

Abstract
The purification of recombinant proteins by affinity chromatography is one of the most efficient strategies due to the high recovery yields and purity achieved. However, this is dependent on the availability of specific affinity adsorbents for each particular target protein. The diversity of proteins to be purified augments the complexity and number of specific affinity adsorbents needed, and therefore generic platforms for the purification of recombinant proteins are appealing strategies. This justifies why genetically encoded affinity tags became so popular for recombinant protein purification, as these systems only require specific ligands for the capture of the fusion protein through a pre-defined affinity tag tail. There is a wide range of available affinity pairs “tag-ligand” combining biological or structural affinity ligands with the respective binding tags. This review gives a general overview of the well-established “tag-ligand” systems available for fusion protein purification and also explores current unconventional strategies under development.
Keywords: Affinity tags, Fusion proteins, Recombinant proteins, Affinity ligands
Highlights
-
•
Current and future trends for the purification of recombinant proteins
-
•
Comparison of affinity ligands for fusion protein purification
-
•
Versatile and unconventional purification strategies for fusion proteins
1. Introduction
The wealth of products and methodologies for recombinant protein production and purification has increased enormously in recent years. This has contributed to the growth in the use of recombinant proteins for academic research and therapeutic and diagnostic applications as well as in industrial settings (Demain and Vaishnav, 2009, Palomares et al., 2004). The production and purification of recombinant proteins are intimately linked. The choice of host for protein production affects not only the amplification and isolation of the protein, but also the way in which the product can be subsequently purified. The advances in genetic engineering have increased the availability of large amounts of recombinant proteins produced in host cells – bacterial, mammalian, insect and yeast – and where Escherichia coli still represents the most widely used platform (Demain and Vaishnav, 2009).
Chromatography is a well-established platform for protein purification, as it is considered economically feasible and yields high recoveries at high purities with very few process steps (Carta and Jungbauer, 2010, Milne, 2011, Walsh, 2003, Walter and Gottschalk, 2010). In affinity chromatography, selectivity towards a specific target protein is introduced through the chemical functionalization of the solid support with desired affinity ligands, which can be divided into three main categories: biological, structural and synthetic (Fig. 1(A)) (Roque and Lowe, 2007). Synthetic affinity ligands have been developed in an attempt to overcome disadvantages of natural and structural ligands, by combining the best of two worlds: Molecular recognition features associated with high resistance to chemical and biological degradation and high scalability as well as low production costs and low toxicity (Clonis et al., 2000, Lowe, 2001, Lowe et al., 2001). These have been tailor-made for the purification of specific biomolecules as antibodies (Haigh et al., 2009, Qian et al., 2012, Roque et al., 2005) although they are not regarded as universal purification adsorbents for fusion proteins, and therefore will not be widely discussed in this review.
Fig. 1.
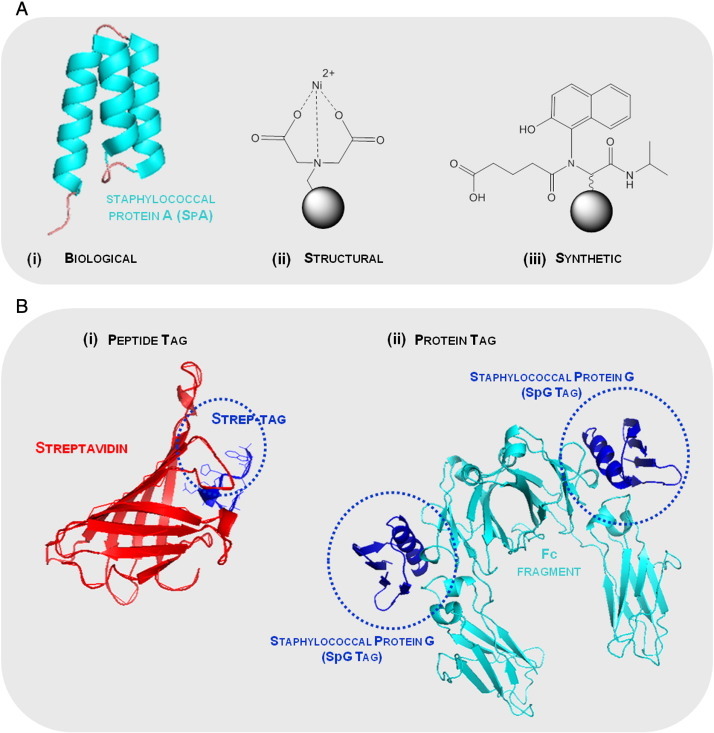
Examples of (A) affinity ligands and (B) peptide and protein affinity tags with their respective biological ligands employed on the purification of fusion proteins based on affinity chromatography. (A) The common affinity ligands can be (i) a biological ligand (staphylococcal protein A domain, PDB: 1DEE), (ii) a structural ligand (metal chelate such as iminodiacetic acid cheated to Ni2 +) and (iii) a synthetic biomimetic ligand (ligand A3C1 specific for immunoglobulins (Haigh et al., 2009). The solid support is representing agarose beads ( ). (B) The (i) peptide tag is the Strep-tag, an eight amino acid sequence, with the affinity for streptavidin protein (PDB: 1RST), whilst the (ii) example of a protein used as an affinity tag is related with the staphylococcal protein G and the respective biological ligand, immunoglobulin G (PDB: 1FCC).
). (B) The (i) peptide tag is the Strep-tag, an eight amino acid sequence, with the affinity for streptavidin protein (PDB: 1RST), whilst the (ii) example of a protein used as an affinity tag is related with the staphylococcal protein G and the respective biological ligand, immunoglobulin G (PDB: 1FCC).
The diversity of proteins and their biochemical properties makes the development of universal purification and capturing strategies difficult. Most proteins of interest lack a suitable, specific and robust affinity ligand for capture on a solid matrix. Genetically encoded affinity tags are a viable and common option for the purification of recombinant proteins and also represent important tools for structural and functional proteomics initiatives. This approach requires the existence and availability of specific ligands for the capture of the fusion protein through an encoded affinity tag tail (Fig. 2 ), which can be denominated as affinity “tag-ligand” pairs. Currently available affinity “tag-ligand pairs” fall within one of these categories: Protein–protein, protein–small biological ligands, peptide–protein or peptide–metal chelating ligands.
Fig. 2.
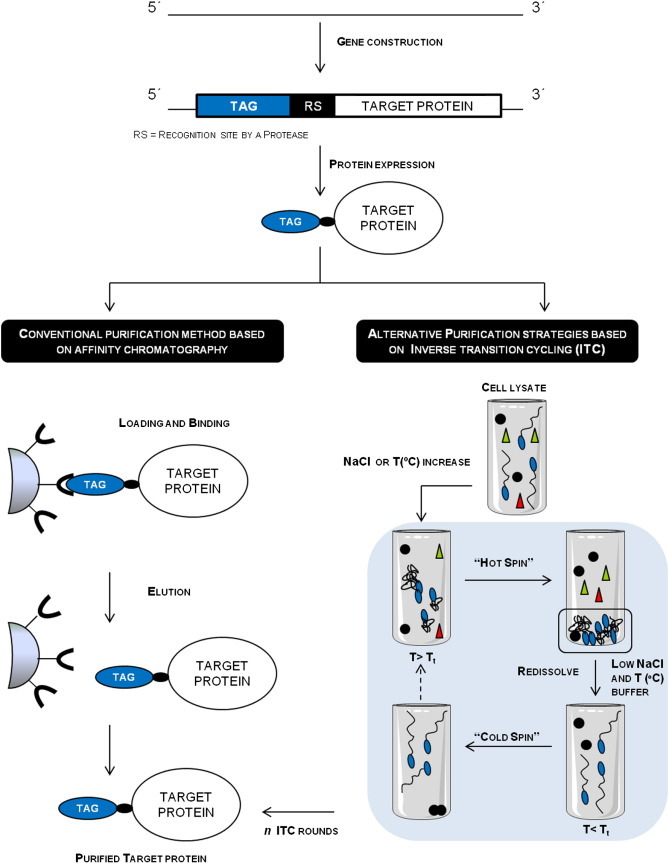
Overview of a recombinant fusion protein purification process through the use of affinity tags fused to the target protein by conventional methods such as affinity chromatography and alternative methods based on inverse transition cycling (ITC). Both processes comprise several stages; (i) fragment DNA construction of the fusion protein, where the fragment DNA which encodes the affinity tag is fused to the nucleotide sequence of the target protein; (ii). The fusion protein is expressed in the selected host and (iii) purified by different methods. The most conventional method involves the use of affinity chromatography, where protein capture is performed through the molecular recognition between the tag and the ligand. An alternative method for the purification of fusion proteins is based on ITC, which exploits the reversible soluble–insoluble phase transition behaviour of the affinity tags and the desired fusion protein yield can be achieved with n ITC rounds. Afterwards, in both methods, the tag can be removed leading to a pure target protein.
Affinity tags display different size ranges from a single amino acid to entire proteins, and can be genetically fused to the N- or C-terminal of the target biomolecule (Arnau et al., 2006, Hedhammar et al., 2005, Waugh, 2005, Young et al., 2012). Apart from facilitating the purification process, affinity tags can also enhance protein solubility and stability, increase expression levels (Hu et al., 2001, Walls and Loughran, 2011) and allow labelling for cellular localization and imaging studies (Malhotra, 2009). An overview of the main advantages and disadvantages associated with different affinity tags has been already thoroughly discussed in the literature (Arnau et al., 2006, Hearn and Acosta, 2001, Hedhammar et al., 2005, Hu et al., 2001, Malhotra, 2009, Terpe, 2003, Walls and Loughran, 2011, Waugh, 2005, Young et al., 2012). In general, shorter affinity tags (peptides) are more attractive as they are less likely to interfere with the expression, structure and function of the target protein, and their removal can be exempt, decreasing thus the overall costs of the purification process (Hearn and Acosta, 2001, Hedhammar et al., 2005, Terpe, 2003).
This review is a comprehensive overview of the well-implemented “tag-ligand” pairs used for the purification of recombinant fusion proteins with a detailed discussion of the lesser known tags with unconventional properties currently under development.
2. Purification of recombinant fusion proteins by “tag-ligand” strategies
2.1. Biological ligands as binding partners of affinity tags
Biological ligands comprise biomolecules obtained from natural sources and from in vitro selection techniques. These are usually associated with high selectivity and affinity for the target, but also with high costs of production and purification, poor stabilization under SIP (sterilization-in-place) and CIP (cleaning-in-place) conditions, and potential leakage and end-product contamination (Clonis et al., 2000, Lowe, 2001, Roque and Lowe, 2006). Examples of common biological affinity ligands include immunoglobulins against a target protein (antigen), bacterial immunoglobulin-binding domains such as Staphylococcal proteins A, G and L (Björck and Kronvall, 1984, Duhamel et al., 1979, Füglistaller, 1989, Lindmark et al., 1983, Nilson et al., 1993), and natural lectins targeting glycoproteins (Vretblad, 1976). Novel biological affinity ligands can be obtained through in vitro selection techniques such as phage, ribosome or yeast display (Smith and Petrenko, 1997), with phage display being very popular for these purposes.
A general overview of the biological ligands and respective tags employed in the purification of tagged recombinant proteins is given in Table 1 and Fig. 1(B). Biological ligands include peptides, proteins and carbohydrates.
Table 1.
Overview of biological ligands employed as binding partners of affinity tags. The “Target protein” is a representative case and the reference listed is for the example target protein.
| Biological ligand | Tag | Tag size | Tag sequence | Elution condition | Yield1 (%) | Purity2 (%) | Representative Target Protein | Application | Reference | |
|---|---|---|---|---|---|---|---|---|---|---|
| Peptides and Proteins | Glutathione | GST | 26 kDa | – | 20 mM reduced glutathione | 100 | 80 | DNA topoisomerase type II | Purification and solubility | Singh et al. (2011) |
| Calmodulin | CBP | 26 aa | KRRWKKNFIAVSAANRFKKISSSGAL | 2 mM EDTA | n.a. | High | Nicotinamide nucleotide transhydrogenase | Purification | Egorov et al. (2004) | |
| S protein (S-fragment RNAse A) |
S-tag | 15 aa | KETAAAKFERGHMDS | Proteolytic cleavage | Fair | High | Recombinant human interleukin-29 |
Purification | Li and He (2006) | |
| Streptavidin | Strep-tag | 9 aa | SAWRHPQFGG | 1 mM iminobiotin | n.a. | High | Fv fragment | Detection and purification | Schmidt and Skerra (1993) | |
| Nano tag | 9–15 aa | DVEAWLGAR | 2 mM d-biotin | 90 | 71 | Bovine heart fatty acid-binding protein (FABP) | Lamla and Erdmann (2004) | |||
| SBP | 38 aa | MDEKTTGWRGGHVVEGLAGELEQLRARLEHHPQGQREP | 2 mM biotin | High | High | Maltose-binding protein | Keefe et al. (2001) | |||
| Strep-Tactin (modified streptavidin) |
Strep tag II | 8 aa | WSHPQFEK | 2.5 mM d-desthiobiotin | n.a | 99 | Human tissue transglutaminase | Schmidt and Skerra (2007) | ||
| NeutraAvidin | AviD-tag | 6 aa | DRATPY | 500 μM biotin | High | High | Green fluorescent protein (GFP) | Gaj et al. (2007) | ||
| IgG | Protein A (SpA) | 14–31 kDa | – | Glycine-HCl pH 3 | 95 | High | β-galactosidase | Purification | Nilsson et al. (1985) | |
| Z domain | 7 kDa | – | 0.3 M acetic acid, pH 3.1 | High | High | Klenow fragment of DNA polymerase I | Nilsson et al. (1994) | |||
| IgG/HAS | Protein G (SpG) | 28 kDa | – | 85 ºC 10 minute incubation | n.a. | 90 | DNA polymerase | Gräslund et al. (1997) | ||
| Monoclonal antibody M1, M2 | FLAG | 8 aa | DYKDDDDK | 150 mM glycine-HCl pH 3.5 | 60 | 95 | Metal response element binding transcription factor-1 (MTF-1) | Detection and purification | Huyck et al. (2012) | |
| Monoclonal antibody 9E10 | c-myc | 10 aa | EQKLISEEDL | Western blot (detection) | – | – | Tobacco etch virus (TEV) protease | Geisbrecht et al. (2006) | ||
| Anti-T7 monoclonal antibody | T7 tag | 11 aa | MASMTGGQQMG | 0.1 M citric acid, pH 2.2, 5 mM glycerophosphate,5 mM KF | High | n.a | SR protein | Cazalla et al. (2005) | ||
| Monoclonal antibody 12 CA5 | HA tag | 9 aa | YPYDVPDYA | 0.85 M KCl | n.a. | High | Transcription factor IID | Carey et al. (2010) | ||
| Polyol responsive monoclonal antibodies | Softag 1 | 13 aa | SLAELLNAGLGGS | 0.7 M NaCl and 30% propylene glycol | n.a | High | GFP | Detection and purification | Thompson et al., 2003a, Thompson et al., 2003b | |
| Softag 2 | 14 aa | PTSPSYSPTSPSYS | 0.5 M ammonium sulphate and 30% ethylene glycol | n.a. | High | RNA polymerase II | Thompson et al. (1990) | |||
| Softag 3 | 8 aa | TKDPSRVG | 75 M ammonium sulphate and 40% propylene glycol | n.a | n.a | GFP | Duellman et al. (2004) | |||
| Carbohydrates | Cross-linked amylose | MBP | 42 kDa | – | 10 mM Maltose | 100 | 75 | Tyrosine hydroxylase | Purification and solubility | Higgins et al. (2012) |
| Cellulose | Cellulose binding protein | 100 kDa | – | Pure water | 80 | High | Red fluorescent protein | Purification | Wan et al. (2011) | |
| Chitin | Chitin binding domain |
5 kDa | – | Thiol induced self-cleavage | n.a. | 99 | Phosphite Dehydrogenase | Purification | Guan et al. (2013) | |
| Starch | Starch binding domain | 133 aa | – | 10 mM glycine-NaOH pH 11 | 86 | 90 | Enhanced GFP | Purification | Lin et al. (2009) | |
1 Yield—Recovery of pure protein; 2 Purity—The purity of eluted proteins was evaluated by SDS-PAGE electrophoresis; GST—Gluthatione-S-transferase; CBP—calmodulin binding peptide; SPB—strepdavidin-binding peptide; IgG—immunoglobulin G; HSA—human serum albumin; HA—hemaglutinin antigen; MPB—maltose binding protein.
2.1.1. Immunoglobulin-based ligands and respective tags
Immunoglobulin-based adsorbents for affinity chromatography are usually very selective for the target proteins but the costs associated tend to be high. Also, as the interaction between ligand and tag is often strong, elution traditionally involves drastic conditions, typically extremes of pH.
The first affinity “tag-ligand” pair was based on the intrinsic selectivity and affinity between the bacterial immunoglobulin-binding domain Staphylococcal protein A (SpA) and the Fc region of mammalian IgG (Nilsson and Abrahmsén, 1990). As an example, alkaline phosphatase was fused to SpA and its purification was performed in a single step using an IgG adsorbent, with elution at acidic pH (Nilsson et al., 1985). It is known that SpA presents five homologous domains (E, D, A, B and C) and that IgG binds preferentially to the B domain (Nilsson and Abrahmsén, 1990). This domain was mutated to improve its resistance towards tag removal by chemical methods and denominated as the Z domain (Nilsson et al., 1987). The bacterial immunoglobulin-binding domain staphylococcal protein G (SpG) has been also studied as a fusion partner due to its bifunctional behaviour – SpG is composed of different domains (A, B, C and D) with affinity for both IgG and human serum albumin (HSA) (Akerström et al., 1987, Nygren et al., 1988) – allowing the purification of SpG tagged proteins through HSA and IgG affinity chromatography (Akerström et al., 1987, Hedhammar et al., 2005, Nilsson et al., 1997, Nygren et al., 1988).
Other affinity tags recognizing immunoglobulin-based adsorbents include the peptide epitopes FLAG, c-myc, T7, hemagglutinin antigen (HA) and Softags (Hedhammar et al., 2005, Young et al., 2012). The c-myc is a product of a proto-oncogene whose epitope presents high affinity for the monoclonal antibody 9E10 (Evan et al., 1985). This affinity pair has been mostly used as a tool for the detection of recombinant proteins through immunoblotting assays rather than for purification processes (Evan et al., 1985, Kipriyanov et al., 1996, Terpe, 2003). The same is observed for the affinity tags T7 and HA (Walls and Loughran, 2011, Young et al., 2012). The T7-tag is a leader peptide of phage T7 with eleven amino acids with affinity for the anti-T7 monoclonal antibody (Jarvik and Telmer, 1998, Studier and Moffatt, 1986), and the HA tag is a peptide epitope of the influenza virus hemagglutinin (Wilson et al., 1984) recognized by the monoclonal antibody 12 CA5 (Field et al., 1988, Foreman and Davis, 1994).
The FLAG tag technology is quite popular for purification purposes, and has been successfully employed on the purification of several recombinant proteins such as immunoglobulins, cytokines and gene-regulatory proteins. The FLAG tag is a hydrophilic peptide with the sequence DYKDDDDK possessing high affinity for the monoclonal antibodies M1 and M2 (Einhauer and Jungbauer, 2001). As the binding between the FLAG tag and M1 adsorbents is calcium dependent, the elution of bound tagged protein can be carried out under mild conditions in the presence of metal chelators such as ethylenediaminetetraacetic acid (EDTA) and ethylene glycol tetraacetic acid (EGTA), at the expense of risking calcium co-elution. On the contrary, elution from M2 resins usually requires a decrease in pH, which in some cases can be detrimental for the protein of interest (Allen et al., 2003, Prickett et al., 1989). An attractive feature of FLAG tag technology is the possibility of simultaneous purification and removal as enterokinase recognizes the DDDDK sequence (Einhauer and Jungbauer, 2001, Hopp et al., 1988). Lately, two additional FLAG sequences were added in tandem to the original FLAG tag, the triple FLAG tag (3 × FLAG), which showed an increased detection limit of the target protein, favoured binding to the M2 antibody, and the possibility to recover the protein by competitive elution with 3 × FLAG peptide (Hernan et al., 2000). The 3 × FLAG technology has been employed in the purification of a bioactive metabolite (Ueda et al., 2011) and full-length human huntingtin (Li et al., 2006).
The epitope tags referred to as Softags are recognized by polyol-responsive monoclonal antibodies (PR-mAb). Proteins tagged with Softags can be eluted under mild conditions employing low molecular weight polyols as competing agents (Burgess and Thompson, 2002, Duellman et al., 2004). This gentle elution protocol, when compared to other immunoaffinity chromatography methods, contributes to the maintenance of the biological activity and structural integrity of the eluted target protein (Thompson et al., 2009). There are three Softags. Softag 1 is a thirteen amino acid sequence near the C-terminal of the β′ subunit of E. coli RNA polymerase which can be recognized by the PR-mAb NT73 (Thompson et al., 2003a, Thompson et al., 2003b). This epitope tag is not conserved in all bacterial species. Other PR-mAb's such as 8RB13 were used for the purification of Shewanella oneidensis core RNA polymerase with high yield and purity (Probasco et al., 2007). Softag 2 is a heptapeptide repeat found on the C-terminal of eukaryotic RNA polymerase II, which is a highly conserved domain from all almost species and presents strong reactivity for PR-mAb8WG16 (Edwards et al., 1990, Thompson et al., 2009); Softag 3 is an epitope tag with an eight amino-acid sequence located near the N-terminal of human transcription factor IIB, and interacts with PR-mAb IIB8, an antibody mapped by phage display and site-directed mutagenesis (Duellman et al., 2004).
2.1.2. Streptavidin-based ligands and respective tags
Another important group of biological affinity ligands includes streptavidin. Here, the tag-ligand system is based on the selective natural binding pair avidin–biotin with a dissociation constant of 10− 15 M (Bayer and Wilchek, 1990). The first reported tag with affinity for streptavidin was Strep-tag, a nine amino acid peptide selected from a random peptide library (Schmidt and Skerra, 1993), generally fused to the C-terminal of the target protein. According to structural studies, this affinity tag binds to the same pocket as biotin (Schmidt et al., 1996). The main advantages of Strep-tag are related to the resistance to proteolysis in vivo, the lack of interference with expression in E. coli and the mild elution conditions employed which include competitive elution with a biotin or analogue compound such as iminobiotin (Schmidt and Skerra, 1993). However, a limitation of the Strep-tag system is the restriction to C-terminal fusion, and an improved version was developed and termed Strep-tag II with an equilibrium dissociation constant of 37 × 10− 6 M (Schmidt et al., 1996). Another engineered streptavidin chromatographic support (Strep-Tactin) was developed for improved binding capacity towards Strep-tag II. This new version of streptavidin was generated from random mutagenesis on the amino acids 44–47 located at the flexible loop region near the binding site (Korndörfer and Skerra, 2002). Although the affinity pair Strep-tag II—Strep-Tactin has been extensively used, affinity tags with higher affinity for streptavidin matrices were still investigated: The streptavidin-binding peptide (SPB) tag comprising a 38 amino acid sequence with an equilibrium dissociation constant of 2.5 × 10− 9 M was selected from a peptide library screened against immobilized streptavidin (Keefe et al., 2001); A Nano-tag combines the small size of Strep-tag II and presents a nanomolar dissociation constant of 4 × 10− 9 M for binding to streptavidin (Lamla and Erdmann, 2004). The latter is a 15 amino acid peptide selected from a synthetic library based on the heart fatty-acid binding protein by ribosome display (Lamla and Erdmann, 2003). The AviD-tag is another affinity tag based on the Strep-tag that displays affinity for neutrAvidin, a neutral form of avidin (Gaj et al., 2007). The AviD-tag is composed by a 6-amino acid cyclic peptide selected through phage display with a dissociation constant of 12 × 10− 6 M for both neutrAvidin and avidin (Gaj et al., 2007).
2.1.3. Other examples of protein/peptide affinity systems
Calmodulin is a small (17 kDa) and stable peptide employed as an affinity ligand on the purification of calmodulin binding peptide (CBP) fusion proteins. CBP is a 26 amino acid sequence derived from the carboxyl-terminal of rabbit skeletal muscle myosin light chain kinase, displaying a dissociation constant of 10− 9 M for calmodulin (Melkko and Neri, 2003, Stofko-Hahn et al., 1992, Vaillancourt et al., 2000). CBP has been fused at the N-terminus of a wide range of recombinant proteins that were further produced at high levels of expression. The size and stability of calmodulin ligands allow the production of calmodulin affinity resins at affordable prices. In addition, as the interaction between calmodulin and CBP is calcium dependent, protein elution can be carried out under mild conditions with calcium chelating agents such as EDTA and EGTA (Melkko and Neri, 2003, Stofko-Hahn et al., 1992, Vaillancourt et al., 2000).
Another example includes the S peptide tag and S-protein derived from the enzymatic cleavage of ribonuclease A by the protease subtilisin (Karpeisky et al., 1994). The S-peptide, a fifteen amino acid sequence, is used as an affinity tag recognized by the S-protein immobilized on the resin. The major advantages are related to the small size of the affinity tag and the nanomolar dissociation affinity constant of the affinity pair (Kim and Raines, 1993).
Glutathione-S-transferase (GST) is a monomeric protein (26 kDa) originating from Schistosoma japonicum which belongs to a family of enzymes that catalyse the reaction between a nucleophile, reduced glutathione, and electrophilic compounds (Boyer, 1989, Smith and Johnson, 1988). Glutathione acts as the ligand and the complementary GST tag acts both as a purification anchoring point and as a solubility/stability enhancer, which justifies the popularity of this affinity pair system (Frangioni and Neel, 1993, Smith, 2000). In addition, GST resins are cost effective and the elution conditions are mild as reduced glutathione can be employed as a competitive agent (Smith and Jonhson, 1988). However, if reducing conditions are not guaranteed, the fusion protein can undergo oxidative aggregation due to the existence of four cysteine residues exposed at the surface of GST tag (Kaplan et al., 1997). Furthermore, fusion proteins with higher molecular weights than 100 kDa can lead to partially or completely insoluble proteins (Frangioni and Neel, 1993).
2.1.4. Carbohydrates as biological ligands for tagged proteins
Biological ligands based on carbohydrates include cross-linked amylose, cellulose, and chitin, among others. The greatest advantages of these affinity ligands are that carbohydrates display high affinity for their respective tags, the affinity matrices are inexpensive and are prepared in a simple manner, which contributes to the ease of process scale-up. In addition, polysaccharides represent extremely interesting adsorbents for affinity based purification as they tend to be non-toxic, inert and stable (Kurek et al., 2009, Luojing et al., 1991, Tomme et al., 1998).
The maltose-binding protein (MBP) is a 42 kDa periplasmic protein engaged in the transport of maltose and maltodextrins across the bacterial cytoplasmic membrane (Kellermann and Ferenci, 1982, Nikaido, 1994). Fusion proteins containing the MPB tag fused on the N-terminus can be purified in one-single step using a cross-linked amylose affinity resin (Guana et al., 1988). The MBP tag presents high affinity towards amylose, a maltose analogue, with a similar dissociation constant (10− 6 M) which is often employed as the competitive elution agent (Kellermann and Ferenci, 1982, Miller et al., 1983). This presents one of the greatest advantages of this affinity pair because the elution of the target fusion protein can be carried out under mild conditions with 10 mM of maltose (Guana et al., 1988). In addition, the MBP tag is an extremely efficient solubilizing agent when compared to GST and thioredoxin (Trx) (Kapust and Waugh, 1999), and does not contain cysteine residues and thus does not interfere with disulfide bonds of the target protein (Guana et al., 1988, Waugh, 2005).
The cellulose and chitin binding proteins present high affinity for the polysaccharides, cellulose, and chitin respectively, which act as affinity ligands for purification purposes. Cellulose binding proteins can be found in a wide range of carbohydrolases, displaying dissociation constants in the low micromolar range for cellulosic supports, and varying in size from small (33–36 amino acids) to large domains with more than 180 amino acids (Tomme et al., 1998). For the particular application of fusion protein expression and purification, smaller domains are preferred. Cellulosic supports are commercially available in a wide range of geometries including fibres, beads, membranes and hydrogels, with a diverse range of porosities and functionalities. Chitin binding domains present high affinity for chitin, the second most abundant polymer after cellulose, and are part of the enzymes involved in the biodegradation of chitin and chitosan (Kurek et al., 2009). Starch binding domains have also been explored as affinity tags. These domains were fused to the N-terminal of β-galactosidase which was then purified by using starch granules (Luojing et al., 1991).
2.2. Structural ligands as binding partners of affinity tags
Structural ligands are produced at affordable prices with relatively facile chemistries, and possess high resistance to harsh conditions. These ligands comprise ion-exchange (Gräslund et al., 2000, Hedhammar et al., 2004, Hedhammar et al., 2006), hydrophobic (Cummins and O'Connor, 2011), metal chelate (Charlton and Zachariou, 2007), thiophilic (Porath, 1987, Roque and Lowe, 2006, Roque and Lowe, 2007), boronate (Borlido et al., 2012, Bouriotis et al., 1981, Liu, 2006, Liu and Scouten, 1994, Liu and Scouten, 2000), and mixed-mode affinity ligands (Brochier et al., 2008, Burton et al., 1997, Mant and Hodges, 2008, Yon, 1981, Zhao et al., 2009). Multimodal affinity chromatography relies on multiple forms of interactions and the respective mixed-mode affinity ligands usually combine a hydrophobic moiety with ionic and hydrophilic groups (Zhao et al., 2009). However, their limited selectivity and low affinity for specific targets usually necessitates the design of multi-step purification schemes.
Structural affinity ligands, mainly cation and anion-exchange, metal chelate and hydrophobic ligands are extremely popular as binding partners of affinity tags and are widely employed for the affinity purification of fusion proteins (Table 2 ).
Table 2.
Overview of the structural ligands involved as binding partners of affinity tags. The “Target protein” is a representative case and the reference listed is for the example target protein.
| Structural ligands | Tag | Tag size | Tag sequence | Elution condition | Yield1 (%) | Purity2 (%) | Representative target protein | Application | Reference | |
|---|---|---|---|---|---|---|---|---|---|---|
| Metal chelate | Ni2 +-NTA/Ni2 +-IDA | Histidine residues (His tag) | 6 aa | HHHHHH | 100 mM imidazole | 95 | 94 | Retroviral zinc finger motif | Purification | Voráčková et al. (2011) |
| Co2 +-CMA | HAT | 18 aa | KDHLIVHLEEHAHAHNK | 150 mM imidazole | 94 | 95 | Chloramphenicol acetyltransferase | Chaga et al. (1999) | ||
| Ni2 +-TACN | Peptide phage display library | 7 aa | HHHNSWD | 250 mM imidazole | n.a. | n.a. | GFP | Mooney et al. (2011) | ||
| Ion exchange (IEX) | Cation-exchange | Arginine residues | 5–15 aa | RRRRRRRR | 1 M NaCl | n.a. | n.a. | RNase A | Purification and refolding | Fuchs and Raines (2005) |
| Z basic | 7 kDa | – | 300 mM NaCl. | n.a. | High | Protease 3C | Hedhammar et al. (2006) | |||
| Anion-exchange | Aspartic acid residues | 5–16 aa | DDDDD | 1 M NaCl | 70 | High | β-glucuronidase | Purification | Zhang et al. (2001) | |
| Glutamic acid residues | 1–8 aa | EEEEEE | 600 mM NaCl | 47 | n.a. | Viral coat protein | Purification | Stubenrauch et al. (2000) | ||
| Z acidic | 7 kDa | – | Linear gradient 0.15–1 M NaCl | n.a. | 90 | GFP | Purification | Hedhammar et al. (2004) | ||
| Multipurpose—IEX, IMAC and HIC | Ni2 +-NTA (IMAC) Phenyl agarose (HIC) |
Multifunctional tag | 6 aa | HYDHYD | Depending on the structural based ligand | n.a. | n.a. | Human haemoglobin | Purification | Becker et al. (2008) |
| Covalent | Chloroalkane linker | Halo tag | 34 kDa | – | TEV protease cleavage | 80–90 | n.a. | Coactivator-associated arginine methyl transferase (CARM1) | Purification | Chumanov et al. (2011) |
1 Yield—Recovery of pure protein; 2 Purity—The purity of eluted proteins was evaluated by SDS-PAGE electrophoresis.
2.2.1. Metal chelators as affinity ligands for His-tagged proteins
The most implemented structural ligands are metal chelates complementary to the His-tag (6 × His). Purification of the fusion protein is performed by immobilized metal affinity chromatography (IMAC) which has been comprehensively reviewed in the literature (Block et al., 2009, Charlton and Zachariou, 2007, Cheung et al., 2012, Gaberc-Porekar and Menart, 2001, Porath, 1992). The concept of IMAC was developed by Porath and co-workers in 1975, through the exploitation of the affinity between proteins and heavy metal ions (Zn2 +, Cu2 +, Ni2 +), in particularly zinc and copper metal which strongly adsorb polypeptides containing histidine and cysteine residues in aqueous solutions (Porath et al., 1975). Based on this principle, the same authors coupled iminodiacetic acid (IDA) on to agarose to bind metal ions and further chelate proteins through histidine and cysteine residues (Porath and Olin, 1983). Later, nitrilotriacetic acid (NTA), another highly stable quadridentate chelating adsorbent, was found to bind strongly to Ni2 + and Cu2 + and to further chelate adjacent histidine residues (Hochuli et al., 1988). NTA technology was then reported for the production and purification of a recombinant protein. The hexapeptide 6 × His was genetically fused to the N-terminal of mouse dihydrofolate reductase protein, produced in E. coli and then purified by IMAC using a Ni2 +-NTA adsorbent. Subsequently, protein elution was attempted by pH gradient (pH 8–5) in the presence of denaturing agents (e.g. guanidine-HCl), and the tag was further removed by carboxypeptidase A cleavage (Hochuli et al., 1988). However, the major drawback of this pioneer purification study was the employment of denaturing conditions, which triggered the improvement of the procedure by conducting binding under native conditions and elution with low concentrations of a competitor imidazole or EDTA (Janknecht et al., 1991, Westra et al., 2001).
The compatibility of His-tag with denaturing agents can be a benefit for recombinant proteins produced as inclusion bodies, as they require steps of solubilization with chaotropic agents prior to binding to the column (Hochuli et al., 1988). The main disadvantage of IMAC adsorbents is the metal leaching that contributes to end-product contamination and additional steps of purification with increased costs. In addition, IMAC adsorbents can bind to native histidine patches in proteins or to other amino acids (glutamic and aspartic acid, tyrosine, cysteine, arginine, and methionine) resulting in low selectivity. Other disadvantages are related to evidence that the imidazoyl side chain groups of histidine can interfere with protein expression and folding and also mask the biological activity of the protein (Hearn and Acosta, 2001). Nonetheless, the universe of applications for the IMAC/His-tag system is widely used for protein purification and is extended to matrix-assisted refolding (Dashivets et al., 2009), detection and immobilization, protein microarrays (Kato et al., 2005, Wegner et al., 2003, Wilson and Nock, 2002), and to the development of other affinity tag technologies.
The HAT tag is one of these examples and comprises a polyhistidine peptide sequence that can be fused on either ends of the target protein. The purification can be carried out under the same conditions as for His-tag; however, the adsorbent used is based on carboxymethylaspartate (CMA) complexed with Co2 + with a very high capacity (Chaga et al., 1999). Novel metal chelating agents based on 1,4,7-triazacyclononane (TACN) complexed with Ni2 + were developed in an attempt to overcome the limitations of conventional IMAC matrices such as leaching, and lack of stability and selectivity (Mooney et al., 2011). The respective affinity tags correspond to random heptapeptide sequences displaying multiple histidine, tryptophan, and/or tyrosine residues selected from a phage display library (Mooney et al., 2011). The selected peptide tag (HHHNSWD) was fused to the N-terminal of green fluorescent protein (GFP), produced in E. coli and then purified under physiological conditions with high salt concentrations. Recovery was executed with imidazole due to the presence of three histidine residues (Mooney et al., 2011).
2.2.2. Ion-exchange adsorbents for tagged protein purification
Ion-exchange chromatographic resins present affinity for several tags comprising of charged amino acids through the establishment of electrostatic interactions with opposite charges. The polyarginine-tag (polyArg) is such an example and consists of six arginine residues with high retention on cationic exchange resins. A great advantage of using cation-exchange chromatography for the purification of recombinant proteins produced in E. coli is that most host proteins are neutral or acidic, which can contribute to the reduction of non-specific interactions at physiological and alkaline pH values (Gräslund et al., 2000, Hearn and Acosta, 2001). The first reported example was the production and two-step purification of human urogastrone fused to the polyArg tail. The tagged protein eluted under a NaCl linear gradient at alkaline pH, with 44% yield and purity higher than 95% (Sassenfeld and Brewer, 1984). The polyArg-tag has also been successfully employed for on-column refolding studies of tagged proteins produced as inclusion bodies (Kweon et al., 2004, Stempfer et al., 1996). However, the polyArg-tag/cation-exchange resins affinity pair shows low selectivity and the tag promotes aggregation of the protein due to charge–charge interactions (Hedhammar et al., 2005). Poly-anionic affinity tails from one to eight glutamic residues were used for the anion-exchange chromatography based purification of functional human growth hormone (Dalboge et al., 1987) and virus-like particles (Stubenrauch et al., 2000) produced in E. coli. Negatively charged affinity tags based on aspartate residues also yielded stable fusion proteins with unaltered activity (Zhang et al., 2001).
The Z domain, a variant of the B domain of SpA, has been also engineered to create a highly charged Z domain – Zbasic – through the introduction of charged residues at specific positions, which allowed the interaction with negatively charged matrices under physiological conditions (Gräslund et al., 2000, Hedhammar et al., 2006). The Z domain proved to be an excellent protein scaffold for engineering as it is soluble, does not display disulfide bonds and presents reversible folding after exposure to chaotropic agents (Hedhammar et al., 2006). Due to its excellent properties, the Zbasic was explored for matrix assisted refolding of fusion proteins solubilised with chaotropic agents after being produced as inclusion bodies. The Z domain was also on the basis of Zacidic, engineered to be negatively charged and therefore to be purified by anion-exchange chromatography (Hedhammar et al., 2004).
2.2.3. Covalent affinity chromatography
The vast majority of the affinity “tag-ligand” pairs exploit the reversible binding interaction between a tag and its respective ligand. Recently, a novel pair based on the covalent linkage between a tag and a synthetic ligand has been developed. This novel system is composed by the Halo-tag and a chloroalkane linker. The Halo-tag was rationally designed based on a bacterial haloalkane dehalogenase (DhaA) derived from a Rhodococcus spp. to bind covalently to synthetic ligands with a chloroalkane linker. Halotag7 appeared as an improved version of Halotag created to enhance protein expression, solubility and structural compatibility but with the same ability to bind covalently and irreversible to a chloroalkane linker (Ohana et al., 2009). The covalent bond formed between the Halo-tags and the linker is highly specific, irreversible, stable and occurs rapidly under physiological conditions, which facilitates the attachment to a variety of molecules or solid surfaces containing diverse functional groups (Los and Wood, 2006, Los et al., 2008, Ohana et al., 2009). These properties were therefore explored in protein labelling technology for cell imaging and protein analysis (Los et al., 2008), immobilization (Taniguchi and Kawakami, 2010) and purification of recombinant proteins (Chumanov et al., 2011, Ohana et al., 2011). However, the formation of covalent bonds between the tag and the ligands makes the re-use of the solid support difficult, which is particularly problematic in protein purification.
3. Tags with versatile and unconventional properties and respective purification approaches
The affinity tags described so far were developed to be the anchoring point for purification by affinity chromatography where the existence of a complementary affinity ligand, immobilized in a separation matrix, is required. However, tags alone can be auto-sufficient for enhanced fusion protein solubility and purification. This section will describe tags with versatile and unconventional properties that can be combined with alternative purification protocols.
3.1. Solubility enhancers
The production of recombinant proteins in bacterial hosts can lead to the formation of protein aggregates with unfolded or partially folded structures (Demain and Vaishnav, 2009, Lange and Rudolph, 2008), termed inclusion bodies (IBs) (García-Fruitós et al., 2012). When these proteins are fused to solubility tags, solubility is improved during protein expression, although the mechanism is not fully understood (Walls and Loughran, 2011, Waugh, 2005, Young et al., 2012). Thioredoxin A (TrxA) (Katti et al., 1990), small ubiquitin-related modifier (SUMO) (Li and Hochstrasser, 1999, Marblestone et al., 2006), and N-utilization substance A (NusA) (Gusarov and Nudler, 2001, Liu and Hanna, 1995) are examples of widely employed tags with the purpose of increasing the solubility of the fusion partner rather than acting as a handle for further purification. As a consequence, a purification tag must be also added to the fusion partner to facilitate purification through affinity chromatography. The TrxA is a small (12 kDa) oxido-reductase protein with intrinsic thermal stability, solubility and robust folding properties that when fused to a target protein contributes to high yields of soluble protein (LaVallie et al., 1993, LaVallie et al., 2000). The SUMO protein (11 kDa) is involved in post-translational modification in eukaryotic cells, enhances solubility and stability of the binding partner when fused to the N-terminal (Marblestone et al., 2006), and allows simultaneous cleavage as it is recognized by the SUMO protease (Saccharomyces cerevisiae Ulp1) at the conserved dipeptide glycine–glycine (Li and Hochstrasser, 1999). However, when used in eukaryotic hosts, the affinity tag can be cleaved due to the presence of SUMO proteases in vivo (LaVallie et al., 2000, Malakhov et al., 2004). The NusA is a 55 kDa transcription elongation and anti-termination factor, also known to increase the solubility of fusion proteins, however due to their large size can contribute to a high metabolic burn for the host cell (Harrison, 2000).
3.2. Versatile fusion partners
In order to combine several functionalities, a multipurpose peptide tag with the sequence HYDHYD which comprises a bi-repetition of the tripeptide constituted by a histidine, tyrosine and aspartate residues was developed (Becker et al., 2008). This peptide can be fused to the N- or C-terminal of the target protein enabling the purification by a combination of several techniques including IMAC, IEX, aqueous two phase system (ATPS) and hydrophobic interaction chromatography (HIC). This versatile and multifunctional affinity tag was employed for the purification of GFP, lactate dehydrogenase and human haemoglobin (Becker et al., 2008).
Novel dual-tagging methods have been also developed and the concept of tandem affinity chromatography (TAP) has emerged as a generic platform for complex protein purification combined with mass spectrometry, for the isolation, identification and characterization of protein partners in multi-protein complexes (Günzl and Schimanski, 2009, Puig et al., 2001).
The original C-terminal TAP tag was developed by Puig et al by using S. cerevisiae hosts and consists of two IgG binding domains of SpA and a CBP, separated by a tobacco etch virus (TEV) protease cleavage site. The N-terminal version was also developed, being an inverse version of C-terminal (Puig et al., 2001). The main advantage related to this strategy is the highly efficient recovery of target protein (Günzl and Schimanski, 2009, Puig et al., 2001, Xu et al., 2010). Nowadays, the TAP strategy is successfully employed in protein–protein interaction studies in prokaryotic and eukaryotic cells (Ahmed et al., 2011, Günzl and Schimanski, 2009, Ma et al., 2012, Müller et al., 1998, Xu et al., 2010).
A wide variety of affinity TAP tags emerged recently by using different combinations or repetitions of single affinity tags, the most commonly used being SpA, CBP, His tag, HA, FLAG, SBP and Strep-tag II (Li, 2010). Examples of combinations of affinity tags comprise a tandem histidine and FLAG epitope tag for the purification of a Candida albicans septin complex, a complex of cytoskeletal proteins (Kaneko et al., 2004). In addition, combinations of S-tag and Strep-tag II separated by HRV 3C protease cleavage were used for the isolation of protein complexes such as immune cell proteins, adhesion and degranulation promoting adapters (ADAP) (Lehmann et al., 2009). Other examples enclose combinations of streptavidin-binding peptide (SBP) and calmodulin-binding peptide as TAP tag to isolate and identify proteins directly from Parkin (ubiquitin-protein ligase (E3)), a protein involved in Parkinson's disease (Davison et al., 2009). A novel TAP tag has also been developed, SF tag, and consists of a combination between a doublet Strep-tag II and a FLAG tag. This TAP tag can offer several advantages over the others such as the four fold size reduction (4.6 kDa) with better purity and recovery yields. The main applications of this are related to isolation of protein complexes from eukaryotic cells (Gloeckner et al., 2007, Gloeckner et al., 2009). More recently, a novel N-terminal TAP tag denominated as CHiC tag was constructed based on a combination between His-tag and a choline binding domain followed by a cleavage site specific for TEV protease with applications on protein purification (Stamsås et al., 2013).
Although TAP strategies can be useful for proteomics research, there is still a demand for new strategies as classical TAP tags were found to interfere with protein biological function, structure and complexes. Moreover, the identification of interactions occurring for short periods or in specific physiological states is still limited (Xu et al., 2010).
3.3. Affinity tags for the purification of membrane proteins
Membrane proteins are embedded in the lipid layer on cell surfaces, playing an important role on several basic cell functions (e.g. signal transduction, cell–cell communication). These proteins are highly relevant for structural and functional studies, however their production and purification strategies are still a challenge. This is related to the fact that membrane proteins are produced with low yield in common hosts (e.g. mammalian and insect cells) (Lin and Guidotti, 2009, Smith, 2011). Due to their hydrophobic character, membrane proteins have the propensity to fold incorrectly and aggregate (Lin and Guidotti, 2009, Smith, 2011). Moreover, the fact that these proteins are within the lipid layer obliges the use of detergents to enhance their solubilization in aqueous systems, which influences the purification step due to detergent interference in ion-exchange and hydrophobic chromatography (Lin and Guidotti, 2009, Smith, 2011). Tagging systems have been studied to overcome the reported limitations. Affinity tags are usually placed at the C-terminal to minimise interference with the membrane targeting process (Lin and Guidotti, 2009). Examples include a combination between the Sumo technology and His-tag for the purification of severe acute respiratory syndrome coronavirus membrane protein (SARS-CoV). This strategy addresses solutions for the improvement of expression yields in bacterial hosts by using a solubility enhancer tag; the His-tag allows the purification of target membrane proteins through the IMAC technique with a high tolerance for the presence of detergents (Zuo et al., 2005).
3.4. Unconventional fusion partners
Non-chromatographic methods emerged as alternatives of conventional chromatography and comprise simple and inexpensive density-based separation or mechanical gravity methods (Wilken and Nikolov, 2012). Unconventional fusion partners facilitate the use of non-chromatographic methods—these include hydrophobic tags (Fexby and Bülow, 2004), oleosin based tags (Bhatla et al., 2010, Wilken and Nikolov, 2012) and protein body forming peptides such as elastin-like polypeptides (ELPS), ZERA or endogenous hydrophobin (Conley et al., 2011, Khan et al., 2012).
3.4.1. Hydrophobic tags
Examples of hydrophobic tags for recombinant protein purification include tags based on tryptophan and tyrosine, exploited for aqueous two phase separation (ATPS) (Fexby and Bülow, 2004), and tags based on repetitions of the dipeptide tryptophan–proline (WP)n (Lienqueo et al., 2008). It has been reported that as the length of the tag increases, a negative effect on cellular growth, stability of the plasmids, secretion efficiency of the target protein and a decrease of recovery yield on purification stage are observed (Lienqueo et al., 2008). According to Collén et al, during protein expression, fully exposed hydrophobic tags can reduce target protein solubility, compromising the secretion pathway and leading to the formation of insoluble protein aggregates (Collén et al., 2001) and the chance that the target protein will be membrane-associated increases (Fexby and Bülow, 2004). Additionally, it was found that tags with higher tryptophan content in the peptide (n = 6) are more prone to proteolytic cleavage when produced in E. coli and fungi hosts (Collén et al., 2001). Therefore, different combinations comprising tyrosine and proline (YP)n have been exploited, as tyrosine presents a less hydrophobic character than tryptophan. Fexby et al have developed a peptide tag with three tyrosine and proline residues fused to GFP which increased protein expression and protein recovery yield by ATPS based purification. The main advantages of using hydrophobic tags with APTS systems rather than with affinity chromatography based methods rely on the combination of rapid separation and high capacity with inexpensive costs (Fexby et al., 2004).
3.4.2. Oil bodies
An alternative affinity system to enable purification of tagged recombinant proteins relies on the interaction between oil bodies (OBs) and oleosin fusion proteins. OBs are spherical structures that contain a triacylglycerol (TAG) matrix surrounded by a phospholipid mono-layer and several proteins, mainly oleosins, caleosins and steroleosins (Bhatla et al., 2010, McLean et al., 2012, Peng et al., 2004, Roberts et al., 2008). Oleosin, the main structural protein of the oil bodies's membrane, acts as an affinity tag. Oleosin fusion proteins have been produced in plants, which also presents several advantages over bacterial hosts such as simple handling, cost-effectiveness and easy scalability, as well as effective polypeptide folding (Bhatla et al., 2010). However the main disadvantages of this host are related to regulatory demands and production time (Wilken and Nikolov, 2012). Oleosin fusion proteins are targeted to naturally occurring OBs in the seed, and purified by several washing/centrifugation steps, as the OBs along with oleosin fusion protein are easily separated by floating (Bhatla et al., 2010, Roberts et al., 2008). Examples of proteins purifed by this technology include recombinant human insulin, revealing a high protein recovery (Nykiforuk et al., 2006). Apart from naturally occurring oil bodies, artificial oil bodies were also successfully employed for the purification of oleosin fusion proteins expressed as insoluble aggregates in E. coli. In these cases, artificial oil bodies were reconstituted by assembling triacylglycerol (TAG) phospholipid and the insoluble recombinant protein. The recombinant fusion protein can be incorporated at the phospholipid monolayer of artificial oil bodies (AOB) through oleosin whilst the target protein is exposed at the AOB surface, where the target insoluble protein undergoes spontaneous refolding. Afterwards, the fusion protein on the surface of AOB can be easily purified by a flotation centrifugation step. The tag can be then cleaved by enzymatic methods or it can be used whilst still fused with oleosin for further applications (Bhatla et al., 2010). This non-chromatographic strategy based on AOB was employed for the purification of GFP (Peng et al., 2004), nattokinase (Chiang et al., 2007) and hydantoinase (Chiang et al., 2005) with high yield and purity.
3.4.3. Elastin-like polypeptides (ELPs)
Elastin-like polypeptides (ELPs) are composed by repetitions of the pentapeptide VPGXG (where X is any amino acid, with the exception of proline) and undergo a reversible phase transition under a certain temperature, which is designated as lower critical solution temperature (LCST), also known as inverse transition temperature (Tt). Below transition temperature (Tt), ELPs are highly water soluble whereas at temperatures higher than Tt, they aggregate and become insoluble (Floss et al., 2010, Hassouneh et al., 2012, Meyer and Chilkoti, 1999). Due to their unique properties, ELPs have been used as tags, and therefore successfully employed on purification of recombinant fusion proteins (Banki et al., 2005, Meyer and Chilkoti, 1999). The purification of ELP fusion proteins can be carried out through a simple method termed inverse transition cycling (ITC), which allows the purification of fusion proteins by cycling the crude lysate through the insoluble and soluble phases (Hassouneh et al., 2012). Factors such as higher ionic strength, polymer concentration and length can decrease LCST as well as significant hydrophobic surface areas on the target protein. On the other hand, a higher charged surface area can increase LCST (Hassouneh et al., 2001).
The ITC method (Fig. 2) is an alternative strategy for the purification of recombinant proteins without the use of chromatographic resins (Meyer and Chilkoti, 1999). This simple strategy comprises cycles of “hot spin” and “cold spin”, differing mainly in the temperature at which the centrifugation step is performed. The “hot spin” is related to the centrifugation step performed after the aggregation of ELP fusion proteins from cell lysate. Aggregation can be triggered by an increase of temperature above Tt or an increase of salt concentration to decrease Tt; the increase of salt concentration is usually preferred due to protein denaturation. This step allows the separation of soluble host protein contaminants and recovery of the pellet containing the insoluble ELP fusion protein which is further used on the following step termed “cold spin”. This step consists of the transition of the ELP fusion proteins to a soluble form by dissolving the pellet at low temperature and ionic strength buffer so that the insoluble contaminants can be further removed by centrifugation. The supernatant will contain the purified ELP fusion protein (Hassouneh et al., 2001, Hassouneh et al., 2012). The number of ITC rounds depends on the properties of the target protein and final purity desired. This sample methodology has been already extended for the purification of several recombinant proteins (Table 3 ).
Table 3.
Purification of Elastin-like polypeptides (ELP) fusion proteins by inverse transition cycling (ITC). The “Target protein” is a representative case and the reference listed is for the example target protein.
| Tag | ELP | Yield (%) | Purity (%) | n ITC round | Tt (°C) | Representative Target Protein | Reference |
|---|---|---|---|---|---|---|---|
| Elastin-like polypeptides (ELP) | V5-78 | 67 | na | 2 | 25 | Organophosphorus hydrolase (OPH) | Shimazu et al. (2003) |
| V5A2G3-90 | 80 | na | 2 | 25 | Chloramphenicol acetyltransferase (CAT) | Trabbic-Carlson et al. (2004) | |
| V5A2G3-90 | 75 | na | 1 | 25 | Blue fluorescent protein (BFP) | Trabbic-Carlson et al. (2004) | |
| V5A2G3-90 | na | na | 6 | 25 | Calmodulin (CalM) | Trabbic-Carlson et al. (2004) | |
| V5A2G3-90 | 75 | na | 1 | 25 | Thioredoxin (Trx) | Trabbic-Carlson et al. (2004) | |
| V5A2G3-90 | 75 | na | 2 | 45 | Green fluorescent protein (GFP) | Chow et al. (2006) | |
| V5-40 | 98 | na | 1 | 17 | Levansucrase. | Kang et al. (2007) | |
| V5A2G3-110 | 18 | na | na | 25 | β-Lactamase | Fong et al. (2009) | |
| V5-80 | 90 | 95 | 1 | 37 | β-Galactosidase | Yu and Liu (2012) | |
| (KV7F)36 | 50 | 95 | 3–4 | 42 | Trx | Lim et al. (2007) |
3.4.4. Hydrophobin
Hydrophobins are small surface proteins (7–12 KDa) enrolled in numerous functions on filamentous fungi life cycle. The unique properties of these small proteins are related to their amphiphilic nature and self-assembly capacity (Mustalahti et al., 2013). There are two classes (I and II) of hydrophobins differing on the solubility of the formed aggregates and the amino acid composition. The hydrophobin classes I and II do not present as much identity in terms of amino acid composition but present the same pattern of eight conserved cysteine residues (Linder, 2009). In terms of aggregates solubility, class I forms more stable aggregates than class II and therefore, in the first case, the aggregates are only able to dissociate under the presence of strong conditions (e.g. formic acid and trifluoroacetic acid) whilst the aggregates formed by class II hydrophobins are much easier to dissolve with aqueous diluted organic solvents (Linder et al., 2005, Mustalahti et al., 2013).
Hydrophobins class II HFBI and HFBII from Trichoderma reesei have been the most exploited hydrophobin. This hydrophobin fusion technology can simultaneously improve the yield of recombinant protein production by protein body formation in filamentous fungi, and allow subsequent protein recovery by simple methods as ATPS. The protein bodies were found to enhance the accumulation of soluble protein, and to stabilize the storage of large amounts of recombinant protein. The formation of protein bodies also presents other advantages such as protection of fusion proteins from proteolysis degradation and possible toxic effects resultant from the accumulation of the target protein. The hydrophobin fusion technology has been reported in literature as a strategy for GFP purification by ATPS with selective recovery up to 91% (Joensuu et al., 2010, Mustalahti et al., 2013).
3.4.5. γ-zein (ZERA)
The γ-zein is a 27 kDa maize storage protein composed of a signal peptide, a N-terminal proline-rich sequence (PPPVHL)8, a Pro-X motif and a hydrophobic cystein rich domain (Llompart et al., 2010). The N-terminal of γ-zein that comprises the repeat domain (PPPVHL)8 and Pro-X motif is also referred to as ZERA, presented to be sufficient to self-assemble and then induce the formation of protein bodies, which are natural endoplasmic reticulum (ER) derived organelles in the maize seed and which provide great stability to accumulated proteins (Khan et al., 2012, Llompart et al., 2010). ZERA has been exploited as a fusion partner for the production of fusion proteins as well as for protein body inducer in order to facilitate further steps of purification (Torrent et al., 2009). This strategy was extended not only to plant based hosts but also in eukaryotic hosts (mammalian and insect cells or filamentous fungi). By taking advantage of the PB properties such as high density, the protein can be easily recovered by density. Examples of this include human growth hormone (Llompart et al., 2010) and enhanced cyan fluorescent protein (Torrent et al., 2009).
4. Alternative schemes for tag removal
For most applications, affinity tags are innocuous and do not interfere with the structure and biological function of the target protein. However, for therapeutic proteins there is a demand for tag cleavage to avoid immunological responses and to guarantee authenticity of the protein structure. The removal of the affinity tag can be performed by enzymatic cleavage or harsh chemical treatments and has been extensively reviewed (Arnau et al., 2006, Hearn and Acosta, 2001, Terpe, 2003, Waugh, 2011). Chemical cleavage methods mostly used involve cyanogen bromide or hydroxylamine treatments, which are an inexpensive option but with low specificity and stringent conditions detrimental to the protein. Therefore, enzymatic methods are usually preferred as they operate under milder conditions and present higher selectivity as endoproteases recognize specific amino acid sequences or motifs. The main endoproteases used for tag removal comprise enterokinase (Choi et al., 2001), tobacco etch virus (TEV) (Dougherty et al., 1988) Factor Xa (Jenny et al., 2003), thrombin (Chang, 1985, Jenny et al., 2003) and SUMO protease (Li and Hochstrasser, 1999). An attractive feature of enterokinase and factor Xa is that after cleavage there is no additional amino acid residues on the protein structure, contributing to an intact end product (Choi et al., 2001, Hearn and Acosta, 2001, Jenny et al., 2003). The main limitations in the use of proteases are related to the tag incompatibility under certain cleavage conditions (e.g. buffers, temperature), thus influencing its operation, and accessibility to the cleavage site (Waugh, 2011). The high costs associated with proteases limit the scalability of the purification process (Fong et al., 2010). Strategies on the reusability of these proteases without efficiency loss have been studied, as recently reported for the immobilization of enterokinase onto magnetic supports (Santana et al., 2012).
On the same basis, alternative strategies have been exploited to overcome the traditional tag cleavage methods. Self-cleaving tags have emerged as an alternative strategy for tag removal as they possess inducible proteolytic activity, thus representing an extremely interesting class of affinity tags (Fong et al., 2010, Li, 2011). When combining with a purification tag, the recombinant proteins can be purified with the successive tag removal in a single step. Moreover, the use of self-cleaving tags can simplify the entire process at reduced costs, considering that the use of additional steps is not required for the separation of the target protein from the protease and the affinity tag (Wang et al., 2010).
The intein-based system is a well-established example where purified fusion protein cleavage can be induced by thiols such as β-mercaptoethanol or 1,4-dithiothreitol (DTT), or by a shift in pH or temperature (Fong et al., 2010, Li, 2011). The pH-induced inteins are usually fused on C-terminal and are preferred in terms of costs and minor interference with the target protein over the N-or C-terminal thiol-induced intein. This is related to the fact that the presence of thiol groups can disrupt disulfide bonds and therefore confine the purification of the target protein (Fong et al., 2010). The pH-induced inteins combined with ELPs have been involved on a non-chromatographic purification scheme of several proteins, among them GFP (Wu et al., 2006), β-lactamase (Shi et al., 2013) and chloramphenicol acetyl transferase (CAT) (Liu et al., 2012) with further tag removal under mild conditions with a pH shift for 6.0–6.5. Apart from ELPs, the pH-induced inteins were also fused to chitin binding domain (CBD) for standard affinity chromatography purification via intein-mediated cleavage under the same mild conditions. This strategy has been employed for different proteins such as human antibody fragments, E. coli maltose binding protein (Wu et al., 2011) and human epidermal growth factor (Esipov et al., 2008). On the other hand, examples of thiol-induced inteins have been reported in the literature as fusion partners of oleosin for purification and removal of xyalanase (Liu et al., 2008) and hydantoinase (Chiang et al., 2007). The oleosin fusion proteins were purified through AOB and then released from oleosin by using 40 μM DTT.
Intein-based protocols are compatible with several expression hosts (e.g. yeast, insect cells, mammalian cells and bacterial), but premature cleaving can occur during expression, lowering the yield of the target protein (Fong et al., 2010). The potential for scale-up is limited in thiol-induced inteins due to the higher costs of reagents required for tag cleavage. Other self-cleaving tags such as sortase A (SrtA), N-terminal protease (Npro), FrpC module and cysteine protease domain (CPD) have been exploited as alternative strategies for tag removal but with less implementation (Fong et al., 2010, Li, 2011).
5. Concluding remarks and future trends
Nowadays there is still a demand to develop processes for recombinant protein purification that can combine selectivity with affordable prices. Affinity chromatography is the most frequent choice for protein purification; however this technique is dependent on the adsorbents' availability for each particular target protein. Within the range of available ligands for affinity chromatography separations, those of biological origin are still the preferred option due to the high selectivity. Structural ligands are produced at low cost but the conditions for their use need to be optimised for each case due to the lack of specificity, often resulting in purification protocols with multiple steps. Synthetic ligands raised to overcome the high costs associated with biological ligands and the lack of selectivity of structural ligands have been so far tailor-made to very specific high value proteins. As each protein presents different biochemical and physical properties, the use of affinity pairs “tag-ligand” emerged as a universal purification strategy.
Currently there is a wide range of affinity “tag-ligand” pairs that can be used for the production and purification of fusion proteins. Commercial purification supports possess either biological or structural ligands and are presented in Table 4, Table 5 , respectively. The choice of the affinity tag should take into account the target protein, the host and production conditions, as well as the respective binding partner.
Table 4.
Examples of commercially available affinity tags and respective purification supports based on biological ligands.
| Biological ligands | Affinity solid support and supplier | Affinity tag | Capacity (mg/ml) |
|---|---|---|---|
| Peptides and proteins B | Glutathione sepharose (GE Healthcare) Glutathione superflow (Clontech) Glutathione HiCap matrix (Qiagen) Pierce glutathione agarose/magnetic beads (Thermo Scientific) Glutathione agarose and magnetic agarose beads(Sigma) |
GST tag | 10 10 20 40/5–10 5–10/15 |
| Calmodulin resin (VWR) MagneZoom™—CAM (bioWorld) Calmodulin resin (G Biosciences) |
CBP | 3 na 3 |
|
| S protein Agarose (Novagen-EMD Millipore) | S-tag | na | |
|
StrepTactin™ (GE Healthcare) Strep-Tactin superflow/magnetic beads (Qiagen) Strep-Tactin superflow (Novagen-EMD Millipore) |
Strep-tag II | 6 9/0.3 3 |
|
| IgG agarose (GE Healthcare) IgG agarose (Sigma) |
Protein A/G tag |
2 1 |
|
| Anti-FLAG M1, M2 resin (Sigma-Aldrich) Anti-FLAG antibody agarose/magnetic beads (Clontech) |
Flag-tag | 0.6 na/1.3 |
|
| EZview™ red Anti-myc resin (Sigma) Anti-c-myc agarose (Sigma-Aldrich) Anti c-myc antibody agarose (Clontech) Anti-Myc-tag mAb-magnetic beads (MBL) Pierce c-myc-Tag IP (Thermo Scientific) |
c-myc tag | 0.05 0.2 0.5 na na |
|
| T7 Tag® antibody agarose (EMD Millipore) T7 tag antibody agarose (Genetex) Anti-T7 tag® antibody agarose (Abcam) |
T7-tag | 0.3 0.5 na |
|
| Pierce Anti-HA agarose (Thermo Scientific) Pierce Anti-HA magnetic beads (Thermo Scientific) Ezview™ anti-HÁ affinity gel (Sigma) Anti-HÁ affinity matrix (3F10) (Roche) Anti-HA-tag mAb-magnetic beads (MBL) |
HA tag | 150 nmol/ml 10 μg/mg 0.4 9 nmol/ml na |
|
| Softag immune affinity resin (Neoclone) | Softag 1/2/3 |
0.3 | |
| Carbohydrates | Amylose resin/magnetic beads (New England Biolabs) | MBP | 7–10/0.01 |
| Cellulose (Sigma) | Cellulose binding domain | 15 | |
| Chitin agarose (New England Biolabs) | Chitin binding domain | 3 |
na—data not available.
Table 5.
Examples of commercially available affinity tags and respective purification supports based on structural ligands.
| Structural ligands | Affinity solid support and supplier | Affinity tag | Capacity (mg/ml) |
|---|---|---|---|
| Metal chelate | Ni-sepharose high performance (GE Healthcare) His Mag sepharose (GE Healthcare) TALON® superflow™ (GE Healthcare) HisPur Ni-NTA resin/magnetic beads (Thermo Scientific) HisPur cobalt superflow agarose (Thermo Scientific) His60 Ni resin/magnetic beads (Clontech) TALON® resin/magnetic beads (Clontech) Ni-NTA agarose (Invitrogen) Dynabeads® His-Tag (Invitrogen) NTA-Agarose/Magnetic Agarose Beads (Qiagen) Profinity™ IMAC Nickel Charged resin (BIORAD) IMAC HyperCel™ (PALL) |
His-tag | 40 50 20 20/0.5 20 60/n.a 5–18/n.a 5–10 0.04 50/2 15 na |
| TALLON® metal affinity resin (Clontech) | HAT tag | 18 | |
| Ion-exchange (IEX) | Anion or cation exchange resins (several sources) | Charged peptides | 50–120 |
| Covalent | HaloLink™ (Promega) | Halo tag | 7 |
na—data not available.
Unconventional fusion partners have emerged as promising affinity tags, which due to their unique properties of hydrophobicity and self-assembly do not require chromatographic methods for the selective protein recovery. Non-chromatographic affinity tags are attractive choices as they can easily overcome the drawbacks associated with the affinity tag and therefore present high potential as large-scale purification platforms. However, the main limitation of unconventional fusion partners is the low purity of the recovered target protein when compared with the standard affinity tags commonly employed.
The application of genetically encoded tags for the affinity purification of recombinant protein is mostly in manufacturing and research. In manufacturing tag strategies are most applicable to situations where the purity of the target protein is less strict as it is the case for industrial enzymes.
When considering therapeutic proteins, the use of affinity pairs is not the preferred choice due to the need to strictly control the presence of the tag in the final product. Moreover, the additional steps of tag removal and further purification contribute to higher associated costs and low yields.
Therefore, the use of affinity pairs has a higher impact on research rather than manufacturing as their use is often employed in a wide range of applications within the scientific community being used not only for purification processes. These applications are related to protein–protein interaction studies in cell signaling, protein immobilization based strategies for sensing and diagnostics, protein labelling and cell imaging. Genetically encoded affinity tags were initially developed to provide universal strategies for recombinant protein purification that could be “protein-independent”. However, time has shown that there is no ideal affinity pair that can be applied to all proteins—the choice of the right tag-ligand (or combinations thereof) is often dependent on the protein's properties and final application as well as the production host.
Acknowledgements
The authors thank the financial support from Fundação para a Ciência e a Tecnologia through grant no. PEst-C/EQB/LA0006/2011, contracts PTDC/BIO/65383/2006,/EBB-BIO/102163/2008, PTDC/EBB-BIO/098961/2008, PTDC/EBB-BIO/118317/2010 and PhD fellowship SFRH/BD/48804/2008 for A.S.P.
References
- Ahmed S., Daulat A., Angers S. Tandem affinity purification and identification of heterotrimeric g protein-associated proteins. In: Luttrell L.M., Ferguson S.S.G., editors. Signal Transduction Protocols. Humana Press; 2011. pp. 357–370. [DOI] [PubMed] [Google Scholar]
- Akerström B., Nielsen E., Björck L. Definition of IgG- and albumin-binding regions of streptococcal protein G. J Biol Chem. 1987;262:13388–13391. [PubMed] [Google Scholar]
- Allen S., Gan Q., Matthews R., Johnson P. Comparison of optimised isotherm models for basic dye adsorption by kudzu. Bioresour Technol. 2003;88:143–152. doi: 10.1016/s0960-8524(02)00281-x. [DOI] [PubMed] [Google Scholar]
- Arnau J., Lauritzen C., Petersen G., Pedersen J. Current strategies for the use of affinity tags and tag removal for the purification of recombinant proteins. Protein Expr Purif. 2006;48:1–13. doi: 10.1016/j.pep.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Banki M., Feng L., Wood D. Simple bioseparations using self-cleaving elastin-like polypeptide tags. Nat Methods. 2005;2:659–662. doi: 10.1038/nmeth787. [DOI] [PubMed] [Google Scholar]
- Bayer E., Wilchek M. Application of avidin–biotin technology to affinity-based separations. J Chromatogr A. 1990;510:3–11. doi: 10.1016/s0021-9673(01)93733-1. [DOI] [PubMed] [Google Scholar]
- Becker K., Alstine J., Büllow L. Multipurpose peptide tags for protein isolation. J Chromatogr A. 2008;1202:40–46. doi: 10.1016/j.chroma.2008.06.045. [DOI] [PubMed] [Google Scholar]
- Bhatla S., Kaushik V., Yadav M. Use of oil bodies and oleosins in recombinant protein production and other biotechnological applications. Biotechnol Adv. 2010;28:293–300. doi: 10.1016/j.biotechadv.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Björck L., Kronvall G. Purification and some properties of streptococcal protein G, a novel IgG-binding reagent. J Immunol. 1984;133:969–974. [PubMed] [Google Scholar]
- Block H., Maertens B., Spriestersbach A., Brinker N., Kubicek J., Fabis R. Immobilized-metal affinity chromatography (IMAC): a review. In: RR B., MP D., editors. Methods in Enzymology: Guide to Protein Purification. 2nd ed. 2009. pp. 439–473. [DOI] [PubMed] [Google Scholar]
- Borlido L., Azevedo A., Sousa A., Oliveira P., Roque A., Aires-Barros M. Fishing human monoclonal antibodies from a CHO cell supernatant with boronic acid magnetic particles. J Chromatogr B. 2012;903:163–170. doi: 10.1016/j.jchromb.2012.07.014. [DOI] [PubMed] [Google Scholar]
- Bouriotis V., Galpin I., Dean P. Applications of immobilised phenylboronic acids as supports for group-specific ligands in the affinity chromatography of enzymes. J Chromatogr A. 1981;210:267–278. [Google Scholar]
- Boyer T. Special article the glutathione S-transferases: an update. Hepatology. 1989;9:486–496. doi: 10.1002/hep.1840090324. [DOI] [PubMed] [Google Scholar]
- Brochier V.B., Schapman A., Santambien P., Britsch L. Fast purification process optimization using mixed-mode chromatography sorbents in pre-packed mini-columns. J Chromatogr A. 2008;1177:226–233. doi: 10.1016/j.chroma.2007.08.086. [DOI] [PubMed] [Google Scholar]
- Burgess R., Thompson N. Advances in gentle immunoaffinity chromatography. Curr Opin Biotechnol. 2002;13:304–308. doi: 10.1016/s0958-1669(02)00340-3. [DOI] [PubMed] [Google Scholar]
- Burton S., Haggarty N., Harding D. One step purification of chymosin by mixed mode chromatography. Biotechnol Bioeng. 1997;56:45–55. doi: 10.1002/(SICI)1097-0290(19971005)56:1<45::AID-BIT5>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Carey M., Peterson C., Smale S. Cold Spring Harbor Protocols; 2010. Purification of epitope-tagged transcription factor IID. (2010:pdb.prot5450) [DOI] [PubMed] [Google Scholar]
- Carta G., Jungbauer A. Protein Chromatography. Wiley-VCH Verlag GmbH & Co. KGaA; 2010. Downstream processing of biotechnology products; pp. 1–55. [Google Scholar]
- Cazalla D., Sanford J., Cáceres J. A rapid and efficient protocol to purify biologically active recombinant proteins from mammalian cells. Protein Expr Purif. 2005;42:54–58. doi: 10.1016/j.pep.2005.03.035. [DOI] [PubMed] [Google Scholar]
- Chaga G., Bochkariov D., Jokhadze G., Hopp J., Nelson P. Natural poly-histidine affinity tag for purification of recombinant proteins on cobalt(II)-carboxymethylaspartate crosslinked agarose. J Chromatogr A. 1999;864:247–256. doi: 10.1016/s0021-9673(99)01008-0. [DOI] [PubMed] [Google Scholar]
- Chang J. Thrombin specificity. Eur J Biochem. 1985;151:217–224. doi: 10.1111/j.1432-1033.1985.tb09091.x. [DOI] [PubMed] [Google Scholar]
- Charlton A., Zachariou M. Affinity Chromatography:Methods and Protocols. 2007. Immobilized metal ion affinity chromatography of native proteins; pp. 25–36. [DOI] [PubMed] [Google Scholar]
- Cheung R., Wong J., Ng T. Immobilized metal ion affinity chromatography: a review on its applications. Appl Microbiol Biotechnol. 2012;96:1411–1420. doi: 10.1007/s00253-012-4507-0. [DOI] [PubMed] [Google Scholar]
- Chiang C., Chen H., Chao Y., Tzen J. Efficient system of artificial oil bodies for functional expression and purification of recombinant nattokinase in Escherichia coli. J Agric Food Chem. 2005;53:4799–4804. doi: 10.1021/jf050264a. [DOI] [PubMed] [Google Scholar]
- Chiang C., Chen H., Chao Y., Tzen J. One-step purification of insoluble hydantoinase overproduced in Escherichia coli. Protein Expr Purif. 2007;52:14–18. doi: 10.1016/j.pep.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Choi S., Song H., Moon J., Seong B. Recombinant enterokinase light chain with affinity tag: expression from Saccharomyces cerevisiae and its utilities in fusion protein technology. Biotechnol Bioeng. 2001;75:718–724. doi: 10.1002/bit.10082. [DOI] [PubMed] [Google Scholar]
- Chow D., Dreher M., Trabbic-Carlson K., Chilkoti A. Ultra-high expression of a thermally responsive recombinant fusion protein in E. coli. Biotechnol Prog. 2006;22:638–646. doi: 10.1021/bp0503742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chumanov R.S., Kuhn P.A., Xu W., Burgess R.R. Expression and purification of full-length mouse CARM1 from transiently transfected HEK293T cells using HaloTag technology. Protein Expr Purif. 2011;76:145–153. doi: 10.1016/j.pep.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clonis Y., Labrou N., Kotsira V., Mazitsos C., Melissis S., Gogolas G. Biomimetic dyes as affinity chromatography tools in enzyme purification. J Chromatogr A. 2000;891:33–44. doi: 10.1016/s0021-9673(00)00577-x. [DOI] [PubMed] [Google Scholar]
- Collén A., Ward M., Tjerneld F., Stålbrand H. Genetic engineering of the Trichoderma reesei endoglucanase I (Cel7B) for enhanced partitioning in aqueous two-phase systems containing thermoseparating ethylene oxide–propylene oxide copolymers. J Biotechnol. 2001;87:179–191. doi: 10.1016/s0168-1656(01)00241-3. [DOI] [PubMed] [Google Scholar]
- Conley A., Joensuu J., Richman A., Menassa R. Protein body-inducing fusions for high-level production and purification of recombinant proteins in plants. Plant Biotechnol J. 2011;9:419–433. doi: 10.1111/j.1467-7652.2011.00596.x. [DOI] [PubMed] [Google Scholar]
- Cummins P., O'Connor B. Hydrophobic interaction chromatography. In: Walls D., Loughran S.T., editors. Protein Chromatography. Humana Press; 2011. pp. 431–437. [Google Scholar]
- Dalboge H., Dahl H., Pedersen J., Hansen J., Christensen T. A novel enzymatic method for purification if authentic hGH from an Escherichia coli produced hGH-precurser. Nat Biotechnol. 1987;5:161–164. [Google Scholar]
- Dashivets T., Wood N., Hergersberg C., Buchner J., Haslbeck M. Rapid matrix-assisted refolding of histidine-tagged proteins. ChemBioChem. 2009;10:869–876. doi: 10.1002/cbic.200800697. [DOI] [PubMed] [Google Scholar]
- Davison E., Pennington K., Hung C., Peng J., Rafiq R., Ostareck-Lederer A. Proteomic analysis of increased Parkin expression and its interactants provides evidence for a role in modulation of mitochondrial function. Proteomics. 2009;9:4284–4297. doi: 10.1002/pmic.200900126. [DOI] [PubMed] [Google Scholar]
- Demain A., Vaishnav P. Production of recombinant proteins by microbes and higher organisms. Biotechnol Adv. 2009;27:297–306. doi: 10.1016/j.biotechadv.2009.01.008. [DOI] [PubMed] [Google Scholar]
- Dougherty W., Carrington J., Cary S., Parks T. Biochemical and mutational analysis of a plant virus polyprotein cleavage site. EMBO J. 1988;7:1281–1287. doi: 10.1002/j.1460-2075.1988.tb02942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duellman S., Thompson N., Burgess R. An epitope tag derived from human transcription factor IIB that reacts with a polyol-responsive monoclonal antibody. Protein Expr Purif. 2004;35:147–155. doi: 10.1016/j.pep.2003.12.011. [DOI] [PubMed] [Google Scholar]
- Duhamel R., Schur P., Brendel K., Meezan E. pH gradient elution of human IgG1, IgG2 and IgG4 from protein A-Sepharose. J Immunol Methods. 1979;31:211–217. doi: 10.1016/0022-1759(79)90133-9. [DOI] [PubMed] [Google Scholar]
- Edwards A., Darst S., Feaver W., Thompson N., Burgess R., Kornberg R. Purification and lipid-layer crystallization of yeast RNA polymerase II. Proc Natl Acad Sci. 1990;87:2122–2126. doi: 10.1073/pnas.87.6.2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egorov M., Tigerström A., Pestov N., Korneenko T., Kostina M., Shakhparonov M. Purification of a recombinant membrane protein tagged with a calmodulin-binding domain: properties of chimeras of the Escherichia coli nicotinamide nucleotide transhydrogenase and the C-terminus of human plasma membrane Ca2 +-ATPase. Protein Expr Purif. 2004;36:31–39. doi: 10.1016/j.pep.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Einhauer A., Jungbauer A. The FLAG™ peptide, a versatile fusion tag for the purification of recombinant proteins. J Biochem Biophys Methods. 2001;49:455–465. doi: 10.1016/s0165-022x(01)00213-5. [DOI] [PubMed] [Google Scholar]
- Esipov R., Stepanenko V., Chupova L., Boyarskikh U., Filipenko M., Miroshnikov A. Production of recombinant human epidermal growth factor using Ssp dnaB mini-intein system. Protein Expr Purif. 2008;61:1–6. doi: 10.1016/j.pep.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Evan G., Lewis G., Ramsay G., Bishop J. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fexby S., Bülow L. Hydrophobic peptide tags as tools in bioseparation. Trends Biotechnol. 2004;22:511–516. doi: 10.1016/j.tibtech.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Fexby S., Nilsson A., Hambraeus G., Tjerneld F., Bülow L. Partitioning and characterization of tyrosine-tagged green fluorescent proteins in aqueous two-phase systems. Biotechnol Prog. 2004;20:793–798. doi: 10.1021/bp034177j. [DOI] [PubMed] [Google Scholar]
- Field J., Nikawa J., Broek D., MacDonald B., Rodgers L., Wilson I. Purification of a RAS-responsive adenylyl cyclase complex from Saccharomyces cerevisiae by use of an epitope addition method. Mol Cell Biol. 1988;8:2159–2165. doi: 10.1128/mcb.8.5.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floss D., Schallau K., Rose-John S., Conrad U., Scheller J. Elastin-like polypeptides revolutionize recombinant protein expression and their biomedical application. Trends Biotechnol. 2010;28:37–45. doi: 10.1016/j.tibtech.2009.10.004. [DOI] [PubMed] [Google Scholar]
- Fong B., Wu W., Wood D. Optimization of ELP-intein mediated protein purification by salt substitution. Protein Expr Purif. 2009;66:198–202. doi: 10.1016/j.pep.2009.03.009. [DOI] [PubMed] [Google Scholar]
- Fong B., Wu W., Wood D. The potential role of self-cleaving purification tags in commercial-scale processes. Trends Biotechnol. 2010;28:272–279. doi: 10.1016/j.tibtech.2010.02.003. [DOI] [PubMed] [Google Scholar]
- Foreman P., Davis R. Cloning vectors for the synthesis of epitope-tagged, truncated and chimeric proteins in Saccharomyces cerevisiae. Gene. 1994;144:63–68. doi: 10.1016/0378-1119(94)90204-6. [DOI] [PubMed] [Google Scholar]
- Frangioni J., Neel B. Solubilization and purification of enzymatically active glutathione S-transferase (pGEX) fusion proteins. Anal Biochem. 1993;210:179–187. doi: 10.1006/abio.1993.1170. [DOI] [PubMed] [Google Scholar]
- Fuchs S., Raines R. Polyarginine as a multifunctional fusion tag. Protein Sci. 2005;14:1538–1544. doi: 10.1110/ps.051393805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Füglistaller P. Comparison of immunoglobulin binding capacities and ligand leakage using eight different protein A affinity chromatography matrices. J Immunol Methods. 1989;124:171–177. doi: 10.1016/0022-1759(89)90350-5. [DOI] [PubMed] [Google Scholar]
- Gaberc-Porekar V., Menart V. Perspectives of immobilized-metal affinity chromatography. J Biochem Biophys Methods. 2001;49:335–360. doi: 10.1016/s0165-022x(01)00207-x. [DOI] [PubMed] [Google Scholar]
- Gaj T., Meyer S., Ghosh I. The AviD-tag, a NeutrAvidin/avidin specific peptide affinity tag for the immobilization and purification of recombinant proteins. Protein Expr Purif. 2007;56:54–61. doi: 10.1016/j.pep.2007.06.010. [DOI] [PubMed] [Google Scholar]
- García-Fruitós E., EVázquez Díez-Gil C., Corchero J., Seras-Franzoso J., Ratera I., Veciana J. Bacterial inclusion bodies: making gold from waste. Trends Biotechnol. 2012;30:65–70. doi: 10.1016/j.tibtech.2011.09.003. [DOI] [PubMed] [Google Scholar]
- Geisbrecht B.V., Bouyain S., Pop M. An optimized system for expression and purification of secreted bacterial proteins. Protein Expr Purif. 2006;46:23–32. doi: 10.1016/j.pep.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Gloeckner C., Boldt K., Schumacher A., Roepman R., Ueffing M. A novel tandem affinity purification strategy for the efficient isolation and characterisation of native protein complexes. Proteomics. 2007;7:4228–4234. doi: 10.1002/pmic.200700038. [DOI] [PubMed] [Google Scholar]
- Gloeckner C., Boldt K., Schumacher A., Ueffing M. Tandem affinity purification of protein complexes from mammalian cells by the Strep/FLAG (SF)-TAP Tag. In: Reinders J., Sickmann A., editors. Proteomics. Humana Press; 2009. pp. 359–372. [DOI] [PubMed] [Google Scholar]
- Gräslund T., Lundin G., Uhlén M., Nygren P., Hober S. Charge engineering of a protein domain to allow efficient ion-exchange recovery. Protein Eng. 2000;13:703–709. doi: 10.1093/protein/13.10.703. [DOI] [PubMed] [Google Scholar]
- Gräslund T., Nilsson J., Lindberg A., Uhlén M., Nygren P. Production of a thermostable DNA polymerase by site-specific cleavage of a heat-eluted affinity fusion protein. Protein Expr Purif. 1997;9:125–132. doi: 10.1006/prep.1996.0674. [DOI] [PubMed] [Google Scholar]
- Guan D., Ramirez M., Chen Z. Split intein mediated ultra-rapid purification of tagless protein (SIRP) Biotechnol Bioeng. 2013;110:2471–2481. doi: 10.1002/bit.24913. [DOI] [PubMed] [Google Scholar]
- Guana C. d, Lib P., Riggsa P., Inouyeb H. Vectors that facilitate the expression and purification of foreign peptides in Escherichia coli by fusion to maltose-binding protein. Gene. 1988;67:21–30. doi: 10.1016/0378-1119(88)90004-2. [DOI] [PubMed] [Google Scholar]
- Günzl A., Schimanski B. Tandem Affinity Purification of Proteins. Current Protocols in Protein Science. 2009;55:19.19.1–19.19.16. doi: 10.1002/0471140864.ps1919s55. [DOI] [PubMed] [Google Scholar]
- Gusarov I., Nudler E. Control of intrinsic transcription termination by N and NusA: the basic mechanisms. Cell. 2001;107:437–449. doi: 10.1016/s0092-8674(01)00582-7. [DOI] [PubMed] [Google Scholar]
- Haigh J., Hussain A., Mimmack M., Lowe C. Affinity ligands for immunoglobulins based on the multicomponent Ugi reaction. J Chromatogr B. 2009;877:1440–1452. doi: 10.1016/j.jchromb.2009.03.010. [DOI] [PubMed] [Google Scholar]
- Harrison R. Expression of soluble heterologous proteins via fusion with NusA protein. Innov. 2000;11:4–7. [Google Scholar]
- Hassouneh W., Christensen T., Chilkoti A. Current Protocols in Protein Science. John Wiley & Sons, Inc.; 2001. Elastin-Like polypeptides as a purification tag for recombinant proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassouneh W., MacEwan S., Chilkoti A. Chapter nine—fusions of elastin-like polypeptides to pharmaceutical proteins. In: Wittrup K.D., Gregory L.V., editors. Methods in Enzymology. Academic Press; 2012. pp. 215–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearn M., Acosta D. Applications of novel affinity cassette methods: use of peptide fusion handles for the purification of recombinant proteins. J Mol Recognit. 2001;14:323–369. doi: 10.1002/jmr.555. [DOI] [PubMed] [Google Scholar]
- Hedhammar M., Alm T., Gräslund T., Hober S. Single-step recovery and solid-phase refolding of inclusion body proteins using a polycationic purification tag. Biotechnol J. 2006;1:187–196. doi: 10.1002/biot.200500023. [DOI] [PubMed] [Google Scholar]
- Hedhammar M., Gräslund T., Hober S. Protein engineering strategies for selective protein purification. Chem Eng Technol. 2005;28:1315–1325. [Google Scholar]
- Hedhammar M., Gräslund T., Uhlén M., Hober S. Negatively charged purification tags for selective anion-exchange recovery. Protein Eng Des Sel. 2004;17:779–786. doi: 10.1093/protein/gzh092. [DOI] [PubMed] [Google Scholar]
- Hernan R., Heuermann K., Brizzard B. Multiple epitope tagging of expressed proteins for enhanced detection. Biotechniques. 2000;28:789–793. doi: 10.2144/00284pf01. [DOI] [PubMed] [Google Scholar]
- Higgins C., Vermeer L., Doorn J., Roman D. Expression and purification of recombinant human tyrosine hydroxylase as a fusion protein in Escherichia coli. Protein Expr Purif. 2012;84:219–223. doi: 10.1016/j.pep.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochuli E., Bannwarth W., Dobeli H., Gentz R., Stuber D. Genetic approach to facilitate purification of recombinant proteins with a novel metal chelate adsorbent. Nat Biotechnol. 1988;6:1321–1325. [Google Scholar]
- Hopp T., Prickett K., Price V., Libby R., March C., Cerretti D. A short polypeptide marker sequence useful for recombinant protein identification and purification. Nat Biotechnol. 1988;6:1204–1210. [Google Scholar]
- Hu S., Wang A., Wang T. Encyclopedia of life sciences. 2001. Expression tags for protein production. [Google Scholar]
- Huyck R., Keightley A., Laity J. Expression and purification of full length mouse metal response element binding transcription factor-1 using Pichia pastoris. Protein Expr Purif. 2012;85:86–93. doi: 10.1016/j.pep.2012.06.018. [DOI] [PubMed] [Google Scholar]
- Janknecht R., Martynoff G.D., Lou J., Hipskind R., Nordheim A., Stunnenberg H. Rapid and efficient purification of native histidine-tagged protein expressed by recombinant vaccinia virus. Proc Natl Acad Sci. 1991;88:8972–8976. doi: 10.1073/pnas.88.20.8972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvik J., Telmer C. Epitope tagging. Annu Rev Genet. 1998;32:601–618. doi: 10.1146/annurev.genet.32.1.601. [DOI] [PubMed] [Google Scholar]
- Jenny R., Mann K., Lundblad R. A critical review of the methods for cleavage of fusion proteins with thrombin and factor Xa. Protein Expr Purif. 2003;31:1–11. doi: 10.1016/s1046-5928(03)00168-2. [DOI] [PubMed] [Google Scholar]
- Joensuu J., Conley A., Lienemann M., Brandle J., Linder M., Menassa R. Hydrophobin fusions for high-level transient protein expression and purification in Nicotiana benthamiana. Plant Physiol. 2010;152:622–633. doi: 10.1104/pp.109.149021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko A., Umeyama T., Hanaoka N., Monk B., Uehara Y., Niimi M. Tandem affinity purification of the Candida albicans septin protein complex. Yeast. 2004;21:1025–1033. doi: 10.1002/yea.1147. [DOI] [PubMed] [Google Scholar]
- Kang H., Kim J., Chang W., Kim E., Koo Y. Heterologous expression and optimized one-step separation of levansucrase via elastin-like polypeptides tagging system. J Microbiol Biotechnol. 2007;17:1751–1757. [PubMed] [Google Scholar]
- Kaplan W., Erhardt J., Sluis-Cremer N., Dirr H., Hüsler P., Klump H. Conformational stability of pGEX-expressed Schistosoma japonicum glutathione S-transferase: a detoxification enzyme and fusion-protein affinity tag. Protein Sci. 1997;6:399–406. doi: 10.1002/pro.5560060216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapust R., Waugh D. Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 1999;8:1668–1674. doi: 10.1110/ps.8.8.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpeisky M., Senchenko V., Dianova M., Kanevsky V. Formation and properties of S-protein complex with S-peptide-containing fusion protein. FEBS Lett. 1994;339:209–212. doi: 10.1016/0014-5793(94)80417-6. [DOI] [PubMed] [Google Scholar]
- Kato K., Sato H., Iwata H. Immobilization of histidine-tagged recombinant proteins onto micropatterned surfaces for cell-based functional assays. Langmuir. 2005;21:7071–7075. doi: 10.1021/la050893e. [DOI] [PubMed] [Google Scholar]
- Katti S., LeMaster D., Eklund H. Crystal structure of thioredoxin from Escherichia coli at 1.68 Å resolution. J Mol Biol. 1990;212:167–184. doi: 10.1016/0022-2836(90)90313-B. [DOI] [PubMed] [Google Scholar]
- Keefe A., Wilson D., Seelig B., Szostak J. One-step purification of recombinant proteins using a nanomolar-affinity streptavidin-binding peptide, the SBP-tag. Protein Expr Purif. 2001;23:440–446. doi: 10.1006/prep.2001.1515. [DOI] [PubMed] [Google Scholar]
- Kellermann O., Ferenci T. Maltose-binding protein from Escherichia coli. In: Willis A.W., editor. Methods in Enzymology – Carbohydrate Metabolism – Part E. Academic Press; 1982. pp. 459–463. [75] [DOI] [PubMed] [Google Scholar]
- Khan I., Twyman R., Arcalis E., Stoger E. Using storage organelles for the accumulation and encapsulation of recombinant proteins. Biotechnol J. 2012;7:1099–1108. doi: 10.1002/biot.201100089. [DOI] [PubMed] [Google Scholar]
- Kim J., Raines R. Ribonuclease S-peptide as a carrier in fusion proteins. Protein Sci. 1993;2:348–356. doi: 10.1002/pro.5560020307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipriyanov S., Kupriyanova O., Little M., Moldenhauer G. Rapid detection of recombinant antibody fragments directed against cell-surface antigens by flow cytometry. J Immunol Methods. 1996;196:51–62. doi: 10.1016/0022-1759(96)00115-9. [DOI] [PubMed] [Google Scholar]
- Korndörfer I., Skerra A. Improved affinity of engineered streptavidin for the Strep-tag II peptide is due to a fixed open conformation of the lid-like loop at the binding site. Protein Sci. 2002;11:883–893. doi: 10.1110/ps.4150102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurek D., Lopatin S., Varlamov V. Prospects of application of the chitin-binding domains to isolation and purification of recombinant proteins by affinity chromatography. Appl Biochem Microbiol. 2009;45:1–8. [PubMed] [Google Scholar]
- Kweon D., Lee D., Han N., Seo J. Solid-phase refolding of cyclodextrin glycosyltransferase adsorbed on cation-exchange resin. Biotechnol Prog. 2004;20:277–283. doi: 10.1021/bp0341895. [DOI] [PubMed] [Google Scholar]
- Lamla T., Erdmann V. Searching sequence space for high-affinity binding peptides using ribosome display. J Mol Biol. 2003;329:381–388. doi: 10.1016/s0022-2836(03)00432-7. [DOI] [PubMed] [Google Scholar]
- Lamla T., Erdmann V. The nano-tag, a streptavidin-binding peptide for the purification and detection of recombinant proteins. Protein Expr Purif. 2004;33:39–47. doi: 10.1016/j.pep.2003.08.014. [DOI] [PubMed] [Google Scholar]
- Lange C., Rudolph R. Protein Folding Handbook. Wiley-VCH Verlag GmbH; 2008. Production of recombinant proteins for therapy, diagnostics, and industrial research by in vitro folding; pp. 1245–1280. [Google Scholar]
- LaVallie E., DiBlasio E., Kovacic S., Grant K., Schendel P., McCoy J. A Thioredoxin gene fusion expression system that circumvents inclusion body formation in the E. coli cytoplasm. Nat Biotechnol. 1993;11:187–193. doi: 10.1038/nbt0293-187. [DOI] [PubMed] [Google Scholar]
- LaVallie E., Lu Z., Diblasio-Smith E., Collins-Racie L., McCoy J. Thioredoxin as a fusion partner for production of soluble recombinant proteins in Escherichia coli. In: Thorner J., Emr S.D., Abelson J.N., editors. Methods in Enzymology—Applications of Chimeric Genes and Hybrid Proteins: Gene expression and protein purification. Academic Press; 2000. pp. 322–340. [21] [DOI] [PubMed] [Google Scholar]
- Lehmann R., Meyer J., Schuemann M., Krause E., Freund C. A novel S3S-TAP-tag for the isolation of T-cell interaction partners of adhesion and degranulation promoting adaptor protein. Proteomics. 2009;9:5288–5295. doi: 10.1002/pmic.200900294. [DOI] [PubMed] [Google Scholar]
- Li M., He S. Purification and characterization of recombinant human interleukin-29 expressed in Escherichia coli. J Biotechnol. 2006;122:334–340. doi: 10.1016/j.jbiotec.2005.11.019. [DOI] [PubMed] [Google Scholar]
- Li S., Hochstrasser M. A new protease required for cell-cycle progression in yeast. Nature. 1999;398:246–251. doi: 10.1038/18457. [DOI] [PubMed] [Google Scholar]
- Li W., Serpell L.C., Carter W.J., Rubinsztein D.C., Huntington J.A. Expression and characterization of full-length human huntingtin, an elongated HEAT repeat protein. J Biol Chem. 2006;281:15916–15922. doi: 10.1074/jbc.M511007200. [DOI] [PubMed] [Google Scholar]
- Li Y. Commonly used tag combinations for tandem affinity purification. Biotechnol Appl Biochem. 2010;55:73–83. doi: 10.1042/BA20090273. [DOI] [PubMed] [Google Scholar]
- Li Y. Self-cleaving fusion tags for recombinant protein production. Biotechnol Lett. 2011;33:869–881. doi: 10.1007/s10529-011-0533-8. [DOI] [PubMed] [Google Scholar]
- Lienqueo M., Salazar O., Calado C., Fonseca L., Cabral J. Influence of tryptophan tags on the purification of cutinase, secreted by a recombinant Saccharomyces cerevisiae, using cationic expanded bed adsorption and hydrophobic interaction chromatography. Biotechnol Lett. 2008;30:1353–1358. doi: 10.1007/s10529-008-9696-3. [DOI] [PubMed] [Google Scholar]
- Lim D., Trabbic-Carlson K., MacKay J., Chilkoti A. Improved non-chromatographic purification of a recombinant protein by cationic elastin-like polypeptides. Biomacromolecules. 2007;8:1417–1424. doi: 10.1021/bm060849t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S., Guidotti G. Chapter 35 purification of membrane proteins. In: Richard R.B., Murray P.D., editors. Guide to Protein Purification. 2nd ed. Academic Press; 2009. pp. 619–629. [Google Scholar]
- Lin S., Lin I., Chou W., Hsieh C., Liu S., Huang R. CBM21 starch-binding domain: a new purification tag for recombinant protein engineering. Protein Expr Purif. 2009;65:261–266. doi: 10.1016/j.pep.2009.01.008. [DOI] [PubMed] [Google Scholar]
- Linder M. Hydrophobins: proteins that self assemble at interfaces. Curr Opin Colloid Interface Sci. 2009;14:356–363. [Google Scholar]
- Linder M., Szilvay G., Nakari-Setälä T., Penttilä M. Hydrophobins: the protein-amphiphiles of filamentous fungi. FEMS Microbiol Lett. 2005;29:877–896. doi: 10.1016/j.femsre.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Lindmark R., Thorén-Tolling K., Sjöquist J. Binding of immunoglobulins to protein A and immunoglobulin levels in mammalian sera. J Immunol Methods. 1983;62:1–13. doi: 10.1016/0022-1759(83)90104-7. [DOI] [PubMed] [Google Scholar]
- Liu F., Tsai S., Madan B., Chen W. Engineering a high-affinity scaffold for non-chromatographic protein purification via intein-mediated cleavage. Biotechnol Bioeng. 2012;109:2829–2835. doi: 10.1002/bit.24545. [DOI] [PubMed] [Google Scholar]
- Liu J., Duan C., Zhao X., Tzen J., Cheng K., Pai C. Cloning of a rumen fungal xylanase gene and purification of the recombinant enzyme via artificial oil bodies. Appl Microbiol Biotechnol. 2008;79:225–233. doi: 10.1007/s00253-008-1418-1. [DOI] [PubMed] [Google Scholar]
- Liu K., Hanna M. NusA contacts nascent RNA in Escherichia coli transcription complexes. J Mol Biol. 1995;247:547–558. doi: 10.1006/jmbi.1994.0161. [DOI] [PubMed] [Google Scholar]
- Liu X. Boronic acids as ligands for affinity chromatography. Chin J Chromatogr. 2006;24:73–80. [PubMed] [Google Scholar]
- Liu X., Scouten W. New ligands for boronate affinity chromatography. J Chromatogr A. 1994;687:61–69. [Google Scholar]
- Liu X., Scouten W. Boronate affinity chromatography. In: Bailon P., Ehrlich G.K., Fung W.J., Berthold B., editors. Affinity Chromatography. Humana Press; 2000. pp. 119–128. [Google Scholar]
- Llompart B., Llop-Tous I., Marzabal P., Torrent M., Pallissé R., Bastida M. Protein production from recombinant protein bodies. Process Biochem. 2010;45:1816–1820. [Google Scholar]
- Los G., Wood K. The HaloTag™. In: Taylor D.L., Haskins J., Giuliano K., editors. High Content Screening. Humana Press; 2006. pp. 195–208. [Google Scholar]
- Los G.V., Encell L.P., McDougall M.G., Hartzell D.D., Karassina N., Zimprich C. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol. 2008;3:373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- Lowe C. Combinatorial approaches to affinity chromatography. Curr Opin Chem Biol. 2001;5:248–256. doi: 10.1016/s1367-5931(00)00199-x. [DOI] [PubMed] [Google Scholar]
- Lowe C., Lowe A., Gupta G. New developments in affinity chromatography with potential application in the production of biopharmaceuticals. J Biochem Biophys Methods. 2001;49:561–574. doi: 10.1016/s0165-022x(01)00220-2. [DOI] [PubMed] [Google Scholar]
- Luojing C., Ford C., Nikolov Z. Adsorption to starch of a β-galactosidase fusion protein containing the starch-binding region of Aspergillus glucoamylase. Gene. 1991;99:121–126. doi: 10.1016/0378-1119(91)90043-b. [DOI] [PubMed] [Google Scholar]
- Ma H., McLean J., Chao L., Mana-Capelli S., Paramasivam M., Hagstrom K. A highly efficient multifunctional tandem affinity purification approach applicable to diverse organisms. Mol Cell Proteomics. 2012;11:501–511. doi: 10.1074/mcp.O111.016246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malakhov M., Mattern M., Malakhova O., Drinker M., Weeks S., Butt T. SUMO fusions and SUMO-specific protease for efficient expression and purification of proteins. J Struct Funct Genomics. 2004;5:75–86. doi: 10.1023/B:JSFG.0000029237.70316.52. [DOI] [PubMed] [Google Scholar]
- Malhotra A. Chapter 16 tagging for protein expression. In: Burgess R.R., Deutscher M.P., editors. Methods in Enzymology. Academic Press; 2009. pp. 239–258. [Google Scholar]
- Mant C., Hodges R. Mixed-mode hydrophilic interaction/cation-exchange chromatography (HILIC/CEX) of peptides and proteins. J Sep Sci. 2008;31:2754–2773. doi: 10.1002/jssc.200800243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marblestone J., Edavettal S., Lim Y., Lim P., Zuo X., Butt T. Comparison of SUMO fusion technology with traditional gene fusion systems: enhanced expression and solubility with SUMO. Protein Sci. 2006;15:182–189. doi: 10.1110/ps.051812706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean M., Chen R., Yu D., Mah K., Teat J., Wang H. Purification of the therapeutic antibody trastuzumab from genetically modified plants using safflower Protein A-oleosin oilbody technology. Transgenic Res. 2012;21:1291–1301. doi: 10.1007/s11248-012-9603-5. [DOI] [PubMed] [Google Scholar]
- Melkko S., Neri D. Calmodulin as an affinity purification tag. In: Vaillancourt P., editor. Methods in Molecular Biology—E coli gene expression protocols. Academic Press; 2003. pp. 69–77. [DOI] [PubMed] [Google Scholar]
- Meyer D., Chilkoti A. Purification of recombinant proteins by fusion with thermally-responsive polypeptides. Nat Biotechnol. 1999;17:1112–1115. doi: 10.1038/15100. [DOI] [PubMed] [Google Scholar]
- Miller D., Olson J., Pflugrath J., Quiocho F. Rates of ligand binding to periplasmic proteins involved in bacterial transport and chemotaxis. J Biol Chem. 1983;258:13665–13672. [PubMed] [Google Scholar]
- Milne J. Scale-up of protein purification: downstream processing issues. In: Walls D., Loughran S.T., editors. Protein Chromatography. Humana Press; 2011. pp. 73–85. [Google Scholar]
- Mooney J., Fredericks D., Hearn M. Use of phage display methods to identify heptapeptide sequences for use as affinity purification ‘tags’ with novel chelating ligands in immobilized metal ion affinity chromatography. J Chromatogr A. 2011;1218:92–99. doi: 10.1016/j.chroma.2010.10.113. [DOI] [PubMed] [Google Scholar]
- Müller K., Arndt K., Bauer K., Plückthun A. Tandem immobilized metal-ion affinity chromatography/immunoaffinity purification of his-tagged proteins —evaluation of two anti-his-tag monoclonal antibodies. Anal Biochem. 1998;259:54–61. doi: 10.1006/abio.1998.2606. [DOI] [PubMed] [Google Scholar]
- Mustalahti E., Saloheimo M., Joensuu J. Intracellular protein production in Trichoderma reesei (Hypocrea jecorina) with hydrophobin fusion technology. N Biotechnol. 2013;30:262–268. doi: 10.1016/j.nbt.2011.09.006. [DOI] [PubMed] [Google Scholar]
- Nikaido H. Maltose transport system of Escherichia coli: an ABC-type transporter. FEBS Lett. 1994;346:55–58. doi: 10.1016/0014-5793(94)00315-7. [DOI] [PubMed] [Google Scholar]
- Nilson B., Lögdberg L., Kastern W., Björck L., Åkerström B. Purification of antibodies using protein L-binding framework structures in the light chain variable domain. J Immunol Methods. 1993;164:33–40. doi: 10.1016/0022-1759(93)90273-a. [DOI] [PubMed] [Google Scholar]
- Nilsson B., Abrahmsén L. Fusions to staphylococcal protein A. Methods Enzymol. 1990;185:144–161. doi: 10.1016/0076-6879(90)85015-g. [DOI] [PubMed] [Google Scholar]
- Nilsson B., Abrahmsén L., Uhlén M. Immobilization and purification of enzymes with staphylococcal protein A gene fusion vectors. EMBO J. 1985;4:1075–1080. doi: 10.1002/j.1460-2075.1985.tb03741.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson B., Moks T., Jansson B., Abrahmsén L., Elmblad A., Holmgren E. A synthetic IgG-binding domain based on staphylococcal protein A. Protein Eng. 1987;1:107–113. doi: 10.1093/protein/1.2.107. [DOI] [PubMed] [Google Scholar]
- Nilsson J., Nilsson P., Williams Y., Pettersson L., Uhlén M., Nygren P. Competitive elution of protein A fusion proteins allows specific recovery under mild conditions. Eur J Biochem. 1994;224:103–108. doi: 10.1111/j.1432-1033.1994.tb20000.x. [DOI] [PubMed] [Google Scholar]
- Nilsson J., Stahl S., Lundeberg J., Uhlén M., Nygren P. Affinity fusion strategies for detection, purification, and immobilization of recombinant proteins. Protein Expr Purif. 1997;11:1–16. doi: 10.1006/prep.1997.0767. [DOI] [PubMed] [Google Scholar]
- Nygren P., Eliasson M., Abrahmsén L., Uhlén M., Palmcrantz E. Analysis and use of the serum albumin binding domains of streptococcal protein G. J Mol Recognit. 1988;1:69–74. doi: 10.1002/jmr.300010204. [DOI] [PubMed] [Google Scholar]
- Nykiforuk C., Boothe J., Murray E., Keon R., Goren H., Markley N. Transgenic expression and recovery of biologically active recombinant human insulin from Arabidopsis thaliana seeds. Plant Biotechnol J. 2006;4:77–85. doi: 10.1111/j.1467-7652.2005.00159.x. [DOI] [PubMed] [Google Scholar]
- Ohana R., Encell L., Zhao K., Simpson D., Slater M., Urh M. HaloTag7: a genetically engineered tag that enhances bacterial expression of soluble proteins and improves protein purification. Protein Expr Purif. 2009;68:110–120. doi: 10.1016/j.pep.2009.05.010. [DOI] [PubMed] [Google Scholar]
- Ohana R.F., Hurst R., Vidugiriene J., Slater M.R., Wood K.V., Urh M. HaloTag-based purification of functional human kinases from mammalian cells. Protein Expr Purif. 2011;76:154–164. doi: 10.1016/j.pep.2010.11.014. [DOI] [PubMed] [Google Scholar]
- Palomares L., Estrada-Moncada S., Ramírez O. Production of recombinant proteins. In: Balbás P., Lorence A., editors. Recombinant Gene Expression. Humana Press; 2004. pp. 15–51. [Google Scholar]
- Peng C., Chen J., Shyu D., Chen M., Tzen J. A system for purification of recombinant proteins in Escherichia coli via artificial oil bodies constituted with their oleosin-fused polypeptides. J Biotechnol. 2004;111:51–57. doi: 10.1016/j.jbiotec.2004.03.013. [DOI] [PubMed] [Google Scholar]
- Porath J. Metal ion—hydrophobic, thiophilic and II-electron governed interactions and their application to salt-promoted protein adsorption chromatography. Biotechnol Prog. 1987;3:14–21. [Google Scholar]
- Porath J. Immobilized metal ion affinity chromatography. Protein Expr Purif. 1992;3:263–281. doi: 10.1016/1046-5928(92)90001-d. [DOI] [PubMed] [Google Scholar]
- Porath J., Carlsson J.A.N., Olsson I., Belfrage G. Metal chelate affinity chromatography, a new approach to protein fractionation. Nature. 1975;258:598–599. doi: 10.1038/258598a0. [DOI] [PubMed] [Google Scholar]
- Porath J., Olin B. Immobilized metal affinity adsorption and immobilized metal affinity chromatography of biomaterials. Serum protein affinities for gel-immobilized iron and nickel ions. Biochemistry. 1983;22:1621–1630. doi: 10.1021/bi00276a015. [DOI] [PubMed] [Google Scholar]
- Prickett K., Amberg D., Hopp T. A calcium-dependent antibody for identification and purification of recombinant proteins. Biotechniques. 1989;7:580–589. [PubMed] [Google Scholar]
- Probasco M., Thompson N., Burgess R. Immunoaffinity purification and characterization of RNA polymerase from Shewanella oneidensis. Protein Expr Purif. 2007;55:23–30. doi: 10.1016/j.pep.2007.03.020. [DOI] [PubMed] [Google Scholar]
- Puig O., Caspary F., Rigaut G., Rutz B., Bouveret E., Bragado-Nilsson E. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001;24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- Qian J., Khoury G.E., Issa H., Al-Qaoud K., Shihab P., Lowe C. A synthetic protein G adsorbent based on the multi-component Ugi reaction for the purification of mammalian immunoglobulins. J Chromatogr B. 2012;898:15–23. doi: 10.1016/j.jchromb.2012.03.043. [DOI] [PubMed] [Google Scholar]
- Roberts N., Scott R., Tzen J. Recent biotechnological applications using oleosins open. Biotechnol J. 2008;2:13–21. [Google Scholar]
- Roque A., Lowe C. Advances and applications of the novo designed affinity ligands in proteomics. Biotechnol Adv. 2006;24:17–26. doi: 10.1016/j.biotechadv.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Roque A., Lowe C. Affinity chromatography: history, perspectives, limitations and prospects. In: Zachariou M., editor. Affiinity Chromatography: Methods and Protocols. Human Press; 2007. pp. 1–23. [PubMed] [Google Scholar]
- Roque A., Taipa M., Lowe C. An artificial protein L for the purification of immunoglobulins and Fab fragments by affinity chromatography. J Chromatogr A. 2005;1064:157–167. doi: 10.1016/j.chroma.2004.11.102. [DOI] [PubMed] [Google Scholar]
- Santana S., Pina A., Roque A. Immobilization of enterokinase on magnetic supports for the cleavage of fusion proteins. J Biotechnol. 2012;161:378–382. doi: 10.1016/j.jbiotec.2012.07.005. [DOI] [PubMed] [Google Scholar]
- Sassenfeld H., Brewer S. A polypeptide fusion designed for the purification of recombinant proteins. Nat Biotechnol. 1984;2:76–81. [Google Scholar]
- Schmidt T., Koepke J., Frank R., Skerra A. Molecular interaction between the strep-tag affinity peptide and its cognate target, streptavidin. J Mol Biol. 1996;255:753–766. doi: 10.1006/jmbi.1996.0061. [DOI] [PubMed] [Google Scholar]
- Schmidt T., Skerra A. The random peptide library-assisted engineering of a C-terminal affinity peptide, useful for the detection and purification of a functional Ig Fv fragment. Protein Eng Des Sel. 1993;6:109–122. doi: 10.1093/protein/6.1.109. [DOI] [PubMed] [Google Scholar]
- Schmidt T., Skerra A. The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat Protoc. 2007;2:1528–1535. doi: 10.1038/nprot.2007.209. [DOI] [PubMed] [Google Scholar]
- Shi C., Meng Q., Wood D. A dual ELP-tagged split intein system for non-chromatographic recombinant protein purification. Appl Microbiol Biotechnol. 2013;97:829–835. doi: 10.1007/s00253-012-4601-3. [DOI] [PubMed] [Google Scholar]
- Shimazu M., Mulchandani A., Chen W. Thermally triggered purification and immobilization of elastin—OPH fusions. Biotechnol Bioeng. 2003;81:74–79. doi: 10.1002/bit.10446. [DOI] [PubMed] [Google Scholar]
- Singh P., Chan P., Hibbs M., Vazquez M., Segura D., Thomas D. High-yield production and characterization of biologically active GST-tagged human topoisomerase IIa protein in insect cells for the development of a high-throughput assay. Protein Expr Purif. 2011;76:165–172. doi: 10.1016/j.pep.2010.08.001. [DOI] [PubMed] [Google Scholar]
- Smith D. Generating fusions to glutathione S-transferase for protein studies. In: Thorner J., Emr S., DAbelson J.N., editors. Methods Enzymol. Academic Press; 2000. pp. 254–270. [17] [DOI] [PubMed] [Google Scholar]
- Smith D.B., Jonhson K.S. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- Smith G., Petrenko V. Phage display. Chem Rev. 1997;97:391–410. doi: 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- Smith S. Strategies for the purification of membrane proteins. In: Walls D., Loughran S.T., editors. Protein Chromatography. Humana Press; 2011. pp. 485–496. [Google Scholar]
- Stamsås G., Håvarstein L., Straume D. CHiC, a new tandem affinity tag for the protein purification toolbox. J Microbiol Methods. 2013;92:59–63. doi: 10.1016/j.mimet.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Stempfer G., Holl-Neugebauer B., Rudolph R. Improved refolding of an immobilized fusion protein. Nat Biotechnol. 1996;14:329–334. doi: 10.1038/nbt0396-329. [DOI] [PubMed] [Google Scholar]
- Stofko-Hahn R., Carr D., Scott J. A single step purification for recombinant proteins. Characterization of a microtubule associated protein (MAP 2) fragment which associates with the type II cAMP-dependent protein kinase. FEBS Lett. 1992;302:274–278. doi: 10.1016/0014-5793(92)80458-s. [DOI] [PubMed] [Google Scholar]
- Stubenrauch K., Bachmann A., Rudolph R., Lilie H. Purification of a viral coat protein by an engineered polyionic sequence. J Chromatogr B. 2000;737:77–84. doi: 10.1016/s0378-4347(99)00392-8. [DOI] [PubMed] [Google Scholar]
- Studier F., Moffatt B. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- Taniguchi Y., Kawakami M. Application of HaloTag protein to covalent immobilization of recombinant proteins for single molecule force spectroscopy. Langmuir. 2010;26:10433–10436. doi: 10.1021/la101658a. [DOI] [PubMed] [Google Scholar]
- Terpe K. Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol. 2003;60:523–533. doi: 10.1007/s00253-002-1158-6. [DOI] [PubMed] [Google Scholar]
- Thompson A., Hart S., Franz C., Barnouin K., Ridley A., Crame R. Characterization of protein phosphorylation by mass spectrometry using immobilized metal ion affinity chromatography with on-resin β-elimination and Michael addition. Anal Biochem. 2003;75:3232–3243. doi: 10.1021/ac034134h. [DOI] [PubMed] [Google Scholar]
- Thompson N., Aronson D., Burgess R. Purification of eukaryotic RNA polymerase II by immunoaffinity chromatography. Elution of active enzyme with protein stabilizing agents from a polyol-responsive monoclonal antibody. J Biol Chem. 1990;265:7069–7077. [PubMed] [Google Scholar]
- Thompson N., Arthur T., Burgess R. Development of an epitope tag for the gentle purification of proteins by immunoaffinity chromatography: application to epitope-tagged green fluorescent protein. Anal Biochem. 2003;323:171–179. doi: 10.1016/j.ab.2003.08.003. [DOI] [PubMed] [Google Scholar]
- Thompson N., Foley K., Stalder E., Burgess R. Chapter 28 identification, production, and use of polyol-responsive monoclonal antibodies for immunoaffinity chromatography. In: Richard R.B., Murray P.D., editors. Methods in Enzymology. Academic Press; 2009. pp. 475–494. [DOI] [PubMed] [Google Scholar]
- Tomme P., Boraston A., McLean B., Kormos J., Creagh A., Sturch K. Characterization and affinity applications of cellulose-binding domains. J Chromatogr B Biomed Sci Appl. 1998;715:283–296. doi: 10.1016/s0378-4347(98)00053-x. [DOI] [PubMed] [Google Scholar]
- Torrent M., Llop-Tous I., Ludevid M. Protein body induction: a new tool to produce and recover recombinant proteins in plants. In: Faye L., Gomord V., editors. Recombinant Proteins From Plants. Humana Press; 2009. pp. 193–208. [DOI] [PubMed] [Google Scholar]
- Trabbic-Carlson K., Liu L., Kim B., Chilkoti A. Expression and purification of recombinant proteins from Escherichia coli: comparison of an elastin-like polypeptide fusion with an oligohistidine fusion. Protein Sci. 2004;13:3274–3284. doi: 10.1110/ps.04931604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda M., Manabe Y., Mukai M. The high performance of 3 × FLAG for target purification of a bioactive metabolite: a tag combined with a highly effective linker structure. Bioorg Med Chem Lett. 2011;21:1359–1362. doi: 10.1016/j.bmcl.2011.01.038. [DOI] [PubMed] [Google Scholar]
- Vaillancourt P., Zheng C., Hoang D., Breister L. Affinity purification of recombinant proteins fused to calmodulin or to calmodulin-binding peptides. In: Thorner J., Emr S., DAbelson J.N., editors. Methods in Enzymology. Academic Press; 2000. pp. 340–362. [22] [DOI] [PubMed] [Google Scholar]
- Voráčková I., Suchanová Š., Ulbrich P., Diehl W.E., Ruml T. Purification of proteins containing zinc finger domains using immobilized metal ion affinity chromatography. Protein Expr Purif. 2011;79:88–95. doi: 10.1016/j.pep.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vretblad P. Purification of lectins by biospecific affinity chromatography. Biochim Biophys Acta. 1976;434:169–176. doi: 10.1016/0005-2795(76)90047-7. [DOI] [PubMed] [Google Scholar]
- Walls D., Loughran S. Tagging recombinant proteins to enhance solubility and aid purification. In: Walls D., Loughran S.T., editors. Protein Chromatography: Methods and protocols. Humana Press; 2011. [Google Scholar]
- Walsh G. The drug manufacturing process. In: Wiley John, Sons L., editors. Biopharmaceuticals Biochemistry and Biotechnology. 2003. pp. 93–185. [Google Scholar]
- Walter J., Gottschalk U. Concepts for disposables in biopharmaceutical manufacture. In: Shire S.J., Gombotz W., Bechtold-Peters K., Andya J., editors. Current Trends in Monoclonal Antibody Development and Manufacturing. Springer; New York: 2010. pp. 87–99. [Google Scholar]
- Wan W., Wang D., Gao X., Hong J. Expression of family 3 cellulose-binding module (CBM3) as an affinity tag for recombinant proteins in yeast. Appl Microbiol Biotechnol. 2011;91:789–798. doi: 10.1007/s00253-011-3373-5. [DOI] [PubMed] [Google Scholar]
- Wang L., Kang J., Kim K., Lee E. Expression of intein-tagged fusion protein and its applications in downstream processing. J Chem Technol Biotechnol. 2010;85:11–18. [Google Scholar]
- Waugh D. Making the most of affinity tags. Trends Biotechnol. 2005;23:316–320. doi: 10.1016/j.tibtech.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Waugh D. An overview of enzymatic reagents for the removal of affinity tags. Protein Expr Purif. 2011;80:283–293. doi: 10.1016/j.pep.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegner G., Lee H., Marriott G., Corn R. Fabrication of histidine-tagged fusion protein arrays for surface plasmon resonance imaging studies of protein–protein and protein–DNA interactions. Anal Chem. 2003;75:4740–4746. doi: 10.1021/ac0344438. [DOI] [PubMed] [Google Scholar]
- Westra D., Welling G., Koedijk D., Scheffer A., The T., Welling-Wester S. Immobilised metal-ion affinity chromatography purification of histidine-tagged recombinant proteins: a wash step with a low concentration of EDTA. J Chromatogr B Biomed Sci Appl. 2001;760:129–136. doi: 10.1016/s0378-4347(01)00261-4. [DOI] [PubMed] [Google Scholar]
- Wilken L., Nikolov Z. Recovery and purification of plant-made recombinant proteins. Biotechnol Adv. 2012;30:419–433. doi: 10.1016/j.biotechadv.2011.07.020. [DOI] [PubMed] [Google Scholar]
- Wilson D., Nock S. Functional protein microarrays. Curr Opin Chem Biol. 2002;6:81–85. doi: 10.1016/s1367-5931(01)00281-2. [DOI] [PubMed] [Google Scholar]
- Wilson I., Niman H., Houghten R., Cherenson A., Connolly M., Lerner R. The structure of an antigenic determinant in a protein. Cell. 1984;37:767–778. doi: 10.1016/0092-8674(84)90412-4. [DOI] [PubMed] [Google Scholar]
- Wu W., Mee C., Califano F., Banki R., Wood D. Recombinant protein purification by self-cleaving aggregation tag. Nat Protoc. 2006;1:2257–2262. doi: 10.1038/nprot.2006.314. [DOI] [PubMed] [Google Scholar]
- Wu W., Miller K., Coolbaugh M., Wood D. Intein-mediated one-step purification of Escherichia coli secreted human antibody fragments. Protein Expr Purif. 2011;76:221–228. doi: 10.1016/j.pep.2010.12.004. [DOI] [PubMed] [Google Scholar]
- Xu X., Song Y., Li Y., Chang J., Zhang H., An L. The tandem affinity purification method: an efficient system for protein complex purification and protein interaction identification. Protein Expr Purif. 2010;72:149–156. doi: 10.1016/j.pep.2010.04.009. [DOI] [PubMed] [Google Scholar]
- Yon R. Versatility of mixed-function adsorbents in biospecific protein desorption: accidental affinity and an improved purification of aspartate transcarbamoylase from wheat germ. Anal Biochem. 1981;113:219–228. doi: 10.1016/0003-2697(81)90070-1. [DOI] [PubMed] [Google Scholar]
- Young C., Britton Z., Robinson A. Recombinant protein expression and purification: a comprehensive review of affinity tags and microbial applications. Biotechnol J. 2012;7:620–634. doi: 10.1002/biot.201100155. [DOI] [PubMed] [Google Scholar]
- Yu S., Liu Y. Expression and one-step purification of a β-galactosidase by fusion with elastin-like polypeptides. Process Biochem. 2012;47:1108–1114. [Google Scholar]
- Zhang C., Love R., Jilka J., Glatz C. Genetic engineering strategies for purification of recombinant proteins from canola by anion exchange chromatography: an example of β-glucuronidase. Biotechnol Prog. 2001;17:161–167. doi: 10.1021/bp000140c. [DOI] [PubMed] [Google Scholar]
- Zhao G., Dong X., Sun Y. Ligands for mixed-mode protein chromatography: principles, characteristics and design. J Biotechnol. 2009;144:3–11. doi: 10.1016/j.jbiotec.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Zuo X., Li S., Hall J., Mattern M., Tran H., Shoo J. Enhanced expression and purification of membrane proteins by SUMO fusion in Escherichia coli. J Struct Funct Genomics. 2005;6:103–111. doi: 10.1007/s10969-005-2664-4. [DOI] [PMC free article] [PubMed] [Google Scholar]