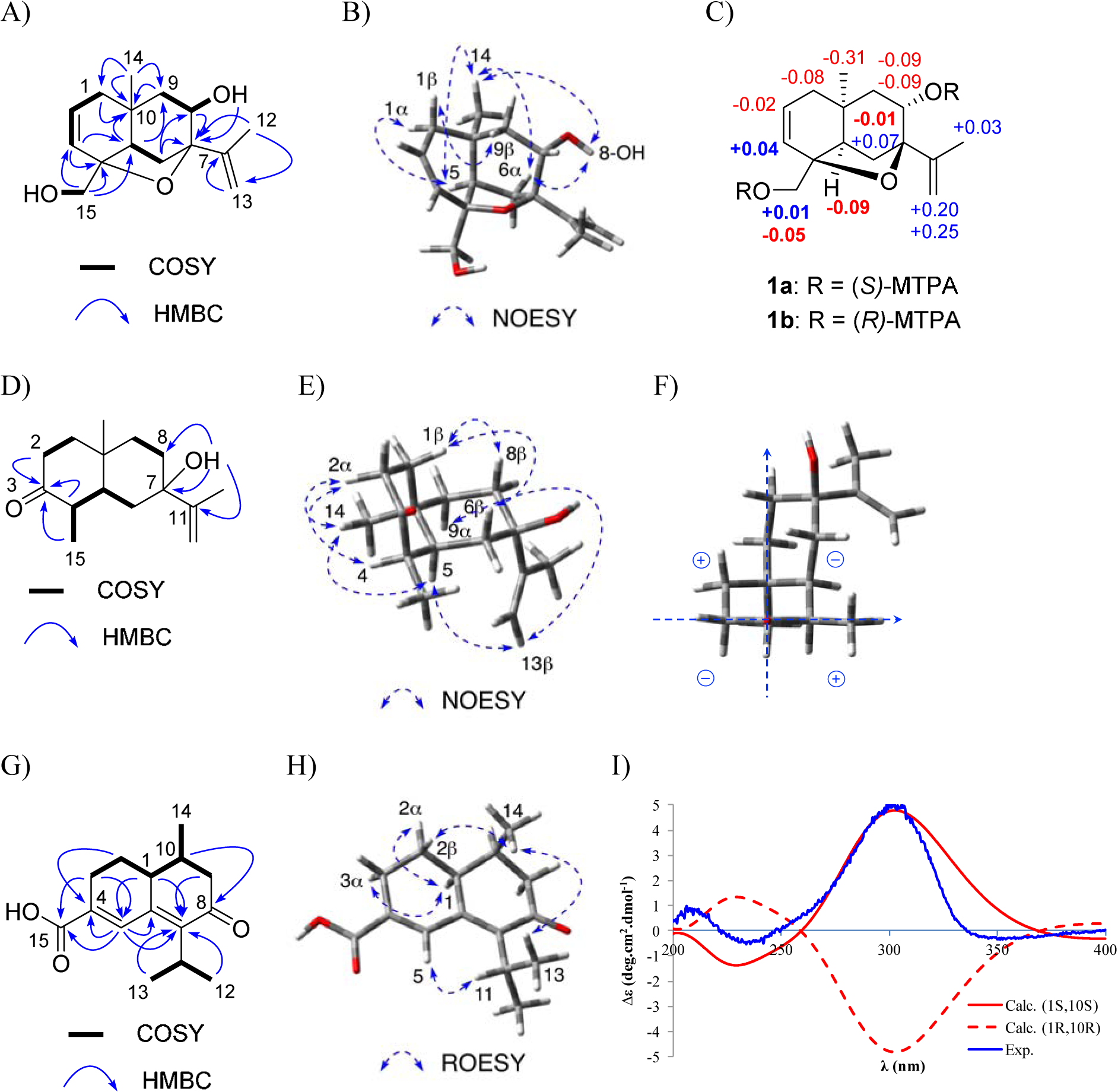
A: Key HMBC and COSY correlations of 1. B: Key NOESY correlations of 1. C: Mosher’s ester analysis of MTPA-1 (irregular ΔδS-R signs in bold). D: Key HMBC and COSY correlations for dictyosporin C (3). E: Key NOESY correlations of 3. F: Octant rules applied for 3. G: Key HMBC and COSY correlations of dictyosporin D (4). H: Key NOESY correlations of 4. I: Experimental ECD spectrum of 4 and calculated ECD spectra of (1S, 10S)-4 and (1R, 10R)-4.
A series of 1D NOESY experiments were employed to define the relative configuration of 1. A 1,3-diaxial NOESY correlation between H-1α and H-5 allowed a half-chair conformation of the cyclohexene to be established. A chair conformation and cis-ring fusion of the A and B rings were assigned from 1,3-diaxial NOESY correlations of H-6α, 8-OH and H3-14; and NOESY correlations from H-5 to H3-14 and H-6α; and from H-1β to H-9β (Fig. 2B). The Mosher’s ester method was employed to establish the absolute configuration of C-8, and by inference, the rest of the stereogenic centers in 1 (Hoye et al., 2007; Cimmino et al., 2017). Treatment of dictyosporin A (1) with (R)-(−)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride [(R)-MTPA-Cl] or (S)-(+)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride [(S)-MTPA-Cl] afforded the bis (S)-MTPA ester (1a) and bis (R)-MTPA ester (1b), respectively. The proton chemical shift differences between 1a and 1b indicated that most protons with ΔδS-R > 0 were located on the left side of the MTPA plane and the ones with ΔδS-R < 0 were on the right side (Fig. 2C). It was noted that ΔδSR signs of H-3, H-5, H-6α and H-15β were inconsistent with those of their neighboring protons. The inconsistent ΔδSR values of H-5 and H-6α were likely due to their lying on the MTPA plane, which causes unreliable results (Ohtani et al., 1991). The protons H-3 and H-15β were too far from the MTPA group at C-8 and were affected by the MTPA group at C-15 which led to their irregular ΔδSR values (Ohtani et al., 1991). Based on proton chemical shift analysis between 1a and 1b, the (S)-configuration was established at C-8, thus allowing the full absolute configuration of (4S, 5R, 7R, 8S, 10S) to be assigned for 1. This assignment was further supported by similar patterns between the experimental ECD spectrum of 1 and the calculated ECD of the (4S, 5R, 7R, 8S, 10S)-stereoisomer using time-dependent density functional theory (TDDFT) methods (Supplementary data Fig. S94A). Therefore, compound 1 was determined as (4S, 5R, 7R, 8S, 10S)-dictyosporin A.
Dictyosporin B (2) was isolated as a white powder and the molecular formula was deduced from HRESIMS data to be C15H22O3, which indicated it was isomeric with 1. The 1H NMR data of 2 (Table 1) differed notably from that of 1 by the absence of two olefinic protons at C-2 and C-3, and a hydroxymethyl group at C-15. In addition, two nonequivalent methylene protons at C-3 and an isolated methyl group at C-15 were observed. The 13C NMR data of 2 revealed one carbonyl carbon which showed HMBC correlations with H-1 and H-3, suggesting this carbonyl was located at C-2. All of the other structural components and connectivities established for 1 were also observed in 2. Detailed ROESY analysis indicated that 2 also had chair-chair conformations and cis-fusion of the A and B rings, similar to those of 1. Mosher’s ester analysis, supported by ECD calculations, (Supplementary data Fig. S94B, S95) gave the same conclusion of (4S, 5R, 7R, 8S, 10R) absolute stereochemistry for dictyosporin B (2). Dictyosporins A (1) and B (2) are the first sesquiterpenes in the eudesmanoid family, consisting of more than 1,000 natural product compounds (Wu et al., 2006), which possess a 4,7-epoxy-11(13)-eudesmen skeleton.
Compound 3 was isolated as a white powder and the molecular formula was established as C15H24O2 based on (+)-HRESIMS measurements. Careful NMR analyses (Table 1) indicated 3 shared the same 11(13)-eudesmene skeleton with 1 and 2, but lacked the tetrahydrofuranoid ring. A ketone (δC 212.3) was assigned at C-3 based on HMBC correlations of H-2/C-3, H-4/C-3, and H-15/C-3 (Fig. 2D). The deshielded resonance of C-7 (δC 72.2) together with HMBC correlations from a singlet hydroxy proton (δH 4.47) to C-6, C-7, C-8, and C-11 allowed the hydroxy group to be located at C-7. The 1,3-diaxial NOESY correlations between H-2α, H-4, and H-14 and correlations between H-1β, H-6β, and H-8β revealed chair conformations for the hexanone and hexane rings in 3. A cis-fusion of the two rings was supported by NOESY correlations between H-5, H-14, H-9α, and H-13b (Fig. 2E). With the relative configuration of 3 established, the ECD spectrum was recorded and it displayed a negative Cotton effect at 288 nm (Supplementary data Fig. S94C). This indicated that ring B resided in a negative CD region (upper right rear octant) on the basis of the ketone octant rule for carbonyl n-π* ECD bands (Fig. 2F) (Murphy, 1975). Thus, the absolute configuration of 3 was revealed as 4R, 5S, 7R, 10R, and this assignment was further supported by quantum chemical ECD calculations which closely matched the experimentally measured ECD spectrum (Fig. S94C, Supplementary data). Therefore, compound 3 was defined as 4R, 5S, 7R, 10R-dictyosporin C.
Compound 4 gave a protonated adduct ion [M + H]+ in the (+)-HRESIMS spectrum at m/z 249.1487 consistent with the molecular formula C15H20O3. Examination of the COSY spectrum of 4 revealed two spin systems including a chain from H-3 to H-9 through contiguous protons H-2, H-1, and H10, and an iso-propyl group from H-12 to H-13 (Fig. 2G). HMBC correlations from a singlet olefinic proton H-5 to C-4, C-6, and C-7 allowed a diene system from C-4 to C-7 to be established. HMBC correlations of H-2/C-4; H-3/C-4 and C-5; H-1/C-5, C-6, and C-7; H-9/C-7 and C-8; and H-10/C-8 facilitated direct connections from C-3 to C-4, and C-1 to C-6; and a connection from C-9 to C7 via a ketone at C-8. The position of the iso-propyl group and a carboxylic acid at C-15 were revealed by HMBC correlations from both H-12 and H-13 to C-7; and from both H-3 and H-5 to C-15. A large 3J1,2β coupling of 13.2 Hz indicated an axial orientation for H-1. The presence of NOESY correlations for H-1/H-2α, H-1/H-3α, and H-2β/H-14 together with the absence of NOESY correlations between H-1/H-14 supported all axial relationships between H-1, H-2β, H-3α, and H-14 (Fig. 2H). The calculated ECD of (1S, 10S)-4 was in good agreement with the experimentally measured ECD spectrum (Fig. 2I), which allowed assignment of 4 as (1S, 10S)-dictyosporin D.
Compound 5 was obtained as a white powder. The (+)-HRESIMS spectrum displayed a sodium adduct ion [M + Na]+ at m/z 263.0536 corresponding to a molecular formula of C11H12O6. 1H and 13C NMR signals characteristic of a tetra-substituted benzene ring were apparent (Table 2). The 7.8 Hz coupling between H-6 and H-7 together with HMBC correlations of H-6/C-4 and C-7a, and H-7/C-3a and C-5 helped establish the substitution pattern of the benzene ring. HMBC correlations of H-7/C-1; 4-OH/C-3a, C-4, and C-5; and 5-OH/C-4, C-5, and C-6 supported placement of an ester carbonyl and two hydroxy groups at C-7a, C4, and C-5, respectively. The position of a 1-methoxy-2-hydroxypropyl group was assigned based on COSY correlations from H-8 to H-9 and 8-OH, and HMBC correlations from H-9 and 3-OCH3 to C-3. The molecular formula requirement of one additional double-bond equivalent and one more oxygen atom enabled establishment of a γ-lactone ring fused to the benzene ring in 5. The deshielded chemical shift of C-3 (δC 110.0) was consistent with a ketal group. The ECD spectrum of 5 showed three positive Cotton Effects (CEs) at 269, 238, and 204 nm and one negative CE at 225 nm which corresponded closely to calculated ECD spectra of the two stereoisomers (3R, 8S)-5 and (3R, 8R)-5 (Fig. S96, Supplementary data). Further DFT calculations for the 1H and 13C chemical shifts of (3R, 8S)-5 and (3R, 8R)-5 indicated that the experimental NMR data of 5 was in agreement with that of the (3R, 8R)-isomer with a high level of confidence (Table S6, Supplementary data) (Smith and Goodman, 2010). Therefore, the structure of 5 was suggested as (3R, 8R)-dictyophthalide A.
Table 2.
1H NMR (600 MHz) and 13C NMR (150 MHz) data for compounds 5-9 in DMSO-d6.
| Pos. | 5 | 6 | 7 | 8 | 9 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δC | δH | δC | δH | δC | δH | δC | δH | δC | δH | |
| 1 | 168.0 | |||||||||
| 2 | 153.5 | 158.2 | 157.0 | 157.9 | ||||||
| 3 | 110.0 | 102.0 | 6.27, s | 108.1 | 6.39, s | 101.8 | 6.98, s | 103.1 | 7.05, s | |
| 3a | 130.1 | 119.9 | 143.1 | 120.2 | 129.6 | |||||
| 4 | 140.6 | 192.1 | 36.4 | 2.91, m | 127.7 | 119.1 | ||||
| 5 | 151.2 | 77.1 | 3.89, dd (4.6, 7.8) | 26.7 | Hα: 2.10, m | 115.3 | 7.25, s | 131.8 | 7.55, d (8.1) | |
| Hβ: 1.82, m | ||||||||||
| 6 | 118.0 | 7.01, d (7.8) | 70.7 | 3.95, m | 36.2 | 2.43, m | 154.6 | 110.4 | 6.73, d (8.1) | |
| 7 | 116.6 | 7.16, d (7.8) | 30.4 | Hα: 3.15, dd (4.8, 16.8) | 184.2 | 103.6 | 7.21, s | 150.5 | ||
| Hβ: 2.80, dd (7.8, 16.8) | ||||||||||
| 7a | 119.5 | 162.4 | 145.9 | 155.7 | 143.5 | |||||
| 8 | 67.3 | 4.26, q (6.6) | 13.1 | 2.27, s | 13.7 | 2.34, s | 13.8 | 2.44, s | 13.8 | 2.47, s |
| 9 | 17.1 | 1.12, d (6.6) | 63.2 | Hα: 3.64, m | 192.4 | 10.07, s | 189.7 | 9.83, s | ||
| Hβ: 3.52, m | ||||||||||
| 3-OCH3 | 50.8 | 2.97, s | ||||||||
| 4-OH | 9.39, s | |||||||||
| 5-OH | 10.51, s | 5.50, d (4.6) | ||||||||
| 6-OH | 5.44, d (4.2) | a | ||||||||
| 7-OH | a | |||||||||
| 8-OH | 5.63, br s | |||||||||
| 9-OH | 4.87, t (5.4) | |||||||||
Not observed
Compound 6 provided a sodium adduct ion [M + Na]+ at m/z 205.0472 in the HRESIMS spectrum that indicated it had a molecular formula of C9H10O4. A proton spin system –C5(OH)–C6(OH)–C7– was deduced from COSY data, while the C-4 carbonyl carbon was assigned by HMBC correlations from H-5 and OH-5 to C-4. HMBC correlations of H-3/C-2, C-3a, and C-7a; and H-8/C-2 and C-3 facilitated assignment of a 2-methylfuran moiety in 6. HMBC correlations of H-6/C-7a; and H-7/C-3a and H-7a allowed the dihydroxy ketone system to be attached to the methylfuran at C-7a. No HMBC correlations from H-5 to C-3a or from H-3 to C-4 were observed in a HMBC experiment with nJHC = 8.3 Hz; however, when the HMBC was optimized for nJHC = 2.0 Hz a week correlation from H-8 to C-4 was observed, which supported a connection between C-3a and C-4. The relative configuration of 6 was established by the 3JHH coupling constant values and selective 1D NOESY data. The large 3J5,6 and 3J6,7β values of 7.8 Hz and the small 3J6,7α value of 4.8 Hz indicated an axial-axial-axial-equatorial relationship for H-5, H-6, H-7β, and H-7α. The 1,3-diaxial orientation of H-5 and H-7β was confirmed by NOESY correlations observed between these two protons. The ECD spectrum calculation for the (5R,6S)-isomer of 6 showed a similar pattern with the experimental ECD spectrum of 6 (Fig. S97, Supplementary data) suggesting the absolute configuration of 6 as (5R,6S)-dictyofuran A.
Compound 7 was obtained as a white powder and gave a sodium adduct ion [M + Na]+ in the (+)-HRESIMS spectrum at m/z 203.0679 consistent with a molecular formula of C10H12O3. Detailed NMR analyses indicated that 7 possessed the same 2-methylfuran moiety as 6. COSY correlations revealed a spin system from H-6 to 9-OH through contiguous H-5, H-4, and H-9 protons. Three-bond HMBC correlations of H-9/C-3a; H-5/C-3a and C-7; and H-6/C-7a supported connections from C-4 to C-3a and from C-6 to C-7a via C-7 forming a planar structure as shown in Fig. 1. The experimental ECD spectrum of 7 matched the calculated ECD spectrum of the (4R)-isomer (Fig. S98, Supplementary data) allowing the assignment of (4R)-dictyofuran B (7).
Dictyofuran C (8) was obtained as white powder. The (−)-HRESIMS spectrum provided a [M - H]− ion at m/z 175.0400 that established a molecular formula of C10H8O3 with seven degrees of unsaturation for 8. HMBC analysis indicated that 8 also had a 2-methylfuran moiety and that this moiety was fused to a tetra-substituted benzene ring at C-3a and C-7a, based on HMBC correlations of H-5/C-3 and C-3a; and H-7/C-3a and C-7a (Table S9, Supplementary data). An aldehyde group was located at C-4 due to HMBC correlations from the aldehyde proton to C-3a, C-4, and C-5. A remaining hydroxy group was substituted at C-6 (δC 154.8) based on its deshielded chemical shift, which completed the structure assignment of dictyofuran C (8).
Dictyofuran D (9) was obtained as white powder and its molecular formula of C10H8O3 was identical to that of 8. The 1H NMR spectrum of 9 was also similar to that of 8, except for replacement of two singlet aromatic protons in 8 by two doublet aromatic protons with a J coupling value of 7.8 Hz in 9 (Table 2). Detailed 2D NMR analysis indicated that structure of 9 only differed from that of 8 by the location of a hydroxy group at C-7. This is the first time that dictyofuran D (9) has been isolated from a natural source, however it was previously reported as a synthetic intermediate in the synthesis of phosphodiesterase type 4 inhibitors, N-(3,5-dichloro-1-oxo-4-pyridyl)-6-difluoromethoxybenzo[4,5]furo[3,2-c]pyridine-9-carboxamide and its derivatives (Gharat et al., 2008).
Compound 10 was isolated as a white powder and (+)-HRESIMS analysis indicated it had a molecular formula of C12H16O4. The 1H and 13C NMR data for 10 were consistent with the molecular formula (Table S11, Supplementary data). The COSY experiment revealed the presence of a propenyl group with an E-configuration at Δ8 due to the J coupling value of 15 Hz for the olefinic protons. The HMBC spectrum showed correlations from two oxygenated methylene protons H-7 (δH 4.79 and 4.82) and a methyl singlet (δH 1.96) to an ester carbonyl C-1’ (δC 170.3) which established an acetate substituent at C-7. A cyclohexenone moiety was deduced based on COSY correlations of H-3/H-4/H-5 and key HMBC correlations of H-3/C-1 and C-2; and H-5/C-6. Long-range HMBC correlations from H-7 to C-2 and C-6; and from H-9 to C-2 supported the connection of C-7 to C-1 and the propenyl group to C-2. Substitution of a hydroxy group at C-4 (δC 64.3) fulfilled the molecular formula requirements of 10. Due to the limited supply of 10, Mosher’s ester analysis to determine the absolute configuration at C-4 was not performed. However, based on the comparison between experimental and calculated ECD spectra (Fig. S99, Supplementary data), the structure of (4R)-dictyosporone A (10) was proposed.
Compound 11 was obtained as a colorless oil. A sodium adduct ion [M + Na]+ at m/z 205.0834 in the (+)-HRESIMS spectrum indicated 11 had a molecular formula of C10H14O3. Two proton spin systems –C2=C3– and –C6–C7(OH)–C8–C9 were assigned based on COSY correlations. HMBC correlations of H-2/C-1, C-4 and H-3/C-1, C-5 together with the requirements of one remaining degree of unsaturation and one additional oxygen atom resulted in the establishment of a disubstituted α-pyrone ring. HMBC correlations of H-7/C-4 and H-3/C-6 allowed the aliphatic chain to be attached to C-4. A relatively deshielded singlet methyl C-10 was assigned as the second substituent on the α-pyrone ring due to a HMBC correlation of H-10/C-5. The 7S absolute configuration for 11 was assigned as a result of Mosher’s ester analysis (Fig. S100, Supplementary data) and thus the compound was established as (7S)-xylariolide E (11).
Compound 12 was obtained as colorless oil and it had the same molecular formula of C10H14O3 as xylariolide E (11). NMR data of 12 was largely analogous to those of 11 (Table 3). The major differences between the 1H of 12 and 11 were that the former had a doublet methyl proton signal H-9 and a relative downfield resonance of the C-8 methine (δH/δC 3.55/64.8) leading to the assignment of a hydroxy group (δH 4.48) at C-8. HMBC correlations from 8-OH to C-7 and C-8 further supported its location. The Mosher’s ester analysis (Fig. S101A, Supplementary data) led to the establishment of (8S)-xylariolide F (12).
Table 3.
1H NMR (600 MHz) and 13C NMR (150 MHz) data for compounds 11-13 in DMSO-d6.
| Position | 11 | 12 | 13 | |||
|---|---|---|---|---|---|---|
| δC | δH | δCa | δH | δC | δH | |
| 1 | 161.7 | 161.6 | 161.5 | |||
| 2 | 111.8 | 6.11, d (9.6) | 112.1 | 6.13, d (9.6) | 112.7 | 6.20, d (9.6) |
| 3 | 149.0 | 7.40, d (9.6) | 147.8 | 7.41, d (9.6) | 144.7 | 7.56, d (9.6) |
| 4 | 113.1 | 115.1 | 119.2 | |||
| 5 | 159.2 | 158.3 | 157.6 | |||
| 6 | 36.4 | Hα: 2.39, dd (4.2, 14.4) | 25.0 | Hα: 2.30, m | 66.2 | 4.47, t (6.6) |
| Hβ: 2.28, dd (8.4, 14.4) | Hβ: 2.36, m | |||||
| 7 | 71.3 | 3.42, m | 38.8 | 1.45, q (7.2) | 39.1 | Hα: 1.41, m |
| Hβ: 1.58, m | ||||||
| 8 | 29.6 | Hα: 1.42, m | 64.8 | 3.55, m | 18.3 | Hα: 1.21, m |
| Hβ: 1.33, m | Hβ: 1.32, m | |||||
| 9 | 10.1 | 0.89, dd (7.2, 7.8) | 23.6 | 1.07, d (6.0) | 13.9 | 0.87, t (7.2) |
| 10 | 17.3 | 2.19, s | 16.8 | 2.19, s | 16.6 | 2.21, s |
| 6-OH | 5.16, br s | |||||
| 7-OH | 4.56, d (5.4) | |||||
| 8-OH | 4.48, d (4.8) | |||||
Determined from HSQC and HMBC spectra
The structure of compound 13 was elucidated as xylariolide D based on NMR and MS analyses. This compound was previously isolated from the endophytic fungal strain Xylaria sp. NCY2; however, its absolute configuration was not determined (Hu et al., 2010). Compound 13 in this study was determined as (6S)-xylariolide D based on the Mosher’s ester analysis (Fig. S101B, Supplementary data). Other known compounds, 2,6-dihydroxy-4-methylacetophenone (14) (Yu et al., 1998), 2,5-dimethyl-7-hydroxychromone (15) (Hu et al., 2014), and emidin (16) (Uno et al., 2001) were assigned by spectroscopic data comparisons (NMR and MS) with the literature values.
3. Concluding Remarks
As part of our ongoing efforts to discover natural product inhibitors of the MALT1 paracaspase enzyme, all of the isolated compounds (1-16) were tested for their ability to inhibit the proteolytic activity of MALT1. These assay results indicated that none of the fungal metabolites were active at a high-test concentration of 100 μM. The sixteen compounds obtained in the current study include a series of variously functionalized sesquiterpenes and benzofurans, a chromenone, a hexanone, an acetophenone, a pyranone, and an anthraquinone. These metabolites possess diverse structural scaffolds and functional group arrays, yet none of the tested compounds were active against the MALT1 enzyme. Our prior work with D. digitatum resulted in the isolation and testing of eleven additional diverse metabolites, but only two compounds with 6H-oxepino[2,3-b]chromen-6-one skeletons were able to inhibit MALT1 activity (Tran et al., 2019). These results suggest that the MALT1 enzyme is not impacted by many classes of natural products and is only subject to inhibition by a relatively narrow subset of compounds. With the identification of various novel sesquiterpenoids and new polyketides, this study broadens our understanding of the metabolic potential of the under explored fungal genus Dictyosporium. These findings reveal that D. digitatum is a prolific source of low molecular weight metabolites with previously undescribed molecular architecture.
4. Experimental section
4.1. General experimental procedures
NMR spectra were acquired on a Bruker Avance III spectrometer equipped with a 3 mm cryogenically cooled probe operating at 600 MHz for 1H and 150 MHz for 13C. 1H and 13C spectra were referenced to the residual deuterated solvent peaks at δH 7.24 and δC 77.2 (CDCl3) and δH 2.50 and δC 39.5 (DMSO-d6). HMBC experiments were optimized for nJCH = 8.3 Hz or 2.0 Hz. All 2D NMR experiments were acquired with nonuniform sampling (NUS) set to 50%, except for HSQC which had NUS set to 25%. HRESIMS data were acquired on an Agilent 6520 Accurate Mass Q-TOF instrument. HPLC purifications were performed using a Varian ProStar 218 solvent delivery module HPLC equipped with a Varian ProStar 325 UV–VIS detector, operating under Star 6.41 chromatography workstation software. All solvents used for extraction and chromatography were HPLC grade and the H2O used was ultrapure water. Optical rotations ([α]D) were measured on a PerkinElmer 241 polarimeter. UV spectra were obtained on an Agilent 8453 UV–VIS spectrophotometer. ECD spectra were measured on a AVIV 420SF Circular Dichroism spectrometer.
4.2. Fungal material and extraction
The fungal isolate was obtained from a soil sample collected in Herod, Illinois, USA (GPS coordinates: 37.53°, −88.97°). The isolate grew slowly on the surfaces of both malt extract and Czapek plates. Mycelium was collected from the isolate and subjected to homogenization in TE buffer (10 mM EDTA HCl, 0.1 mM EDTA, pH 8.0) with zirconium oxide beads in a Bullet Blender Storm (MidSci #BBY24M). The isolate was identified as Dictyosporium digitatum based on analysis of gene sequence data for tef-1 (translation elongation factor 1 alpha gene) (Genbank accession number MN496466), as well as the ribosomal internal transcribed spacer region (1TS1F-5.8S-ITS4) (GenBank accession number MH882417). BLASTn analysis revealed sequence similarity values of >98% for tef-1 and >99% for 1TS1F-5.8S-ITS4 between the fungal isolate and several samples in the NCBI database corresponding to Dictyosporium digitatum. Voucher specimens of the fungus have been retained at the University of Illinois Fungarium, Illinois Natural History Survey (specimen ILLS00121147) and the University of Oklahoma Citizen Science Soil Collection, Fungal Repository (specimen 61B2, ANM413). Additionally, the fungus was deposited in the CBS Collection under specimen number ILLS00121147. For chemical studies, the fungus was grown on Cheerios breakfast cereal supplemented with a 0.3% sucrose solution and 0.005% chloramphenicol in three large mycobags (Unicorn Bags, Plano, TX, USA). The fungus was grown for 6 weeks, whereupon it had achieved complete colonization of the solid substrate. The fungal biomass was extracted by soaking overnight in 8 L of ethyl acetate. The organic extract was subjected to partitioning three times against water (1:1, vol/vol). The ethyl acetate layer was retained, and the solvent removed by evaporation in vacuo, yielding 5.5 g of vibrant orange-red organic-soluble material.
4.3. Isolation and characterization data
The extract (5 g) was fractionated on a CombiFlash Diol-MPLC column (Teledyne ISCO, Licoln, NE, USA) eluted with a flow rate of 85 mL/min using a series of organic solvents: (1) 90% hexanes/10% CH2Cl2 in 10 min; (2) 95% CH2Cl2/5% EtOAc in 10 min; (3) 100% EtOAc in 10 min; (4) 83% EtOAc/17% MeOH in 10 min and (5) 100% MeOH in 20 min, to give 14 fractions A-N. Fraction D was purified on Phenominex Luna C18 HPLC column, 5 μm, 21.4 × 250 mm, (Phenomenex, Torrance, CA, USA) at a flow rate of 9 mL/min with a contiguous series of CH3CN/H2O: a linear gradient from 15% CH3CN/85% H2O to 50% CH3CN/50% H2O in 20 min; isocratic elution at 50% CH3CN/50% H2O for 10 min and then a linear gradient from 50% CH3CN/50% H2O to 100% CH3CN in 30 min to yield compounds 1 (1.2 mg, tR = 21.3 min), 2 (1.0 mg, tR = 25.5 min) and 3 (0.4 mg, tR = 34.0 min). Fraction F was purified on a similar C18 HPLC column (5 μm, 250 × 10 mm) at a flow rate of 4 mL/min with a linear gradient from 20% CH3CN/80% H2O to 80% CH3CN/20% H2O in 25 min and then from 80% CH3CN/20% H2O to 100% CH3CN in 5 min to afford 13 (3.2 mg, tR = 10.4 min), 14 (0.9 mg, 12.5 min), 8 (2.0 mg, tR = 15.3 min) and 16 (1.8 mg, 25.1 min). Fraction G was chromatographed on a C18 HPLC column (5 μm, 250 × 10 mm) with a similar elution program used for fraction F to yield 11 (1.5 mg, tR = 8.4 min), 13 (1.5 mg, tR = 10.4 min), 9 (0.4 mg, tR = 13.3 min) and 8 (0.8 mg, tR = 15.4 min).
Fraction I was separated by C18 HPLC (5 μm, 250 × 10 mm) at a flow rate of 4 mL/min with a linear gradient from 5% CH3CN/95% H2O to 50% CH3CN/50% H2O in 20 min and then from 50% CH3CN/50% H2O to 100% CH3CN in 10 min to obtain compounds 6 (0.3 mg, tR = 11.5 min), 12 (0.2 mg, tR = 13.4 min), 11 (1.5 mg, tR = 14.5 min) and 4 (1.0 mg, tR = 25.2 min). Fraction J was injected into a C18 HPLC column (5 μm, 250 × 10 mm) with a similar program used for fraction I to give 7 (0.4 mg, tR = 12.4 min), 5 (0.5 mg, tR = 14.5 min) and 10 (0.5 mg, tR = 15.6 min). Fraction K was separated by C18 HPLC (5 μm, 250 × 10 mm) with a similar program used for fraction I to give 15 (0.8 mg, tR = 18.4 min).
Dictyosporin A (1):
white powder; [α]22D +68 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 220 (3.27) nm; ECD (c 2,988 × 10−6 M, MeOH) λmax (Δε) 216 (−0.22) and 202 (+2.03) nm; 1H and 13C NMR data in Table 1. (+) HRESIMS m/z 273.1464 [M + Na]+ (calcd for C15H22O3Na, 273.1461).
Dictyosporin B (2):
white powder; [α]22D +17 (c 0.07, MeOH); UV (MeOH) λmax (log ε) 236 (3.94) and 280 (3.40) nm; ECD (c 2,074 × 10−6 M, MeOH) λmax (Δ) 320 (+0.21), 244 (+0.68) and 214 (−0.17) nm; 1H and 13C NMR data in Table 1. (+) HRESIMS m/z 273.1452 [M + Na]+ (calcd for C15H22O3Na, 273.1461).
Dictyosporin C (3):
white powder; [α]22D −34 (c 0.03, MeOH); UV (MeOH) λmax (log ε) 225 (3.57) and 284 (3.40) nm; ECD (c 2,119 × 10−6 M, MeOH) λmax (Δε) 288 (−0.62) nm; 1H and 13C NMR data, Table 1; (+) HRESIMS m/z 259.1670 [M + Na]+ (calcd for C15H24O2Na, 259.1669).
Dictyosporin D (4):
white powder; [α]22D +148 (c 0.09, MeOH); UV (MeOH) λmax (log ε) 302 (4.15) nm; ECD (c 1,512 × 10−6 M, MeOH) λmax (Δε) 350 (−0.31), 303 (+5.06), 234 (−0.47) and 209 (+0.85) nm; 1H and 13C NMR data in Table 2; (+) HRESIMS m/z 249.1487 [M + H]+ (calcd for C15H21O3, 249.1485).
Dictyophthalide A (5):
white powder; [α]22D +9 (c 0.04, MeOH); UV (MeOH) λmax (log ε) 205 (4.12), 222 (3.99), 268 (3.68) and 298 (3.58) nm; ECD (c 1,852 × 10−6 M, MeOH) λmax (Δε) 269 (+0.39), 238 (+0.86), 225 (−1.84) and 204 (+2.04) nm; 1H and 13C NMR data in Table 3; (+) HRESIMS m/z 263.0536 [M + Na]+ (calcd for C11H12O6Na, 263.0526).
Dictyofuran A (6):
white powder; [α]22D +59 (c 0.02, MeOH); UV (MeOH) λmax (log ε) 203 (4.04) and 270 (3.41) nm; ECD (c 1,099 × 10−6 M, MeOH) λmax (Δε) 303 (+0.33), 282 (−0.29), 254 (+0.44) and 212 (+1.65) nm; 1H and 13C NMR data in Table S7 (Supplymentary data); (+) HRESIMS m/z 205.0472 [M + Na]+ (calcd for C9H10O4Na, 205.0471).
Dictyofuran B (7):
white powder; [α]22D +67 (c 0.03, MeOH); UV (MeOH) λmax (log ε) 234 (3.74) and 283 (4.31) nm; ECD (c 926 × 10−6 M, MeOH) λmax (Δε) 307 (−1.58), 278 (+3.01) and 235 (+3.47) nm; 1H and 13C NMR data in Table S8 (Supplymentary data); (+) HRESIMS m/z 203.0677 [M + Na]+ (calcd for C10H12O3Na, 203.0679).
Dictyofuran C (8):
white powder; UV (MeOH) λmax (log ε) 229 (3.54) and 284 (3.26) nm; 1H and 13C NMR data in Table S9 (Supplymentary data); (−) HRESIMS m/z 175.0397 [M - H]− (calcd for C10H7O3, 175.0401).
Dictyofuran D (9):
white powder; UV (MeOH) λmax (log ε) 217 (4.46), 300 (4.29) and 347 (4.47) nm; 1H and 13C NMR data in Table S10 (Supplymentary data); (+) HRESIMS m/z 177.0547 [M + H]+ (calcd for C10H9O3, 177.0446).
Dictyosporone A (10):
white powder; [α]22D +2.0 (c 0.05, MeOH); UV (MeOH) λmax (log ε) 279 (3.66); ECD (c 2,480 × 10−6 M, MeOH) λmax (Δε) 322 (+0.04), 283 (−0.11) and 226 (−0.17) nm; 1H and 13C NMR data in Table S11 (Supplymentary data); (+) HRESIMS m/z 247.0939 [M + Na]+ (calcd for C12H16O4Na, 247.0941).
Xylariolide E (11):
colorless oil; [α]22D +25 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 222 (3.61) and 306 (3.81) nm; 1H and 13C NMR data in Table S12 (Supplymentary data); (+) HRESIMS m/z 205.0834 [M + Na]+ (calcd for C10H14O3Na, 205.0835).
Xylariolid F (12):
colorless oil; [α]22D +21 (c 0.01, MeOH); UV (MeOH) λmax (log ε) 220 (3.29) and 304 (3.14) nm; 1H and 13C NMR data in Table S13 (Supplymentary data); (+) HRESIMS m/z 205.0835 [M + Na]+ (calcd for C10H14O3Na, 205.0835).
4.4. Preparation of the Mosher’s ester derivatives of compounds 1, 2, 11, 12 and 13
Compounds 1, 2, 11, 12, and 13 (0.1 mg each) were individually dissolved in deuterated pyridine (250 μL), transferred into separate NMR tubes and (R)-MTPA-Cl (2 μL) was added to each tube. The NMR tubes were then shaken carefully and kept in the dark at room temperature for 24 h to afford the (S)-MTPA ester derivatives. A similar procedure was performed using the (S)-MTPA-Cl to produce the (R)-MTPA ester derivatives of each compound. The 1H NMR spectra of the R and S Mosher’s ester derivatives were individually recorded and COSY and HSQC experiments were used to confirm the 1H NMR assignments.
4.4.1. 1-(S)-MTPA ester
1H NMR (pyridine-d5, 600 MHz) δH 6.00 (1H, d, J = 9.6 Hz, H-3), 5.81 (1H, m, H-2), 5.47 (1H, br s, H-8), 5.04 (1H, s, Hα−13), 5.02 (1H, s, Hβ−13), 4.62 (1H, d, J = 10.8 Hz, Hα−15), 4.19 (1H, d, J = 10.8 Hz, Hβ−15), 2.40 (1H, d, J = 12.0 Hz, Hα−6), 2.33 (1H, d, J = 16.2 Hz, Hβ−9), 2.16 (1H, d, J = 12.0 Hz, Hβ−6), 1.94 (3H, s, H-12), 1.93 (1H, m, H-5), 1.65 (2H, s, H-1), 1.33 (1H, d, J = 15.6 Hz, Hα−9), 0.80 (3H, s, H-14); (+) HRESIMS m/z 705.2263 [M + Na]+ (calcd for C35H36F6O7Na, 705.2258).
4.4.2. 1-(R)-MTPA ester
1H NMR (pyridine-d5, 600 MHz) δH 5.96 (1H, d, J = 9.0 Hz, H-3), 5.83 (1H, m, H-2), 5.54 (1H, br s, H-8), 4.84 (1H, s, Hα−13), 4.77 (1H, s, Hβ−13), 4.61 (1H, d, J = 10.8 Hz, Hα−15), 4.24 (1H, d, J = 10.8 Hz, Hβ−15), 2.42 (1H, m, Hβ−9), 2.41 (1H, m, Hα−6), 2.09 (1H, d, J = 12.6 Hz, Hβ−6), 2.02 (1H, br s, H-5), 1.91 (3H, s, H-12), 1.73 (2H, s, H-1), 1.42 (1H, d, J = 15.6 Hz, Hα−9), 1.11 (3H, s, H-14); (+) HRESIMS m/z 705.2250 [M + Na]+ (calcd for C35H36F6O7Na, 705.2258).
4.4.3. 2-(S)-MTPA ester
1H NMR (pyridine-d5, 600 MHz) δH 5.39 (1H, m, H-8), 5.02 (1H, s, Hα-13), 5.01 (1H, s, Hβ-13), 2.99 (1H, d, J = 12.0 Hz, Hβ-3), 2.51 (1H, d, J = 12.0 Hz, Hα-3), 2.42 (1H, d, J = 12.6 Hz, Hα-6), 2.25 (2H, s, H-1), 2.25 (1H, m, Hβ-6), 1.99 (1H, m, H-5), 1.98 (1H, m, Hβ-9), 1.92 (3H, s, H-12), 1.46 (1H, d, J = 16.8 Hz, Hα-9), 1.23 (3H, s, H-14), 0.85 (3H, s, H-15); (+) HRESIMS m/z 489.1863 [M + Na]+ (calcd for C25H29F3O5Na, 489.1860).
4.4.4. 2-(R)-MTPA ester
1H NMR (pyridine-d5, 600 MHz) δH 5.46 (1H, m, H-8), 4.84 (1H, s, Hα−13), 4.78 (1H, s, Hβ−13), 3.01 (1H, d, J = 12.0 Hz, Hβ−3), 2.53 (1H, d, J = 12.0 Hz, Hα−3), 2.44 (1H, d, J = 12.6 Hz, Hα−6), 2.32 (2H, s, H-1), 2.18 (1H, m, Hβ−6), 2.06 (1H, m, Hβ−9), 2.03 (1H, m, H-5), 1.90 (3H, s, H-12), 1.55 (1H, d, J = 16.8 Hz, Hα−9), 1.23 (3H, s, H-14), 1.15 (3H, s, H-15); (+) HRESIMS m/z 489.1868 [M + Na]+ (calcd for C25H29F3O5Na, 489.1860).
4.4.5. 11-(S)-MTPA ester
1H NMR (pyridine-d5, 600 MHz) δH 7.31 (1H, d, J = 9.0 Hz, H-3), 6.29 (1H, d, J = 9.6 Hz, H-2), 5.27 (1H, m, H-7), 2.69 (1H, dd, J = 8.4, 14.4 Hz, Hα−6), 2.55 (1H, dd, J = 4.8, 14.4 Hz, Hβ−6), 2.09 (3H, s, H-10), 1.64 (2H, pent, J = 7.2 Hz, H-8), 0.84 (3H, t, J = 7.2 Hz, H-9); (+) HRESIMS m/z 399.1411 [M + H]+ (calcd for C20H22F3O5, 399.1414).
4.4.6. 11-(R)-MTPA ester
1H NMR (pyridine-d5, 600 MHz) δH 7.18 (1H, d, J = 9.0 Hz, H-3), 6.18 (1H, d, J = 9.0 Hz, H-2), 5.27 (1H, m, H-7), 2.58 (1H, dd, J = 9.0, 15.0 Hz, Hα−6), 2.47 (1H, dd, J = 4.2, 15.0 Hz, Hβ−6), 1.88 (3H, s, H-10), 1.67 (2H, pent, J = 7.2 Hz, H-8), 0.93 (3H, t, J = 7.2 Hz, H-9); (+) HRESIMS m/z 399.1433 [M + H]+ (calcd for C20H22F3O5, 399.1414).
4.4.7. 12-(S)-MTPA ester
1H NMR (pyridine-d5, 600 MHz) δH 7.16 (1H, d, J = 9.6 Hz, H-3), 6.28 (1H, d, J = 9.0 Hz, H-2), 5.26 (1H, m, H-8), 2.30 (1H, m, Hα−6), 2.25 (1H, m, Hβ−6), 2.04 (3H, s, H-10), 1.77 (1H, m, Hα−7), 1.67 (1H, m, Hβ−7), 1.28 (3H, d, J = 6.0 Hz, H-9). (+) HRESIMS m/z 399.1417 [M + H]+ (calcd for C20H22F3O5, 399.1414).
4.4.8. 12-(R)-MTPA ester
1H NMR (pyridine-d5, 600 MHz) δH 7.05 (1H, d, J = 10.2 Hz, H-3), 6.25 (1H, d, J = 10.2 Hz, H-2), 5.26 (1H, m, H-8), 2.13 (1H, m, Hα−6), 2.12 (1H, m, Hβ−6), 1.93 (3H, s, H-10), 1.68 (1H, m, Hα−7), 1.62 (1H, m, Hβ−7), 1.35 (3H, d, J = 6.0 Hz, H-9). (+) HRESIMS m/z 399.1419 [M + H]+ (calcd for C20H22F3O5, 399.1414).
4.4.9. 13-(S)-MTPA ester
1H NMR (pyridine-d5, 600 MHz) δH 7.54 (1H, d, J = 9.6 Hz, H-3), 6.43 (1H, d, J = 9.6 Hz, H-2), 6.00 (1H, t, J = 7.2 Hz, H-6), 2.36 (3H, s, H-10), 1.85 (1H, m, Hα-7), 1.61 (1H, m, Hβ-7), 1.23 (1H, m, Hα-8), 1.09 (1H, m, Hβ-8), 0.79 (3H, t, J = 7.2 Hz, H-9); (+) HRESIMS m/z 399.1421 [M + H]+ (calcd for C20H22F3O5, 399.1414).
4.4.10. 13-(R)-MTPA ester
1H NMR (pyridine-d5, 600 MHz) δH 7.18 (1H, d, J = 9.6 Hz, H-3), 6.29 (1H, d, J = 9.6 Hz, H-2), 5.92 (1H, t, J = 7.2 Hz, H-6), 2.33 (3H, s, H-10), 1.87 (1H, m, Hα−7), 1.63 (1H, m, Hβ−7), 1.31 (1H, m, Hα−8), 1.26 (1H, m, Hβ−8), 0.85 (3H, t, J = 7.2 Hz, H-9); (+) HRESIMS m/z 399.1419 [M + H]+ (calcd for C20H22F3O5, 399.1414).
4.5. Computational Details
4.5.1. TDDFT ECD calculations for 1–7 and 10
ECD calculations of were performed at 298 K using Maestro and Gaussian 09. Molecular mechanics calculations were performed using Macromodel interfaced to the Maestro program (Version 2015.3, Schrödinger) (Schrödinger Release 2015–3: MacroModel). All conformational searches used the OPLS_2005 force field. Conformers having internal relative energies within 3 kcal/mol were subjected to geometry optimization on Gaussian 09 (Frisch et al., 2013) at the DFT level with the B3LYP functional and the 6–31G(d,p) basis set. Optimized conformers were then subjected to TDDFT calculations in MeOH on Gaussian 09 using the B3LYP functional, and the 6–31+G(d,p) basis set for 1, 3-7 and 10, and the 6–31G(d,p) basis set for 2. All calculations were performed in MeOH solvent. For each conformer, all of the resultant rotational strengths were converted into Gaussian distributions and summed to obtain the final calculated ECD spectrum based on the Boltzmann distribution of each conformer. ECD spectra were generated using the SpecDis program (Bruhn et al., 2015).
4.5.2. DFT NMR calculations for (3R, 8R)-5 and (3R, 8S)-5
Molecular mechanics and quantum chemical calculations for (3R, 8R)-5 and (3R, 8S)-5 were performed in DMSO with the same procedure as above. Single point calculations in DMSO with the B3LYP functional and the 6–311+G(d,p) basis set were then employed to provide the shielding constants of carbon and proton nuclei. Exchangeable protons were excluded in the calculation (Kwan and Liu, 2015). Meanwhile, the same procedure was applied on TMS (tetramethylsilane). Final 1H and 13C chemical shifts were obtained as the result of the Boltzmann weighted average. The theoretical chemical shifts were calculated according to the equation
where is the calculated shift for nucleus × (in ppm); σx is the shielding constant for nucleus x; σTMS is the shielding constant of TMS computed at the same level of theory. The mean absolute error (MAE) was defined as after removing systematic errors during the chemical shift calculations. DP4 parameters were calculated using the online applet at http://www-jmg.ch.cam.ac.uk/tools/nmr/.
4.6. MALT1 assay
Enzymatic expression and the MALT1 assay procedure were similar to those described previously (Tran et al., 2019).
Supplementary Material
Highlights.
Twelve undescribed compounds were identified from the soil-derived fungus Dictyosporium digitatum.
Absolute configurations of the isolated compounds were established using Mosher’s ester analysis and quantum chemical ECD calculations.
Two sesquiterpenes, dictyosporins A (1) and B (2), have a unique carbon-oxygen structural framework.
Acknowledgments
We thank H. Bokesch for HRESIMS data collection. This research utilized the computational resources of the NIH HPC Biowulf cluster and it was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E. The research was also supported in part by a grant from the National Cancer Institute (UO1CA182740). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors declare no conflicts of interest.
References
- Schrödinger Release 2015–3: MacroModel. Schrödinger, LLC, New York. [Google Scholar]
- Blackwell M, 2011. The fungi: 1, 2, 3… 5.1 million species? Am. J. Bot 98, 426–438. [DOI] [PubMed] [Google Scholar]
- Bruhn T, Schaumlöffel A, Hemberger Y, 2015. SpecDis. University of Wuerzburg, Germany. [Google Scholar]
- Cimino A, Masi M, Evidente M, Superchi S, Evidente A, 2017. Application of Mosher’s method for absolute configuration assignment to bioactive plants and fungi metabolites. J. Pharm. Biomed. Anal 144, 59–89. [DOI] [PubMed] [Google Scholar]
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ, 2013. Gaussian 09. Gaussian, Inc., Wallingford CT. [Google Scholar]
- Gharat LA, Gajera JM, Farande AV, Kadam SM, Chapala K, Bhadane S, 2008. Process for the synthesis of N9-(3,5-dichloro-4-pyridyl)-6-difluoromethoxybenzo[4,5] furo[3,2-c]pyridine-9-carboxamide and salts thereof. Int. Appl PCT/IB2007/004045, Int. Pub. WO 2008/081282 A2, 20080710. [Google Scholar]
- Goh T-K, Hyde KD, Ho WH, Yanna, 1999. A revision of the genus Dictyosporium, with descriptions of three new species. Fungal Divers. 2, 65–100. [Google Scholar]
- Gonzalez-Medina M, Owen JR, El-Elimat T, Pearce CJ, Oberlies NH, Figueroa M, Medina-Franco JL, 2017. Scaffold diversity of fungal metabolites. Front. Pharmacol 8, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoye TR, Jeffrey CS, Shao F, 2007. Mosher ester analysis for the determination of absolute configuration of stereogenic (chiral) carbinol carbons. Nat. Protoc 2, 2451–2458. [DOI] [PubMed] [Google Scholar]
- Hu L, Chen N-N, Hu Q, Yang C, Yang Q-S, Wang F-F, 2014. An Unusual Piceatannol Dimer from Rheum austral D. Don with Antioxidant Activity. Molecules 19, 11453–11464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu ZY, Li YY, Lu CH, Lin T, Hu P, Shen YM, 2010. Seven Novel Linear Polyketides from Xylaria sp NCY2. Helv. Chim. Acta 93, 925–933. [Google Scholar]
- Kwan EE, Liu RY, 2015. Enhancing NMR Prediction for Organic Compounds Using Molecular Dynamics. J. Chem. Theory Comput 11, 5083–5089. [DOI] [PubMed] [Google Scholar]
- Murphy WS, 1975. The Octant Rule. Its Place in Organic Chemistry. J. Chem. Educ 52, 774–776. [Google Scholar]
- Ohtani I, Kusumi T, Kashman Y, Kakisawa H, 1991. High-Field FT NMR Application of Mosher’s Method. The Absolute Configurations of Marine Terpenoids. J. Am. Chem. Soc 113, 4092–4096. [Google Scholar]
- Prasher IB, Verma RK, 2015. Two new species of Dictyosporium from India. Phytotaxa 204, 193–202. [Google Scholar]
- Rédou V, Navarri M, Cladière LM, Barbier G, Burgaud G, 2015. Species Richness and Adaptation of Marine Fungi from Deep-Subseafloor Sediments. Appl. Environ. Microbiol 81, 3571–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SG, Goodman JM, 2010. Assigning Stereochemistry to Single Diastereoisomers by GIAO NMR Calculation: The DP4 Probability. J. Am. Chem. Soc 132, 12946–12959. [DOI] [PubMed] [Google Scholar]
- Spiteller P, 2015. Chemical ecology of fungi. Nat. Prod. Rep 32, 971–993. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Hirayama K, Yonezawa H, Sato G, Toriyabe A, Kudo H, Hashimoto A, Matsumura M, Harada Y, Kurihara Y, Shirouzu T, Hosoya T, 2015. Revision of the Massarineae (Pleosporales, Dothideomycetes). Stud. Mycol 82, 75–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran TD, Wilson BAP, Henrich CJ, Staudt LM, Krumpe LRH, Smith EA, King J, Wendt KL, Stchigel AM, Miller AN, Cichewicz RH, O’Keefe BR, Gustafson KR, 2019. Secondary Metabolites from the Fungus Dictyosporium sp. and Their MALT1 Inhibitory Activities. J. Nat. Prod 82, 154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uno H, Masumoto A, Honda E, Nagamachi Y, Yamaoka Y, Ono N, 2001. Intramolecular aldol-type condensation between side chains of naphthoquinones: biomimetic synthesis of 1,6- and 1,8-dihydroxyanthraquinones. J. Chem. Soc., Perkin Trans 1 0, 3189–3197 [Google Scholar]
- Wu Q-X, Shi Y-P, Jia Z-J, 2006. Eudesmane sesquiterpenoids from the Asteraceae family. Nat. Prod. Rep 23, 699–734. [DOI] [PubMed] [Google Scholar]
- Yu T-W, Shen Y, McDaniel R, Floss HG, Khosla C, Hopwood DA, Moore BS, 1998. Engineered Biosynthesis of Novel Polyketides from Streptomyces Spore Pigment Polyketide Synthases. J. Am. Chem. Soc 120, 7749–7759. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.