

Graphical abstract

Keywords: Hepatitis C virus, HCV, Antiviral agent, Nucleoside
Abstract
On the basis of our previous study on antiviral agents against the severe acute respiratory syndrome (SARS) coronavirus, a series of nucleoside analogues whose 5′-hydroxyl groups are masked by various protective groups such as carboxylate, sulfonate, and ether were synthesized and evaluated to develop novel anti-hepatitis C virus (HCV) agents. Among these, several 5′-O-masked analogues of 6-chloropurine-2′-deoxyriboside (e.g., 5′-O-benzoyl, 5′-O-p-methoxybenzoyl, and 5′-O-benzyl analogues) were found to exhibit effective anti-HCV activity. In particular, the 5′-O-benzoyl analogue exhibited the highest potency with an EC50 of 6.1 μM in a cell-based HCV replicon assay. Since the 5′-O-unmasked analogue (i.e., 6-chloropurine-2′-deoxyriboside) was not sufficiently potent (EC50 = 47.2 μM), masking of the 5′-hydroxyl group seems to be an effective method for the development of anti-HCV agents. Presently, we hypothesize two roles for the 5′-O-masked analogues: One is the role as an anti-HCV agent by itself, and the other is as a prodrug of its 5′-O-demasked (deprotected) derivative.
1. Introduction
The hepatitis C virus (HCV), a member of the family Flaviviridae, is an enveloped, positive-sense, single-strand RNA virus.1 The virus is a major causative agent of non-A, non-B hepatitis and infects an estimated 170 million people worldwide.2 In most cases, acute phase infection with HCV is asymptomatic; however, the virus frequently establishes chronic hepatitis in up to 80% of the infected individuals and persists for decades with a substantially high risk of developing liver cirrhosis and hepatocellular carcinoma.3 Currently, there is no vaccine available against HCV, and the approved drugs are combinations of interferon-α (IFN-α) and ribavirin (1-β-d-ribofuranosyl-1H-1,2,4-triazole-3-carboxamide). Although the use of pegylated IFN-α instead of the unmodified one has resulted in improved therapeutic effectiveness, the sustained virological response is still poor (41–55%); moreover, in some cases, significant side effects can be caused.4 Therefore, more efficacious therapies are urgently required for ensuring public health.5
To date, a number of nucleoside analogues, including ribavirin, have been synthesized and tested for anti-HCV activity.6, 7 Among these, some 2′-modified ribonucleoside analogues such as 2′-C-methyl analogues 1,6a 2′-fluoro analogues 2,6b and 2′-O-methyl analogue 3 6c exhibit potent anti-HCV activities in a cell-based HCV replicon assay (Fig. 1 ). Particularly, Valopicitabine, a prodrug of 1c, is currently in phase II clinical trials.8 Recently, the anti-HCV properties of 4′-substituted ribonucleosides such as 4 have also been reported by the Roche group.6d Concerning the antiviral mechanism of common nucleoside analogues, it is known that most of these analogues are converted to their corresponding 5′-triphosphates by cellular and/or viral enzymes and then compete with natural triphosphates as substrates for incorporation into viral nucleic acids during viral replication.9 Therefore, the 5′-hydroxyl group is indispensable to the anti-HCV activity of these nucleoside analogues.10
Figure 1.
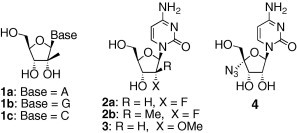
Examples of anti-HCV nucleoside analogues.
Recently, we have reported that a carbocyclic oxetanocin analogue 5, one of whose hydroxyl groups (that corresponds to the 5′-hydroxyl group of the ribonucleoside) was masked by a benzoyl group, showed antiviral activity against the severe acute respiratory syndrome (SARS) coronavirus (Fig. 2 ).11 Interestingly, the antiviral activity of 5 (EC50 = 14.5 μM) was much more efficacious than that of the unmasked 6 (almost no activity, EC50 > 300 μM), which seems to indicate an inconsistency with the necessity of the 5′-hydroxyl group to this compound.
Figure 2.
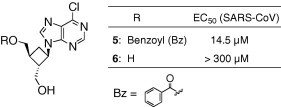
Anti-SARS-coronavirus (CoV) activities of carbocyclic oxetanocin analogues.
This result attracted our interest and led us to expect that this unique trend could contribute to developing anti-HCV agents as well. Thus, several nucleoside analogues, including 5 and 6, were designed as candidate compounds for the agents (Fig. 3 ). In this paper, we report the syntheses of these nucleoside analogues and their biological evaluations as anti-HCV agents.
Figure 3.
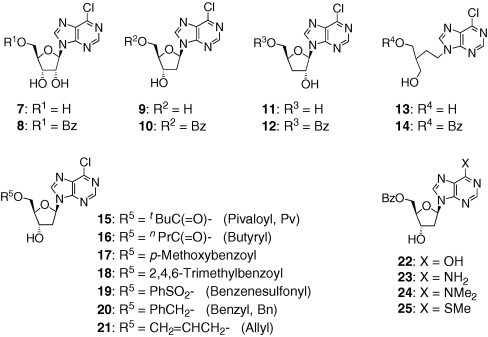
The candidate compounds for anti-HCV agents.
2. Results and discussion
2.1. Chemistry
Compounds 7–9, 11, 13, 14, 22, and 23 were prepared based on the previous reports.12 The syntheses of compounds 10, 12, and 15–19 were carried out by the treatment of diol 9 or 11 with the corresponding acyl or sulfonyl chloride (i.e., benzoyl chloride for 10 and 12; pivaloyl chloride, butyryl chloride, p-methoxybenzoyl chloride, 2,4,6-trimethylbenzoyl chloride, and benzenesulfonyl chloride for 15–19, respectively), as shown in Scheme 1 . In the case of compound 18, conventional conditions (trimethylbenzoyl chloride, DMAP, and pyridine) required a long reaction time and led to the gradual decomposition of 9 and 18, probably because the 6-chloropurine moiety reacted with the amine bases by degrees due to its electrophilic nature. This side reaction was slightly suppressed by the use of a low-nucleophilic base, N,N-diisopropylethylamine instead of pyridine and DMAP.
Scheme 1.
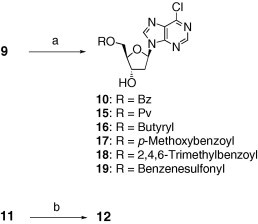
Reagents and conditions: (a) for 10, BzCl, pyr, 0 °C, 85%; for 15, PvCl, DMAP, pyr, 0 °C to rt, 81%; for 16, butyryl chloride, pyr, 0 °C to rt, 68%; for 17, p-methoxybenzoyl chloride, pyr, 0 °C, 72%; for 18, 2,4,6-trimethylbenzoyl chloride, N,N-diisopropylethylamine, MeCN, rt, 21%; for 19, benzenesulfonyl chloride, pyr, 0 °C to rt, 40%; (b) BzCl, pyr, CH2Cl2, 0 °C, 59%.
Compound 10 was also prepared via an alternative route illustrated in Scheme 2 A. Regioselective protection of the 5′-hydroxyl group of 2′-deoxyadenosine (26) yielded the benzoate 23 in 65% yield,12c which was subsequently converted to the silyl ether 27 in 93% yield. After transformation to the 6-chloropurine derivative 28 with a 58% yield,13 the TBS group was cleaved to afford 10 in 72% yield. Compound 28 was also used as an intermediate in the synthesis of 25, as shown in Scheme 2B. Exposure of 28 to sodium methylthiolate resulted in the formation of the debenzoylated 6-SMe analogue 29 with a 67% yield, which was reacylated into 30 in 97% yield. Unfortunately, the use of thiourea in place of sodium methylthiolate led to the decomposition of the resultant products. Finally, the removal of the TBS group afforded 25 in 88% yield. The synthesis of 24 is illustrated in Scheme 2C. The diacetate 31, which was prepared as described in a previous report,11 was treated with dimethylamine to furnish 32 in 62% yield.14 Subsequently, regioselective benzoylation of the 5′-hydroxyl group in 32 afforded 24 in 77% yield.
Scheme 2.
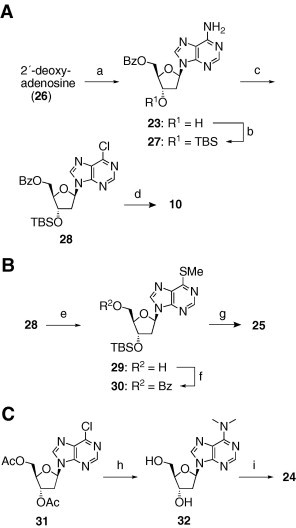
Reagents and conditions: (a) BOPDC, BzOH, pyr, rt, 65% (Ref. 12c); (b) TBSCl, imidazole, pyr, rt, 93%; (c) tBuONO, Et4NCl, CCl4–CH2Cl2, 0–50 °C, 58%; (d) TBAF, AcOH, THF, rt, 72%; (e) MeSNa, water–DMF, rt, 67%; (f) BzCl, Et3N, DMAP, CH2Cl2, 0 °C, 97%; (g) TBAF, THF, 0 °C, 88%; (h) Me2NH, water–dioxane, rt, 62%; (i) BzCl, pyr, 0 °C to rt, 77%.
Meanwhile, the syntheses of 20 and 21 turned out to be unexpectedly difficult because of the instability of these molecules under both strongly basic and acidic conditions; for example, the exposure of 9 to BnBr–NaH or BnOC( NH)CCl3–CF3SO3H afforded a complex mixture. Thus, a stepwise protection–deprotection process was required, which is illustrated in Scheme 3 (3A: synthesis of 20, 3B: synthesis of 21). The monoacetate 33, which was prepared from 26 by employing a partly modified Santaniello’s procedure,15 was converted to the silyl ether 34 in 94% yield. The use of a mixed solvent (pyridine–DMF) effectively reduced the competitive silylation of the 6-amino group. Protection of the amino group with pivaloyl chloride followed by the removal of the acetyl group in 35 afforded 36 with a 98% yield in two steps. The addition of DMAP and MeOH in the N-pivaloylation step was effective in converting the N,N-dipivaloylated by-product to the desired 35. Chemoselective benzylation of the 5′-hydroxyl group in 36 was successfully carried out by using 2.5 equiv of potassium tert-butoxide and 1.1 equiv of benzyl bromide in THF to provide 37 in 97% yield. After the removal of the pivaloyl group (76% yield), the resultant amino group of 38 was substituted with a chloro group to provide 39, which was subsequently deprotected to afford 20 with a 34% yield in two steps. Compound 21 was synthesized employing almost the same method as that for 20 described above (Scheme 3B, 20% overall yield from 36).
Scheme 3.
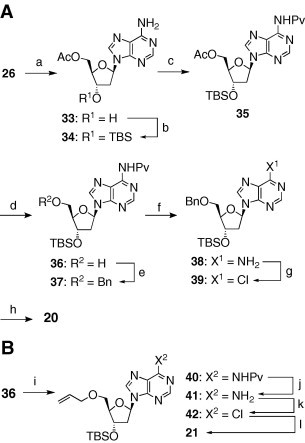
Reagents and conditions: (a) lipase (CAL), vinyl acetate, MS4A, THF, 60 °C, 89%; (b) TBSCl, DMAP, pyr–DMF, rt, 94%; (c) PvCl, Et3N, CH2Cl2, rt, and then DMAP, MeOH, rt; (d) K2CO3, MeOH, 0 °C, 98% from 34; (e) BnBr, tBuOK, THF, 0 °C, 97%: (f) K2CO3, MeOH, rt, 76%, (g) tBuONO, Et4NCl, CCl4–CH2Cl2, 0–50 °C (h) TBAF, AcOH, THF, rt, 34% from 38; (i) allyl bromide, tBuOK, THF, 0 °C; (j) K2CO3, MeOH, rt, 73% from 36; (k) tBuONO, Et4NCl, CCl4–CH2Cl2, 0–50 °C (l) TBAF, AcOH, THF, rt, 28% from 41.
2.2. Biological evaluation
The above synthesized nucleoside analogues were assayed for their ability to inhibit HCV RNA replication in a subgenomic replicon Huh7 cell line (LucNeo#2),16 and the result is shown in Table 1 . These cells contain a HCV subgenomic replicon RNA encoding a luciferase reporter gene as a marker. The potency of the analogues against the HCV replicon is expressed as EC50, which was quantified by a luciferase assay after a two-day incubation period with the corresponding compound. In addition, the associated cytotoxicity, which is expressed as CC50 in Table 1, was evaluated in a tetrazolium (XTT)-based assay according to the manufacturer’s protocol.
Table 1.
Inhibitory potency (EC50) and cytotoxicity (CC50) of compounds 5–25 and ribavirin (positive control) in HCV replicon assaya
| Compound | EC50b (μM) | CC50c (μM) | Antiviral indexd |
|---|---|---|---|
| 5 | 6.4 | 20.0 | 3.1 |
| 6 | 46.1 | >200 | 4.3 |
| 7 | 31.0 | >200 | >6.5 |
| 8 | 41.0 | 97.3 | 2.4 |
| 9 | 47.2 | 173 | 3.7 |
| 10 | 6.1 | 111 | 18.2 |
| 11 | 64.1 | >200 | >3.1 |
| 12 | 38.1 | 70.5 | 1.9 |
| 13 | 80.8 | >200 | >2.5 |
| 14 | 13.3 | 108 | 8.1 |
| 15 | 28.0 | 177 | 6.3 |
| 16 | 30.3 | >200 | >6.6 |
| 17 | 8.4 | 76.4 | 9.1 |
| 18 | >50 | 91.0 | <1.8 |
| 19 | 28.8 | 146 | 5.1 |
| 20 | 17.7 | 178 | 10.1 |
| 21 | 24.5 | 140 | 5.7 |
| 22 | >100 | — | — |
| 23 | 105 | >200 | >1.9 |
| 24 | 22.3 | >200 | >9.0 |
| 25 | >100 | — | — |
| Ribavirin | 54.8 | >200 | >3.7 |
The same experiment was performed at least three times independently.
Average of 50% effective concentrations.
Average of 50% cytotoxic concentrations.
Antiviral index was defined as the 50% toxic dose divided by the 50% effective dose.
As expected, the benzoylated carbocyclic oxetanocin analogue 5 exhibited a stronger anti-HCV activity than that shown by the unprotected 6 (EC50 = 6.4 μM and 46.1 μM, respectively). This trend seems to be consistent with that of the anti-SARS-coronavirus activity, as mentioned above (Fig. 2; EC50 = 14.5 μM and >300 μM, respectively). However, compound 5 displayed a cytotoxic effect at a level close to its EC50 value (CC50 = 20.0 μM; antiviral index = 3.1); therefore, further evaluation of this compound was not undertaken.
2.2.1. Structure–activity relationship (SAR) of the sugar moiety
To evaluate the effect of the sugar moiety, several 6-chloropurine derivatives that possessed the ribofuranosyl structure (7 and 8), or deoxyribofuranosyl structure (2′-deoxy: 9 and 10; 3′-deoxy: 11 and 12), or an acyclic backbone (13 and 14) were tested. Among them, the 2′- deoxyribonucleoside derivative 10 exhibited the highest potency against the HCV replicon with an EC50 of 6.1 μM, which is ninefold potent over that of ribavirin, and a CC50 of 111 μM. Additionally, the 5′-O-benzoyl analogue 10 was more potent than the corresponding unprotected analogue 9 (EC50: 6.1 μM (5′-OBz, 10) vs 47.2 μM (5′-OH, 9)). This trend was also observed in the acyclic analogues 13 and 14 (EC50: 13.3 μM (−OBz, 14) vs 80.8 μM (−OH, 13)). In contrast, in case of the ribofuranosyl structure, the 5′-hydroxyl analogue 7 was slightly more potent as compared with the 5′-O-benzoyl analogue 8 (EC50 = 31.0 and 41.0 μM, respectively). These results indicate the importance of the masked 5′-hydroxyl group in the 2′-deoxyribofuranosyl structure. It is also interesting to note that the 2′-deoxyribonucleoside derivative 10 was more potent than the corresponding ribonucleoside derivatives such as 7 and 8 in spite of the fact that HCV is an RNA virus and most of the anti-HCV agents bear ribofuranosyl structures (or their bioisosteres).6, 7
2.2.2. SAR of the 5′-O-moiety
As a next step, we evaluated the inhibitory activities of compounds 15–21, whose 5′-hydroxyl groups were masked with various protective groups (i.e., pivaloyl, butyryl, trimethylbenzoyl, benzenesulfonyl, benzyl, and allyl groups) to examine the effect of the substituent group at the 5′-position. Among these compounds, the EC50 value of the close structural analogue 17 (p-methoxybenzoyl analogue) was comparable to that of 10 (EC50 = 8.4 and 6.1 μM, respectively); however, 17 led to an undesirable increase in cytotoxicity (CC50 = 76.4 μM). Notably, other types of protective groups such as ether groups (compounds 20 and 21) and a sulfonate group (compound 19) also exhibited good anti-HCV activity (EC50 = 17.7, 24.5, and 28.8 μM, respectively). Overall, it appears that protective groups containing a phenyl ring are preferable for the antiviral activity.
2.2.3. SAR at position 6 of the purine base
The effect of the substituents at the purine 6-position was investigated by the evaluation of compounds 22–25. Among these analogues, the 6-dimethylamino derivative 24 exhibited a good potency with an EC50 of 22.3 μM and low cytotoxicity (CC50 > 200 μM), while the 6-amino derivative 23 showed a weak potency (EC50 = 105 μM). Unfortunately, the others (i.e., 6-hydroxyl (hypoxanthine) analogue 22 and 6-methylthio analogue 25) did not show any significant potency (EC50 > 100 μM). Accordingly, the chloro group at the purine 6-position was considered to be important for the anti-HCV activity.17
The luciferase assay described above revealed that compound 10 exhibited the maximum potency among the analogues. Next, in order to confirm the anti-HCV activity of 10, the replicon RNA levels were quantified by performing real-time RT-PCR analysis.16 Fig. 4 shows the result obtained with 10 and ribavirin (positive control). Compound 10 reduced the replicon RNA amount up to approximately 45% at 12.5 μM and 6% at 25 μM, which is almost consistent with the result of the luciferase assay (EC50 = 6.1 μM).
Figure 4.
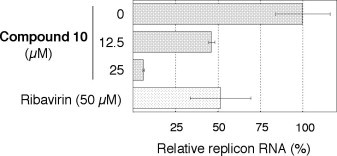
The quantity of HCV subgenomic replicon RNA. The RNA amounts were determined by real-time RT-PCR analysis after a two-day incubation period with compound 10 or ribavirin.
Among these molecules, two compounds can be hypothesized to be species with real antiviral activity. One is the 5′-O-masked 2′-deoxyadenosine analogue itself such as 10, and the other is the deprotected 9 (or its activated form, 5′-triphosphate) since carboxylic ester bonds are often hydrolyzed in cultured cells, as reported earlier.18 However, the chemically stable 5′-O-masked analogues 20 and 21 (Bn and allyl ether, respectively) showed anti-HCV potency to some extent. Moreover, the deoxyadenosine derivative 23, which would be transformed to the inactive 2′-deoxyadenosine after the hydrolysis, also showed potency. Thus, taking these results into consideration, it would be reasonable to consider that some 5′-O-masked analogues themselves possess the anti-HCV potency. On the other hand, it would also be reasonable to consider that compound 9 (or its 5′-triphosphate) is one of the real active species since 9 also exhibited anti-HCV activity, though only moderately; in other words, compound 10 operates as a prodrug of 9.19 Therefore, at this stage, we believe that both compounds (e.g., 9 and 10) would function as the species with antiviral activity in the cells.
3. Conclusion
A series of nucleoside analogues 5–25 were synthesized and their abilities to inhibit HCV RNA replication were evaluated. Among these, several 5′-O-masked analogues of 6-chloropurine-2′-deoxyriboside, such as 10, 17, and 20, exhibited effective anti-HCV activity. In particular, 5′-O-benzoyl analogue 10 exhibited the highest activity. Presently, we hypothesize two roles for these 5′-O-masked analogues: One is the role as an anti-HCV agent by itself, and the other is as a prodrug of its 5′-O-demasked (deprotected) derivative.
There are two notable structural features of these potent compounds: one is the masked 5′-hydroxyl group, and the other is the 2′-deoxyribofuranosyl structure. In relation to the substituent group at position 6 of the purine base, the chloro group seems to be preferable. These unique features are rarely seen in common anti-HCV agents; therefore, although some issues such as improvement of the antiviral potency remain to be resolved, we hope that the present study will contribute to developing a new class of HCV therapeutic agents.
4. Experimental
4.1. Chemistry
4.1.1. General methods
The chemicals used were of commercial origin (Wako chemicals, TCI, Kanto Kagaku, nacalai tesque, and Aldrich) and were employed without further purification. The progress of all reactions was monitored by TLC (silica gel 60F254, Merck). The column chromatography was performed with a CombiFlash Companion system (Teledyne Isco, Inc.). The melting points were measured on a Yanaco melting point apparatus and are uncorrected. The 1H NMR and 13C NMR spectra were recorded on a Bruker AV400M spectrometer. The solvent 1H and 13C signals were used as internal standards (DMSO-d 6: 2.50 and 39.5 ppm; acetone-d 6: 2.05 and 29.8 ppm; CD3OD: 3.31 and 49.0 ppm; CDCl3: 7.26 and 77.0 ppm, respectively). The mass spectra were recorded on a Bruker Apex-Qe FT-ICR MS spectrometer.
4.1.2. Synthesis of 9-(5-O-benzoyl-β-d-2-deoxyribofuranosyl)-6-chloropurine (10)
To a stirred solution of 9 (50 mg, 0.18 mmol) in pyridine (0.5 mL) was added benzoyl chloride (32 μL, 0.28 mmol) at ice-water temperature, and the mixture was stirred at the same temperature for 1.5 h. Subsequent to the addition of saturated NaHCO3 solution, the mixture was extracted with ethyl acetate. The combined organic layer was washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The resultant residue was purified by silica gel column chromatography (ethyl acetate/hexane, 2:1 to 3:1) to give 10 (59 mg, 85%) as a white solid. Mp 111.0–111.5 °C. 1H NMR (DMSO-d 6) δ: 2.47 (1H, m), 2.99 (1H, dt, J = 13.6, 6.4 Hz), 4.19 (1H, q-like, J = 4.4 Hz), 4.44 (1H, dd, J = 12.0, 6.0 Hz), 4.56 (1H, dd, J = 12.0, 4.0 Hz), 4.68 (1H, quintet-like, J = 4.8 Hz), 5.61 (1H, br d, J = 2.8 Hz), 6.50 (1H, t, J = 6.4 Hz), 7.48 (2H, br t, J = 7.6 Hz), 7.64 (1H, br t, J = 7.6 Hz), 7.85 (2H, br d, J = 7.6 Hz), 8.74 (1H, s), 8.86 (1H, s); 13C NMR (DMSO-d 6) δ: 38.4, 64.1, 70.2, 84.2, 84.3, 128.7, 129.1, 129.3, 131.5, 133.4, 146.2, 149.3, 151.2, 151.6, 165.4; HRMS (ESI) calcd for C17H15ClN4NaO4 (M+Na+) 397.0680, found 397.0664.
4.1.3. 6-Chloro-9-(5-O-pivaloyl-β-d-2-deoxyribofuranosyl)purine (15)
To a stirred solution of 9 (63 mg, 0.23 mmol) and DMAP (3 mg, 10 mol%) in pyridine (0.5 mL) was added pivaloyl chloride (43 μL, 0.35 mmol) at ice-water temperature, and the mixture was stirred at the same temperature for 30 min and then at room temperature for 15 min. The work-up process was performed in the same manner as described for 10. The crude material was purified by silica gel column chromatography (ethyl acetate/hexane, 1:1 to 2:1) to give 15 (67 mg, 81%) as a white solid. Mp 111.5–112.0 °C. 1H NMR (DMSO-d 6) δ: 1.06 (9H, s), 2.45 (1H, m), 2.91 (1H, dt, J = 13.6, 6.0 Hz), 4.04 (1H, m), 4.16 (1H, dd, J = 12.0, 6.0 Hz), 4.26 (1H, dd, J = 12.0, 4.4 Hz), 4.52 (1H, m), 5.54 (1H, br d, J = 2.8 Hz), 6.47 (1H, t, J = 6.4 Hz), 8.80 (1H, s), 8.84 (1H, s); 13C NMR (DMSO-d 6) δ: 26.7, 38.1, 38.4, 63.6, 70.1, 84.1, 84.2, 131.5, 146.0, 149.3, 151.2, 151.6, 177.2; HRMS (ESI) calcd for C15H19ClN4NaO4 (M+Na+) 377.0993, found 377.0978.
4.1.4. 9-(5-O-Butyryl-β-d-2-deoxyribofuranosyl)-6-chloro-purine (16)
To a stirred solution of 9 (100 mg, 0.37 mmol) in pyridine (1.0 mL) was added butyryl chloride (61 μL, 0.59 mmol) at ice-water temperature, and the mixture was stirred at the same temperature for 4.5 h. The work-up process was performed in the same manner as described for 10. The crude material was purified by silica gel column chromatography (ethyl acetate/hexane, 1:1 to 3:2) to give 16 (97 mg, 68%) as a colorless oil. 1H NMR (acetone-d 6) δ: 0.88 (3H, t, J = 7.6 Hz), 1.56 (2H, sextet, J = 7.6 Hz), 2.26 (2H, t, J = 7.6 Hz), 2.61 (1H, m), 3.03 (1H, dt, J = 13.6, 6.4 Hz), 4.21 (1H, q-like, J = 4.4 Hz), 4.31 (1H, dd, J = 12.4, 5.6 Hz), 4.34 (1H, dd, J = 12.4, 4.8 Hz), 4.70–4.75 (2H, m), 6.59 (1H, t, J = 6.4 Hz), 8.66 (1H, s), 8.73 (1H, s); 13C NMR (acetone-d 6) δ: 13.7, 18.9, 36.2, 40.3, 64.3, 72.0, 85.8, 85.9, 133.0, 145.9, 150.9, 152.4, 173.4; HRMS (ESI) calcd for C14H17ClN4NaO4 (M+Na+) 363.0836, found 363.0821.
4.1.5. 6-Chloro-9-(5-O-p-methoxybenzoyl-β-d-2-deoxyribofuranosyl)purine (17)
To a stirred solution of 9 (100 mg, 0.37 mmol) in pyridine (1.0 mL) was added p-methoxybenzoyl chloride (61 μL, 0.44 mmol) at ice-water temperature, and the mixture was stirred at the same temperature for 1 h. The work-up process was performed in the same manner as described for 10. The crude material was purified by silica gel column chromatography (ethyl acetate/hexane, 3:1 to 1:0) to give 17 (108 mg, 72%) as a white solid. Mp 148.5–149.0 °C. 1H NMR (DMSO-d 6) δ: 2.47 (1H, m), 2.98 (1H, dt, J = 13.2, 6.4 Hz), 3.83 (3H, s), 4.17 (1H, dt, J = 6.0, 4.0 Hz), 4.38 (1H, dd, J = 12.0, 6.0 Hz), 4.52 (1H, dd, J = 12.0, 4.4 Hz), 4.67 (1H, quintet, J = 5.2 Hz), 5.59 (1H, br d, J = 4.4 Hz), 6.49 (1H, t, J = 6.4 Hz), 6.98 (2H, br d, J = 8.8 Hz), 7.79 (2H, br d, J = 8.8 Hz), 8.76 (1H, s), 8.84 (1H, s); 13C NMR (DMSO-d 6) δ: 38.4, 55.5, 63.7, 70.2, 84.3, 84.4, 113.9, 121.5, 131.2, 131.5, 146.1, 149.3, 151.2, 151.6, 163.2, 165.1; HRMS (ESI) calcd for C18H17ClN4NaO5 (M+Na+) 427.0785, found 427.0767.
4.1.6. 6-Chloro-9-(5-O-(2,4,6-trimethylbenzoyl)-β-d-2-deoxyribofuranosyl)purine (18)
To a stirred solution of 9 (100 mg, 0.37 mmol) and N,N-diisopropylethylamine (478 μL, 2.96 mmol) in acetonitrile (4.0 mL) was added 2,4,6-trimethylbenzoyl chloride (303 μL, 1.85 mmol) at ice-water temperature, and the mixture was stirred at room temperature for 3 h. The work-up process was performed in the same manner as described for 10. The crude material was purified by preparative thin layer chomatography (Merck, 113895) (ethyl acetate/hexane, 2:1) to give 18 (33 mg, 21%) as a white semi-solid. 1H NMR (acetone-d 6) δ: 2.14 (6H, s), 2.25 (3H, s), 2.62 (1H, ddd, J = 13.6, 6.8, 4.0 Hz), 3.11 (1H, dt, J = 13.6, 6.0 Hz), 4.34 (1H, m), 4.56 (1H, dd, J = 12.0, 4.4 Hz), 4.59 (1H, dd, J = 12.0, 6.0 Hz), 4.79–4.86 (2H, m), 6.61 (1H, t, J = 6.4 Hz), 6.84 (2H, br s), 8.60 (1H, s), 8.66 (1H, s); 13C NMR (acetone-d 6) δ: 19.7, 21.1, 40.0, 65.1, 72.4, 85.9, 86.0, 129.0, 131.8, 133.0, 135.6, 140.0, 146.1, 150.9, 152.3, 152.4, 169.9; HRMS (ESI) calcd for C20H21ClN4NaO4 (M+Na+) 439.1149, found 439.1127.
4.1.7. 9-(5-O-Benzenesulfonyl-β-d-2-deoxyribofuranosyl)-6-chloropurine (19)
To a stirred solution of 9 (100 mg, 0.37 mmol) in pyridine (1.0 mL) was added benzenesulfonyl chloride (94 μL × 4 times at intervals of 30 min, 0.74 × 4 mmol) at room temperature, and the mixture was stirred at the same temperature for 3 h in total. The work-up process was performed in the same manner as described for 10. The crude material was purified by silica gel column chromatography (methanol/chloroform, 1:20) to give 19 (60 mg, 40%) as a white semi-solid. 1H NMR (acetone-d 6) δ: 2.57 (1H, ddd, J = 14.0, 6.8, 4.4 Hz), 3.00 (1H, dt, J = 14.0, 6.4 Hz), 4.21 (1H, m), 4.33 (1H, dd, J = 10.8, 6.0 Hz), 4.40 (1H, dd, J = 10.8, 4.0 Hz), 4.73 (1H, m), 4.81 (1H, br d, J = 4.0 Hz), 6.56 (1H, t, J = 6.8 Hz), 7.56 (2H, t, J = 8.0 Hz), 7.68 (1H, t, J = 8.0 Hz), 7.85 (2H, d, J = 8.0 Hz), 8.57 (1H, s), 8.66 (1H, s); 13C NMR (acetone-d 6) δ: 39.8, 70.8, 72.0, 85.5, 86.0, 128.5, 130.2, 133.1, 134.8, 136.7, 146.1, 150.9, 152.3, 152.3; HRMS (ESI) calcd for C16H15ClN4NaO5S (M+Na+) 433.0349, found 433.0330.
4.1.8. 9-(5-O-Benzoyl-β-d-3-deoxyribofuranosyl)-6-chloropurine (12)
To a stirred solution of 11 (600 mg, 2.22 mmol) in CH2Cl2 (15 mL) were added pyridine (3.5 mL) and benzoyl chloride (268 μL, 2.31 mmol) at ice-water temperature, and the mixture was stirred at the same temperature for 2 h. Subsequent to the addition of saturated NaHCO3 solution, the mixture was extracted with ethyl acetate. The combined organic layer was washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The resultant residue was purified by silica gel column chromatography (ethyl acetate/hexane, 1:1 to 2:1) to give 12 (488 mg, 59%) as a white solid. Mp 160.5–161.0 °C. 1H NMR (DMSO-d 6) δ: 2.12 (1H, ddd, J = 13.2, 6.0, 2.0 Hz), 2.47 (1H, m), 4.50 (1H, dd, J = 12.0, 5.6 Hz), 4.57 (1H, dd, J = 12.0, 2.8 Hz), 4.73 (1H, m), 4.87 (1H, m), 5.85 (1H, br s), 6.06 (1H, d, J = 2.0 Hz), 7.47–7.51 (2H, m), 7.66 (1H, m), 7.85–7.87 (2H, m), 8.75 (1H, s), 8.80 (1H, s); 13C NMR (DMSO-d 6) δ: 34.6, 65.4, 74.1, 78.3, 91.7, 128.7, 129.1, 129.3, 131.4, 133.4, 145.6, 149.2, 151.1, 151.6, 165.5; HRMS (ESI) calcd for C17H16ClN4O4 (M+H+) 375.0860, found 375.0867.
4.1.9. 5′-O-Benzoyl-3′-O-(tert-butyldimethylsilyl)-2′-deoxyadenosine (27)
To a stirred solution of 23 (915 mg, 2.45 mmol) in pyridine (12 mL) were added imidazole (1.00 g, 14.7 mmol) and tert-butyldimethylsilyl chloride (1.11 g, 7.36 mmol) at room temperature, and the mixture was stirred overnight at the same temperature. Subsequent to the addition of water, the mixture was stirred for 15 min. After concentration under reduced pressure, the residue was dissolved in ethyl acetate. The ethyl acetate solution was washed with water, 1 M aqueous HCl, saturated NaHCO3 solution and brine, dried over Na2SO4, and concentrated under reduced pressure. The resultant residue was purified by silica gel column chromatography (ethyl acetate/hexane, 4:1 to 1:0) to give 27 (1.07 g, 93%) as a white solid. Mp 129.5–130.0 °C. 1H NMR (CDCl3) δ: 0.14 (6H, s), 0.93 (9H, s), 2.49 (1H, ddd, J = 13.2, 6.4, 4.8 Hz), 2.97 (1H, dt, J = 13.2, 6.0 Hz), 4.28 (1H, q-like, J = 4.0 Hz), 4.50 (1H, dd, J = 12.4, 4.8 Hz), 4.62 (1H, dd, J = 12.4, 4.4 Hz), 4.80 (1H, m), 5.75 (2H, br s), 6.39 (1H, t, J = 6.4 Hz), 7.42 (2H, t, J = 8.0 Hz), 7.56 (1H, t, J = 8.0 Hz), 7.94 (1H, s), 7.97 (2H, d, J = 8.0 Hz), 8.32 (1H, s); 13C NMR (CD3OD) δ: −4.7, −4.5, 18.8, 26.3, 40.5, 64.5, 73.4, 86.0, 86.2, 120.7, 129.6, 130.5, 130.9, 134.4, 141.5, 150.2, 153.8, 157.3, 167.6; HRMS (ESI) calcd for C23H31N5NaO4Si (M+Na+) 492.2043, found 429.2021.
4.1.10. 9-[5-O-Benzoyl-3-O-(tert-butyldimethylsilyl)-β-d-2-deoxyribofuranosyl]-6-chloropurine (28)
To a stirred solution of 27 (245 mg, 0.52 mmol) in CCl4 (10 mL) were added a solution of tetraethylammonium chloride (346 mg, 2.08 mmol) in CH2Cl2 (2.5 mL) and then tert-butyl nitrite (313 μL, 2.60 mmol) at ice-water temperature. The mixture was stirred for 1 h at the same temperature, warmed to room temperature for 0.5 h, and stirred at 50 °C for 2 h. The solvent was removed under reduced pressure, and the resultant residue was purified by silica gel column chromatography (ethyl acetate/hexane, 1:3 to 1:2) to give 28 (147 mg, 58%) as a pale yellow oil. 1H NMR (DMSO-d 6) δ: 0.12 (3H, s), 0.13 (3H, s), 0.90 (9H, s), 2.47 (1H, m), 3.09 (1H, dt, J = 13.2, 6.0 Hz), 4.19 (1H, q-like, J = 4.8 Hz), 4.42 (1H, dd, J = 12.0, 5.2 Hz), 4.57 (1H, dd, J = 12.0, 4.8 Hz), 4.92 (1H, q, J = 5.6 Hz), 6.50 (1H, t, J = 6.4 Hz), 7.46–7.50 (2H, m), 7.64 (1H, m), 7.82–7.85 (2H, m), 8.74 (1H, s), 8.87 (1H, s); 13C NMR (acetone-d 6) δ: −4.7, −4.6, 18.5, 26.1, 40.1, 64.4, 73.1, 85.9, 85.9, 129.4, 130.1, 130.7, 133.2, 134.0, 146.5, 151.0, 152.3, 152.3, 166.4; HRMS (ESI) calcd for C23H29ClN4NaO4Si (M+Na+) 511.1544, found 511.1517.
4.1.11. An alternative synthesis of compound 10
To a stirred solution of 28 (330 mg, 0.67 mmol) in THF (5 mL) was added 1 M THF solution of TBAF–AcOH (1:1, 810 μL, 0.81 mmol) at ice-water temperature, and the mixture was stirred at the same temperature for 5 h and then at room temperature for 1 h. The solvent was removed under reduced pressure, and the resultant residue was purified by silica gel column chromatography (ethyl acetate/hexane, 2:1 to 3:1) to give 10 (181 mg, 72%) as a white solid.
4.1.12. 3′-O-(tert-Butyldimethylsilyl)-2′-deoxy-6-S-methyl-6-thioinosine (29)
To a stirred solution of 28 (225 mg, 0.46 mmol) in DMF (3.5 mL) was added a solution of methyl mercaptan sodium salt (15% in water, 860 μL, 1.84 mmol) at ice-water temperature, and the mixture was stirred at room temperature for 3 h. After dilution of the mixture with ethyl acetate, the organic layer was washed with water, saturated NaHCO3 solution and brine, dried over Na2SO4, and concentrated under reduced pressure. The resultant residue was purified by silica gel column chromatography (ethyl acetate/hexane, 1:3 to 1:2) to give 29 (122 mg, 67%) as a white solid. Mp 96.0–96.5 °C. 1H NMR (DMSO-d 6) δ: 0.12 (6H, s), 0.90 (9H, s), 2.33 (1H, ddd, J = 13.2, 6.4, 3.2 Hz), 2.67 (3H, s), 2.87 (1H, ddd, J = 13.2, 7.6, 6.0 Hz), 3.51 (1H, m), 3.61 (1H, dt, J = 11.6, 5.2 Hz), 3.88 (1H, m), 4.62 (1H, m), 5.04 (1H, t, J = 5.6 Hz), 6.42 (1H, t, J = 6.8 Hz), 8.67 (1H, s), 8.74 (1H, s); 13C NMR (acetone-d 6) δ: −4.7, −4.6, 11.6, 18.5, 26.1, 41.5, 63.0, 74.0, 86.5, 90.0, 133.2, 143.7, 148.6, 152.0, 162.3; HRMS (ESI) calcd for C17H28N4NaO3SSi (M+Na+) 419.1549, found 419.1558.
4.1.13. 5′-O-Benzoyl-3′-O-(tert-butyldimethylsilyl)-2′-deoxy-6-S-methyl-6-thioinosine (30)
To a stirred solution of 29 (122 mg, 0.31 mmol) in CH2Cl2 (4 mL) were added triethylamine (255 μL, 1.86 mmol), DMAP (4 mg, 10 mol%), and benzoyl chloride (71 μL, 0.62 mmol) at ice-water temperature, and the mixture was stirred at the same temperature for 2 h. Subsequent to the addition of saturated NaHCO3 solution, the mixture was extracted with ethyl acetate. The combined organic layer was washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The resultant residue was purified by silica gel column chromatography (ethyl acetate/hexane, 1:4) to give 30 (150 mg, 97%) as a colorless oil. 1H NMR (DMSO-d 6) δ: 0.12 (3H, s), 0.12 (3H, s), 0.90 (9H, s), 2.40–2.55 (1H, overlapping with DMSO-d 6), 2.66 (3H, s), 3.10 (1H, dt, J = 13.2, 6.4 Hz), 4.17 (1H, q-like, J = 4.4 Hz), 4.41 (1H, dd, J = 12.0, 5.2 Hz), 4.55 (1H, dd, J = 12.0, 4.8 Hz), 4.91 (1H, m), 6.46 (1H, t, J = 6.4 Hz), 7.46–7.50 (2H, m), 7.65 (1H, m), 7.84–7.87 (2H, m), 8.64 (1H, s), 8.68 (1H, s); 13C NMR (acetone-d 6) δ: −4.7, −4.6, 11.5, 18.5, 26.1, 40.1, 64.5, 73.2, 85.5, 85.7, 129.3, 130.2, 130.8, 133.0, 134.0, 143.7, 148.8, 152.3, 161.9, 166.5; HRMS (ESI) calcd for C24H32N4NaO4SSi (M+Na+) 533.1811, found 533.1818.
4.1.14. 5′-O-Benzoyl-2′-deoxy-6-S-methyl-6-thioinosine (25)
To a stirred solution of 30 (150 mg, 0.30 mmol) in THF (3 mL) was added 1 M THF solution of TBAF (360 μL, 0.36 mmol) at ice-water temperature, and the mixture was stirred at the same temperature for 1.5 h. The solvent was removed under reduced pressure, and the resultant residue was purified by silica gel column chromatography (ethyl acetate/hexane, 1:1 to 2:1) to give 25 (102 mg, 88%) as a white solid. Mp 148.0–148.5 °C. 1H NMR (DMSO-d 6) δ: 2.46 (1H, m), 2.66 (3H, s), 2.99 (1H, dt, J = 13.6, 6.4 Hz), 4.17 (1H, m), 4.43 (1H, dd, J = 12.0, 6.0 Hz), 4.55 (1H, dd, J = 12.0, 4.4 Hz), 4.68 (1H, m), 5.58 (1H, br d, J = 4.4 Hz), 6.47 (1H, t, J = 6.4 Hz), 7.49 (2H, t, J = 7.6 Hz), 7.65 (1H, t, J = 7.6 Hz), 7.87 (2H, d, J = 7.6 Hz), 8.62 (1H, s), 8.69 (1H, s); 13C NMR (DMSO-d 6) δ: 11.5, 40.0, 65.1, 72.2, 85.5, 85.7, 129.4, 130.2, 130.9, 132.9, 134.0, 143.3, 148.9, 152.4, 161.9, 166.5; HRMS (ESI) calcd for C18H18N4NaO4S (M+Na+) 409.0946, found 409.0929.
4.1.15. N6,N6-Dimethyl-2′-deoxyadenosine (32)
To a stirred solution of 31 (200 mg, 0.56 mmol) in dioxane (1 mL) was added 50% dimethylamine–water solution (8 mL) at room temperature, and the mixture was stirred overnight at the same temperature. The solvent was removed under reduced pressure, and the resultant residue was washed with ether several times, and purified by silica gel column chromatography (methanol/chloroform, 1:10) to give 32 (98 mg, 62%) as a white solid. The 1H and 13C NMR spectra and the mass spectrum were identical to the reported values.14
4.1.16. 5′-O-Benzoyl-N6,N6-dimethyl-2′-deoxyadenosine (24)
To a stirred solution of 32 (290 mg, 1.04 mmol) in pyridine (5 mL) was added benzoyl chloride (181 μL, 1.56 mmol) at ice-water temperature, and the mixture was stirred at room temperature for 4 h. Subsequent to the addition of saturated NaHCO3 solution, the mixture was extracted with ethyl acetate. The combined organic layer was washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The resultant residue was purified by silica gel column chromatography (ethyl acetate/hexane, 3:2 to 4:1) to give 24 (306 mg, 77%) as a white semi-solid. 1H NMR (acetone-d 6) δ: 2.53 (1H, ddd, J = 13.6, 6.8, 4.0 Hz), 3.05 (1H, dt, J = 13.6, 6.8 Hz), 3.49 (6H, br s), 4.30 (1H, q-like, J = 4.4 Hz), 4.54 (1H, dd, J = 12.0, 5.6 Hz), 4.62 (1H, dd, J = 12.0, 4.4 Hz), 4.70 (1H, br d, J = 4.0 Hz), 4.85 (1H, m), 6.50 (1H, t, J = 6.8 Hz), 7.49 (2H, t, J = 8.0 Hz), 7.64 (1H, t, J = 8.0 Hz), 7.99 (2H, d, J = 8.0 Hz), 8.14 (1H, s), 8.20 (1H, s); 13C NMR (acetone-d 6) δ: 38.4, 40.1, 65.3, 72.3, 72.4, 85.1, 85.5, 121.3, 129.4, 130.2, 130.9, 134.0, 138.4, 151.2, 152.9, 155.7, 166.6; HRMS (ESI) calcd for C19H21N5NaO4 (M+Na+) 406.1491, found 406.1475.
4.1.17. 5′-O-Acetyl-2′-deoxyadenosine (33)
2′-Deoxyadenosine (26) (540 mg, 2.01 mmol), vinyl acetate (417 μL, 4.5 mmol), molecular sieves 4 A (500 mg), and lipase acrylic resin from Candida antarctica (300 mg) purchased from SIGMA were suspended in THF (20 mL), and the mixture was stirred at 60 °C for 1.5 h. The enzyme was filtered off and washed with MeOH, and the solvents were removed under reduced pressure. The resultant residue was purified by silica gel column chromatography (methanol/chloroform, 1:20 to 1:10) to give 33 (523 mg, 89%) as a white solid. The 1H NMR spectrum was identical to the reported values.15
4.1.18. 5′-O-Acetyl-3′-O-(tert-butyldimethylsilyl)-2′-deoxyadenosine (34)
To a stirred solution of 33 (670 mg, 2.29 mmol) in pyridine-DMF (1:1, 12 mL) were added tert-butyldimethylsilyl chloride (860 mg, 5.71 mmol) and DMAP (140 mg, 1.15 mmol) at room temperature, and the mixture was stirred overnight at the same temperature. Subsequent to the addition of water, the mixture was stirred for 20 min and then extracted with ethyl acetate. The combined organic layer was washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The resultant residue was purified by silica gel column chromatography (ethyl acetate) to give 34 (940 mg, 94%) as a white solid. Mp 145–146 °C. 1H NMR (DMSO-d 6) δ: 0.12 (6H, s), 0.90 (9H, s), 1.98 (3H, s), 2.32 (1H, ddd, J = 13.6, 6.8, 4.0 Hz), 2.97 (1H, dt, J = 13.6, 6.4 Hz), 3.99 (1H, q-like, J = 3.6 Hz), 4.12 (1H, dd, J = 12.0, 6.0 Hz), 4.24 (1H, dd, J = 12.0, 5.2 Hz), 4.69 (1H, m), 6.34 (1H, t, J = 6.8 Hz), 7.29 (2H, br s), 8.14 (1H, s), 8.32 (1H, s); 13C NMR (DMSO-d 6) δ: −5.1, −4.9, 17.6, 20.5, 25.6, 38.4, 63.4, 72.4, 83.3, 84.1, 119.2, 139.7, 149.0, 152.5, 156.1, 170.0; HRMS (ESI) calcd for C18H30N5O4Si (M+H+) 408.2067, found 408.2082.
4.1.19. 3′-O-(tert-Butyldimethylsilyl)-2′-deoxy-N-(2,2-dimethyl-1-oxopropyl)adenosine (36)
To a stirred solution of 34 (300 mg, 0.74 mmol) and triethylamine (408 μL, 2.94 mmol) in CH2Cl2 (6 mL) was added pivaloyl chloride (207 μL, 1.70 mmol) at ice-water temperature, and the mixture was stirred at room temperature for 1 h. Subsequent to the addition of DMAP (9 mg, 10 mol%) and methanol (2 mL), the mixture was stirred overnight at room temperature. After dilution of the mixture with ethyl acetate, the organic layer was washed with water, 5% KHSO4 solution, saturated NaHCO3 solution and brine, dried over Na2SO4, and concentrated under reduced pressure to leave the crude compound 35 (385 mg), which was employed in the next reaction without purification.
To a stirred solution of 35 (385 mg) in methanol (5 mL) was added potassium carbonate (203 mg, 1.48 mmol) at ice-water temperature, and the mixture was stirred at the same temperature for 0.5 h. Subsequent to the addition of acetic acid (150 μL) and water (3 mL), the organic solvent was removed under reduced pressure. The resultant mixture was extracted with ethyl acetate, and the organic layer was washed with saturated NaHCO3 solution and brine, dried over Na2SO4, and concentrated under reduced pressure. The resultant residue was purified by silica gel column chromatography (ethyl acetate/hexane, 3:1 to 4:1) to give 36 (324 mg, 98% from 34) as a white solid. Compound 35: 1H NMR (DMSO-d 6) δ: 0.13 (6H, s), 0.91 (9H, s), 1.28 (9H, s), 1.98 (3H, s), 2.40 (1H, ddd, J = 13.2, 6.4, 4.0 Hz), 3.01 (1H, dt, J = 13.2, 6.4 Hz), 4.03 (1H, m), 4.14 (1H, dd, J = 12.0, 6.0 Hz), 4.25 (1H, dd, J = 12.0, 4.8 Hz), 4.73 (1H, m), 6.46 (1H, t, J = 6.8 Hz), 8.61 (1H, s), 8.69 (1H, s), 10.14 (1H, br s). Compound 36: Mp 86–87 °C. 1H NMR (DMSO-d 6) δ: 0.12 (6H, s), 0.91 (9H, s), 1.28 (9H, s), 2.34 (1H, ddd, J = 13.6, 6.4, 3.6 Hz), 2.89 (1H, ddd, J = 13.6, 7.2, 6.0 Hz), 3.52 (1H, dt, J = 12.0, 5.2 Hz), 3.61 (1H, dt, J = 12.0, 5.2 Hz), 3.89 (1H, m), 4.63 (1H, m), 5.05 (1H, br t, J = 5.2 Hz), 6.45 (1H, dd, J = 7.2, 6.4 Hz), 8.64 (1H, s), 8.69 (1H, s), 10.14 (1H, br s); 13C NMR (acetone-d 6) δ: −4.7, −4.6, 18.5, 26.1, 27.5, 40.7, 41.4, 63.0, 74.0, 86.5, 90.0, 126.1, 143.7, 151.3, 152.2, 176.0; HRMS (ESI) calcd for C21H35N5NaO4Si (M+Na+) 472.2356, found 472.2363.
4.1.20. 5′-O-Benzyl-3′-O-(tert-butyldimethylsilyl)-2′-deoxy-N-(2,2-dimethyl-1-oxopropyl)adenosine (37)
To a stirred solution of 36 (340 mg, 0.76 mmol) in THF (7.5 mL) was added potassium tert-butoxide (210 mg, 1.89 mmol) at ice-water temperature, and the mixture was stirred at the same temperature for 1 min. Subsequent to the addition of benzyl bromide (99 μL, 0.83 mmol) at ice-water temperature, the mixture was stirred at the same temperature for further 1 h. After dilution of the mixture with water, the aqueous layer was extracted with ethyl acetate. The organic layer was washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The resultant residue was purified by silica gel column chromatography (ethyl acetate/hexane, 1:1 to 2:1) to give 37 (397 mg, 97%) as a colorless oil. 1H NMR (DMSO-d 6) δ: 0.10 (3H, s), 0.10 (3H, s), 0.89 (9H, s), 1.28 (9H, s), 2.37 (1H, ddd, J = 13.2, 6.4, 4.0 Hz), 2.94 (1H, dt, J = 13.2, 6.4 Hz), 3.57 (1H, dd, J = 10.4, 4.8 Hz), 3.68 (1H, dd, J = 10.8, 4.8 Hz), 4.01 (1H, q-like, J = 4.4 Hz), 4.51 (2H, s), 4.68 (1H, m), 6.46 (1H, t, J = 6.4 Hz), 7.27–7.34 (5H, m), 8.58 (1H, s), 8.67 (1H, s), 10.14 (1H, br s); 13C NMR (acetone-d 6) δ: −4.7, −4.6, 18.5, 26.1, 27.5, 40.7, 41.0, 70.6, 73.8, 73.8, 85.1, 87.4, 125.6, 128.3, 128.4, 129.1, 139.2, 143.0, 151.0, 152.5, 175.9; HRMS (ESI) calcd for C28H42N5O4Si (M+H+) 540.3006, found 540.3017.
4.1.21. 5′-O-Benzyl-3′-O-(tert-butyldimethylsilyl)-2′-deoxyadenosine (38)
To a stirred solution of 37 (390 mg, 0.72 mmol) in methanol (7 mL) was added potassium carbonate (299 mg, 2.17 mmol) at ice-water temperature, and the mixture was stirred at room temperature for 7 h. After dilution with CH2Cl2, the mixture was filtrated. Subsequent to the condensation of the filtrate in vacuum, the resultant residue was diluted with ethyl acetate. The ethyl acetate layer was washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The resultant residue was purified by silica gel column chromatography (ethyl acetate/hexane, 3:2 to 3:1) to give 38 (250 mg, 76%) as a white solid. Mp 134–135 °C. 1H NMR (DMSO-d 6) δ: 0.09 (3H, s), 0.10 (3H, s), 0.88 (9H, s), 2.30 (1H, ddd, J = 13.2, 6.8, 3.6 Hz), 2.88 (1H, dt, J = 13.2, 6.8 Hz), 3.57 (1H, dd, J = 10.8, 5.2 Hz), 3.67 (1H, dd, J = 10.8, 5.2 Hz), 3.98 (1H, q-like, J = 4.4 Hz), 4.51 (2H, s), 4.64 (1H, m), 6.33 (1H, t, J = 6.8 Hz), 7.26–7.35 (7H, m), 8.12 (1H, s), 8.26 (1H, s); 13C NMR (CD3OD) δ: −4.7, −4.6, 18.8, 26.3, 42.0, 70.7, 74.1, 74.5, 85.7, 88.1, 120.3, 128.9, 129.0, 129.5, 139.2, 140.8, 150.2, 153.8, 157.3; HRMS (ESI) calcd for C23H34N5O3Si (M+H+) 456.2431, found 456.2444.
4.1.22. 9-(5-O-Benzyl-β-d-2-deoxyribofuranosyl)-6-chloropurine (20)
To a stirred solution of 38 (250 mg, 0.55 mmol) in CCl4 (10 mL) were added a solution of tetraethylammonium chloride (364 mg, 2.20 mmol) in CH2Cl2 (2.5 mL) and then tert-butyl nitrite (326 μL, 2.75 mmol) at ice-water temperature. The mixture was stirred for 1 h at the same temperature, warmed to room temperature for 0.5 h, and stirred at 50 °C for 2 h. The solvent was removed under reduced pressure, and the resultant residue was purified by silica gel column chromatography (ethyl acetate/hexane, 1:3 to 1:2) to give 39 (108 mg) with inseparable by-products, which was employed in the next reaction without further purification.
To a stirred solution of 39 (108 mg) in THF (2 mL) was added 1 M THF solution of TBAF–AcOH (1:1, 300 μL, 0.30 mmol) at ice-water temperature, and the mixture was stirred at the same temperature for 1 h and then at room temperature for 5 h. The solvent was removed under reduced pressure, and the resultant residue was purified by preparative thin layer chromatography (Merck, 113895) (methanol/chloroform, 1:10) to give 20 (181 mg, 34% from 38) as a colorless oil. Compound 39: 1H NMR (acetone-d 6) δ: 0.14 (6H, s), 0.93 (9H, s), 2.54 (1H, m), 2.97 (1H, m), 3.71 (1H, dd, J = 10.8, 4.0 Hz), 3.80 (1H, dd, J = 10.8, 4.0 Hz), 4.14 (1H, m), 4.59 (2H, s), 4.84 (1H, m), 6.58 (1H, t, J = 6.4 Hz), 7.27–7.36 (5H, m), 8.66 (1H, s), 8.70 (1H, s). Compound 20: 1H NMR (DMSO-d 6) δ: 2.41 (1H, ddd, J = 13.2, 6.4, 4.0 Hz), 2.83 (1H, dt, J = 13.2, 6.4 Hz), 3.59 (1H, dd, J = 10.8, 5.2 Hz), 3.69 (1H, dd, J = 10.8, 4.4 Hz), 4.04 (1H, m), 4.47–4.52 (3H, m), 5.45 (1H, br s, J = 4.4 Hz), 6.48 (1H, t, J = 6.4 Hz), 7.23–7.33 (5H, m), 8.78 (1H, s), 8.80 (1H, s); 13C NMR (acetone-d 6) δ: 41.2, 71.0, 72.6, 73.8, 85.7, 87.6, 128.4, 128.5, 129.1, 132.8, 139.2, 145.7, 150.7, 152.4, 152.5; HRMS (ESI) calcd for C17H18ClN4O3 (M+H+) 361.1067, found 361.1077.
4.1.23. 5′-O-Allyl-3′-O-(tert-butyldimethylsilyl)-2′-deoxyadenosine (41)
To a stirred solution of 36 (300 mg, 0.67 mmol) in THF (20 mL) was added potassium tert-butoxide (188 mg, 1.68 mmol) at ice-water temperature, and the mixture was stirred at the same temperature for 1 min. Subsequent to the addition of allyl bromide (203 μL, 2.35 mmol) at ice-water temperature, the mixture was stirred at the same temperature for further 2.5 h. After dilution of the mixture with saturated NaHCO3 solution, the aqueous layer was extracted with ethyl acetate. The organic layer was washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The resultant residue was purified by silica gel column chromatography (ethyl acetate/hexane, 1:2 to 1:1) to give 40 (300 mg) with inseparable by-products, which was employed in the next reaction without further purification.
To a stirred solution of 40 (300 mg) in methanol (7 mL) was added potassium carbonate (253 mg, 1.84 mmol) at ice-water temperature, and the mixture was stirred at room temperature for 7 h. After dilution with CH2Cl2, the mixture was filtrated. Subsequent to the condensation of the filtrate in vacuum, the resultant residue was diluted with ethyl acetate. The ethyl acetate layer was washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The resultant residue was purified by silica gel column chromatography (ethyl acetate/hexane, 1:1 to 4:1) to give 41 (198 mg, 73% from 36) as a white solid. Compound 40: 1H NMR (acetone-d 6) δ: 0.16 (6H, s), 0.95 (9H, s), 1.38 (9H, s), 2.49 (1H, m), 2.94 (1H, m), 3.64–3.75 (2H, m), 4.05–4.10 (3H, m), 4.80 (1H, m), 5.15 (1H, d, J = 10.4 Hz), 5.28 (1H, d, J = 17.2 Hz), 5.93 (1H, m), 6.55 (1H, t, J = 6.4 Hz), 8.45 (1H, s), 8.60 (1H, s), 8.98 (1H, br s). Compound 41: Mp 100.0–100.5 °C. 1H NMR (DMSO-d 6) δ: 0.11 (6H, s), 0.90 (9H, s), 2.30 (1H, ddd, J = 13.2, 6.4, 3.6 Hz), 2.87 (1H, dt, J = 13.2, 7.2 Hz), 3.52 (1H, dd, J = 10.8, 5.2 Hz), 3.61 (1H, dd, J = 10.8, 5.2 Hz), 3.94–3.98 (3H, m), 4.63 (1H, m), 5.14 (1H, dq, J = 10.4, 2.0 Hz), 5.23 (1H, dq, J = 17.2, 2.0 Hz), 5.87 (1H, ddt, J = 17.2, 10.4, 5.6 Hz), 6.33 (1H, t, J = 6.8 Hz), 7.26 (2H, br s), 8.14 (1H, s), 8.29 (1H, s); 13C NMR (CD3OD) δ: −4.7, −4.6, 18.8, 26.3, 42.0, 70.8, 73.3, 74.0, 85.7, 88.0, 117.5, 120.3, 135.7, 140.9, 150.2, 153.8, 157.3; HRMS (ESI) calcd for C19H32N5O3Si (M+H+) 406.2274, found 406.2289.
4.1.24. 9-(5-O-Allyl-β-d-2-deoxyribofuranosyl)-6-chloropurine (21)
To a stirred solution of 41 (235 mg, 0.58 mmol) in CCl4 (11 mL) were added a solution of tetraethylammonium chloride (384 mg, 2.32 mmol) in CH2Cl2 (2.6 mL) and then tert-butyl nitrite (345 μL, 2.90 mmol) at ice-water temperature. The mixture was stirred for 1 h at the same temperature, warmed to room temperature for 0.5 h, and stirred at 50 °C for 2 h. The solvent was removed under reduced pressure, and the resultant residue was purified by silica gel column chromatography (ethyl acetate/hexane, 1:6) to give 42 (95 mg) with inseparable by-products, which was employed in the next reaction without further purification.
To a stirred solution of 42 (95 mg) in THF (1.5 mL) was added 1 M THF solution of TBAF–AcOH (1:1, 330 μL, 0.33 mmol) at ice-water temperature, and the mixture was stirred at the same temperature for 1 h and then at room temperature for 6 h. The solvent was removed under reduced pressure, and the resultant residue was purified by silica gel column chromatography (methanol/chloroform, 0:1 to 1:20) to give 21 (50 mg, 28% from 41) as a colorless oil. Compound 42: 1H NMR (acetone-d 6) δ: 0.16 (6H, s), 0.94 (9H, s), 2.54 (1H, m), 2.95 (1H, dt, J = 13.2, 6.0 Hz), 3.67 (1H, dd, J = 10.8, 4.0 Hz), 3.74 (1H, dd, J = 10.8, 4.0 Hz), 4.05 (2H, d, J = 5.6 Hz), 4.12 (1H, m), 4.82 (1H, m), 5.15 (1H, d, J = 10.4 Hz), 5.27 (1H, d, J = 17.2 Hz), 5.91 (1H, m), 6.59 (1H, t, J = 6.4 Hz), 8.69 (1H, s), 8.72 (1H, s). Compound 21: 1H NMR (DMSO-d 6) δ: 2.40 (1H, ddd, J = 13.6, 6.8, 4.4 Hz), 2.81 (1H, dt, J = 13.6, 6.4 Hz), 3.54 (1H, dd, J = 10.4, 5.2 Hz), 3.63 (1H, dd, J = 10.4, 4.0 Hz), 3.95 (2H, dt, J = 5.2, 1.2 Hz), 4.00 (1H, q-like, J = 4.8 Hz), 4.47 (1H, m), 5.12 (1H, dq, J = 10.4, 2.0 Hz), 5.20 (1H, dq, J = 17.2, 2.0 Hz), 5.46 (1H, br d, J = 4.4 Hz), 5.84 (1H, ddt, J = 17.2, 10.4, 5.6 Hz), 6.48 (1H, t, J = 6.4 Hz), 8.81 (1H, s), 8.82 (1H, s); 13C NMR (DMSO-d 6) δ: 39.0, 69.8, 70.5, 71.3, 84.1, 85.9, 116.5, 131.3, 134.9, 145.7, 149.2, 151.3, 151.6; HRMS (ESI) calcd for C13H16ClN4O3 (M+H+) 311.0911, found 311.0918.
4.2. Biological evaluation
Cell culture, luciferase assay, and real-time RT-PCR analysis were performed as described previously.16 The cytotoxicity was evaluated in a tetrazolium (XTT)-based assay according to the manufacturer’s protocol (Cell Proliferation Kit II (XTT), Roche Diagnostics, Cat. No. 1465015).
Acknowledgment
This research was partially supported by a Grant-in-Aid for Young Scientists (B), No. 18790093, from the Japan Society for the Promotion of Science (JSPS).
Footnotes
Supplementary information (1H and 13C NMR spectra of compounds 10, 12, 15–21, 24, 25, 27–30, 34, 36–38, and 41) is available online at www.sciencedirect.com. Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmc.2007.08.025.
Supplementary data
References and notes
- 1.Murphy F.A., Fauquet C.M., Bishop D.H.L., Ghabrial S.A., Jarvis A.W., Martelli G.P., Mayo M.A., Summers M.D. Classification and Nomenclature of Viruses: Sixth Report of the Internal Committee on Taxonomy of Viruses. Springer-Vielag; Vienna Austria: 1995. pp. 424–426. [Google Scholar]
- 2.Choo Q.L., Kuo G., Weiner A.J., Overby L.R., Bradley D.W., Houghton M. Science. 1989;244:359. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 3.Goodman Z.D., Ishak K.G. Semin. Liver Dis. 1995;15:70. doi: 10.1055/s-2007-1007264. [DOI] [PubMed] [Google Scholar]
- 4.Manns M.P., McHutchison J.G., Gordon S.C., Rustgi V.K., Shiffman M., Reindollar R., Goodman Z.D., Koury K., Ling M., Albrecht J.K. Lancet. 2001;358:958. doi: 10.1016/s0140-6736(01)06102-5. [DOI] [PubMed] [Google Scholar]; Fried M.W., Shiffman M.L., Reddy K.R., Smith C., Marinos G., Goncales F.L., Jr., Haussinger D., Diago M., Carosi G., Dhumeaux D., Craxi A., Lin A., Hoffman J., Yu J. N. Eng. J. Med. 2002;347:975. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 5.(a) Gordon C.P., Keller P.A. J. Med. Chem. 2005;48:1. doi: 10.1021/jm0400101. [DOI] [PubMed] [Google Scholar]; (b) De Francesco R., Migliaccio G. Nature. 2005;436:953. doi: 10.1038/nature04080. [DOI] [PubMed] [Google Scholar]
- 6.(a) Stuyver L.J., McBrayer T.R., Tharnish P.M., Hassan A.E.A., Chu C.K., Pankiewicz K.W., Watanabe K.A., Schinazi R.F., Otto M.J. J. Virol. 2003;77:10689. doi: 10.1128/JVI.77.19.10689-10694.2003. For example: [DOI] [PMC free article] [PubMed] [Google Scholar]; Eldrup A.B., Allerson C.R., Bennett C.F., Bera S., Bhat B., Bhat N., Bosserman M.R., Brooks J., Burlein C., Carroll S.S., Cook P.D., Getty K.L., MacCoss M., McMasters D.R., Olsen D.B., Prakash T.P., Prhavc M., Song Q., Tomassini J.E., Xia J. J. Med. Chem. 2004;47:2283. doi: 10.1021/jm030424e. [DOI] [PubMed] [Google Scholar]; Eldrup A.B., Prhavc M., Brooks J., Bhat B., Prakash T.P., Song Q., Bera S., Bhat N., Dande P., Cook P.D., Bennett C.F., Carroll S.S., Ball R.G., Bosserman M., Burlein C., Colwell L.F., Fay J.F., Flores O.A., Getty K., LaFemina R.L., Leone J., MacCoss M., McMasters D.R., Tomassini J.E., Von Langen D., Wolanski B., Olsen D.B. J. Med. Chem. 2004;47:5284. doi: 10.1021/jm040068f. [DOI] [PubMed] [Google Scholar]; (b) Stuyver L.J., McBrayer T.R., Whitaker T., Tharnish P.M., Ramesh M., Lostia S., Cartee L., Shi J., Hobbs A., Schinazi R.F., Watanabe K.A., Otto M.J. Antimicrob. Agents Chemother. 2004;48:651. doi: 10.1128/AAC.48.2.651-654.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; Clark J.L., Hollecker L., Mason J.C., Stuyver L.J., Tharnish P.M., Lostia S., McBrayer T.R., Schinazi R.F., Watanabe K.A., Otto M.J., Furman P.A., Stec W.J., Patterson S.E., Pankiewicz K.W. J. Med. Chem. 2005;48:5504. doi: 10.1021/jm0502788. [DOI] [PubMed] [Google Scholar]; (c) Carroll S.S., Tomassini J.E., Bosserman M., Getty K., Stahlhut M.W., Eldrup A.B., Bhat B., Hall D., Simcoe A.L., LaFemina R., Rutkowski C.A., Wolanski B., Yang Z., Migliaccio G., De Francesco R., Kuo L.C., MacCoss M., Olsen D.B. J. Biol. Chem. 2003;278:11979. doi: 10.1074/jbc.M210914200. [DOI] [PubMed] [Google Scholar]; (d) Smith D.B., Martin J.A., Klumpp K., Baker S.J., Blomgren P.A., Devos R., Granycome C., Hang J., Hobbs C.J., Jiang W.-R., Laxton C., Le Pogan S., Leveque V., Ma H., Maile G., Merrett J.H., Pichota A., Sarma K., Smith M., Swallow S., Symons J., Vesey D., Najera I., Cammack N. Bioorg. Med. Chem. Lett. 2007;17:2570. doi: 10.1016/j.bmcl.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 7.Smith K.L., Lai V.C.H., Prigaro B.J., Ding Y., Gunic E., Girardet J.-L., Zhong W., Hong Z., Lang S., An H. Bioorg. Med. Chem. Lett. 2004;14:3517. doi: 10.1016/j.bmcl.2004.04.067. For example: [DOI] [PubMed] [Google Scholar]; Wang P., Du J., Rachakonda S., Chun B.-K., Tharnish P.M., Stuyver L.J., Otto M.J., Schinazi R.F., Watanabe K.A. J. Med. Chem. 2005;48:6454. doi: 10.1021/jm058223t. [DOI] [PubMed] [Google Scholar]; Shi J., Du J., Ma T., Pankiewicz K.W., Patterson S.E., Tharnish P.M., McBrayer T.R., Stuyver L.J., Otto M.J., Chu C.K., Schinazi R.F., Watanabe K.A. Bioorg. Med. Chem. 2005;13:1641. doi: 10.1016/j.bmc.2004.12.011. [DOI] [PubMed] [Google Scholar]; Naka K., Ikeda M., Abe K., Dansako H., Kato N. Biochem. Biophys. Res. Commun. 2005;330:871. doi: 10.1016/j.bbrc.2005.03.062. [DOI] [PubMed] [Google Scholar]; Clark J.L., Mason J.C., Hollecker L., Stuyver L.J., Tharnish P.M., McBrayer T.R., Otto M.J., Furman P.A., Schinazi R.F., Watanabe K.A. Bioorg. Med. Chem. Lett. 2006;16:1712. doi: 10.1016/j.bmcl.2005.12.002. [DOI] [PubMed] [Google Scholar]; Yoo B.N., Kim H.O., Moon H.R., Seol S.K., Jang S.K., Lee K.M., Jeong L.S. Bioorg. Med. Chem. Lett. 2006;16:4190. doi: 10.1016/j.bmcl.2006.05.089. [DOI] [PubMed] [Google Scholar]; Hocek M., Šilhár P., Shih I.-h., Mabery E., Mackman R. Bioorg. Med. Chem. Lett. 2006;16:5290. doi: 10.1016/j.bmcl.2006.07.092. [DOI] [PubMed] [Google Scholar]; Lee J.A., Kim H.O., Tosh D.K., Moon H.R., Kim S., Jeong L.S. Org. Lett. 2006;8:5081. doi: 10.1021/ol061959f. [DOI] [PubMed] [Google Scholar]; Butora G., Olsen D.B., Carroll S.S., McMasters D.R., Schmitt C., Leone J.F., Stahlhut M., Burlein C., MacCoss M. Bioorg. Med. Chem. 2007;15:5219. doi: 10.1016/j.bmc.2007.05.020. [DOI] [PubMed] [Google Scholar]
- 8.Pierra C., Amador A., Benzaria S., Cretton-Scott E., D’Amours M., Mao J., Mathieu S., Moussa A., Bridges E.G., Standring D.N., Sommadossi J.-P., Storer R., Gosselin G. J. Med. Chem. 2006;49:6614. doi: 10.1021/jm0603623. [DOI] [PubMed] [Google Scholar]; Afdhal N., Godofsky E., Dienstag J., Rustgi V., Schick L., McEniry D., Zhou X.J., Chao G., Fang C., Fielman B., Myers M., Brown N. Hepatology. 2004;40:726A. [Google Scholar]
- 9.Arimilli M.N., Dougherty J.P., Cundy K.C., Bischofberger N. In: De Clercq E., editor. Vol. 3. Jai Press Inc.; Stamford Connecticut: 1999. pp. 69–91. (Advances in Antiviral Drug Design). and also see Ref. 5a and references therein. [Google Scholar]
- 10.Prakash T.P., Prhave M., Eldrup A.B., Dan Cook P., Carroll S.S., Olsen D.B., Stahlhut M.W., Tomassini J.E., MacCoss M., Galloway S.M., Hilliard C., Bhat B. J. Med. Chem. 2005;48:1199. doi: 10.1021/jm0495172. A prodrug approach involving 5′-monophosphate derivatives is also effective in increasing the antiviral potency. For example: [DOI] [PubMed] [Google Scholar]; Ding Y., Girardet J.-L., Hong Z., Lai V.C.H., An H., Koh Y.-h., Shaw S.Z., Zhong W. Bioorg. Med. Chem. Lett. 2005;15:709. doi: 10.1016/j.bmcl.2004.11.020. [DOI] [PubMed] [Google Scholar]; Gunic E., Chow S., Rong F., Ramasamy K., Raney A., Li D.Y., Huang J., Hamatake R.K., Hong Z., Girardet J.-L. Bioorg. Med. Chem. Lett. 2007;17:2456. doi: 10.1016/j.bmcl.2007.02.029. [DOI] [PubMed] [Google Scholar]; Perrone P., Luoni G.M., Kelleher M.R., Daverio F., Angell A., Mulready S., Congiatu C., Rajyaguru S., Martin J.A., Levêque V., Le Pogam S., Najera I., Klumpp K., Smith D.B., McGuigan C. J. Med. Chem. 2007;50:1840. doi: 10.1021/jm0613370. [DOI] [PubMed] [Google Scholar]
- 11.Ikejiri M., Saijo M., Morikawa S., Fukushi S., Mizutani T., Kurane I., Maruyama T. Bioorg. Med. Chem. Lett. 2007;17:2470. doi: 10.1016/j.bmcl.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Ref. 11 and references therein. A racemic mixture of 14 was employed.; (b) Webb R.R., II, Wos J.A., Martin J.C., Brodfuehrer P.R. Nucleosides Nucleotides. 1988;7:147. [Google Scholar]; (c) Liguori A., Perri E., Sindona G., Uccela N. Tetrahedron. 1988;44:229. [Google Scholar]
- 13.Česnek M., Holý A., Masojídková M. Tetrahedron. 2002;58:2985. [Google Scholar]
- 14.Ueda T., Nomoto Y., Matsuda A. Chem. Pharm. Bull. 1985;33:3263. Another synthesis of 32: [Google Scholar]
- 15.Ciuffreda P., Casati S., Santaniello E. Tetrahedron. 2000;56:3239. [Google Scholar]
- 16.Watashi K., Hijikata M., Hosaka M., Yamaji M., Shimotohno K. Hepatology. 2003;38:1282. doi: 10.1053/jhep.2003.50449. [DOI] [PubMed] [Google Scholar]; Murata T., Hijikata M., Shimotohno K. Virology. 2005;340:105. doi: 10.1016/j.virol.2005.06.015. [DOI] [PubMed] [Google Scholar]; Goto K., Watashi K., Murata T., Hishiki T., Hijikata M., Shimotohno K. Biochem. Biophys. Res. Commun. 2006;343:879. doi: 10.1016/j.bbrc.2006.03.059. [DOI] [PubMed] [Google Scholar]
- 17.A similar SAR trend was observed in our previous study (Ref. 11), and some comments are mentioned therein.
- 18.Parang K., Knaus E.E., Wiebe L.I. Nucleosides Nucleotides. 1998;17:987. doi: 10.1080/07328319808004216. and references therein. [DOI] [PubMed] [Google Scholar]
- 19.Several 5′-O-acyl nucleoside analogues have been reported as prodrugs of the corresponding deacylated analogues. See Ref. 18.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.