

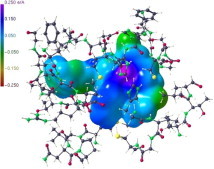
Graphical abstract
Electrostatic potential on an electron density iso-surface (0.0067 eÅ−3) of an APE substrate in the active site pocket of the SARS main protease Mpro. Geometry data from PDB entry 2A5I.
Keywords: Biological recognition, Electron density, Molecular surfaces, Electrostatic potential
Abstract
To contribute to an understanding of biological recognition and interaction, an easy-to-use procedure was developed to generate and display molecular surfaces and selected electron density based surface properties. To overcome the present limitations to derive electron densities of macromolecules, the considered systems were reduced to appropriate substructures around the active centers. The combination of experimental X-ray structural information and aspherical atomic electron density data from theoretical calculations resulted in properties like the electrostatic potential and the Hirshfeld surface which allowed a study of electronic complementarity and the identification of sites and strengths of drug-receptor interactions. Applications were examined for three examples. The anilinoquinazoline gefitinib (Iressa®) belongs to a new class of anticancer drugs that inhibit the tyrosine kinase activity of the epidermal growth factor receptor (EGFR). In the second example, the interaction of epoxide inhibitors with the main protease of the SARS coronavirus was investigated. Furthermore, the progesterone receptor complex was examined. The quantitative analysis of hydrogen bonding in the chosen substructure systems follows a progression elaborated earlier on the basis of accurate small molecule crystal structures. This finding and results from modified substructures suggest that also the surface properties seem robust enough to provide stable information about the recognition of interacting biomolecular species although they are obtained from medium molecular weighted subfragments of macromolecular complexes, which consist of no more than ∼40 residues.
1. Introduction
Since the pharmacological action of most drugs is promoted by a recognition process enabling a macromolecular target protein and a low molecular weight substrate to form a complex, the understanding of mutual drug-receptor recognition on an atomic scale is a key challenge in that field. Biological recognition is dominated by intermolecular interactions and it is generally accepted that not only steric but also complementary electronic properties of the involved species play a distinctive role in this process.1, 2 Whereas steric information can be provided by various methods, for example, by the determination of the molecular structure from a conventional X-ray diffraction experiment using the spherical independent atom model (IAM †), electronic properties can be obtained from high-resolution X-ray data at low temperatures followed by an aspherical electron density modelling of the structure. For aspherical atom modelling, the Hansen and Coppens multipole formalism is commonly used that describes the asphericity of an atomic electron density by a sum of spherical harmonic density functions.3 In addition to intramolecular atomic and bonding properties, experimental electron density (ED) distributions contain information on weak intermolecular interactions in the crystal. For example, the electrostatic potential can be derived, being one of the most important properties in the study of molecular reactivity and the analysis of molecular association, which follows the so called principle of electrostatic complementarity.1, 4, 5, 6 ED properties in the crystal can serve in a first approximation as a model for the interactions under biological conditions, since the same types of effects as the ligand-target interactions should exist in this environment (e.g., steric, electrostatic, hydrogen bonding, van-der-Waals effects), and these are expected to be comparable in size.2, 7 In this respect, experimental ED properties are much better suited to simulate physiological conditions than those from theoretical gas phase calculations on isolated molecules.2
Several experimental advances have taken place in ED research in the last decade.8 Since the ED of a chemical structure consists of a large spherical and a very small aspherical contribution, located mainly in covalent bonds and lone pair regions, a very precise X-ray diffraction experiment at high-resolution (d ⩽ 0.5 Å) and low temperature (T <100 K) has to be carried out to make these small aspherical effects visible. To meet these demanding experimental conditions the appearance of CCD area detectors in the mid-1990s and the availability of an increasing number of highly intense synchrotron beamlines of the third generation have contributed substantially.9 Nevertheless, there still exist some limitations for a broad application of experimental ED work in the life sciences, where at least two major problems have to be addressed.
-
(i)
In medicinal chemistry, especially in drug development, it is common practise to screen over a large number of compounds to identify their potential activity. This makes also ED studies on series of molecules in short time periods necessary.
-
(ii)
Important biological molecules are macromolecules (proteins, polynucleotides), of which ED studies are known only in extremely rare cases.10, 11, 12, 13
The above mentioned experimental advances have led to a dramatic reduction of X-ray exposure time to a few hours or even minutes per data collection so that in combination with methodical and computational developments the examination and quantitative analysis of series of ED distribution seems feasible already at present.14
For macromolecules, a generally lower crystal quality, ubiquitous disorder, large amount of solvent in the crystal structure, cause altogether limited diffraction properties, so that these compounds are still an experimental challenge. As one approach to overcome these shortcomings, various activities have been initiated in the last few years to establish data bases of aspherical ED fragments obtained either from experimental or theoretical investigations.15, 16, 17 One concept is to replace the IAM by the invariom formalism,18 an aspherical scattering model which assigns fixed multipole populations from theoretical calculations (stored in an invariom library19) to each atom of a structure to calculate non-spherical scattering factors. Following the transferability and additivity concept expressed in Bader’s Qtaim theory,20 the data base entries are then applied to the building blocks of a given macromolecule to generate additively the ED of the entire molecule.
Making use in parts of this concept, we describe in this study an easy-to-use procedure to derive ED information at least in the active site regions of drug-receptor complexes from a combination of macromolecular X-ray data and theoretical calculations. Applications were considered for three examples. The anilinoquinazoline gefitinib (Iressa®) belongs to a new class of anticancer drugs that inhibit the tyrosine kinase activity of the epidermal growth factor receptor (EGFR). In the second example, the interaction of epoxide inhibitors with the main protease of the SARS coronavirus was investigated. Furthermore we analyzed the progesterone receptor complex.
2. The applied procedure
A procedure to obtain ED information for drug-receptor complexes was elaborated as follows (see also schematic representation in the flow chart of Fig. 1 ):
-
(1)
The X-ray structure atomic coordinates of the considered protein or protein complex were extracted from the protein data bank (PDB).21
-
(2)
The protein was reduced to an appropriate substructure in a region around the active site, hydrogens were added to free valences, their positions were set according to steric considerations using GaussView.22
-
(3)
The substructure obtained this way was entered into a single point ab initio density-functional calculation with the B3LYP/6-31G∗∗ basis set, using gaussian03.22
-
(4)
The wave functions were entered into the program tonto,23 which generates a pseudo periodic structure, so that a theoretical crystallographic structure factor file (hkl-file) could be generated.
-
(5)
This hkl-file was used as input into the program xd,24 which is normally used to refine an aspherical atomic electron density model against experimental structure factors. Aspherical scattering factors (multipole populations) from our invariom library are assigned to the atoms of the protein substructure and, if present, also to the substrate molecule. For atoms not in the invariom library multipoles are refined against the theoretical structure factors, while invariom multipoles, positional and thermal parameters are kept fixed.
-
(6)
The obtained electron density of the subfragment can then be analyzed by the corresponding subprograms of the xd program suite.
Figure 1.
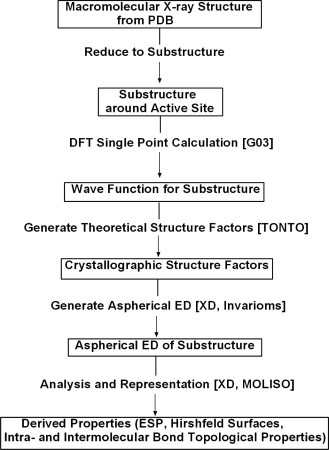
Flow chart of the applied procedure to derive ED based surfaces.
We note that all substrate-in-protein structures are compared to optimized geometries of the corresponding isolated substrate molecules at the same level of theory.
The procedure proposed above has a couple of shortcomings. One open question is the accuracy of the protein structure. Proteins in the PDB are frequently not determined up to atomic resolution. It is common practise to apply a restraint refinement with the aim to approach an idealized geometry of the contributing amino acids as introduced by Engh and Huber.25 Since in our procedure atomic positions are not further refined, a potential inaccuracy remains. In the examples detailed below, the accuracy of bonds in terms of rms deviations from ideality was reported in the range of some hundreds of Å, which is about one order of magnitude larger than for small molecules, where bond length errors are generally smaller than 0.005 Å.
A second point is the choice of the substructure in the active site region, that means whether it is appropriate or not. It has to be admitted, that the choice is somewhat ambiguous, however, care was taken to cover this region as complete as necessary on one hand but as small as possible on the other hand to allow affordable computing time in the subsequent calculations.
The advantage of this procedure is that it is rather fast and can be applied with reasonable effort, because the considered molecular structure is much smaller than the entire protein or protein receptor complex and that due to the support of invariom parameters only moderate least-squares refinements are necessary. Moreover, the procedure provides an easy access to molecular surfaces and surfaces properties. Since recognition of interaction processes takes place via surface properties, their inspection is considered as a valuable source of information. Supported by corresponding graphical software developed in our lab (Moliso)26 we concentrate in this study on two types of surfaces:
-
(i)
The electrostatic potential (ESP), calculated in the XDPROP routine of XD24 after the method of Volkov et al.27 mapped as colour code on an ED iso-surface of 0.0067 eÅ−3 (corresponding to 0.001 au). For the consideration of the reactive behaviour of a chemical system, the three-dimensional distribution of its ESP is very helpful in that negative regions can be regarded as nucleophilic centers, whereas regions with positive electrostatic potential are potential electrophilic sites. Moreover, the electrostatic potential makes the polarization of the electron density visible.
-
(ii)
The Hirshfeld surface,28, 29, 30 which provides a computationally simple and elegant partitioning procedure for molecules in a crystal or another appropriate environment. It is defined by the ratio of the molecular density over the crystal density which should be a constant c, where c = 0.5 is normally chosen. We calculate the Hirshfeld surface according to this condition and map the non-spherical ED on this surface by a colour code. This allows the amount of density at a given site to be visualized. This way the Hirshfeld surface makes ED concentrations and hence sites and strengths of intermolecular interactions visible.
The example in Figure 2 shows the experimentally obtained Hirshfeld surface of the recently examined indolylquinazoline derivative LA1810 (1),31, 32 a further potential EGFR tyrosine kinase inhibitor. This compound exists as a dimer in the crystal structure formed by a pair of N–HO hydrogen bonds. Further strong intermolecular contacts are not seen. Consequently the Hirshfeld surface shows only two strongly coloured regions, the N–H donor and the oxygen acceptor sites of the hydrogen bond.
Figure 2.
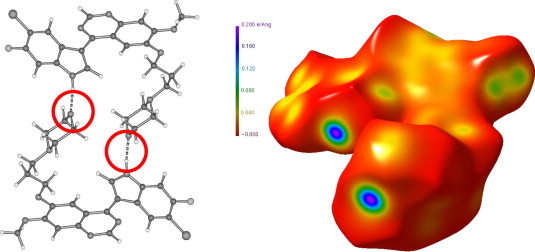
Dimeric structure (left)32 and Hirshfeld surface of the indolylquinazoline derivative (1) LA1810 (right). The two deeply coloured regions indicate the donor and acceptor sites of the dimer forming N–H⋯O hydrogen bond.
As the chemical information provided by the presented procedure is dependent on a proper construction of the three-dimensional ED distribution within the active site substructure of the proteins, it was necessary to evaluate the stability of the results by repeating the procedure for different substructures of the same active site. To meet the two questions raised before (accuracy of the protein structure, choice of the substructure), this quality test was applied to the progesterone receptor as it provides the most complete packing around the substrate molecule. On the one hand, a random generator was used to ‘jiggle’ the atomic positions. This was done in two steps. First, the amount of displacement in atomic coordinates was chosen to be in the range of the experimental uncertainties of the protein structure. In a second step, the atomic displacements were chosen significantly beyond the experimental errors. On the other hand, the substructure was reduced to half of its size, carrying then 20 amino acids in the protein pocket. For the jiggled structure the multipole populations of the original combined assignment/refinement were applied. For the new smaller substructure, the above presented procedure (Fig. 1) was followed from its second step, which means, a DFT single point calculation was performed, followed by a calculation of theoretical structure factors and subsequent assignment/refinement of multipoles. For these modifications of the progesterone receptor substructures, reactive surfaces were generated and the topology of the intermolecular interactions was derived for the smaller substructure. The results are described in Section 3.4. and in parts in the Supplementary data.
3. Results and discussion
3.1. EGFR tyrokinase inhibitor
The EGFR is a transmembrane glycoprotein with an extracellular ligand-binding domain and an intracellular domain with tyrosine kinase activity for signal transduction. Ligand binding activates the receptor and its signalling pathways leading to the activation or modulation of cellular processes.33, 34, 35 The receptor is expressed on healthy cells (40–100 EGFR/cell) as well as on malignant tissues (more than 1 000 000 EGFR/cell).36 The epidermal growth factor (EGF) and the transforming growth factor-α (TGF-α) are the most important stimulatory ligands for EGFR. Overexpression of EGFR has been demonstrated in a wide variety of malignant cells. Thus, therapeutic strategies to inhibit EGFR and EGFR-related pathways have been pursued, including the development of ATP-competitive small molecule inhibitors of the intracellular tyrosine kinase domain of the receptor like gefitinib (Iressa®) (2).
The macromolecular structure of an EGFR tyrosine kinase domain complexed with the anilinoquinazoline derivative (2) is available at a resolution of 2.6 Å from the PDB, code 1M17.37 We have extracted a subfragment consisting of 34 amino acids around the active site and also the structure of the anilinoquinazoline substrate. Making use of the procedure described above, the ESP of the isolated substrate (Fig. 3 above), the ESP of the substrate in the substructure pocket (Fig. 3 below) and the ESP of the substructure pocket (Fig. 4 ) were generated. Whereas Figure 3 (above) shows only a moderate polarization of the isolated anilinoquinazoline gefitinib (2), the polarization increases considerably, if the substrate ESP is calculated in the environment of the pocket (Fig. 3 below). Moreover, we note that positively polarized regions dominate the substrate molecular surface. In accordance with these findings, the receptor pocket (Fig. 4) shows an ESP where negative regions dominate so that the principle of electrostatic complementarity is properly realized. Figure 5 shows the Hirshfeld surface of (2) where interactions with the receptor pocket are clearly visible at sites with strong colour code. In contrast to LA1810 (1) (Fig. 2 right) several sites of interaction are observed for the anilinoquinazoline gefitinib (2), indicating its increased potency for forming contacts with the protein (Scheme 1 ).
Figure 3.
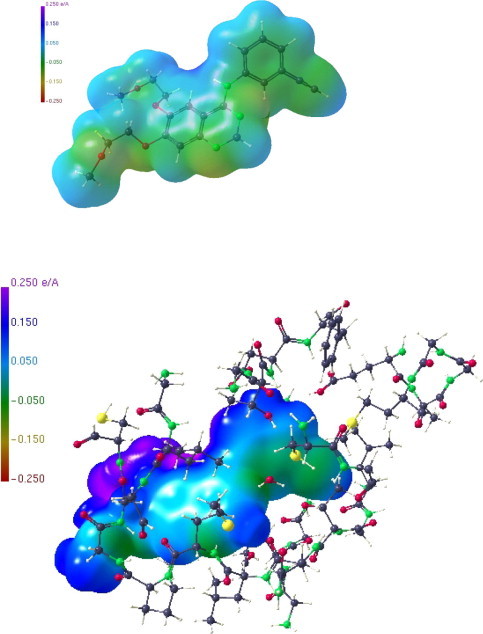
ESP of the isolated anilinoquinazoline derivative (2) (above); ESP of the anilinoquinazoline substrate in the receptor substructure pocket (below).
Figure 4.
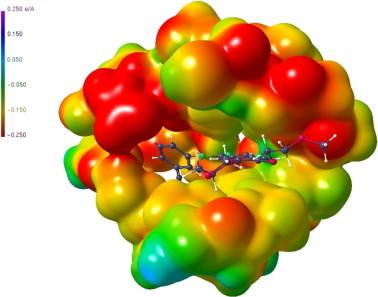
ESP of the receptor pocket. The substrate in the pocket is displayed as a ball-and-stick representation.
Figure 5.
Hirshfeld surface of the anilinoquinazoline substrate (2). Strong interactions with the active site environment are indicated by deeply coloured regions.
Scheme 1.
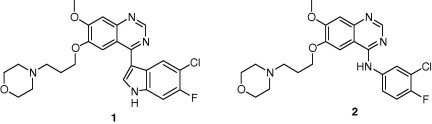
The indolylquinazoline LA1810 (1) and the anilinoquinazoline gefitinib (Iressa®) (2).
3.2. Main protease of the SARS coronavirus
SARS (severe acute respiratory syndrome) became apparent in fall 2002 and was distributed within a few weeks over China and other countries with the highest number of almost 4000 people infected in May 2003.38, 39 SARS is caused by a previously unknown coronavirus, SARS-CoV. For the replication of this virus, its main cysteine protease Mpro (molecular mass 33.8 kDa per protomer) is essential since it releases the key proteins in viral replication. As a therapeutic approach towards anti-SARS drugs, the inhibition of Mpro by aziridine or epoxides was considered. The mechanism is displayed schematically in Figure 6 . A nucleophilic attack from the cysteinic sulphur of Mpro takes place at a carbon atom of the epoxide inhibitor. This causes a ring opening and a covalent bond is formed between the cystein Sγ and the attacked epoxide carbon atom, so that an irreversible inhibition of Mpro results.
Figure 6.
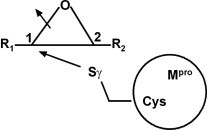
Schematic representation of the nucleophilic interaction of the Mpro cysteinic sulphur with a potential epoxide inhibitor.
For the application of the procedure described above to the Mpro inhibition procedure, we have made use of two corresponding macromolecular structures reported by Lee et al.39 One is the unbound Mpro structure (PDB code 2A5A, resolution 2.08 Å), the second one is a complex of Mpro with an azapeptide epoxide (APE) substrate (R1 = Cbz-Leu-Phe-AGln, R2 = COOEt, (3), see Fig. 6), PDB code 2A5I (resolution 1.88 Å). After extraction of the active site subfragment (41 residues) we have obtained ESP’s in these regions, which are shown in Figure 7 (top left) for the case of the unbound protein and in Figure 7 (top right) for the active site of the complex where the interaction with the substrate is taken into account. In addition, Figure 7 (bottom left and right) shows the ESP of the substrate before and after ring opening has taken place. While in the unbound active site pocket a more or less balanced distribution of positive and negative ESP sites is seen, the interior of the pocket is dominated by negative ESP in case of the complex which is in accordance with the mostly positive ESP surface of the substrate.
Figure 7.
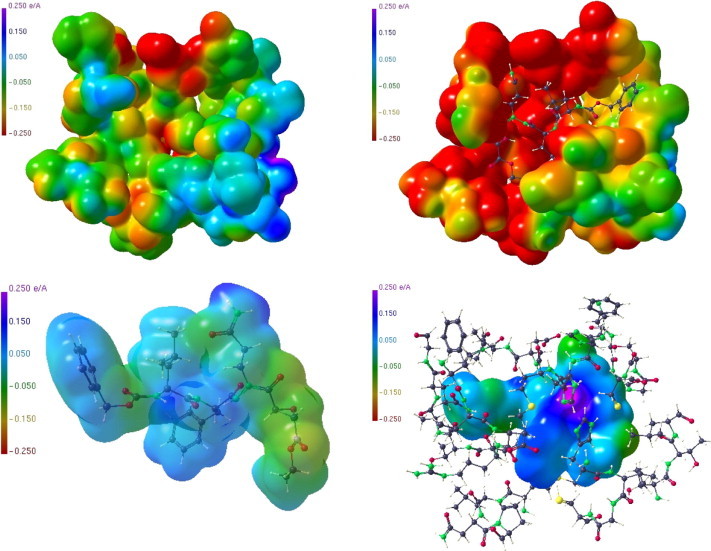
ESP of the active site substructure of the unbound Mpro, 2A5A data used (top left); corresponding ESP in the active site region of the Mpro complexed with an APE substrate, 2A5I data used (top right); ESP of the isolated APE substrate, epoxide ring still intact (bottom left); ESP of the APE substrate in the Mpro pocket after ring opening (bottom right).
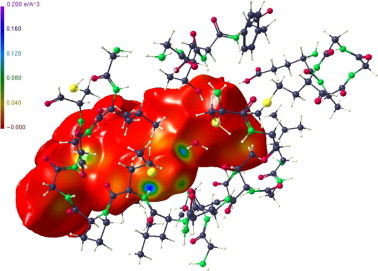
Figure 8 shows the Hirshfeld surface of the APE substrate with the electron density mapped as colour code on this surface. As mentioned before, deeply coloured regions show high electron density concentrations being generally indicative for strong intra or intermolecular interactions. We note that the strongest ED is found just at the site where the covalent bond with the Sγ has been established.
Figure 8.
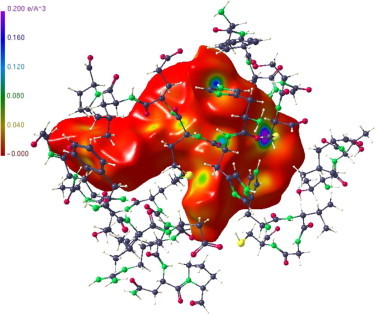
Hirshfeld surface of the APE substrate. The strongest ED (deep blue colour code) is visible at the site where the covalent bond to the cystenic Sγ was established.
3.3. Progesterone receptor complex
The steroid hormone progesterone is a key component in the complex regulation of normal female reproductive function. The major physiological roles of progesterone in the mammal are (1) in the uterus and ovary: release of mature oocytes, facilitation of implantation, and maintenance of pregnancy, by promotion of uterine growth and suppression of myometrial contractility; (2) in the mammary gland: lobular-alveolar development in preparation for milk secretion and suppression of milk protein synthesis before parturition; and (3) in the brain: mediation of signals required for sexually responsive behaviour. Recent evidence also supports a role for progesterone in modulation of bone mass. Progesterone effects are mediated by its nuclear receptor, which is a dimer composed of two receptor progesterone binding proteins, PR A and PR B.40, 41, 42
The crystal structure of a progesterone-bound ligand-binding domain of the human progesterone receptor was published at 1.8 Å resolution (PDB code 1A28) by Williams and Sigler.43 The interactions of the hormone with the ligand-binding domain (LBD) were discussed in details based on steric considerations. Strong interactions were identified from the protein to the 3-keto group and less pronounced to the methyl-ketone substituent at the C17 carbon of the progesterone (Scheme 2 ).
Scheme 2.
The steroid hormone progesterone (4).
We have generated a 40 residue subfragment around the hormone binding pocket and calculated ESP’s for progesterone in the isolated state and bound to the pocket (Fig. 9 ). In both cases the hydrocarbon skeleton region exhibits a more or less uniform positive ESP, whereas the 3-keto region and the methyl-ketone substituent are negatively polarized, being less pronounced in the methyl-ketone region. Hence, the interacting protein region should provide the opposite charge at the corresponding sites. Figure 10 shows a selected region of ESP in the hormone binding pocket of the LBD, where a rather strong positive region is next to the 3-keto oxygen, while otherwise mostly negative ESP faces parts of the positive region of the hydrocarbon section of the hormone.
Figure 9.
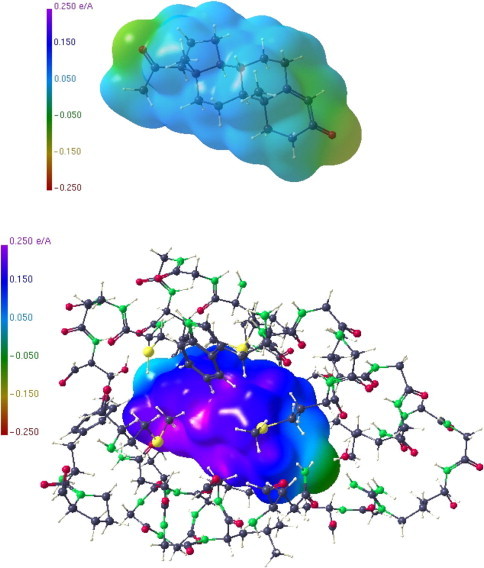
ESP of the progesterone molecule (4) in the isolated state (above) and bound to the pocket of the LBD (below).
Figure 10.
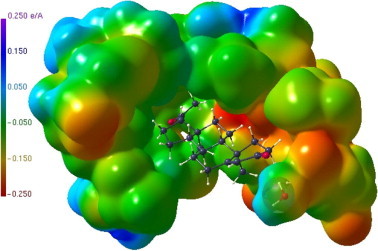
ESP region in the hormone binding pocket of the LBD, progesterone (4) as ball-and-stick representation. A water molecule which also interacts with the hormone is displayed in transparent mode.
In Figure 11 a further Hirshfeld surface representation is given, showing the hormone (4) in the same orientation as in Figure 9 (lower part) in its pocket of the LBD. It can easily be recognized that the most coloured region and hence the strongest interaction is at the 3-keto group followed by the oxygen of the methyl-ketone. However, further less intense sites of interactions are visible, so that a close inspection of this surface can help to identify additional interactions of relevance. The strength of the electron density at a considered site can serve as a quantitative measure for the interaction in question.
Figure 11.
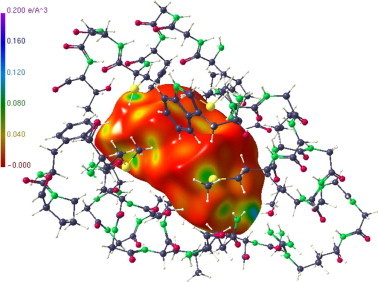
Hirshfeld representation of the hormone (4) in its pocket of the LBD.
3.4. Tests on the stability of the procedure
For evaluation of the stability of the results, three modifications of the progesterone receptor substructure were created and analyzed. In one approach, using a random generator, the original coordinates were displaced (‘jiggled’) randomly in two steps. In a first step an average displacement of atomic coordinates of 0.05 Å was applied which is slightly above the reported experimental uncertainty of this progesterone receptor complex of ∼0.01 Å.43 In a second step, the random displacement was enlarged to 0.5 Å, far beyond the experimental error. For both models the ESP’s were generated for the substrate molecule and the protein pocket.
For the 0.05 Å randomization the ESP’s are indistinguishable to the results for the original substructure, see Figure 10 and upper part of Figure 12 (see also Figs. S1 and S2, Supplementary data). This does no longer hold for the stronger 0.5 Å randomization (Fig. 12 below and Fig. S3). Especially the ESP features in the pocket displayed in Figure 12 (below) differ strongly from the original substructure. This finding can be supported quantitatively making use of the parameters of the Politzer analysis44 which examines the distribution of positive/negative ESP values on a given surface. Table 1 shows the Politzer parameter π, which gives the average deviation from the overall average potential.45 For both the substrate and the pocket, the π values are identical for the original and the jiggle 0.05 model but change significantly when going to the jiggle 0.5 model. Hence, if the uncertainty of the model is close to or slightly above the experimental error, the procedure described here is unaffected but becomes questionable if the randomization is extended to the large value of 0.5 Å.
Figure 12.
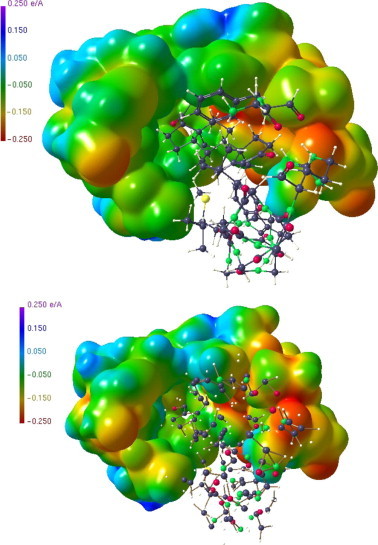
Above: ESP region in the ‘jiggle 0.05’ substructure of the hormone binding pocket. Part of the substructure not contributing to the ESP calculation shown as ball-and-stick representation. There is practically no difference to the ESP of the original substructure displayed in Figure 10. Below: Corresponding ESP in the ‘jiggle 0.5’ substructure.
Table 1.
Politzer π-parameter for the original and randomized progesterone receptor models
| Model | Substrate (e/Å) | Pocket (e/Å) |
|---|---|---|
| Original | 0.0249 | 0.0581 |
| Jiggle 0.05 | 0.0246 | 0.0582 |
| Jiggle 0.5 | 0.0461 | 0.0653 |
Alternatively, a smaller substructure of 20 amino acid residues was generated (see Fig. S4) for which density-functional calculation, calculation of theoretical structure factors, and subsequent multipole assignment/refinement was performed. Although the results for the ESP’s are somewhat different (Fig. 13 ), which is expected for a different substructure, the overall agreement with the major features of the original 40 residue fragment is good. Especially the extended negative region neighbouring the positive hydrocarbon section of progesterone is clearly visible (compare Fig. 10). The positive region next to the 3-keto oxygen is less pronounced. This is confirmed by the Hirshfeld surface (Fig. 14 ) where sites of intermolecular interactions are in most cases comparably distributed as for the 40 residue substructure (see Fig. 11).
Figure 13.
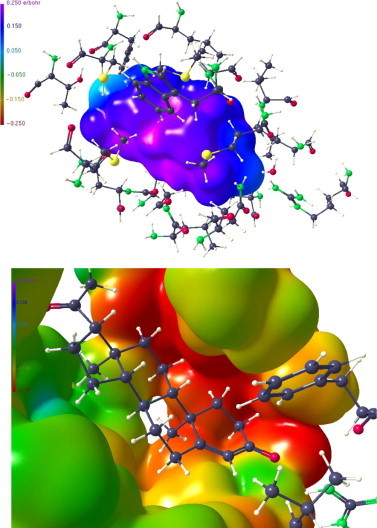
Above: ESP of progesterone in the 20 residue substructure. There are small differences to the representations in the lower part of Figure 9. Below: ESP in a smaller section of the 20 residue substructure close to the progesterone substrate.
Figure 14.
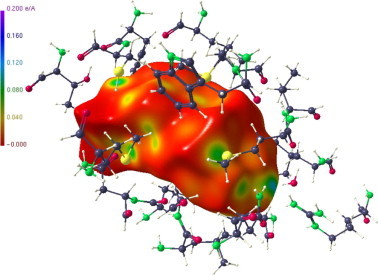
Hirshfeld surface of the progesterone in the 20 residue substructure pocket.
3.5. Quantitative analysis of hydrogen bonding
The more qualitative discussion on intermolecular interactions between substrates and active sites based on reactive surfaces is complemented by a topological analysis of the ED at the bond critical points (bcp) found between hydrogen bonded atoms.20 Espinosa and co-workers investigated the dependence of various topological parameters,46, 47, 48 such as the electron density ρ(r bcp), the Laplacian ρ(r bcp) of the ED, the Hessian Eigenvalues λ 1–λ 3 of the Laplacian, and the kinetic and potential energy densities, G(r bcp) and V(r bcp), respectively, on different geometrical quantities for a characterization of the strength and nature of intermolecular interactions. Amongst others, they found systematical relations between the H···O distances and ρ(r bcp), ρ(r bcp), and λ 3, respectively, for an analysis of 83 X–H···O (X = C, N, O) contacts.47 The functions obtained by this statistical approach were applied to the 60 hydrogen bonds found in this work for all three protein substructures including the substrate molecules. These hydrogen bonds appear between substrate and protein as well as between different amino acids of the protein site. Figure 15 displays the values for ρ(r bcp), ρ(r bcp), and λ 3 in relation to the H···Y (Y = O, N, S) distances.
Figure 15.
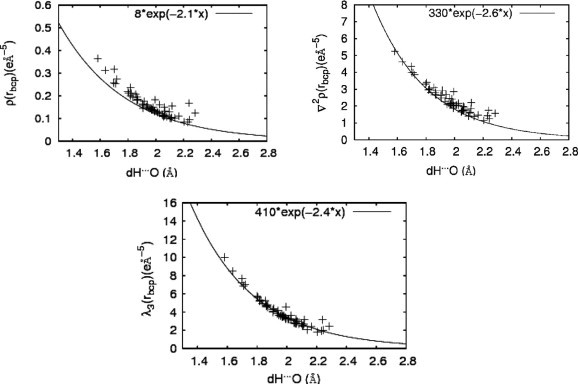
Representation of topological descriptors ρ(rbcp) (top left), ρ(rbcp) (top right), λ3 (bottom) at bond critical points (bcp’s) on hydrogen bonds versus H⋯O distances for all hydrogen bonds considered in this study. The curves elaborated by Espinosa et al.47 (solid lines) are also shown.
Although the X–H (X = C, N, O, S) distances of the protein structures are not fixed to values obtained by neutron diffraction, an approach which is commonly used in ED studies, the overall agreement between the functions taken from Ref. 47 (details see Fig. 15) and the topological properties obtained by the above described procedure is very good. Especially λ 3 matches nearly perfectly the expected progression. On one hand, these results confirm the quality of the ED distribution of the whole substructure, on the other hand this procedure enables the detailed analysis of the hydrogen bonding network within these large systems. Hydrogen bonds are one source of weak intra and intermolecular interactions and play an important role in molecular association and in ligand-target interactions in which hydrogen bond donors face hydrogen bond acceptors; hence the topological analysis described above provides a tool for a quantitative study of these important contacts. A corresponding hydrogen bond analysis for the reduced 20 residue progesterone substructure, introduced in Section 3.4 was also carried out. For results, see ‘Supplementary data’, Figure S5.
4. Conclusion
For biological processes the understanding of intermolecular recognition has to be understood on an atomic scale. Considering the fact that in a first step the mutual recognition takes place via the surfaces of the involved molecules, we have developed a procedure to generate and display molecular surfaces and selected ED surface properties of biomolecules. The present limitations to obtain ED information for macromolecules are diminished by reducing the macromolecular protein structure to a region around the active site, so that the computational effort becomes affordable. The use of two data bases is needed. Experimental X-ray structural information is provided by the protein data base (PDB), while atomic electron density data are supplied by the Invariom data base which allows then the additive generation of the ED of the entire system following Bader’s transferability and additivity concept.20
The procedure is easy to apply with the presently existing software and provides surface properties that allow the identification of sites and strengths of intermolecular interactions. This was shown for the ESP surfaces in protein active site pockets and corresponding substrates and on Hirshfeld surfaces, on which interactions directly become visible in terms of ED concentrations.
We believe that the joint information from the ESP (to indicate nucleophilic/electrophilic sites) and from the Hirshfeld surface (to identify sites and strengths of preferred interactions), which in this combination can only be obtained from the ED, provides various options to enter electronic information into the examination of drug-receptor interactions with acceptable effort. If, for example, docking or fitting techniques for substrate-target systems are carried out based on steric considerations, the obtained geometry can then be entered into the procedure described here to examine also the electronic complementarity. In addition to the possibilities to determine drug-receptor interactions, also potential sites at the receptor can be identified which can preferably be attacked by drugs. These findings can be helpful for the design of chemical structures as new agonists/antagonists on a specific receptor.
One shortcoming should not be overlooked. It is the choice and accuracy of the medium molecular weighted substructure which should mimic the active site region. However, the considered examples have shown that even the quantitative analysis of hydrogen bonding in the chosen substructure systems follows the progression developed by Espinosa et al.47 on the basis of accurate small molecule crystal structures. Furthermore, the analysis of the additional model systems introduced for the progesterone receptor active site (see Section 3.4), the jiggled structure on the one hand and the smaller pocket on the other hand, reveal that the results presented in this study are stable to a high degree. The ESP is virtually identical for the original and the jiggle 0.05 Å pockets and only slightly different for the smaller 20 residue model. The latter one also shows minor changes in the Hirshfeld surface as well as in the topology of the hydrogen bonds (see Supplementary data). So we are confident that as long as fine details of the atomic structure are not considered, the surface properties are robust enough to provide reliable hints for the recognition and interaction of biomolecular species.
Acknowledgement
The authors thank the Deutsche Forschungsgemeinschaft (DFG) for financial support within the special priority program SPP1178, grant Lu 222/29-2.
Footnotes
Abbreviations: EGFR, epidermal growth factor receptor; EGF, epidermal growth factor; SARS, severe acute respiratory syndrome; SARS-CoV, SARS coronavirus; IAM, independent atom model; ED, electron density; CCD, charge coupled device; QTAIM, quantum theory of atoms in molecules; PDB, protein data bank; ESP, electrostatic potential; TGF-α, transforming growth factor α; ATP, adenosine triphosphate; MPro, main protease of the SARS coronavirus; APE, azapeptide epoxide; LBD, ligand-binding domain; bcp, bond critical point.
Supplementary data (additional description of the modified substructures of the progesterone receptor complex and illustration of further results in Figures S1–S5) associated with this article can be found, in the online version, at doi:10.1016/j.bmc.2010.06.080.
Supplementary data
Additional description of the modified substructures of the progesterone receptor complex and illustration of further results in Figures S1-S5.
References and notes
- 1.Naray-Szabo G., Ferenczy G.G. Chem. Rev. 1995;95:829. [Google Scholar]
- 2.Mladenovic M., Arnone M., Fink R.F., Engels B. J. Phys. Chem. 2009;B113:5072. doi: 10.1021/jp809537v. [DOI] [PubMed] [Google Scholar]
- 3.Hansen N.K., Coppens P. Acta Crystallogr., A. 1978;34:909. [Google Scholar]
- 4.Gruber J., Zawaira A., Saunders R., Barrett C.P., Noble M.E.M. Acta Crystallogr., D. 2007;63:50. doi: 10.1107/S0907444906046762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muzet N., Guillot B., Jelsch C., Howard E., Lecomte C. Proc. Natl. Acad. Sci. U.S.A. 2003;100:8742. doi: 10.1073/pnas.1432955100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grabowsky S., Pfeuffer T., Morgenroth W., Paulmann C., Schirmeister T., Luger P. Org. Biomol. Chem. 2008;6:2295. doi: 10.1039/b802831a. [DOI] [PubMed] [Google Scholar]
- 7.Luger P. Org. Biomol. Chem. 2007;5:2529. doi: 10.1039/b706235d. [DOI] [PubMed] [Google Scholar]
- 8.Coppens P. Angew. Chem., Int. Ed. 2005;44:6810. doi: 10.1002/anie.200501734. [DOI] [PubMed] [Google Scholar]
- 9.Koritsanszky T., Flaig R., Zobel D., Krane H.-G., Morgenroth W., Luger P. Science. 1998;279:356. doi: 10.1126/science.279.5349.356. [DOI] [PubMed] [Google Scholar]
- 10.Jelsch C., Teeter M.M., Lamzin V., Pichon-Pesme V., Blessing R.H., Lecomte C. Proc. Natl. Acad. Sci. U.S.A. 2000;97:3171. doi: 10.1073/pnas.97.7.3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Housset D., Benabicha F., Pichon-Pesme V., Jelsch C., Maierhofer A., David S., Fontecilla-Camps J.C., Lecomte C. Acta Crystallogr., D. 2000;56:151. doi: 10.1107/s0907444999014948. [DOI] [PubMed] [Google Scholar]
- 12.Jelsch C., Guillot B., Lagoutte A., Lecomte C. J. Appl. Crystallogr. 2005;38:38. [Google Scholar]
- 13.Volkov A., Messerschmidt M., Coppens P. Acta Crystallogr., D. 2007;63:160. doi: 10.1107/S0907444906044453. [DOI] [PubMed] [Google Scholar]
- 14.Luger P., Wagner A., Hübschle C.B., Troyanov S.I. J. Phys. Chem. A. 2005;109:10177. doi: 10.1021/jp053505q. [DOI] [PubMed] [Google Scholar]
- 15.Pichon-Pesme V., Lecomte C., Lachekar H. J. Phys. Chem. 1995;99:6242. [Google Scholar]
- 16.Zarychta B., Pichon-Pesme V., Guillot B., Lecomte C., Jelsch C. Acta Crystallogr., A. 2007;63:108. doi: 10.1107/S0108767306053748. [DOI] [PubMed] [Google Scholar]
- 17.Dominiak P.M., Volkov A., Li X., Messerschmidt M., Coppens P. J. Chem. Theory Comput. 2007;3:232. doi: 10.1021/ct6001994. [DOI] [PubMed] [Google Scholar]
- 18.Dittrich B., Koritsánszky T., Luger P. Angew. Chem., Int. Ed. 2004;43:2718. doi: 10.1002/anie.200353596. [DOI] [PubMed] [Google Scholar]
- 19.Hübschle, C. B.; Luger, P.; Dittrich, B. J. Appl. Crystallogr.2007, 40, 623. Software available from: http://wwwuser.gwdg.de/~chuebsc/dittrich/dev.html.
- 20.Bader R.F.W. Clarendon Press; Oxford: 1994. Atoms in Molecules—A Quantum Theory. For transferability see Chapter 6.4.2, p 209 ff. [Google Scholar]
- 21.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. Protein Data Bank, Nucleic Acids Res. 2002;28:235. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. gaussian 03 and GaussView3.0 (revision E.01), Gaussian Inc., Wallingford CT, USA, 2004.
- 23.Jayatilaka, D.; Grimwood, D. J. TONTO: A Fortran Based Object-Oriented System for Quantum Chemistry and Crystallography, User Manual, 2003. Software available from: http://www.theochem.uwa.edu.au/tonto/download.
- 24.Volkov, A.; Macchi, P.; Farrugia, L. J.; Gatti, C.; Mallinson, P. R.; Richter, T.; Koritsanszky, T. 2006, XD2006. University at Buffalo, NY, USA; University of Milano, Italy; University of Glasgow, UK; CNRISTM, Milano, Italy; Middle Tennessee State University, TN, USA. Software available from: http://xd.chem.buffalo.edu/download.html.
- 25.Engh R., Huber R. Acta Crystallogr., A. 1991;47:392. [Google Scholar]
- 26.Hübschle, C. B.; Luger, P. J. Appl. Crystallogr.2006, 39, 901. Software available from: http://www.moliso.de/download.html.
- 27.Volkov A., King H.F., Coppens P., Farrugia L.J. Acta Crystallogr., A. 2006;62:400. doi: 10.1107/S0108767306026298. [DOI] [PubMed] [Google Scholar]
- 28.Spackman M.A., McKinnon J.J., Jayatilaka D. Cryst. Eng. Commun. 2008;10:377. [Google Scholar]
- 29.McKinnon J.J., Mitchel S., Spackman M.A. Chem. Eur. J. 1998;4:2136. [Google Scholar]
- 30.Spackman M.A., Byrom P.G. Chem. Phys. Lett. 1997;267:215. [Google Scholar]
- 31.Lüth, A. Ph. D. Thesis, Freie Universität: Berlin, 2006.
- 32.Mebs S., Lüth A., Löwe W., Paulmann C., Luger P. Z. Krist. 2008;223:502. [Google Scholar]
- 33.Schlessinger J. Cell. 2000;103:211. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 34.Prenzel N. Endocr. Rel. Cancer. 2001;8:11. doi: 10.1677/erc.0.0080011. [DOI] [PubMed] [Google Scholar]
- 35.Hou T., Zhu L., Chen L., Xu X. J. Chem. Inf. Comput. Sci. 2003;43:273. doi: 10.1021/ci025552a. [DOI] [PubMed] [Google Scholar]
- 36.Pedersen M.W., Poulsen H.S. Sci. Med. 2002:206. [Google Scholar]
- 37.Stamos J., Sliwkowski M.X., Eigenbrot C. J. Biol. Chem. 2002;277:46265. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 38.Martina E., Stiefl N., Degel B., Schulz F., Breuning A., Schiller M., Vicik R., Baumann K., Ziebuhr J., Schirmeister T. Bioorg. Med. Chem. Lett. 2005;15:5365. doi: 10.1016/j.bmcl.2005.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee T.-W., Cherney M.M., Huitema C., Liu J., James K.E., Powers J.C., Eltis L.D., James M.N.G. J. Mol. Biol. 2005;353:1137. doi: 10.1016/j.jmb.2005.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu Z., Li Y., Fang M., Wai M.S. Curr. Med. Chem. 2009;16:1418. doi: 10.2174/092986709787846523. [DOI] [PubMed] [Google Scholar]
- 41.Boonyaratanakornkit V., Bia Y., Rudda M., Edwards D.P. Steroids. 2009;73:922. doi: 10.1016/j.steroids.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 42.Ellmann S., Sticht H., Thiel F., Beckmann M.W., Strick R., Strissel P.L. Cell Mol. Life Sci. 2009;66:2405. doi: 10.1007/s00018-009-0017-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williams S.P., Sigler P.B. Nature. 1998;393:392. doi: 10.1038/30775. [DOI] [PubMed] [Google Scholar]
- 44.Politzer P., Murray J.S., Preralta-Inga Z. Int. J. Quantum Chem. 2001;85:676. [Google Scholar]
- 45.Hübschle C.B., Dittrich B., Grabowsky S., Messerschmidt M., Luger P. Acta Crystallogr., B. 2008;64:363. doi: 10.1107/S0108768108005776. [DOI] [PubMed] [Google Scholar]
- 46.Espinosa E., Molins E., Lecomte C. Chem. Phys. Lett. 1998;285:170. [Google Scholar]
- 47.Espinosa E., Souhassou M., Lachekar H., Lecomte C. Acta Crystallogr., B. 1999;55:563. doi: 10.1107/s0108768199002128. [DOI] [PubMed] [Google Scholar]
- 48.Espinosa E., Lecomte C., Molins E. Chem. Phys. Lett. 1999;300:745. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional description of the modified substructures of the progesterone receptor complex and illustration of further results in Figures S1-S5.