

Abstract
Expression of the Gag-Pol polyprotein of Rous sarcoma virus (RSV) requires a −1 ribosomal frameshifting event at the overlap region of the gag and pol open reading frames. The signal for frameshifting is composed of two essential mRNA elements; a slippery sequence (AAAUUUA) where the ribosome changes reading frame, and a stimulatory RNA structure located immediately downstream. This RNA is predicted to be a complex stem-loop but may also form an RNA pseudoknot. We have investigated the structure of the RSV frameshift signal by a combination of enzymatic and chemical structure probing and site-directed mutagenesis. The stimulatory RNA is indeed a complex stem-loop with a long stable stem and two additional stem-loops contained as substructures within the main loop region. The substructures are not however required for frameshifting. Evidence for an additional interaction between a stretch of nucleotides in the main loop and a region downstream to generate an RNA pseudoknot was obtained from an analysis of the frameshifting properties of RSV mutants translated in the rabbit reticulocyte lysate in vitro translation system. Mutations that disrupted the predicted pseudoknot-forming sequences reduced frameshifting but when the mutations were combined and should re-form the pseudoknot, frameshifting was restored to a level approaching that of the wild-type construct. It was also observed that the predicted pseudoknot-forming regions had reduced sensitivity to cleavage by the single-stranded probe imidazole. Overall, however, the structure probing data indicate that the pseudoknot interaction is weak and may form transiently. In comparison to other characterised RNA structures present at viral frameshift signals, the RSV stimulator falls into a novel group. It cannot be considered to be a simple hairpin-loop yet it is distinct from other well characterised frameshift-inducing RNA pseudoknots in that the overall contribution of the RSV pseudoknot to frameshifting is less dramatic.
Keywords: Rous sarcoma virus, retrovirus, frameshifting, RNA structure probing, pseudoknot
Abbreviations: RSV, Rous sarcoma virus; ORF, open reading frame; RRL, rabbit reticulocyte lysate; DMS, dimethyl sulphate; AMV, avian myeloblastosis virus; DEPC, diethylpyrocarbonate; CMCT, 1-cyclohexyl-3-(2-morpholinoethyl) carbodiimide metho-p-toluenesulphonate
Introduction
The process of −1 ribosomal frameshifting on viral RNAs was first described as the mechanism by which expression of the Gag-Pol polyprotein of the retrovirus Rous sarcoma virus (RSV) occurs from the overlapping gag and pol open reading frames (ORFs; Jacks & Varmus, 1985). Since this inaugural report, related frameshift signals have been characterised in several other retroviruses, a number of eukaryotic positive-strand RNA viruses, a double-stranded RNA virus of yeast, some plant RNA viruses and certain bacteriophage (for reviews, see Brierley 1995, Farabaugh 1996). In most of the systems studied to date, frameshifting is involved in the expression of replicases. In retroviruses, it allows the synthesis of the Gag-Pol and Gag-Pro-Pol polyproteins from which protease and reverse transcriptase are derived, and for most other viruses frameshifting is required for expression of RNA-dependent RNA polymerases. Much of our understanding of the basic composition of −1 frameshift signals is derived from an in vitro translation analysis of the RSV gag/pol frameshift signal (Jacks et al., 1988). The coding sequence of pol lies downstream of gag and overlaps it in the −1 reading frame. Synthetic mRNAs containing the RSV frameshift signal, when translated in the rabbit reticulocyte lysate system (RRL), produced a translation product corresponding to ribosomes which terminated at the gag stop codon and additionally, a Gag-Pol fusion protein at about one twentieth of the level of Gag alone. This product corresponded to ribosomes which frameshifted prior to encountering the gag stop codon and continued to translate pol in the −1 reading frame. Further analysis using specific point mutations and deletions identified two essential components of the RSV frameshift signal; a homopolymeric “slippery” sequence of nucleotides (AAAUUUA), where the frameshift takes place, and a region of RNA secondary structure located a few nucleotides downstream (Jacks et al., 1988).
Such an arrangement of a slippery sequence and stimulatory RNA structure is found at almost all of the viral −1 frameshift sites identified to date. The slippery sequences conform in general to the motif XXXYYYN (where X is any nucleotide, Y is A or U and N is A, C or U) and frameshifting is thought to occur by simultaneous slippage of two ribosome-bound tRNAs at this sequence (from X XXY YYN to XXX YYY; Jacks et al., 1988). A variety of stimulatory RNA structures are predicted to be present downstream of the slippery sequence, although most of these have yet to be confirmed by secondary structure mapping. At some sites, a stem-loop structure is evident but more often, the downstream stimulator is an RNA pseudoknot, a structure formed when nucleotides in the loop region of a hairpin-loop base-pair with a region elsewhere in the mRNA to create a quasi-continuous double-helix joined by single-stranded connecting loops (see ten Dam et al., 1992). Within each general category of stem-loop or pseudoknot, at least two classes are present. The human astrovirus serotype 1 (HAst-1) 1a/1b frameshift signal for example is perhaps the most simple, possessing a short stem and loop (Marczinke et al., 1994). However, more complex stem loops have been predicted; the stimulatory RNA at the red clover necrotic mottle virus p27/p57 frameshift site is thought to comprise a very long stem containing several mismatches and bulges (Kim & Lommel, 1994) and the RSV stimulator is highly complex (see below). Two distinct classes of RNA pseudoknot structure have also been recognised. At the frameshift sites of the corona-, toro- and arteriviruses (all members of the Nidovirales; Cavanagh, 1997) the pseudoknots possess a long stem 1 of 11–14 bp and usually, a long loop 2 (30–164 nucleotides). The second class are typified by the structures present at the simian retrovirus 1 gag-pro (SRV-1; Ten et al 1994, Ten et al 1995, Du et al 1997, Sung and Kang 1998) and the retrovirus mouse mammary tumour virus (MMTV) gag-pro overlap regions Chen et al 1995, Chen et al 1996, Shen and Tinoco 1995, Kang et al 1996, Kang and Tinoco 1997. These pseudoknots contain stems of 5 to 7 bp, shorter loops and have an unpaired, intercalated A residue at the junction between the two stems which introduces a pronounced bend. This characteristic bent conformation appears to be essential for efficient frameshifting Chen et al 1996, Kang and Tinoco 1997.
Here, we describe an investigation of the secondary structure of the RSV frameshift signal using enzymatic and chemical structure probing and site-directed mutagenesis. Only limited information exists as to the precise folding of the downstream stimulator present at the RSV gag/pol overlap. Computer-derived secondary structure predictions (Jacks et al., 1988) suggest that the RNA folds into a stem-loop structure containing a long stem (14 bp) and a large loop (71 nucleotides) containing sub-structures (Figure 1). Mutational analysis of the RSV signal (Jacks et al., 1988) has confirmed that the long stem region is essential for efficient frameshifting and in addition, a region downstream of the stem-loop was also shown to contribute to frameshifting, raising the possibility that the stimulatory element is in fact an RNA pseudoknot. A stretch of eight nucleotides within the downstream region with complementarity to bases in the loop region had been noticed previously (Brierley et al., 1989; boxed nucleotides in Figure 1), but base-pair formation between these sequences would generate an unusual pseudoknot, quite different from those seen at other well characterised frameshift sites. Here we show that the RNA folds into a structure very similar to that predicted from computer-assisted folding studies. Significantly, the proposed pseudoknot interaction does contribute to the efficiency of the RSV frameshift signal. However, the structure probing data generated from wild-type and mutant variants of the RSV frameshift region indicate that this interaction is weak and may form transiently.
Figure 1.

A computer-derived secondary structure prediction of the RSV gag/pol frameshift region as proposed by Jacks et al. (1988). The slippery sequence is underlined and bases with the potential to interact and form stem 2 of an RNA pseudoknot (PK2 and PK4; Brierley et al., 1989) are highlighted in black.
Results
Structure probing of the RSV frameshift signal by primer extension
Structure probing of the RSV frameshift region was performed initially within the context of a longer transcript using the primer extension approach (Ehresmann et al., 1987). For this analysis, a 1.5 kb transcript containing the frameshift region was prepared by T7 transcription of plasmid pRSV1 cut with PvuII (Figure 2). In the first step, RNA was either untreated or subjected to limited RNase hydrolysis or chemical modification using structure-specific probes. After stopping the reactions, the RNA was phenol:chloroform extracted and hybridised with oligonucleotides complementary to either RSV nucleotides 2692–2676, 2639–2623 or 2600–2584, which were used as primers for avian myeloblastosis virus (AMV) reverse transcriptase. In order to determine the sites of cleavage/modification produced by the various treatments, the labelled primer extension products were analysed on 8% denaturing polyacrylamide gels alongside size markers prepared by dideoxy sequencing of pRSV1/PvuII RNA using AMV reverse transcriptase. For each structure probe used, between five and ten primer extension reactions were carried out with each primer and on at least three independent preparations of modified RNA. A representative selection of the individual biochemical analyses are shown in Figure 3(a), (b) and (c) along with a diagrammatic summary (Figure 3(d)). The single-strand specific probes employed were: RNase T1, which cleaves internucleotide bonds 3′ of unpaired G residues; 2-keto-3-ethoxybutyraldehyde (kethoxal), which reacts with unpaired G residues at N-1 and N-2; dimethyl sulphate (DMS), which methylates unpaired A at N-1 and unpaired C at N-3; diethylpyrocarbonate (DEPC), which reacts with the N-7 group of unpaired A residues (and may also react with unpaired C and U under the conditions of pH and KCl concentration employed here; see Materials and Methods) and 1-cyclohexyl-3-(2-morpholinoethyl) carbodiimide metho-p-toluenesulphonate (CMCT), which modifies the N-3 group of unpaired uridines and weakly, N-1 of unpaired guanine bases. To probe double-stranded regions, RNase V1 was employed, which cleaves internucleotide bonds in helical regions. RNase V1 is not base-specific but cleaves RNA that is in helical conformation, whether base-paired (a minimum of 4–6 bp are required) or single-stranded and stacked. The picture that emerged from the biochemical probing data supports in general the stem-loop model proposed by Jacks et al. (1988), though there were some ambiguities and deviations.
Figure 2.

Construction of plasmid pRSV1. A 266 bp AvrII-BssHII fragment encompassing the RSV frameshift region was isolated from plasmid pATV8 (which harbours a proviral clone of RSV Prague C; Schwartz et al., 1983) and subcloned into the PB1 reporter gene of plasmid pPSO.1, a derivative of pPSO (Somogyi et al., 1993) with unique AvrII and BssHII sites. The open reading frames in pRSV1 are such that the upstream portion of the PB1 reporter gene is in frame with the gag gene and the downstream portion, the pol gene. Analysis of the secondary structure of the RNA at the RSV frameshift site by primer extension was on mRNAs prepared by linearisation of pRSV1 with PvuII and subsequent transcription with T7 RNA polymerase. For structural analysis of end-labelled RNA, related plasmids containing a T3 promoter were prepared (see the text), linearised with DdeI and transcribed with T3 RNA polymerase. In ribosomal frameshift assays, capped mRNAs were prepared by SP6 transcription from AvaII linearised template. The predicted non-frameshifted (45 kDa) and frameshifted (70 kDa) species generated upon in vitro translation of such mRNAs in RRL are shown.
Figure 3.




Primer extension analysis of the RSV frameshift region. RNA derived by T7 transcription of pRSV1/PvuII was subjected to limited RNase hydrolysis or chemical modification using structure-specific probes. Sites of cleavage or modification were mapped by primer extension using AMV RT and a range of oligonucleotide primers. Products were analysed on 8% acrylamide-7 M urea gels alongside sequencing ladders prepared by dideoxy sequencing of the RNA using the relevant primer and AMV RT. For most treatments, three independent primer extension reactions are shown at the optimal probe concentration (determined empirically). (a) Primer extension with oligonucleotide BM14 (complementary to nucleotides 2600–2584). Uniquely modified nucleotides were identified by their absence in untreated control lanes (0). Treatments were as follows: RNase V1 (units/μg RNA), RNase T1 (units/μg RNA), kethoxal (KET, mg/ml), DMS (% [v/v]), DEPC (% [v/v]) and CMCT (mg/ml) under non-denaturing conditions. (b) Primer extension with oligonucleotide BM5 (complementary to nucleotides 2639–2623). Treatments were as detailed in (a) except (∗) indicates semi-denaturing conditions. (c) Primer extension with oligonucleotide BM4 (complementary to nucleotides 2692–2676). Treatments were as in (b). (d) Summary of the probing results. The sensitivity of bases in the RSV frameshift region to the various probes is shown. Only those reactivities observed under non-denaturing conditions are shown. The assignment of signals in the slippery sequence was hindered to some extent by non-specific reactivities (see the text). The size of the symbols is approximately proportional to the intensity of cleavage/modification at that site. In this drawing, the PK4 sequence is represented as part of a short hairpin-loop structure downstream of the main stem.
Slippery sequence
The assignment of signals in this region, which is thought to be single-stranded, was hampered by non-specific reactivities (Figure 3(a), top). Bases A-2477 and A-2478, which form part of the slippery sequence, did react weakly with DEPC and DMS (lanes 14–20) supporting the idea that these bases are single-stranded. U-2481 of the slippery sequence reacted with single-strand reagents DMS, DEPC and CMCT. Although somewhat blurred, an RNase V1 signal (lanes 5 to 9) was also seen at this base; this is probably indicative of base stacking rather than base-pair formation.
Stem 1
Specific RNase V1 signals were seen in both the 5′-arm (C-2493 to C-2495, Figure 3(a), lanes 1–4) and the 3′-arm (U-2579 and C-2580, Figure 3(c), lanes 1–4). Weak single-stranded signals were occasionally observed from bases at the bottom of the predicted stem (5′-arm: A-2484 and G-2485), raising the possibility that these base-pairs may breathe. All other bases in the predicted stem 1 region were unreactive against any of the single-stranded probes, even under semi-denaturing conditions, which suggests strongly that this is a stable stem region.
Loop 1
This loop is proposed to be 19 nucleotides in length and contains eight bases (G-2509 to A-2516), here referred to as the PK2 sequence, that could putatively pair with a region outside the stem-loop (U-2595 to C-2602; the PK4 sequence) to form a pseudoknot. From the primary sequence, it is also possible that the PK2 sequence could be extended on its 5′-side by four nucleotides if U-2603 of PK4 is bulged out. However, the primer extension data did not support strongly the involvement of PK2 residues in a pseudoknot as they almost all emitted single-stranded signals. In most cases however, the signals were weak, with the exception of A-2514, C-2515 and A-2516 with DMS, where stronger signals were obtained (Figure 3(a), lanes 13–16). The other bases of the loop (U-2498 to U-2508) displayed weak reactivity against single-stranded probes, and C-2500 was found to be cleaved consistently by RNase V1.
Stem-loop 2
According to the proposed model, stem-loop 2 consists of an 11 bp long stem with two bulged nucleotides in the 5′-arm and is closed by a six-membered loop. The probing data confirmed this structure in general. Bases in the 3′-arm of the stem were insensitive to single-stranded probes with the exception of U-2536 at the top of the predicted stem (Figure 3(b), lanes 19–28) which showed weak reactivity with DEPC and CMCT, and also A-2539 and A-2540, which are proposed to be in close proximity to the bulged residues (U-2523 and A-2524) on the opposite strand. These bases showed weak reactivity to DMS and DEPC (Figure 3(b), lanes 14–23). Even under semi-denaturing conditions, only A-2546 at the bottom of the 3′-arm displayed a faint single-stranded signal. Complementary bases in the 5′-arm of the predicted stem (Figure 3(b)) displayed a corresponding reactivity pattern, but RNase V1 signals were more pronounced. G-2529, located opposite the single-strand reactive U-2536 of the 3′-arm, reacted moderately with kethoxal and weakly with RNase T1, implying that the proposed G-U base-pair at the top of the stem does not form and instead both residues are part of the highly reactive single-stranded loop. Base U-2525 adjacent to the bulged nucleotides and juxtaposed to the single-strand-reactive A residues in the 3′-arm, produced RNase V1 as well as single-stranded signals. Both of the residues predicted to be bulged out (U-2523 and A-2524) reacted with single-stranded probes. These bulged residues might distort the regularity of the helical region, and render surrounding base-pairs more prone to breathing.
Stem-loop 3
According to the model shown in Figure 1, stem-loop 3 (C-2547 to G-2562) forms immediately adjacent to stem-loop 2. Although the probing data confirm that this region folds into a stem-loop, the stem is two base-pairs shorter than originally predicted. Residues belonging to the 3′-arm of the predicted stem reacted mainly with the double-strand specific RNase V1 but the bottom two bases G-2561 and G-2562 invariably reacted strongly with RNase T1 and kethoxal (Figure 3(b)). A weak kethoxal signal was seen for base G-2557 at the top of the 3′-arm, as might be expected for a residue at the end of a stem where breathing can occur. Under semi-denaturing conditions this G residue also became susceptible to RNase T1 cleavage, whereas all the other bases unreactive against single-stranded probes under non-denaturing conditions remained unreactive. Bases predicted to form the complementary 5′-arm of stem 3 displayed ambiguous reactivity, producing both double- and single-stranded signals (C-2548 to A-2551). As C-2548 was predicted to pair to the clearly single-stranded G-2561 in the 3′-arm of the stem; the reactivity observed with C-2548 against both single-stranded probes and RNase V1 could well be accounted for by stacking of the base at the junction of stem 2 and stem 3. In discrepancy to the predicted single-stranded character of the loop residues, U-2556 was consistently reactive to RNase V1 and gave only weak signals with CMCT, which became more intense under semi-denaturing conditions. Rather faint single-stranded signals were seen at the C-doublet in the loop (C-2554 and C-2555) but strong RNase T1 and kethoxal reactivity was observed at G-2553, suggesting that it does not pair with U-2556 across the loop. Altogether the reactivity pattern of the predicted loop bases is consistent with unpaired nucleotides. Nucleotide U-2556 might be stacked, explaining the reactivity of this base against RNase V1.
Loop 4
In agreement with the predicted single-stranded character of this region, residues A-2563 to C-2568 reacted only with single-stranded reagents and in particular G-2566 reacted strongly with RNase T1 and kethoxal (Figure 3(b), lanes 6–13).
Downstream region
Deletion analysis has established that the 3′-boundary of the region essential for efficient frameshifting in RSV is located somewhere between the bottom of the major stem-loop and base C-2606 some 23 nucleotides downstream (Jacks et al., 1988). This region contains the PK4 sequence (U-2595 to C-2602) which could potentially pair with nucleotides in loop 1 (G-2509 to A-2516, PK2) to form an RNA pseudoknot. The reactivity of the PK4 bases (Figure 3(c)) is consistent with their involvement in a base-pairing interaction in that they were only weakly reactive against single-stranded reagents and in addition, half of the bases were consistently cleaved with RNase V1. However, several additional bases downstream were also reactive against RNase V1 and it is possible that the putative pseudoknot forming sequences in fact pair with the downstream nucleotides to generate a small stem-loop structure (as drawn in Figure 3(d)). This is addressed overleaf.
Site-directed mutagenesis of the RSV frameshift region
Although the structure probing revealed the basic organisation of the RSV frameshift site, it did not provide unambiguous evidence for or against pseudoknot formation, nor did it address the potential role of the various substructures in frameshifting. To further investigate these issues, a reporter plasmid pRSV1 (Figure 2) was constructed to allow measurement of RSV wild-type and mutant frameshift site efficiencies by in vitro transcription and translation. In pRSV1, a region of the RSV genome encompassing the frameshift signal (RSV sequence information from position 2459 to 2725 bp; Schwartz et al., 1983) was inserted into the influenza A/PR8/34 PB1 gene (Young et al., 1983) at position 1167 bp, such that the upstream and downstream PB1 sequences were in frame with the RSV gag and pol open reading frames (ORFs), respectively. Linearisation of the plasmid with AvaII (position 1642 in the PB1 gene) and subsequent in vitro transcription with SP6 RNA polymerase generated an approximately 1970 nt mRNA which upon translation in RRL was predicted to produce a 45 kDa non-frameshift product and a 70 kDa frameshift product. As can be seen in Figure 4, both the 45 kDa and 70 kDa species were produced and the frameshift efficiency was calculated to be 4%; in good agreement with previous values Jacks and Varmus 1985, Jacks et al 1988. Also shown in Figure 4 are the RSV mutants tested. These fell into three categories which examined: (1) the potential involvement of the RNA pseudoknot, (2) the role of stem-loops 2 and 3 (together described as the “rabbit ears”), and (3) loop 1 length and nucleotide composition. The pseudoknot analysis involved complementary and compensatory base-pair changes. In pRSV2, we changed the eight nucleotides of the PK2 sequence to the complementary bases, which should disrupt the pseudoknot interaction. This mutation was found to reduce frameshifting some two to threefold. The same was also true when the equivalent changes were made in PK4 (pRSV4). In pRSV5, both changes were made which should be compensatory and allow the pseudoknot to reform. In support of the pseudoknot hypothesis, frameshifting in this double mutant, pseudo-wild-type construct occurred at a much greater level than seen for either single mutant, although was not restored fully to the wild-type level. To confirm further the contribution of base-pairing between these two regions, the same mutations were tested in the background of an RSV construct (pRSV1.1) in which the RSV slippery sequence, AAAUUUA, had been replaced by that of the coronavirus IBV, UUUAAAC, a more efficient slippery sequence (Brierley et al., 1992). In pRSV1.1, frameshifting occurred at an efficiency of 9%, about two to threefold more than the wild-type signal. The results of the pseudoknot changes were essentially the same; complementary changes in either region (pRSV8, 9) reduced frameshifting two to threefold and frameshifting was restored to some extent in the double mutant construct (pRSV10). In their analysis of RSV frameshifting, Jacks et al. (1988) demonstrated that a maximum of 23 nucleotides downstream of stem 1 were needed for fully efficient frameshifting. In order to test that the contribution of the downstream region to frameshifting is entirely accounted for by the PK4 sequence, the 24 nucleotides immediately downstream of stem 1 were deleted within the background of pRSV1 or pRSV1.1. In each case (pRSV6, 1.6%; pRSV11, 3.1%), the deletion reduced frameshifting to the same level as that seen with the PK4 mutation alone, supporting the idea that the key element in the downstream region is the PK4 sequence. The structure probing analysis described above raised the possibility that this sequence may also interact with a region a few nucleotides downstream to form a small stem-loop. Such an interaction, if it occurred, could compete with the formation of the PK2-PK4 stem of the putative pseudoknot. In construct pRSV12, we addressed this by deleting 12 nucleotides immediately downstream of the PK4 sequence; a deletion which would remove those bases which may compete for PK4. In this construct, frameshifting was unaffected suggesting either that the downstream sequence does not pair with PK4, or that such pairing does not impair pseudoknot formation.
Figure 4.

Analysis of the RSV frameshift signal by mutagenesis. A series of mutations were created in the RSV frameshift region of plasmid pRSV1 (Figure 2) and the effect on frameshifting measured by in vitro transcription and translation in RRL, as described in the legend to Figure 2. The upper portion of the figure shows the RRL translation products synthesized in response to mRNA derived from AvaII-digested pRSV1 and mutant derivatives. Products were labelled with [35S]methionine, separated on a 10% SDS/polyacrylamide gel and detected by autoradiography. The frameshifted (fs) and non-frameshifted (stop) species are marked with arrows. Track M was loaded with [14C]high molecular weight markers (Amersham). The lower part of the Figure shows the location of the various mutations within the RSV frameshift region and the frameshift efficiency measured for each mutation. The frameshift efficiencies quoted are the average of three independent measurements which varied by less than 10%, i.e. a measurement of 4% frameshift efficiency was between 3.6% and 4.4%.
The location of stems 2 and 3 immediately downstream of the PK2 sequence is intriguing in that the formation of a standard hairpin-loop type pseudoknot (H-type; see ten Dam et al., 1992) would position these structures between the stacked stems of the pseudoknot. Only limited information is available on the effect on frameshifting of inserting nucleotides between the component stems of H-type pseudoknots, but in general, such insertions reduce frameshifting Brierley et al 1991, Chen et al 1995. It was feasible therefore that stems 2 and 3 were inhibiting frameshifting. To test this, both stems were deleted in construct pRSV7, and in pRSV3, a complementary change was created in a block of six nucleotides in the 5′-arm of stem 2, which was anticipated to destabilise the stem. In each case, a small increase in frameshifting was observed, hence the stems are not only dispensable for frameshifting but are in fact, slightly inhibitory.
The final set of mutations were created in loop 1. This region reacted only weakly with single-stranded probes, suggesting that it may be inaccessible to the probes employed. Indeed, at least one of the bases may be paired; nucleotide C-2500 reacted consistently with RNase V1. Four mutants were made (within the background of pRSV1.1; efficiency 9%); C-2500 was changed to G (pRSV13), three nucleotides were deleted (pRSV14) or added (pRSV15) or a 12-nucleotide insertion was made (pRSV16). Surprisingly, changing C-2500 to G had a stimulatory effect on frameshifting, increasing the efficiency from 9% to 15%. Deletion or insertion of three nucleotides had a more modest effect, increasing (to 11%) or decreasing (to 7%) frameshifting, respectively. The larger insert reduced frameshifting by twofold (pRSV16). Thus mutations in loop 1 can also influence frameshifting.
Structure probing of the RSV frameshift signal by end-labelling
So far, the evidence for involvement of a pseudoknot in RSV frameshifting was mixed. The mutagenesis data are entirely consistent with pseudoknot formation in that mutations in the component sequences (PK2+PK4) reduce frameshifting efficiency, yet in double mutant, pseudo-wild-type constructs, the frameshift efficiency is restored to a value approaching that of the wild-type constructs. However, the structure probing analysis by primer extension was not strongly supportive of a base-pairing interaction between these regions. In an attempt to resolve this issue, we probed further the structure of the RSV frameshift region and two mutant variants, using an end-labelling procedure Van Belkum et al 1988, Wyatt et al 1990, Polson and Bass 1994 and included two alternative structure mapping reagents, imidazole (Vlassov et al., 1995) and lead acetate Krzyzosiak et al 1988, Kolchanov et al 1996. We began by inserting (by site-directed mutagenesis) a bacteriophage T3 promoter some 15 nucleotides upstream of the slippery sequences of constructs pRSV1.1 (wild-type structure), pRSV4 (PK4 mutant) and pRSV7 (rabbit ears deleted) to create pRSV17, 19 and 18, respectively. Linearisation of these plasmids with DdeI and T3 transcription generated RNAs of 193, 151 and 193 nucleotides, respectively, which were subsequently end-labelled with [γ-32P]ATP, gel purified and subjected to limited chemical and enzymatic digestion prior to analysis on denaturing polyacrylamide gels. We were particularly interested to look for changes in the susceptibility of PK2 and PK4 to cleavage by the single-strand-specific chemical probes imidazole and lead acetate in constructs where base-pair formation between these sequences was predicted to be weaker (pRSV19) or perhaps stronger (pRSV18). As the wild-type frameshift region was present on a much shorter RNA in this case (193 nucleotides as opposed to 1500 nucleotides), we also employed RNases T1, V1 and U2 (which cleaves 3′ of single-stranded A or G, with a preference for A) to allow the overall conformation of the RSV frameshift region to be confirmed. In these experiments, the Mg2+ level was kept at 2 mM, which is the approximate concentration of this ion in RRL (Jackson & Hunt, 1983).
A selection of the biochemical analyses and diagrammatic summaries are shown in Figure 5, Figure 6. Sites of cleavage were scored only from those reactions where 90% or more of the full-length RNA remained intact. In terms of the overall structure of the RSV frameshift signal, the data obtained from the analysis of end-labelled pRSV17 RNA (Figure 5) were in good agreement with those from the primer extension studies described earlier. There were some changes regarding the bases cleaved by RNase V1 however, perhaps as a consequence of the increased reaction temperature (25°C as opposed to 0°C) or the lower MgCl2 and KCl concentrations employed in the end-labelling assay. We found that the distribution of RNase V1 cuts within stem 1, stem 2 and the downstream region was slightly different, although fully consistent with the formation of the predicted base-paired regions, and also that the loop 1 bases were more reactive (Figure 5(a), (b), (c) and (d)). The cleavage patterns observed with RNases T1 and U2 were entirely consistent with the structure predicted from the primer extension data (Figure 5(a) and (c)). Structure probing with either lead or imidazole generated a very similar pattern of bands which were in effect footprints highlighting the major regions of single- and double-stranded RNA. In the case of the wild-type structure (pRSV17; Figure 5(b)), the intensity of the imidazole ladder appeared to be a little weaker in the PK2 and PK4 regions, supporting the idea that base-pairing is occurring between the two sequences. This was more apparent when a direct comparison was made between the wild-type structure and the PK4 mutant (pRSV19), where such base-pairing should be less stable (Figure 5(d); compare track I/17 with I/19). The overall conformation of the wild-type and mutant structures was very similar except in the regions of PK2 and PK4, where some protection against imidazole cleavage was seen in the wild-type but not the mutant structure. For PK2, protection was most apparent for six of the eight residues (G-2509 to A-2514). The noticeably enhanced imidazole cleavages seen at bases C-2501 and C-2515 in the I/17 lane may be artifactual; these bases appear to be particularly susceptible to spontaneous cleavage, as can be seen in the adjacent water control lane W/17 (and on other gels, for example, Figures 5(a) and (b) and data not shown) and this was especially marked in this particular track. The protection against cleavage by single-strand-specific chemical probes afforded by PK2 and PK4 was less obvious in samples probed with lead acetate. However, this probably reflects the nature of the probe; lead is highly reactive and the less reactive imidazole probe is presumably a better indicator of weaker or more transient pairing interactions. We did not see a reduction in the number of RNase V1 cuts in PK2 or PK4 of the mutant structure however; this is considered in Discussion. Structure probing of a transcript derived from pRSV18 which contains the rabbit ears deletion is shown in Figure 6(a) and summarised in Figure 6(b). This structure elicits slightly greater than wild-type frameshift efficiency in RRL and we speculated that this may be a consequence of a more stable PK2-PK4 interaction. As can be seen (Figure 6(a) track I/4) some protection against imidazole cleavage was observed in the PK2 and PK4 regions. In the case of pRSV18, all of the PK2 bases (G-48 to A-55) showed this protection. A surprising observation however was the behaviour of base G50, which should form part of the predicted stem 2 region of the RSV18 pseudoknot (formed by PK2-PK4). This base was extremely sensitive to RNase T1, yet other G residues in the loop region were far less reactive. Whether this hypersensitivity reflects a natural hot-spot in the loop or is a consequence of transient pseudoknot pairing and a subsequent unusual conformation of G-50 in a G-50·C-97 base-pair remains to be seen. Perhaps G-50 marks the first unpaired residue in a shorter, 5 bp PK2:PK4 stem and is especially reactive.
Figure 5.

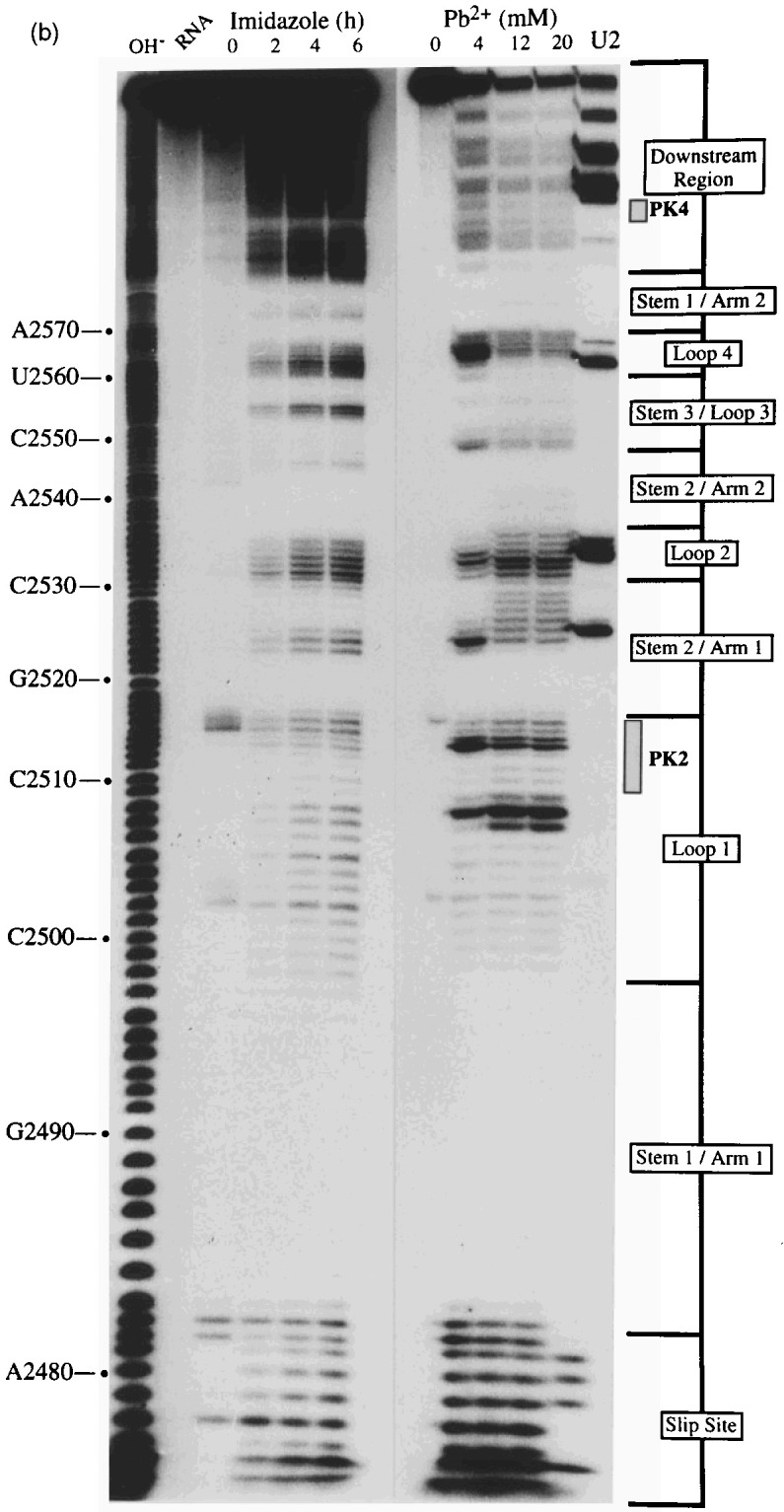


Structure probing of the RSV frameshift signal by end-labelling. RNA derived by T3 transcription of pRSV17/Pvu II (all panels) or pRSV19/Pvu II (d) was 5′ end-labelled with [γ-32P]ATP and subjected to limited RNase or chemical cleavage using structure-specific probes. Sites of cleavage were identified by comparison with a ladder of bands created by limited alkaline hydrolysis of the RNA (OH−) and the position of known RNase T1 and U2 cuts, determined empirically. Products were analysed on 10% acrylamide-7 M urea gels run for varying lengths of time. Data were also collected from 6% and 15% gels (gels not shown). (a) Structure probing of pRSV17 RNA with RNases T1, V1 and U2. Uniquely cleaved nucleotides were identified by their absence in untreated control lanes (0). The number of units of enzyme added to each reaction is indicated. (b) Structure probing with imidazole (hours) or lead acetate (Pb2+; mM concentration in reaction). A U2 lane was included as a marker. RNA represents an aliquot of the purified RNA loaded directly onto the gel without incubation in a reaction buffer. (c) Summary of the RSV17 probing results. The sensitivity of bases in the RSV frameshift region to the various probes is shown. The size of the symbols is approximately proportional to the intensity of cleavage at that site. The lead acetate probing data are omitted for clarity, but were similar to that seen with imidazole. (d) A comparison of the cleavage patterns of pRSV17 and 19 RNAs. Structure probing with imidazole (I, four hours), lead acetate (Pb2+; mM concentration in reaction) and RNase V1 (units per reaction) is shown. U2 reactions were included as markers (however the U2/19 reaction did not work for an unknown reason). The water (W) lanes represent RNA which was dissolved in water, incubated for four hours and processed in parallel to the imidazole-treated samples.
Figure 6.


Structure probing of RNA derived from the rabbit ears deletion mutant, pRSV18. (a) RNA was prepared for analysis as in the legend to Figure 5 and probed with RNase U2, V1 and T1(units per reaction are shown) and the chemicals imidazole (I, 4 hours) and lead acetate (Pb2+; mM concentration in reaction). The water lane (W) represents RNA which was dissolved in water, incubated for four hours and processed in parallel to the imidazole-treated sample. (b). Summary of the RSV18 probing results. The sensitivity of bases in the RSV18 frameshift region to the various probes is shown. The size of the symbols is approximately proportional to the intensity of cleavage at that site. The lead acetate probing data are omitted for clarity, but were similar to that seen with imidazole.
Discussion
Here, the secondary structure of the RSV Prague C gag/pol ribosomal frameshifting signal has been determined by enzymatic and chemical probing, either as a short transcript or within the context of a longer mRNA. The RSV frameshift signal folds into a structure which resembles closely that proposed by Jacks et al. (1988) on the basis of computer-generated predictions and mutagenesis studies. Indeed, the only discrepancies observed related to the length of stems 2 and 3, which are somewhat shorter than predicted; the last base-pair of stem 2 (G-2529:U-2536) actually forms part of loop 2 and stem 3 is only four base-pairs in length, rather than six. A key issue we wished to address was whether the RSV stimulator was a simple hairpin-loop (albeit a loop containing substructures) or a more complex pseudoknot structure as we had predicted (Brierley et al., 1989). Two lines of experimental evidence were obtained that support the pseudoknot hypothesis. Firstly, mutations which disrupted base-pairing between the sequences proposed to form stem 2 of the pseudoknot structure (PK2 and PK4) gave reduced frameshift efficiencies, yet frameshifting was restored to some extent in compensatory mutants in which the pseudoknot structure was predicted to reform. The second line of evidence concerned the reactivity of the PK2 and PK4 sequences to the single-strand-specific probe imidazole. In two constructs where a pseudoknot interaction was proposed, (pRSV17 and 18), some protection from cleavage of the pseudoknot forming sequences was seen. In a mutant construct where the interaction between PK2 and PK4 was expected to be weaker (pRSV19), increased imidazole cleavage was observed.
In general, the PK2 and PK4 sequences were only weakly reactive with single-stranded enzymatic probes, but they were not especially reactive with RNase V1 either, a nuclease which cleaves double-helical stretches of four to six nucleotides in length. The paucity of RNase V1 cleavage sites, particularly in PK2, perhaps reflects the transient nature of the interaction between these sequences, or a weak interaction which is melted upon binding of the enzyme to other parts of the RNA (loop 1 would be a candidate). It may be that the PK2-PK4 interaction involves only a sub-set of the eight proposed base-pairs and that recognition of the shorter helix by RNase V1 is poor. In the wild-type construct, bases A-2514 to A-2516 of PK2 were accessible to DMS, and C-2515 and A-2516 were less protected from imidazole cleavage than other PK2 bases. It may be that the presence of the rabbit ears hinders the formation of those PK2-PK4 base-pairs immediately adjacent. In RNAs where the interaction between PK2 and PK4 was predicted to be weaker (RSV19) or perhaps stronger (RSV18), no great differences in the pattern of RNase V1 cleavage were seen however. Perhaps the differences in stability of the PK2-PK4 interaction predicted for the various mutants is insufficient to affect recognition by RNase V1 but does influence frameshifting efficiency and the sensitivity to cleavage by imidazole. Whatever the case, the data overall support the idea that a pseudoknot forms at the RSV frameshift site and contributes to the frameshift process.
The presence of sub-structures in the RSV loop region, although dispensable for frameshifting, raises questions as to the precise conformation of the RSV pseudoknot. The formation of a classic H-type pseudoknot with coaxially stacked stems would place the rabbit ears (along with loop 4) at the junction of the two stem regions, a configuration one would expect to inhibit frameshifting (Brierley et al., 1991). A mutation which removed stems 2 and 3 increased frameshifting by 50%, but in this mutant, the loop 4 nucleotides were still present between the two stems. It may be that a complete removal of the rabbit ears and loop 4 would result in a more efficient structure. Whatever the case, the analysis presented here and related studies on the virus itself suggest that the stimulatory RNA structure is probably more complex. For example, frameshifting was sensitive to insertions in loop 1, indicating that the precise distance between the stem regions is important. Furthermore, a point mutation which changed the putatively unpaired loop 1 base C-2500 to G almost doubled the frameshift efficiency; perhaps this base is involved in an interaction across the loop. These in vitro observations are supported to some extent by in vivo studies. Two mutants created in an RSV infectious clone which increased the loop 1 length by three or six nucleotides prevented ribosomal frameshifting in vivo (Stewart & Vogt, 1994). We are currently employing a related approach to investigate the role of the RSV pseudoknot in virus replication.
In comparison to other characterised RNA structures present at viral frameshift signals, the RSV stimulator falls into a novel group. It cannot be considered to be a member of the simple hairpin-loop class (e.g.: HAst-1), yet it is distinct from other characterised frameshifter pseudoknots (e.g. IBV, MMTV) in that the overall contribution of the RSV pseudoknot to frameshifting is far less dramatic. However, it seems unlikely that the RSV stimulator is the only member of this group. Within the avian-leukosis sarcoma genus, the frameshift sequences present at the gag/pol overlap region of two sequenced avian leukosis virus (ALV) strains (one subgroup A, Bieth & Darlix, 1992; one subgroup J [ALV103 J], Bai et al., 1995) are closely conserved and PK2 and PK4 are identical. The same is also true for the gag/pol overlap region of two proviral clones (pSRA2, De Lorbe et al., 1980; pRCAS, Hughes et al., 1987) of the related Schmidt-Ruppin subgroup A strain which we have sequenced (data not shown). A number of other retroviruses appear to use a frameshift signal similar in organisation to that employed by RSV. The pro/pol overlap regions of the closely related simian retroviruses 1, 2 Power et al 1986, Thayer et al 1987 and Mason-Pfizer monkey virus (Sonigo et al., 1986) possess the slippery sequence AAAUUUU followed by a predicted stable stem-loop structure with a large loop. The MMTV pro/pol overlap is similar, although the slippery sequence is more divergent (GGAUUUA). Other sites with this kind of organisation include the pro/pol overlaps of the type D retrovirus SMRV-H (Oda et al., 1988) and the lymphoproliferative disease virus of turkeys (Sarid et al., 1994), a type C retrovirus. The latter two viruses have the slippery sequence AAAUUUA, like RSV. The nucleotide sequences of the viral frameshift sites described above contain stretches of between seven and ten nucleotides in the loop region which are complementary to bases downstream of the predicted stem-loop (within 50 nucleotides; data not shown). It seems likely therefore that pseudoknots of the RSV class can also form at these sites.
Materials and methods
Site-directed mutagenesis
Site-specific mutagenesis was carried out by a procedure based on that of Kunkel (1985) as described previously (Brierley et al., 1989).
Construction of plasmids
The basic plasmids used here are detailed in Figure 2. Plasmids pATV8 and pSRA-2, which contain full-length cDNA copies of the Rous sarcoma virus Prague C and Schmidt Ruppin A2 genomes, respectively, were obtained from the American Type Culture Collection. Plasmid pATV8 was digested with AvrII and BssHII and a 266 bp DNA fragment containing the RSV gag/pol frameshift region isolated (RSV sequence information from position 2459 bp to 2725 bp; Schwartz et al., 1983). The fragment was inserted into AvrII and BssHII digested plasmid pPS0.1 (see below) to create plasmid pRSV1. In this plasmid, the RSV sequence information is inserted at nucleotide 1167 of the influenza A/PR8/34 PB1 gene (Young et al., 1983) such that the upstream and downstream PB1 sequences are in frame with the RSV gag and pol open reading frames (ORFs), receptively. The ensemble is under the control of both the bacteriophage SP6 and T7 promoters. Plasmid pRSV1 and mutant derivatives contain the bacteriophage f1 intergenic region (Dotto et al., 1981) in a non-essential region of the vector and may therefore be converted to a single-stranded form by superinfection with bacteriophage R408 (Russel et al., 1986). Plasmid pPSO.1 was constructed by inserting by site-directed mutagenesis, a short polylinker containing unique AvrII and BssHII restriction endonuclease sites into the PB1 reporter gene of plasmid pPS0 (Somogyi et al., 1993) at position 1167 bp. All plasmid junctions were confirmed by dideoxy sequencing (Sanger et al., 1977) of single-stranded templates rescued from Escherichia coli JM101 (Yanisch-Perron et al., 1985). Plasmids pRSV17, 18 and 19 were prepared from pRSV1.1, pRSV7 and pRSV4, respectively, by inserting the sequence 5′ AATTAACCCTCACTAAA 3′ at position 2461 (with respect to the inserted RSV sequences) just upstream of the RSV slippery sequence. This introduced a unique bacteriophage T3 RNA polymerase promoter.
In vitro transcription and translation
Plasmid preparations for in vitro transcription were prepared as described previously (Brierley et al., 1989). In vitro transcription reactions employing the bacteriophage SP6 RNA polymerase were carried out essentially as described by Melton et al. (1984) and included the synthetic cap structure me7GpppG (New England Biolabs) to generate capped mRNA. Product RNA was recovered by a single extraction with phenol:chloroform (50:50, v/v) followed by ethanol precipitation in the presence of 2 M ammonium acetate. The RNA pellet was dissolved in water, and remaining unincorporated nucleotide triphosphates removed by Sephadex G-50 chromatography. RNA was recovered by ethanol precipitation, dissolved in water and checked for integrity by electrophoresis on 1.5% (w/v) agarose gels containing 0.1% (w/v) sodium dodecyl sulphate (SDS). In ribosomal frameshift assays, serial dilutions of purified mRNAs were translated in RRL as described (Brierley et al., 1989). Translation products were analysed on SDS-10% (w/v) polyacrylamide gels according to standard procedures (Hames, 1981). The relative abundance of non-frameshifted or frameshifted products on the gels was determined by direct measurement of [35S]methionine incorporation using a Packard Instant Imager 2024 and adjusted to take into account the differential methionine content of the products. Frameshift efficiencies were calculated from those dilutions of RNA where translation was highly processive (RNA concentrations of 10 to 25 μg RNA/ml of reticulocyte lysate).
RNA structure mapping by primer extension
RNA structure mapping by primer extension was carried out by the general procedure of Stern et al. (1988) largely as described (Marczinke et al., 1994). RNAs for secondary structure probing were prepared by in vitro transcription of PvuII-cut pRSV1 plasmid template using bacteriophage T7 RNA polymerase. Transcription reactions were on a 200 μl scale and contained 20 μg plasmid DNA, 2.5 mM of each rNTP and 500 units T7 RNA polymerase (NEB) in a buffer containing 40 mM Tris (pH 8), 15 mM MgCl2 and 5 mM DTT. After one hour at 37°C, 100 units DNase I was added and the incubation continued for a further 30 minutes. Nucleic acids were harvested by extraction with phenol/chloroform (1:1) and ethanol precipitation. DNA fragments were removed by Sephadex G50 chromatography and the RNA transcripts concentrated by ethanol precipitation. The RNA was quantified by spectrophotometry and its integrity checked by electrophoresis on a 1% agarose gel containing 0.1% SDS. Prior to biochemical analysis, the RNA was heated at 55°C for ten minutes and cooled slowly to room temperature over a 30–45 minutes period in the relevant structure mapping buffer (see below). RNA modification reactions were carried out as described by Christiansen et al. (1990). The following RNA modifications were made, with approximately 1 μg RNA per reaction mixture: RNase V1 (0 to 0.5 units), RNase T1 (0 to 40 units), DMS (0 to 0.3%), kethoxal (0 to 3.5 mg/ml), CMCT (0 to 12.5 mg/ml) and DEPC (0 to 2.5%). The RNase treatments were carried out at 0°C for 30 minutes in 20 μl reaction volumes under non-denaturing conditions (30 mM Tris (pH 7.8), 20 mM MgCl2, 300 mM KCl, 1 mM DTT). For digestion under semi-denaturing conditions, the RNA was heated to 95°C for one minute in 30 mM Tris (pH 7.8), 1 mM EDTA, 300 mM KCl, 1 mM DTT and immediately flash frozen at −70°C. The RNA was thawed on ice prior to addition of the relevant RNase. All enzyme reactions were terminated by extraction with phenol:chloroform and the digested RNA precipitated with three volumes of ethanol in the presence of 0.1 M sodium acetate (pH 4.8) and 10 μg yeast tRNA carrier. Chemical modifications were carried out at 30°C for 20 minutes in 200 μl reaction volumes under non-denaturing conditions (70 mM Hepes-KOH (pH 7.8), 10 mM MgCl2, 270 mM KCl, 1 mM DTT) except for CMCT, where the reaction volume was 40 μl. For digestion under semi-denaturing conditions, the reaction buffer contained EDTA (1 mM) and MgCl2 was omitted. The RNA was heated to 95°C for one minute, the chemicals added and incubation continued at 55°C for five minutes. Chemically modified RNA was precipitated directly by addition of three volumes ethanol and 10 μg carrier tRNA. However, in the case of DMS, the reaction was first terminated by addition of 0.2 volumes DMS stop buffer (1 M Tris-acetate (pH 7.5), 1 M 2-mercaptoethanol, 1.5 M sodium acetate, 0.1 mM EDTA) prior to ethanol precipitation. Sites of RNA modification were mapped by primer extension using avian myeloblastosis virus reverse transcriptase (AMV RT) and oligonucleotide primers complementary to RSV nucleotides 2692–2676 (BM4), 2639–2623 (BM5) and 2600–2584 (BM14) as described by Christiansen et al. (1990). RNA sequencing ladders were prepared by the di-deoxy chain termination method of Sanger et al. (1977) using pRSV1/PvuII RNA template and AMV RT.
Structure probing of end-labelled RNAs
RNAs for secondary structure probing by the end-labelling procedure were prepared by in vitro transcription of DdeI-cut pRSV17, 18 or 19 plasmid templates using bacteriophage T3 RNA polymerase. Transcription reactions were on a 200 μl scale and contained 20 μg plasmid DNA, 2.5 mM of each rNTP and 500 units T3 RNA polymerase (NEB) in a buffer containing 40 mM Tris (pH 8), 15 mM MgCl2 and 5 mM DTT. After three hours at 37°C, 100 units DNase I was added and the incubation continued for a further 30 minutes. Nucleic acids were harvested by extraction with phenol/chloroform (1:1) and ethanol precipitation. DNA fragments were removed by Sephadex G50 chromatography and the RNA transcripts concentrated by ethanol precipitation. The RNA was quantified by spectrophotometry and its integrity checked by electrophoresis on a 2% agarose gel containing 0.1% SDS. Transcripts (10 μg) were 5′-end labelled with [γ-32P]ATP using a standard dephosphorylation-rephosphorylation strategy (Ten Dam et al., 1994), purified from 15% acrylamide-urea gels and dissolved in water. The structure probing experiments followed the general principles outlined by others Van Belkum et al 1988, Wyatt et al 1990, Polson and Bass 1994. Prior to analysis, the RNA was heated at 55°C for ten minutes and cooled slowly to room temperature over a 30–45 minute period. All reactions contained 10,000–50,000 cpm 5′-end-labelled RNA transcript and 10 μg baker’s yeast tRNA as carrier. RNase probing reactions were carried out in 50 μl reaction volumes. Probing with RNase T1 (Amersham) was on ice for 20 minutes in 50 mM sodium cacodylate (pH 7), 2 mM MgCl2 and 0–1 unit T1; RNase V1 (Pharmacia) at 25°C for 20 minutes in 10 mM Tris (pH 8), 2 mM MgCl2, 0.1 M KCl and 0–0.35 unit V1; RNase U2 (USB) on ice for 20 minutes in 20 mM sodium acetate (pH 4.8), 2 mM MgCl2, 100 mM KCl and 0–0.2 unit U2. Enzymatic reactions were stopped by addition of 150 μl ethanol and the RNA recovered by centrifugation. RNAs were dissolved in water, mixed with an equal volume of formamide gel loading buffer (95% (v/v) formamide, 10 mM EDTA, 0.1% bromophenol blue, 0.1% xylene cyanol), boiled for two minutes and analysed on 10 or 15% polyacrylamide-7 M urea sequencing-type gels. Chemical probing experiments were performed with lead acetate and imidazole in 10 μl reaction volumes. Lead probing was at 25°C for five minutes in 20 mM Hepes-NaOH (pH 7.5), 5 mM Mg acetate, 50 mM K acetate and 1–20 mM Pb acetate. Reactions were stopped by addition of EDTA to 33 mM, the RNA recovered by ethanol precipitation, redissolved in water and prepared for gel loading as above. For imidazole probing, the end-labelled RNA was mixed with 10 μg carrier tRNA, dried in a desiccator and redissolved in 10 μl 2 M imidazole (pH 7) containing 40 mM NaCl and 10 mM MgCl2. After incubation at 37°C for two to six hours, the reaction was stopped by addition of 10 μl of a fresh solution of 2% (w/v) lithium perchlorate in acetone. The RNA was recovered by centrifugation, washed with acetone, dried, dissolved in water and prepared for gel loading as above. All structure probing gels included an alkaline hydrolysis ladder of the relevant RNA as a size marker, prepared by dissolving the dried pellet from 3 μl of end-labelled RNA and 10μg carrier tRNA in 3 μl 22.5 mM NaHCO3, 2.5 mM Na2CO3 and boiling for one to 2.5 minutes. After addition of an equal volume of formamide gel loading buffer, the sample was boiled for a further two minutes and loaded immediately onto the gel.
Acknowledgements
This work was supported by the Medical Research Council, UK, the Biotechnology and Biological Sciences Research Council, UK and the Wellcome Trust, UK. We thank Dr Victoria Perreau for her advice on structure probing and Dr Paul Digard for critical reading of the manuscript.
Footnotes
Edited by D. E. Draper
References
- Bai J, Payne L.N, Skinner M.A. HPRS-103 (exogenous avian-leukosis virus, subgroup-J) has an env gene related to those of endogenous elements EAV-0 and E51 and an E-element found previously only in sarcoma viruses. J. Virol. 1995;69:779–784. doi: 10.1128/jvi.69.2.779-784.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieth E, Darlix J.L. Complete nucleotide sequence of a highly infectious avian-leukosis virus. Nucl. Acids Res. 1992;20:367. doi: 10.1093/nar/20.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley I. Ribosomal frameshifting on viral RNAs. J. Gen. Virol. 1995;76:1885–1892. doi: 10.1099/0022-1317-76-8-1885. [DOI] [PubMed] [Google Scholar]
- Brierley I, Digard P, Inglis S. Characterisation of an efficient coronavirus ribosomal frameshifting signal: requirement for an RNA pseudoknot. Cell. 1989;57:537–547. doi: 10.1016/0092-8674(89)90124-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley I, Rolley N.J, Jenner A.J, Inglis S.C. Mutational analysis of the RNA pseudoknot component of a coronavirus ribosomal frameshifting signal. J. Mol. Biol. 1991;220:889–902. doi: 10.1016/0022-2836(91)90361-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley I, Jenner A.J, Inglis S.C. Mutational analysis of the “slippery sequence” component of a coronavirus ribosomal frameshifting signal. J. Mol. Biol. 1992;227:463–479. doi: 10.1016/0022-2836(92)90901-U. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D. Nidovirales, a new order comprising Coronaviridae and Arteriviridae. Arch. Virol. 1997;142:629–633. [PubMed] [Google Scholar]
- Chen X, Chamorro M, Lee S.I, Shen L.X, Hines J.V, Tinoco I, Jr, Varmus H.E. Structural and functional studies of retroviral RNA pseudoknots involved in ribosomal frameshifting: nucleotides at the junction of the two stems are important for efficient ribosomal frameshifting. EMBO J. 1995;14:842–852. doi: 10.1002/j.1460-2075.1995.tb07062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.Y, Kang H.S, Shen L.X, Chamorro M, Varmus H.E, Tinoco I. A characteristic bent conformation of RNA pseudoknots promotes −1 frameshifting during translation of retroviral RNA. J. Mol. Biol. 1996;260:479–483. doi: 10.1006/jmbi.1996.0415. [DOI] [PubMed] [Google Scholar]
- Christiansen J, Egebjerg J, Larsen N, Garrett R.A. Analysis of rRNA structure: experimental and theoretical considerations. In: Spedding G, editor. Ribosomes and Protein Synthesis, A Practical Approach. Oxford University Press; Oxford: 1990. pp. 229–252. [Google Scholar]
- De Lorbe W.J, Luciw P.A, Goodman H.M, Varmus H.E, Bishop J.M. Molecular cloning and characterization of avian sarcoma virus circular DNA molecules. J. Virol. 1980;36:50–61. doi: 10.1128/jvi.36.1.50-61.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotto G.P, Enea V, Zinder N.D. Functional analysis of bacteriophage f1 intergenic region. Virology. 1981;114:463–473. doi: 10.1016/0042-6822(81)90226-9. [DOI] [PubMed] [Google Scholar]
- Du Z.H, Holland J.A, Hansen M.R, Giedroc D.P, Hoffman D.W. Base-pairings within the RNA pseudoknot associated with the simian retrovirus-1 gag-pro frameshift site. J. Mol. Biol. 1997;270:464–470. doi: 10.1006/jmbi.1997.1127. [DOI] [PubMed] [Google Scholar]
- Ehresmann C, Baudin F, Mougel M, Romby P, Ebel J-P, Ehresmann B. Probing the structure of RNAs in solution. Nucl. Acids Res. 1987;15:9109–9128. doi: 10.1093/nar/15.22.9109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farabaugh P.J. Programmed translational frameshifting. Microbiol. Rev. 1996;60:103–134. doi: 10.1128/mr.60.1.103-134.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hames B.D. An introduction to polyacrylamide gel electrophoresis. In: Hames B.D, Rickwood D, editors. Gel Electrophoresis of Proteins – A Practical Approach. IRL Press; Oxford: 1991. pp. 1–91. [Google Scholar]
- Hughes S.H, Greenhouse J.J, Petropoulos C.J, Sutrave P. Adaptor plasmids simplify the insertion of foreign DNA into helper-independent retroviral vectors. J. Virol. 1987;61:3004–3012. doi: 10.1128/jvi.61.10.3004-3012.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Varmus H.E. Expression of the Rous sarcoma virus pol gene by ribosomal frameshifting. Science. 1985;230:1237–1242. doi: 10.1126/science.2416054. [DOI] [PubMed] [Google Scholar]
- Jacks T, Madhani H.D, Masiarz F.R, Varmus H.E. Signals for ribosomal frameshifting in the Rous sarcoma virus gag-pol region. Cell. 1988;55:447–458. doi: 10.1016/0092-8674(88)90031-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson R.J, Hunt T. Preparation and use of nuclease-treated rabbit reticulocyte lysates for the translation of eukaryotic messenger RNA. Methods Enzymol. 1983;96:50–74. doi: 10.1016/s0076-6879(83)96008-1. [DOI] [PubMed] [Google Scholar]
- Kang H.S, Tinoco I. A mutant RNA pseudoknot that promotes ribosomal frameshifting in mouse mammary tumor virus. Nucl. Acids Res. 1997;25:1943–1949. doi: 10.1093/nar/25.10.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H.S, Hines J.V, Tinoco I. Conformation of a non-frameshifting RNA pseudoknot from mouse mammary tumor virus. J. Mol. Biol. 1996;259:135–147. doi: 10.1006/jmbi.1996.0308. [DOI] [PubMed] [Google Scholar]
- Kim K.H, Lommel S.A. Identification and analysis of the site of -1 ribosomal frameshifting in red clover necrotic mosaic virus. Virology. 1994;200:574–582. doi: 10.1006/viro.1994.1220. [DOI] [PubMed] [Google Scholar]
- Kolchanov N.A, Titov I.I, Vlassova I.E, Vlassov V.V. Chemical and computer probing of RNA structure. Prog. Nucl. Acid Res. Mol. Biol. 1996;53:131–196. doi: 10.1016/S0079-6603(08)60144-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzyzosiak W.J, Marciniec T, Wiewiorowski M, Romby P, Ebel J.-P, Giege R. Characterisation of the lead (II)-induced cleavages in tRNAs in solution and effect of the Y-base removal in yeast tRNA Phe. Biochemistry. 1988;27:5771–5777. doi: 10.1021/bi00415a056. [DOI] [PubMed] [Google Scholar]
- Kunkel T.A. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl Acad. Sci. USA. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marczinke B, Bloys A.J, Brown T.D.K, Willcocks M.M, Carter M.J, Brierley I. The human astrovirus RNA-dependent RNA polymerase coding region is expressed by ribosomal frameshifting. J. Virol. 1994;68:5588–5595. doi: 10.1128/jvi.68.9.5588-5595.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melton D.A, Krieg P.A, Robagliati M.R, Maniatis T, Zinn K, Green M.R. Efficient in vitro synthesis of biologically active RNA and RNA hybridisation probes from plasmids containing a bacteriophage SP6 promoter. Nucl. Acids Res. 1984;12:7035–7056. doi: 10.1093/nar/12.18.7035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda T, Ikeda S, Watanabe S, Hatsushika M, Akiyama K, Mitsunobu F. Molecular cloning, complete nucleotide sequence and gene structure of the provirus genome of a retrovirus produced in a human lymphoblastoid cell line. Virology. 1988;167:468–476. [PubMed] [Google Scholar]
- Polson A.G, Bass B.L. Preferential selection of adenosines for modification by double-stranded RNA adenosine deaminase. EMBO J. 1994;13:5701–5711. doi: 10.1002/j.1460-2075.1994.tb06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power M.D, Marx P.A, Bryant M.L, Gardner M.B, Barr P.J, Luciw P.A. Nucleotide-sequence of SRV-1, a type-D simian acquired immune-deficiency syndrome retrovirus. Science. 1986;231:1567–1572. doi: 10.1126/science.3006247. [DOI] [PubMed] [Google Scholar]
- Russel M, Kidd S, Kelley M.R. An improved filamentous helper phage for generating single-stranded plasmid DNA. Gene. 1986;45:333–338. doi: 10.1016/0378-1119(86)90032-6. [DOI] [PubMed] [Google Scholar]
- Sanger F, Nicklen S, Coulson A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl Acad. Sci. USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarid R, Chajut A, Gak E, Kim Y, Hixson C.V, Oroszlan S, Tronick S.R, Gazit A, Yaniv A. Genome organisation of a biologically-active molecular clone of the lymphoproliferative disease virus of turkeys. Virology. 1994;204:680–691. doi: 10.1006/viro.1994.1584. [DOI] [PubMed] [Google Scholar]
- Schwartz D.E, Tizard R, Gilbert W. Nucleotide sequence of Rous sarcoma virus. Cell. 1983;32:853–869. doi: 10.1016/0092-8674(83)90071-5. [DOI] [PubMed] [Google Scholar]
- Shen L.X, Tinoco I. The structure of an RNA pseudoknot that causes efficient frameshifting in mouse mammary tumor virus. J. Mol. Biol. 1995;247:963–978. doi: 10.1006/jmbi.1995.0193. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Jenner A.J, Brierley I, Inglis S.C. Ribosomal pausing during translation of an RNA pseudoknot. Mol. Cell. Biol. 1993;13:6931–6940. doi: 10.1128/mcb.13.11.6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonigo P, Barker C, Hunter E, Wain-Hobson S. Nucleotide-sequence of Mason-Pfizer monkey virus – an immunosuppressive D-type retrovirus. Cell. 1986;45:375–385. doi: 10.1016/0092-8674(86)90323-5. [DOI] [PubMed] [Google Scholar]
- Stern S, Moazed D, Noller H.F. Analysis of RNA structure using chemical and enzymatic probing monitored by primer extension. Methods Enzymol. 1988;164:481–489. doi: 10.1016/s0076-6879(88)64064-x. [DOI] [PubMed] [Google Scholar]
- Stewart L, Vogt V.M. Proteolytic cleavage at the Gag-Pol junction in avian leukosis virus: differences in vitro and in vivo. Virology. 1994;204:45–59. doi: 10.1006/viro.1994.1509. [DOI] [PubMed] [Google Scholar]
- Sung D, Kang H. Mutational analysis of the RNA pseudoknot involved in efficient ribosomal frameshifting in simian retrovirus 1. Nucl. Acids Res. 1998;26:1369–1372. doi: 10.1093/nar/26.6.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ten Dam E, Pleij K, Draper D. Structural and functional aspects of RNA pseudoknots. Biochemistry. 1992;31:11665–11676. doi: 10.1021/bi00162a001. [DOI] [PubMed] [Google Scholar]
- Ten Dam E, Brierley I, Inglis S.C, Pleij C. Identification and analysis of the pseudoknot-containing gag-pro ribosomal frameshift signal of simian retrovirus-1. Nucl. Acids Res. 1994;22:2304–2310. doi: 10.1093/nar/22.12.2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ten Dam E, Verlaan P, Pleij C. Analysis of the role of the pseudoknot component in the SRV-1 gag-pro ribosomal frameshift signal: loop lengths and stability of the stem regions. RNA. 1995;1:146–154. [PMC free article] [PubMed] [Google Scholar]
- Thayer R.M, Power M.D, Bryant M.L, Gardner M.B, Barr P.J, Luciw P.A. Sequence relationships of type-D retroviruses which cause simian acquired immunodeficiency syndrome. Virology. 1987;157:317–329. doi: 10.1016/0042-6822(87)90274-1. [DOI] [PubMed] [Google Scholar]
- Van Belkum A, Verlaan P, Bing KunJ, Pleij C, Bosch L. Temperature dependent chemical and enzymatic probing of the tRNA-like structure of TYMV RNA. Nucl. Acids Res. 1988;16:1931–1950. doi: 10.1093/nar/16.5.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlassov V.V, Zuber G, Felden B, Behr J-P, Giege R. Cleavage of tRNA with imidazole and spermine imidazole constructs: a new approach for probing RNA structure. Nucl. Acids Res. 1995;23:3161–3167. doi: 10.1093/nar/23.16.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt J.R, Puglisi J.D, Tinoco I. RNA pseudoknots; stability and loop size requirements. J. Mol. Biol. 1990;214:455–470. doi: 10.1016/0022-2836(90)90193-P. [DOI] [PubMed] [Google Scholar]
- Yanish-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: nucleotide sequence of the M13 mp18 and pUC19 vectors. Gene. 1985;33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- Young J.F, Desselberger U, Graves P, Palese P, Shatzman A, Rosenberg M. Cloning and expression of influenza virus genes. In: Laver W.G, editor. The Origin of Pandemic Influenza Viruses. Elsevier Science; Amsterdam: 1983. pp. 129–138. [Google Scholar]