

Abstract
Both full-length and subgenomic negative-strand RNAs are initiated at the 3′ terminus of the positive-strand genomic RNA of the arterivirus, simian hemorrhagic fever virus (SHFV). The SHFV 3′(+) non-coding region (NCR) is 76 nts in length and forms a stem loop (SL) structure that was confirmed by ribonuclease structure probing. Two cell proteins, p56 and p42, bound specifically to a probe consisting of the SHFV 3′(+)NCR RNA. The 3′(+)NCR RNAs of two additional members of the arterivirus genus specifically interacted with two cell proteins of the same size. p56 was identified as polypyrimidine tract-binding protein (PTB) and p42 was identified as fructose bisphosphate aldolase A. PTB binding sites were mapped to a terminal loop and to a bulged region of the SHFV 3′SL structure. Deletion of either of the PTB binding sites in the viral RNA significantly reduced PTB binding activity, suggesting that both sites are required for efficient binding of this protein. Changes in the top portion of the SHFV 3′SL structure eliminated aldolase binding, suggesting that the binding site for this protein is located near the top of the SL. These cell proteins may play roles in regulating the functions of the genomic 3′ NCR.
Keywords: Stem loop, simian hemorrhagic fever virus, RNA, Polypyrimidine tract binding protein, Aldolase, RNA–protein interactions
1. Introduction
Simian hemorrhagic fever virus (SHFV) is a member of the family Arteriviridae, which also includes equine arteritis virus (EAV), lactate dehydrogenase-elevating virus (LDV) and porcine respiratory and reproductive syndrome virus (PRRSV). The families Arteriviridae, Coronaviridae and Roniviridae make up the order Nidovirales. The arterivirus genome is a polycistronic, single-stranded RNA of positive polarity with a 5′ type I cap (Sagripanti et al., 1986), and a 3′ poly(A) tract (Brinton et al., 1986, Contag et al., 1986, Sagripanti, 1985, van Berlo et al., 1982). Arterivirus genomes range in length from 12.7 to 15.7 kb with SHFV being the longest (accession numbers: EAV, NC002532; LDV, NC002534; PRRSV, NC001961; SHFV, NC003092). The viral non-structural genes are encoded by ORF1a/ORF1b, located at the 5′ end of the genome and are translated from the genomic RNA. The structural genes are encoded at the 3′ end of the genome and are expressed from a nested set of 3′ and 5′ co-terminal subgenomic mRNAs that are generated by a discontinuous transcription process (Sawicki and Sawicki, 1995, van Marle et al., 1999, Snijder and Meulenberg, 2001). The 5′NCRs of the various arterivirus genomic and subgenomic RNAs range in length from 156 to 212 nts, while the 3′NCRs vary from 62 to 150 nts. The lengths of the SHFV 5′ and 3′NCRs are 209 and 76 nts, respectively.
The cis-acting sequences required for RNA replication have not yet been precisely mapped. However, previous studies with defective interfering (DI) RNA genomes of the coronavirus, mouse hepatitis virus (MHV), have shown that the 5′ and 3′NCRs are required for plus-strand RNA synthesis (Kim et al., 1993, Lin and Lai, 1993) and that the 3′ terminal nucleotides of the MHV 3′NCR as well as the poly(A) tract are required for minus-strand RNA synthesis (Lin et al., 1994). An arterivirus DI RNA that contained approximately 300 nts from both the 5′ and 3′ ends replicated in cells co-infected with helper virus, indicating that these regions contain sufficient cis-acting signals for RNA replication (Molenkamp et al., 2000).
Formation of RNA structures by the 3′ sequences of coronaviruses has been previously reported. For MHV, a bulged secondary structure located within the 3′NCR adjacent to the nucleocapsid (N) gene stop codon was demonstrated to be required for viral replication (Hsue and Masters, 1997, Hsue et al., 2000). The 3′ side of the bottom portion of this structure was subsequently shown to form a pseudoknot with another 3′NCR SL located downstream (Goebel et al., 2004). These two structures were found to be mutually exclusive and could potentially function as a molecular switch during viral replication. In the bovine coronavirus (BCV) genome, a pseudoknot was also reported to form immediately downstream of a SL similar to the one reported in the MHV 3′NCR and this tertiary interaction was required for coronavirus defective genome replication (Williams et al., 1999). For the arterivirus, PRRSV, a SL was predicted near the 5′ end of ORF7 and some evidence suggested that nucleotides within the top loop of this structure formed a tertiary interaction (kissing interaction) with nucleotides in the top loop of a predicted SL in the 3′NCR (Verheije et al., 2002).
Four cell proteins with molecular masses of 90, 70, 58 and 40 kDa were previously reported to bind to the 3′ terminal 42 nts of the genomic RNA of MHV. The 90 kDa cell protein was subsequently identified as mitochondrial aconitase (Nanda and Leibowitz, 2001). Heterogeneous nuclear RNP A1 (hnRNP A1) has also been reported to bind to the MHV genomic 3′NCR and to mediate a genomic 3′-5′ interaction (Huang and Lai, 2001). In a separate study, the 73 kDa cytoplasmic poly(A)-binding protein (PABP) was shown to bind the BCV poly(A68) tract (Spagnolo and Hogue, 2000). Studies with cell mRNAs indicated that PABP facilitates 3′-5′ interactions (Wells et al., 1998).
We report here the first structure probing analysis of the 3′NCR of an arterivirus genomic RNA. This analysis confirmed the predicted secondary structure in this region. Also, this is the first report of cell proteins binding to the 3′(+) NCR of an arterivirus RNA. Two MA104 cellular proteins (p56 and p42) were shown to interact specifically with the SHFV 3′(+)NCR genomic RNA as well as with the 3′(+)NCR RNAs of two additional arteriviruses, EAV and PRRSV. The two cell proteins were identified as polypyrimidine tract-binding protein (PTB) and aldolase.
2. Materials and methods
2.1. Cells and virus
MA104, an embryonic rhesus monkey kidney cell line (Trousdale et al., 1975), was obtained from O. Nainin, Centers for Disease Control and Prevention, and was cultured in Minimal Essential Medium with 10% fetal bovine serum at 37 °C in a CO2 atmosphere. Stock virus pools were prepared in MA104 cells by infecting confluent MA104 monolayers with SHFV, strain LVR 42-0/M6941 (American Type Culture Collection, passage 2), the prototype strain of SHFV, at a MOI of 0.2. Culture media harvested 32 h after infection contained titers of approximately 5 × 108 PFU/ml.
2.2. Construction of DNA templates for in vitro RNA transcription
SHFV genomic RNA was extracted from virions with Trizol (Gibco BRL). To construct pSHFV3′NCR, cDNA was first synthesized from viral RNA by reverse transcription using SuperScript II RT (Gibco BRL) and primer P1 (Table 1 ). The cDNA was ethanol precipitated and then amplified by PCR using Taq DNA-dependent DNA polymerase (Roche) and primers P1 and P2 (Table 1). The PCR product was immediately TA-cloned into pCRII (Invitrogen) and the inserts of selected clones were sequenced to confirm that no mutations had been introduced.
Table 1.
Sequences of oligonucleotides and primers
| Oligo number | Oligonucleotide sequencea,b |
|---|---|
| O1 | 5′-AATTTAATACGACTCACTATAGGCTAAGGACTAACTGGTATATACCATAATTAAATGCATTGCA-3′ |
| O2 | 5′-ATGCATTTAATTATGGTATATACCAGTTAGTCCTTA GCCTATAGTGAGTCGTATTA-3′ |
| O3 | 5′-AATTTAATACGACTCACTATAGGCCTTCCCTAGGCTAAGGACTAACTGGTATATACCATGCATTGCA-3′ |
| O4 | 5′-ATGCATGGTATATACCAGTTAGTCCTTAGCCTAGGGAAGGCCTATAGTGAGTCGTATTA-3′ |
| O5 | 5′-AATTTAATACGACTCACTATAGGGCCAGACACTGATTATATGGTTCATATGGGTAATTATGCATTGCA-3′ |
| O6 | 5′-ATGCATAATTACCCATATGAACCATATAATCAGTGTCTGGCCCTATAGTGAGTCGTATTA-3′ |
| O7 | 5′-AATTTAATACGACTCACTATAGGGGGTAATTACCTTCCCTAGGCTAAGGAC TAATGCATTGCA-3′ |
| O8 | 5′-ATGCATTAGTCCTTAGCCTAGGGAAGGTAATTACCCCCTATAGTGAGTCGTATTA-3′ |
| O9 | 5′-GCCAGACACTGATTATATGGTTCATATGGGTCCTTCCCCCGGCTAAGGACTGG-3′ |
| O10 | 5′-TTTAATTATGGTGGATA CCAGTCCTTAGCCGGGGGAAGGACCC-3′ |
| O11 | 5′-AGTCGACTAATACGAC TCACTATAGGATGGTTCATATGGGTCC-3′ |
| O12 | 5′-ATGGTATATACCAGTCCTTAGCCTAGGGAAGGACCCATATGAACCATCCTATA-3′ |
| O13 | 5′-GCCAGACACTGATTATATGGTTCATATGGGTAAT TACCTAAAAGGCTAAGGAC-3′ |
| O14 | 5′-TTTAATTATGGTATATACCAGTTAG TCCTTAGCCTTTTAGGTAATTACCC-3′ |
| O15 | 5′-GCCAGACACTGATTATATGGTTCATATGGGTAATTACCTTCCCTAGGCTAAGGACTG-3′ |
| O16 | 5′-TTTAATTA TGGTATATACCAGTCCTTAGCCTAGGGAAGGTAATTACCC-3′ |
| Primer number | Primer sequencea |
|---|---|
| P1 | 5′-TTTAATTATGGTATATACCAGTTAGTCC-3′ |
| P2 | 5′-AAAAGTCGACTAATACGACTCACTATAGGGCCAGACACTGATTATATGGTTC-3′ |
| P3 | 5′-AAGTCGACAATAATACGACTCACTATAGGAATCAATTTTGCTGCTATCATC-3′ |
| P4 | 5′-ACCCATATGAACCATATAATCAGTGTC-3′ |
| P5 | 5′-AGTCGACTAATACGACTCACTATAGGGCCAGACACTGATTATA-3′ |
| P6 | 5′-TTTAATTATGGTGGATACCAGTCCTTAGC-3′ |
| P7 | 5′-AGTCGACTAATACGACTCAC-3′ |
| P8 | 5′-ATGGTATATACCAGTCCTTAG-3′ |
| P9 | 5′-AATTTAATACGACTCACTATAGGGAGATCTTCTGCTCTGCACAACC-3′ |
| P10 | 5′-AGTATCCTGTGTTCTCGCAC-3′ |
| P11 | 5′-GAGCGGCCGCCAGTGTG-3′ |
| P12 | 5′-TTTAATTATGGTATATACCAGTCCTTAGCCTAGGGAAGGTAATTACCC-3′ |
| P13 | 5′-AAAAGTCGACTAATACGACTCACTATAGGGACGTGGATATTCTCCTGTG-3′ |
| P14 | 5′-TTTGGTTCCTGGGTGGCTAAT-3′ |
| P15 | 5′-AAAAGTCGACTAATACGACTCACTATAGGGTTTGACAGTCAGGTGAATGG-3′ |
| P16 | 5′-TTTAATTTCGGTCACATGG-3′ |
The T7 promoter sequence is underlined.
The NsiI restriction site is in boldface type and the overhangs are italicized.
Additional plasmids containing various truncated SHFV 3′(+)NCR sequences were constructed by annealing pairs of complementary synthetic oligonucleotides (Table 1, Table 2 ). One oligonucleotide of each pair was longer than its complement to generate a four nucleotide overhang on each end after annealing of the two oligonucleotides that mimicked the overhangs generated by ApoI or PstI restriction enzyme digestion. The annealed oligonucleotide pairs were incubated with polynucleotide kinase (10 U; NEB) and inserted into pUC19 DNA that had been digested with ApoI and PstI (New England BioLabs). Selected clones were sequenced. Prior to use as templates for in vitro transcription, plasmid DNAs (ptruncA, ptruncB, ptruncC and ptruncG) were linearized by digestion with NsiI (New England BioLabs) so that the in vitro synthesized RNAs would have an exact viral 3′ sequence.
Table 2.
List of oligonucleotides, primers and plasmid templates used to generate RNA probes
| Plasmid template | Sense oligo | Antisense oligo | Corresponding RNA |
|---|---|---|---|
| ptruncA | O1 | O2 | RNA-A |
| ptruncB | O3 | O4 | RNA-B |
| ptruncC | O5 | O6 | RNA-C |
| ptruncG | O7 | O8 | RNA-G |
| pmutE | O9 | O10 | – |
| pmutF | O11 | O12 | – |
| pmutH | O13 | O14 | – |
| pmutI | O15 | O16 | – |
| Plasmid template | Sense primer | Antisense primer | Corresponding RNA |
|---|---|---|---|
| pmutE | P5 | P6 | RNA-E |
| pmutF | P7 | P8 | RNA-F |
| pmutH | P2 | P1 | RNA-H |
| pmutI | P2 | P12 | RNA-I |
| pSHFVORF93NCR | P3 | P4 | RNA-D |
| pM115128 | P13 | P14 | EAV3′(+)NCR RNA |
| pTVRORF7 | P15 | P16 | PRRSV3′(+)NCR RNA |
| pWNV3′(+)SL | P9 | P10 | WNV3′(+)SL RNA |
| pSHFV3NCR | P2 | P11 | 3NCRpl RNA |
Additional plasmids were also constructed by annealing pairs of oligonucleotides that had complementary 3′ ends. The annealed oligonucleotides were made double stranded and amplified with Taq DNA polymerase (Roche) and TA-cloned into pCRII (Invitrogen). These plasmids (pmutE, pmutF, pmutH, and pmutI) were used as templates to generate PCR products that were in turn used as templates for in vitro transcription of mutant RNAs (Table 2).
Construction of pSHFVORF9-3′NCR (Godeny et al., 1995) and pWNV3′(+)SL (Blackwell and Brinton, 1995) was described previously. pM115128, which contained the EAV 3′(+)NCR, was generously provided by Dr. Eric Snijder (Leiden University Medical Center, The Netherlands) and pTVRORF7, which contained the PRRSV 3′(+)NCR was kindly provided by Dr. Kay Faaberg (University of Minnesota, St. Paul, MN).
2.3. In vitro RNA synthesis
Radiolabeled RNA probes were generated by in vitro transcription with T7 RNA polymerase in a 20 μl reaction volume. The reactions contained linearized plasmid or PCR product DNA as template, 80 mM HEPES, 16 mM MgCl2, 2 mM spermidine, 10 mM DTT, 1 mM ATP, CTP, and UTP, 50 μM GTP, 50 μCi [α-32P]GTP (3000 Ci/mmol; NEN), and 1 μl T7 RNA polymerase. The T7 RNA polymerase was expressed and purified from a plasmid that was a gift from Dr. William McAllister, State University of New York, Brooklyn, NY (He et al., 1997). The reactions were incubated at 37 °C for 2 h. RNase-free DNase I (10 units; Roche) was added and after incubation for an additional 20 min at 37 °C, the RNA probes were gel purified as previously described (Hwang and Brinton, 1998), resuspended in gel-shift buffer (GSB) (14 mM HEPES, pH 7.5, 6 mM Tris–Cl, pH 7.5, 1 mM EDTA, 1 mM DTT, 60 mM KCl) and stored at −80 °C until use. The probes were quantified using a scintillation counter (Beckman, LS 6500). The specific activities of the probes were calculated as previously described (Blackwell and Brinton, 1997) and were approximately 1.5 × 106 cpm/pmol.
Competitor RNAs [e.g. unlabeled SHFV 3′(+)NCR and WNV 3′(+)SL] were synthesized in 20 μl reactions, RNA used for ribonuclease structure probing was synthesized in 50 μl reactions and RNA used for RNA affinity chromatography was synthesized in 2 ml reactions. These reactions were performed as described above except that 1 mM GTP was used. The synthesized, unlabeled RNA was ethanol precipitated, resuspended in 1× denaturing loading buffer (7 M urea, TBE buffer, 0.025% bromophenol blue), and resolved by 6% denaturing polyacrylamide gel electrophoresis (PAGE). The RNA was visualized by placing the gel on a TLC plate and briefly irradiating with a 254 nm UV lamp (Ultra-violet Products Inc.). The band was excised and RNA was eluted from the gel slice by electrophoresis at 200 V for 2 h at room temperature using an Elutrap electroeluter (Schleicher & Schuell), ethanol precipitated and resuspended in either GSB for competition gel mobility shift assays, in annealing buffer (AB) (10 mM Tris–HCl, pH 7.5, 0.1 M NaCl, 1 mM DTT, 1 mM EDTA, pH 8) for ribonuclease structure probing experiments or in water for RNA affinity chromatography. RNA concentrations were determined by spectrophotometry using an Ultrospec2000 UV/Visible Spectrophotometer (Pharmacia Biotech).
2.4. Preparation of SHFV-infected and mock-infected MA104 cytoplasmic extracts
Cytoplasmic extracts were prepared as previously described with some modifications (Hwang and Brinton, 1998). MA104 cells were grown to confluency in five T-150 flasks. Cultures were infected with SHFV at a MOI of 0.5 and then the cells were manually harvested at 7 h after infection. Cells were pelleted by centrifugation at 150 × g for 5 min at 4 °C and resuspended in cytolysis buffer (CB) (10 mM HEPES, pH 7.4, 100 mM NaCl, 1% Triton X-100, 5 mM DTT, 0.1 M PMSF, 10 μg/ml leupeptin) at 5 × 107 cells/ml either immediately or after storage at −80 °C, vortexed for 30 s, and then kept on ice for 30 min. Nuclei in the lysate were pelleted by centrifugation at 2200 × g for 5 min. The supernatant was centrifuged at 100,000 × g for 1 h at 4 °C and the resulting supernatant (S100) was aliquoted and stored at −80 °C until use. The total protein concentration of each extract was determined using the bicinchoninic acid (BCA) protein assay (Pierce).
2.5. Gel mobility shift assays
An MA104 S100 cytoplasmic extract (250 ng) was incubated with poly(I)–poly(C) (400 ng) and a 32P-labeled RNA probe (2700 cpm; 1.8 fmol) in GSB in a total volume of 10 μl for 15 min at room temperature. For competition gel mobility shift assays, various amounts of unlabeled RNAs were added to the reaction 10 min prior to the addition of the 32P-labeled RNA probe. The RNA–protein complexes (RPCs) were analyzed on 10% non-denaturing polyacrylamide gels containing 2.5% glycerol that were prerun for 10 min at 150 V and then electrophoresed at 150 V at room temperature. The gel was dried and analyzed by autoradiography.
2.6. UV-induced cross-linking assays
An MA104 S100 cytoplasmic extract (1 μg) was incubated with poly(I)–poly(C) (1 μg or 600 ng) and a 32P-labeled RNA probe (35,000 cpm; 23.3 fmol) in GSB in a total volume of 30 μl at room temperature for 15 min. The reactions were then placed in an ice bath and irradiated in a UV chamber (GS Gene Linker, BioRad) at a wavelength of 254 nm, which corresponds to 125 mJ/s, for 30 min. RNase A (20 μg) was then added and the reaction was incubated for 15 min at 37 °C to digest unbound RNA. Cross-linked RPCs were precipitated with acetone/methanol (1:1), resuspended in 1× SDS sample buffer, boiled for 2 min, analyzed by 10% SDS–PAGE and detected by autoradiography.
2.7. Thermodynamic prediction of RNA secondary structure
The secondary structures of the wildtype, truncated, and deleted SHFV 3′(+)NCR RNAs were thermodynamically predicted using Mfold (Mathews et al., 1999, Zuker et al., 1999) accessible at http://www.bioinfo.rpi.edu/∼zukerm/.
2.8. Ribonuclease probing
3NCRpl RNA, consisting of the SHFV 3′(+)NCR, three A residues of the poly(A) tract and 43 nts from pCR2.1 (Invitrogen) at the 3′ end, was subjected to limited RNase digestion followed by primer extension as previously described by Blackwell and Brinton (1997) with some modifications. 3NCRpl RNA (0.5 pmol) and a 17-mer primer (P11) (20 fmol), that was complementary to the 3′ terminal plasmid sequence, were annealed in 10 μl of AB by heating to 70 °C for 1 min and gradually cooling to 30 °C over a 15 min period. A single-strand specific ribonuclease, RNase A (10−7 or 10−6 units), RNase T2 (10−2 or 10−1 units), or RNase T1 (10−3 or 10−2 units), was then added and the mixture was incubated at room temperature for 10 min followed by 10 min at 30 °C. The partially digested RNA and annealed primer were phenol:chloroform:isoamyl extracted, ethanol precipitated, resuspended in 7.7 μl of water and then used as template for reverse transcription with SuperScript II RT (Gibco BRL). The 20 μl reactions contained 50 μCi of 32P-dATP (NEN), 0.4 μM dATP and 40 μM dCTP, dGTP, and dUTP, were incubated at 42 °C and were terminated after 40 min by the addition of 80 μl of stop solution (95% deionized formamide, 20 mM EDTA, 0.05% bromophenol blue, and 0.05% xylene cyanol FF). The reactions were then diluted 1:10 in stop solution, heated for 3 min at 80 °C and analyzed by 9% denaturing-PAGE and autoradiography. In a parallel reaction, undigested 3NCRpl RNA was sequenced as previously described (Nainan et al., 1991).
2.9. RNA affinity chromatography
An RNA affinity column was prepared as previously described (Blyn et al., 1996, Blyn et al., 1997). The 3′ end of the SHFV 3′(+)NCR RNA was covalently linked to an agarose-adipic acid matrix (Amersham Pharmacia Biotech). An MA104 S100 cytoplasmic extract prepared from five T150 flasks in 5 ml of CB was incubated on ice for 30 min with a mixture of non-specific RNA competitors [poly (I)–poly (C) (2 mg), total yeast RNA (1 mg), and WNV 3′(+)SL RNA (300 μg)]. The ratio of competitor RNA to protein used in the cell extract applied to the RNA affinity column was three times less than that used in the UV-induced cross-linking assays because higher concentrations of competitor RNAs caused aggregation problems. The extract was then diluted 1:1 in GSB and passed over the RNA affinity column three times. Fractions were collected in tubes on ice. The column was subsequently washed with four column volumes of GSB and then with one column volume of 0.2 M NaCl, 0.05% Triton X-100. Bound proteins were eluted with one column volume of 3 M NaCl, 0.05% Triton X-100. Both the 0.2 M NaCl fraction and the 3 M NaCl fraction were concentrated and buffer exchanged into storage buffer (SB) (20 mM sodium phosphate, pH 7.4, 1 mM EDTA, 100 mM NaCl, 20% glycerol) using a Centricon-30 concentration cell (Millipore) and stored at 4 °C until analysis. A mock column was prepared without RNA and treated in an identical manner.
2.10. Western blotting analysis
Proteins were resolved by 9% SDS–PAGE and electrophoretically transferred to a PVDF membrane (0.45 μm pore size; Osmonics). The membrane was blocked with 5% non-fat, dry milk in PBS/0.05% Tween-20, incubated first with either rabbit anti-PTB antibody (1:2000, Intronn LLC, Durham, NC) or goat anti-aldolase antibody (1:2000, Chemicon), washed with PBS/0.05% Tween-20 and then incubated with either a goat anti-rabbit IgG (1:2000) or a donkey anti-goat IgG (1:2000) secondary antibody (Santa Cruz). The membrane was washed twice with PBS/0.05% Tween-20, once with PBS and then incubated with SuperSignal West Pico chemiluminescence substrate (Pierce).
2.11. Immunoprecipitation of UV-induced cross-linked proteins
Protein A-sepharose or Protein G-sepharose was diluted 1:1 in dilution buffer (DB) (0.01 M Tris–Cl, pH 8, 0.1 M NaCl, 0.025% NaN3, 0.1% Triton X-100, 0.5% non-fat, dry milk). Rabbit anti-PTB antibody (5 μl) was conjugated to Protein A-sepharose and goat anti-aldolase antibody (5 μl) was conjugated to Protein G-sepharose (Amersham Pharmacia Biotech) according to the manufacturer's instructions. Rabbit anti-hnRNP A1 antibody (a gift from Dr. Gideon Dreyfuss, Howard Hughes Medical Institute, University of Pennsylvania, Philadelphia, PA) was conjugated to Protein A-Sepharose and was used as a control antibody. 32P-labeled SHFV 3′(+)NCR RNA was cross-linked to proteins in an MA104 S100 cytoplasmic extract and incubated with RNase A as described above except that the reaction volume was 90 μl. The reactions were then incubated with the Protein A (or G)-Sepharose-antibody complexes for 1 h at 4 °C with constant rotation. The immune complexes were washed three times with DB, once with TSA (0.01 M Tris–Cl, pH 8, 0.1 M NaCl, 0.025% NaN3), and once with 0.05 M Tris–Cl (pH 6.8). The complexes were pelleted after each wash via low speed centrifugation. The final immunoprecipitates were heated to 100 °C for 5 min in SDS sample buffer and analyzed by 10% SDS–PAGE.
2.12. Partial purification of recombinant PTB
Bacteria containing the plasmid pETPTB (Patton et al., 1991) were kindly provided by Dr. James Patton, Vanderbilt University, Nashville, TN. This plasmid encodes PTB with an N-terminal hexahistidine tag. PTB expression was induced with IPTG (1 mM). The bacteria were pelleted, resuspended in purification buffer (PB) (20 mM Tris–HCl, pH 7.9, 500 mM NaCl, 0.05% Tween-20), supplemented with protease inhibitor cocktail (50 μl, Sigma) and lysed with a French pressure cell press (SLM Instruments Inc.). The cell extract was clarified by centrifugation and the resulting supernatant was filtered (0.2 μm pore size, Gelman), diluted in PB, and passed through a nickel column (His-Bind, Novagen). The column was first washed with PB (50 ml), then with PB + 20 mM imidazole (5 ml), and finally with PB + 60 mM imidazole (1 ml). Bound proteins were eluted with PB + 500 mM imidazole (1 ml) and the eluate was buffer exchanged into SB. His-PTB was further purified from the eluate with a Ni-NTA spin column (Qiagen) according to the manufacturer's instructions.
2.13. Preparation of oligonucleotides
Oligonucleotides were synthesized at the Georgia State University DNA Core Facility using a DNA synthesizer, Model 392 (Applied Biosystems Inc.). Oligos were either purified with butanol or dried by centrifugation under vacuum and resuspended in distilled water. All oligos were quantified by spectrophotometry (OD260).
2.14. Preparation of figures
Gels were analyzed either by autoradiography or with a FUJIFILM BAS-2500 Imaging Analyzer along with FUJIFILM Image Reader 1.8 and Image Gauge 3.0 software. Autoradiographs were scanned with an Arcus Agfa scanner using Adobe Photoshop 5.5 software and figures were generated using Microsoft Powerpoint.
3. Results
3.1. Detection of host cell proteins that specifically interact with the SHFV 3′(+)NCR RNA
Arteriviruses and coronaviruses have similar genome structures and replication strategies. Previous studies have shown that the coronavirus 3′(+)NCR contains cis-acting replication signals and binds to cell proteins. To investigate whether the 3′(+)NCR of an arterivirus also interacts with cell proteins, gel mobility shift assays were used. In vitro transcribed and gel purified 32P-labeled SHFV 3′(+)NCR RNA was incubated with an S100 cytoplasmic extract from either SHFV-infected or mock-infected MA104 cells. A single RPC was detected using either type of cytoplasmic extract, suggesting that this RPC consists only of cellular proteins (Fig. 1A).
Fig. 1.
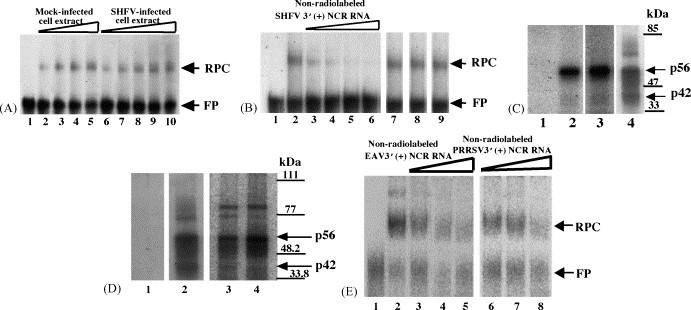
Analysis of the interaction between proteins in MA104 cell extracts and 32P-labeled arterivirus 3′(+)NCR RNAs. (A) Gel mobility shift assay. Radiolabeled SHFV 3′(+)NCR RNA was incubated with an S100 cytoplasmic extract from either SHFV-infected or mock-infected MA104 cells. The RPCs were resolved on a 10% non-denaturing polyacrylamide gel and visualized by autoradiography. (Lane 1) free probe; (lanes 2–5) increasing amounts of mock-infected MA104 S100 cytoplasmic extract (100, 200, 300, and 400 ng); (lanes 6–10) increasing amounts of SHFV-infected MA104 S100 cytoplasmic extract (100, 200, 300, 400, and 500 ng). The locations of the RNA–protein complex and free probe are indicated by arrows. (B) Competition gel mobility shift assay. Different amounts of non-radiolabeled competitor RNAs were incubated with an MA104 S100 cytoplasmic extract before addition of the 32P-labeled SHFV 3′(+)NCR RNA. The RPCs were resolved on a 10% non-denaturing polyacrylamide gel and visualized by autoradiography. (Lane 1) free probe; (lane 2) no competitor; (lanes 3–6) increasing amounts of non-radiolabeled SHFV 3′(+)NCR RNA (5-, 10-, 20-, and 30-fold molar excess); (lane 7) 250-fold molar excess of yeast tRNA; (lane 8) 150-fold molar excess of WNV 3′(+)SL RNA; (lane 9) 250-fold molar excess of poly(I)–poly(C). The locations of the RNA–protein complex and free probe are indicated by arrows. (C) UV-induced cross-linking assay. MA104 S100 cytoplasmic extracts were incubated with radiolabeled SHFV 3′(+)NCR RNA and then were exposed to UV-irradiation. The unprotected RNA was digested with RNase A and the cross-linked proteins were resolved by 10% SDS–PAGE and visualized by autoradiography. (Lane 1) free probe; (lane 2) mock-infected MA104 S100 cytoplasmic extract (1 μg) and poly(I)–(C) (1 μg); (lane 3) SHFV-infected MA104 S100 cytoplasmic extract (1 μg) and poly(I)–(C) (1 μg); (lane 4) mock-infected MA104 S100 cytoplasmic extract (1 μg) and poly(I)–(C) (600 ng). Standard protein markers are indicated by lines and the positions of p56 and p42 are indicated by arrows. (D) UV-induced cross-linking assay. MA104 S100 cytoplasmic extracts and different arterivirus 32P-labeled RNA probes were cross-linked by UV-irradiation in the presence of 600 ng of poly(I)–(C). (Lane 1) free probe; (lane 2) SHFV 3′(+)NCR RNA; (lane 3) EAV 3′(+)NCR RNA; (lane 4) PRRSV 3′(+)NCR RNA. The gels shown in lanes 1 and 2 were analyzed by autoradiography and the gel shown in lanes 3 and 4 was analyzed using the FUJI Bio Imaging Analyzer. The positions of protein standard markers are indicated by lines on the right. The positions of the p56 and p42 bands are indicated by arrows. (E) Competition gel mobility shift assay. MA104 S100 cytoplasmic extracts were incubated with different amounts of non-radiolabeled arterivirus RNAs before addition of the 32P-labeled SHFV 3′(+)NCR RNA. (Lane 1) free probe; (lane 2) no competitor; (lanes 3–5) increasing amounts of unlabeled EAV 3′(+)NCR RNA (25-, 50-, and 75-fold molar excess); (lanes 6–8) increasing amounts of unlabeled PRRSV 3′(+)NCR RNA (25-, 50-, and 75-fold molar excess). The gels were analyzed using the FUJI Bio Imaging Analyzer. The locations of the RNA–protein complex and free probe are indicated by arrows.
The specificity of the interactions between proteins in MA104 S100 cytoplasmic extracts and the 32P-labeled SHFV 3′(+)NCR was investigated using competition gel mobility shift assays. Significant inhibition of binding was observed with a five-fold molar excess of unlabeled SHFV 3′(+)NCR RNA as the specific competitor and complete inhibition was observed with a 30-fold molar excess (Fig. 1B, lanes 3 and 6, respectively). No inhibition was observed when non-specific competitors, such as poly(I)–poly(C), yeast tRNA, or WNV 3′(+)SL RNA, were added in a 150 or 250 molar excess (Fig. 1B, lanes 7–9). The formation of RPCs in the presence of high concentrations of various non-specific competitors indicated that the viral RNA–cell protein interactions detected were specific.
UV-induced cross-linking assays were utilized to determine the molecular masses of the host cell proteins that bound specifically to the SHFV 3′(+)NCR. A strong (56 kDa) and a weak (42 kDa) protein band were detected with MA104 S100 cytoplasmic extracts (Fig. 1C). No bands were detected when the cell extract was omitted (Fig. 1C, lane 1). Two proteins with the same molecular masses (56 and 42 kDa) were also detected in the SHFV-infected extract suggesting that the two proteins that make up the single RPC are cellular, not viral proteins (Fig. 1C, lane 3).
When less stringent conditions were used for the UV-induced cross-linking assays (600 ng instead of 1 μg of non-specific competitor), additional protein bands were detected (Fig. 1C, lane 4). In addition, when an SHFV 3′(+)NCR RNA probe with a poly(A20) tract was used in a UV-induced cross-linking assay (under stringent conditions), an additional protein was detected with a molecular mass of 73 kDa (data not shown). It is likely that the 73 kDa band is PABP (Spagnolo and Hogue, 2000).
3.2. Detection of cell proteins that interact with the 3′(+)NCRs of other arteriviruses
If the interactions between the 3′(+)NCR RNA and a set of cell proteins was relevant for virus replication, then these interactions would be expected to be conserved among divergent arteriviruses. This hypothesis further suggests that the cell proteins involved would be ones that are evolutionarily conserved among different mammalian species. In support of this hypothesis, it has been shown that the host range of individual arteriviruses is restricted at the level of virus attachment and that a single productive replication cycle occurs after transfection of viral genomic RNA into cells from a non-permissive host (Inada et al., 1993, Meulenberg et al., 1998, Kreutz, 1998). To determine whether other arterivirus 3′(+)NCR RNAs show cell protein binding patterns similar to that of the SHFV 3′(+)NCR RNA, UV-induced cross-linking assays were performed with these RNAs. The initial assays were done with either 32P-labeled EAV 3′(+)NCR RNA or PRRSV 3′(+)NCR RNA and MA104 S100 extracts in the presence of 600 ng of non-specific competitor. Four proteins with similar molecular masses (85, 77, 56 and 42 kDa) were detected by the SHFV, EAV, and PRRSV 3′(+)NCR RNAs (Fig. 1D). While the 85 kDa protein appeared to bind the EAV and PRRSV RNAs more efficiently than the SHFV RNA, the other three proteins appeared to bind all three of the viral 3′RNAs with similar efficiency. When 1 μg of non-specific competitor was used, the 56 and 42 kDa proteins, but not the 85 and 77 kDa proteins, were detected (data not shown). The EAV and PRRSV RNAs used in these assays did not contain a poly (A) tract.
Competition gel mobility shift assays were performed using the SHFV 3′(+)NCR RNA as the probe and either unlabeled EAV or PRRSV 3′(+)NCR RNA as the competitor (Fig. 1E). Although competition was observed with all three of the unlabeled RNA competitors, the competition observed with the unlabeled SHFV RNA was the strongest. Some competition was observed when a 25-fold or 50-fold molar excess of either unlabeled EAV or PRRSV 3′(+)NCR RNA was used (Fig. 1E, lanes 3 and 4, and lanes 6 and 7, respectively), and significant competition was observed with a 75-fold molar excess of these competitors (Fig. 1E, lanes 5 and 8). In comparison, complete competition was observed when a 30-fold molar excess of unlabeled SHFV 3′(+)NCR RNA was used (Fig. 1B, lane 6). These data suggest that the 3′(+)NCR RNAs of the three different arteriviruses bind the same cell proteins in the MA104 extracts.
3.3. Localization of the protein binding sites on the SHFV 3′(+)NCR RNA
To determine whether the two cell proteins utilized overlapping or distinct binding sites, a set of sequentially truncated RNA probes were used in gel mobility shift mobility assays as an initial means of identifying the region(s) within the SHFV 3′(+)NCR RNA that interact with p56 and p42. The truncated RNA probes were synthesized in vitro and tested for their ability to bind proteins in MA104 S100 extracts in UV-induced cross-linking assays (Fig. 2 ). To keep the specific activities of the different probes similar, the full-length 3′NCR and RNA-D were labeled with [32P]GTP, while RNAs A, B, and C were labeled with [32P]ATP. The binding of RNA-A, RNA-B, and RNA-C to p56 was markedly reduced as compared to that of the full-length SHFV 3′(+)NCR probe. In contrast, no binding was detected with RNA-D. p42 was not detected with any of the truncated probes.
Fig. 2.
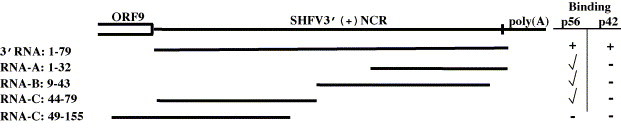
Schematic representation of the truncated 32P-labeled SHFV 3′(+)NCR RNAs used in UV-induced cross-linking assays. Numbering was from the 3′ end. Three adenines of the poly(A) tract were included. The plus symbol (+) indicates wildtype binding observed with the full-length SHFV 3′(+)NCR. The symbol (✓) indicates various degrees of reduced binding and the minus symbol (–) indicates no detectable binding.
3.4. Analysis of the secondary structure of the SHFV 3′(+)NCR
The data obtained with the serially truncated probes suggested that the structure of the 3′NCR was important for protein binding and that multiple binding sites might be present. Functionally relevant SL structures have been reported in the 3′NCRs of coronaviruses (Goebel et al., 2004, Williams et al., 1999). The secondary structure of the SHFV 3′NCR was therefore investigated.
Thermodynamic analysis of the SHFV 3′(+)NCR RNA predicted a structure consisting of a large 3′SL followed by a small SL (Fig. 3B). To determine if the predicted structure of the SHFV 3′(+)NCR RNA was consistent with the structure formed by this RNA in solution, 3′NCRpl RNA, consisting of the 3′(+)NCR sequence plus an additional 43 3′ nts from the plasmid, was used as the template for primer extension structure probing. The presence or absence of the additional 3′nucleotides did not change the Mfold-predicted secondary structure of the SHFV 3′(+)NCR RNA. 3′NCRpl RNA and primer P11, which was complementary to 3′ nts −26 to −42 (Fig. 3B), were annealed. The 3′NCRpl RNA was first partially digested with one of three single-strand specific ribonucleases and then subjected to reverse transcription. At least three experiments were performed with each RNase and each RNase was used at two or more concentrations. A representative gel for each RNase is shown in Fig. 3A. Undigested 3′NCRpl RNA was sequenced as previously described (Nainan et al., 1991) (Fig. 3A, first four lanes). A summary of the data obtained is shown in Fig. 3B. Only strong intensity cleavage bands that were consistently observed in multiple experiments are indicated. All of these strong RNase cleavages were at nucleotides predicted to be single-stranded with only a couple of exceptions. RNase A cleaved nts U24, and U31. Although these nucleotides were predicted to be base-paired, each was located adjacent to a single stranded region which would increase its accessibility to single-strand specific RNase cleavage, especially during RNA structure “breathing”. The same was true for nucleotides U15, U24, A29, U31, U41, U49, U53, U59 and G69, which were cleaved by RNase T2, and nts A29, A35 and U59, cleaved by RNase T1. Other exceptions (U8, A17, A30, G34, C42, U45, U55 and U70) were predicted to be double-stranded and were not directly adjacent to a loop or bulge, but were cleaved. The data obtained from the ribonuclease probing experiments were consistent with the secondary structure predicted for the SHFV 3′(+)NCR.
Fig. 3.
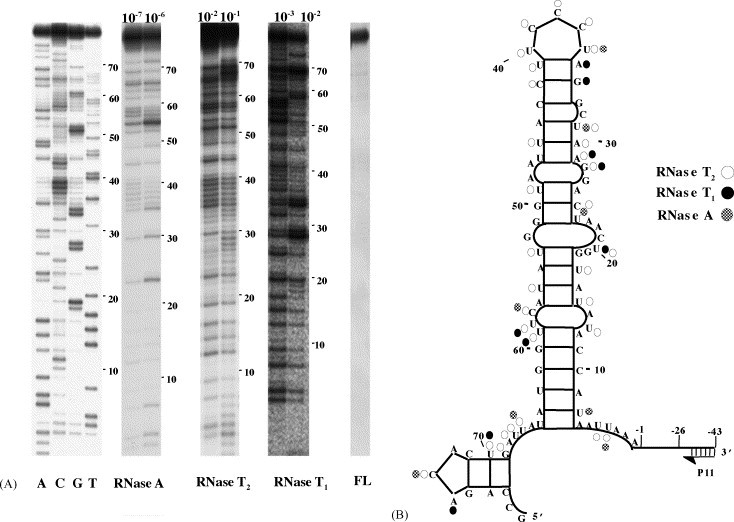
Ribonuclease probing of the SHFV 3′(+)NCR RNA. (A) Autoradiograph of primer extension products. SHFV 3′(+)NCR RNA was partially digested under native conditions with either RNase A (10−7 or 10−6 units), RNase T2 (10−2 or 10−1 units) or RNase T1 (10−3 or 10−2 units) and then used as a template for reverse transcription. cDNAs synthesized by primer extension were resolved by 9% denaturing-PAGE. Representative gels are shown. RNA sequencing reactions were performed as previously described (Nainan et al., 1991) and are shown on the left of the gel. FL (full length), 32P-labeled RNA template prior to treatment. (B) Summary of the ribonuclease probing data. Only strong cleavages that were consistently observed in multiple experiments are indicated.
3.5. Mapping the protein binding sites on the SHFV 3′(+)NCR RNA
When the truncated RNAs used in the initial mapping experiments were folded, their predicted structures differed from that of the full-length SHFV 3′(+)NCR (data not shown) and they bound less efficiently to the proteins (Fig. 2). To gain a further understanding of the protein recognition elements in the SHFV 3′(+)NCR, a deletion/mutation strategy was used. Mutations were designed based on the structural information obtained so that they caused only local or minimal perturbations of the structure of the 3′(+)NCR RNA. Mfold analyses were done to predict the effect of each mutation on the SL structure (Fig. 4A). In RNA-E, the top loop was mutated (5′UCCCU3′ was replaced by 5′AAA3′). Part of the large 3′ internal loop sequence (nts 20–23, 5′AACU3′) was deleted in RNA-F. RNA-G contained a deletion of nts 23–26 and nts 44–48 (5′ACUA3′ and 5′AUUAA3′, respectively). This deletion was thermodynamically predicted to change the sequence of the top loop from 5′UCCCU3′ to 5′CUA3′, to change the bulge sequence in the top portion of the SL and to stabilize the stem below this bulge by removing the internal loop. UV-induced cross-linking assays indicated that RNA-E bound p56 much less efficiently than the wildtype 3′NCR RNA (Fig. 4B, lane 3). Reduced binding of p56 to RNA-F was also observed (Fig. 4B, lane 4) and p56 binding to RNA-G was further reduced (Fig. 4B, lane 5). p42 binding to the RNA-E and RNA-F but not to RNA-G was detected. These data suggest that both the top loop sequence and the sequence of the internal loop in the upper part of the stem are needed for efficient binding of p56. However, even with both of these sites eliminated, a low amount of residual p56 binding was detected.
Fig. 4.
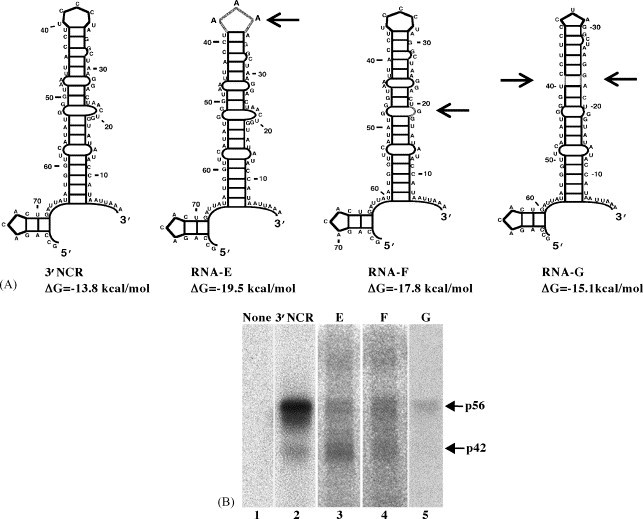
Thermodynamically predicted secondary structures of the full length and deleted SHFV 3′(+)NCR RNAs and UV-induced cross-linking assays with these RNAs. (A) Thermodynamically predicted secondary structures of the indicated RNAs generated with Mfold (Mathews et al., 1999, Zuker et al., 1999). ΔG, the free energy values calculated for each structure. Locations of mutations within the RNAs are indicated by gray lines and arrows. (B) UV-induced cross-linking assays with an MA104 S100 cytoplasmic extract and a 32P-labeled SHFV RNA probe in the presence of 1 μg of poly(I)–(C). (Lane 1) free probe; (lane 2) full-length SHFV 3′(+)NCR RNA; (lane 3) RNA-E; (lane 4) RNA-F; (lane 5) RNA-G. The positions of the p56 and p42 bands are indicated by arrows.
With the data obtained, the binding site for p42 could not be precisely mapped but it appears to be localized to the top portion of the 3′SL. Two additional proteins (∼77 and 85 kDa) were observed to bind to RNA-E and RNA-F at low levels. Proteins of similar sizes were found to bind to the wildtype RNA in the presence of reduced amounts of non-specific competitors (Fig. 1C).
3.6. Partial purification of host proteins that interact with the SHFV 3′(+)NCR RNA by RNA affinity chromatography
As the next step in further analyzing the cell proteins found to interact with the SHFV 3′(+)NCR RNA, the viral RNA binding proteins were enriched by affinity column chromatography. The RNA affinity column was prepared by covalently linking the SHFV 3′(+)NCR RNA to an agarose-adipic acid hydrazide matrix (Blyn et al., 1996, Blyn et al., 1997). A mock column, that contained no RNA linked to the matrix, was also prepared. An S100 cytoplasmic extract prepared from MA104 cells was first incubated with a mixture of various non-specific competitor RNAs and then sequentially passed over an RNA affinity column three times. After washing the column, bound proteins were eluted, concentrated and exchanged into storage buffer as described in Section 2. Aliquots of the 0.2 M wash and 3 M eluate fractions were analyzed by 10% SDS–PAGE and stained with GoldBlot (Fig. 5A). Two protein bands with estimated molecular masses of about 56 kDa and a single protein band of about 42 kDa were consistently detected in samples that also contained bands of the same sizes in UV-induced cross-linking assays (Fig. 5B). The protein purification protocol was repeated several times with similar results. The binding activity of p42 in the 3 M eluate was comparable to that of p56 (Fig. 5B, lane 3) even though the relative amount of p42 in the 3 M eluate was less than that of p56, as indicated by GoldBlot staining (Fig. 5A, lane 2), suggesting that naturally occurring competitors that affect the relative binding activity of p42 to the viral 3′RNA in the unfractionated cell extract may have been eliminated during the purification process. No viral RNA binding activity was detected in the eluate from the mock column (data not shown).
Fig. 5.
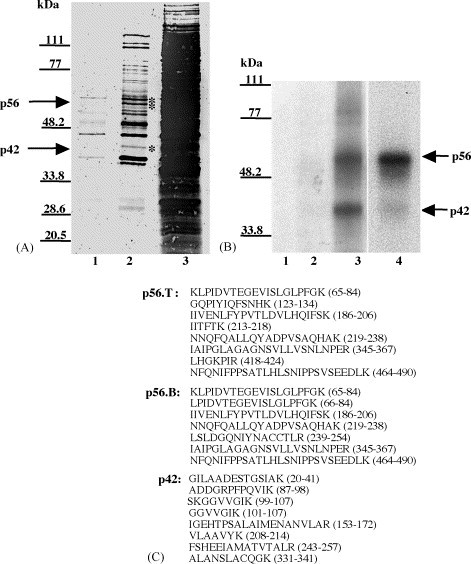
Partial purification and identification of p56 and p42. (A) GoldBlot analysis of eluates of an RNA affinity column. (Lane 1) 0.2 M NaCl wash (4 μl of 300 μl); (lane 2) 3 M NaCl eluate (4 μl of 300 μl); (lane 3) original S100 extract (4 μl of 5 ml). The positions of the p56 and p42 bands are indicated by asterisks. (B) UV-induced cross-linking analysis of eluates of the RNA affinity column. (Lane 1) free probe; (lane 2) 0.2 M NaCl wash (1 μl of 300 μl); (lane 3) 3 M NaCl eluate (1 μl of 300 μl); (lane 4) original S100 extract (1 μl of 5 ml). The positions of the p56 and p42 bands are indicated by arrows. The positions of standard protein markers are indicated by lines on the left side of the gels. (C) Amino acid sequences of peptides derived by tryptic digestion of the purified proteins. p56.T and p56.B represent the top and bottom bands of the p56 doublet, respectively. The numbers to the right of each peptide denote its location within the full-length protein with which it matched.
3.7. Identification of p56 and p42
To determine the identity of proteins in the bands migrating at the positions expected for the two RNA binding proteins, the two p56 and the single p42 Coomassie blue-stained bands were individually excised from gels and sent to the Beckman Research Institute of the City of Hope (Duarte, CA) where the proteins were subjected to endoproteolytic cleavage with trypsin. The resulting peptides were separated by HPLC and selected peptides were sequenced by automated liquid chromatography-tandem mass spectrometry using a Thermo-Finnigan LCQ Classic quadrupole ion trap mass spectrometer (Davis and Lee, 1997, Davis and Lee, 1998, Swiderek et al., 1998). Sequences were obtained for eight peptides derived from the top p56 band, for seven peptides from the bottom p56 band, and for eight peptides from p42 (Fig. 5C). The peptide sequences obtained from both of the p56 bands showed 100% identity with polypyrimidine tract-binding protein. Three isoforms of human PTB with predicted molecular masses of 59 kDa (PTB1/hnRNP I) (Gil et al., 1991), 57.2 kDa (PTB2) (Patton et al., 1991) and 42.8 kDa (PTB3) (Ghetti et al., 1992) are produced by alternative pre-mRNA splicing. PTB1 contains 26 additional amino acids and PTB2 contains 19 additional amino acids not present in PTB3. Since none of the peptides analyzed contained sequences that were unique to PTB1 or PTB2, the identity of the isoforms in the p56 doublet could not be confirmed. However, PTB1 and PTB2 are the closest in size to the two proteins that were purified. The sequences obtained from the p42 band showed 100% identity with fructose 1,6 bisphosphate aldolase A. Three isoforms of aldolase (A, B and C) have been previously identified and are expressed from separate genes on three different chromosomes (16, 9 and 17, respectively). The three aldolase isoforms are identical in size and their sequences are highly conserved (70–82% identical) (Tolan et al., 1987).
3.8. Confirmation of the identity of p56 and p42
Two experimental approaches were used to confirm that the proteins sequenced were actually the viral RNA binding proteins. Both anti-PTB and anti-aldolase antibodies were available and were used to analyze by Western blotting the relative levels of these proteins in the cytoplasmic fractions before and after RNA affinity chromatography. Proteins from the 0.2 NaCl M wash, 3 NaCl M eluate, original S100 extract, and mock column eluate were resolved by 9% SDS–PAGE (Fig. 6A and B). His-tagged PTB (purified as described in Section 2) and aldolase A (Calbiochem) were used as positive controls. After blocking, the membranes were incubated either with rabbit anti-PTB antibody and then anti-rabbit IgG (Fig. 6A) or with goat anti-aldolase antibody followed by anti-goat IgG (Fig. 6B).
Fig. 6.
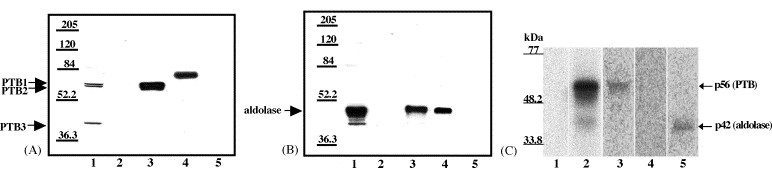
Confirmation of the identity of p56 and p42. (A) Western blotting analysis of RNA affinity chromatography eluate fractions using anti-PTB antibody. (Lane 1) original S100 extract (10 μl of 5 ml); (lane 2) 0.2 M NaCl wash (10 μl of 300 μl); (lane 3) 3 M NaCl eluate (10 μl of 300 μl); (lane 4) recombinant His-PTB (36 ng); (lane 5) mock column 3 M NaCl eluate (10 μl of 300 μl). (B) Western blotting analysis of RNA affinity chromatography eluate fractions using anti-aldolase antibody. (Lane 1) original S100 extract (10 μl of 5 ml); (lane 2) 0.2 M NaCl wash (10 μl of 300 μl); (lane 3) 3 M NaCl eluate (10 μl of 300 μl); (lane 4) purified aldolase (33 ng); (lane 5) mock column 3 M NaCl eluate (10 μl of 300 μl). (C) Immunoprecipitation of proteins cross-linked to SHFV 3′(+)NCR RNA. Scaled up UV-induced cross-linking reactions (90 μl) using cytoplasmic extracts were subjected to immunoprecipitation with various antibodies. (Lane 1) free probe (only one-third of reaction volume was loaded onto the gel); (lane 2), UV-induced cross-linking reaction (only one-third of the reaction volume was loaded onto the gel); (lane 3) immunoprecipitation with anti-PTB antibody; (lane 4) immunoprecipitation with anti-hnRNP A1 antibody; (lane 5) immunoprecipitation with anti-aldolase antibody. The precipitate from the entire reaction volume was loaded onto the gel in lanes 3–5. Standard protein markers are indicated on the left.
Low levels of all three isoforms of PTB were detected in the MA104 S100 cytoplasmic extract with the anti-PTB antibody. No PTB bands were detected in the 0.2 M NaCl wash. PTB1 (59 kDa) and PTB2 (57.2 kDa) but no PTB3 (42.8 kDa) were detected in the 3 M NaCl eluate at higher relative concentrations than in the original S100 extract. The data show that the SHFV 3′(+)NCR RNA affinity column bound and enriched PTB1 and PTB2 but not PTB3 and rule out the possibility that the p42 band detected in the UV-induced cross-linking assays was PTB3. Purified recombinant His-PTB migrated slower than cellular PTB due to the presence of an additional 34 aa at its N-terminus. No PTB was detected in the eluate from the mock column.
As expected, aldolase was present at a higher concentration than PTB in the MA104 S100 cytoplasmic extracts. The anti-aldolase antibody cross reacts with all three isoforms of aldolase and since all three isoforms have the same molecular mass, they cannot be distinguished after PAGE. Aldolase was not detected in the 0.2 M NaCl wash but was detected in the 3 M NaCl eluate. Although aldolase bound efficiently to the RNA affinity column, the relative amount of aldolase in the 3 M NaCl eluate was less than that in the original S100 extract. This may be due to the binding capacity of the RNA on the column or to competition by PTB bound to the column or to preferential binding of one of the aldolase isoforms.
As an additional means of confirming that the p56 and p42 bands detected by UV-induced cross-linking were PTB and aldolase, respectively, immunoprecipitation of UV-induced cross-linked RPCs derived from cytoplasmic extracts with anti-PTB or anti-aldolase antibody after RNase treatment was carried out. p56 cross-linked to the 32P-labeled SHFV 3′(+)NCR RNA was immunoprecipitated by the anti-PTB antibody, while p42 cross-linked to the 32P-labeled SHFV 3′(+)NCR RNA was immunoprecipitated by the anti-aldolase antibody (Fig. 6C, lanes 3 and 5, respectively). When anti-hnRNP A1 antibody was used in this assay, neither cross-linked protein was immunoprecipitated (Fig. 6C, lane 4).
3.9. Further characterization of the specificity of the interactions between the two cell proteins and the SHFV 3′(+)NCR RNA
The specificities of the interactions between the SHFV 3′(+)NCR RNA and purified PTB and aldolase were determined by competition gel mobility shift assays. Recombinant His-PTB which was partially purified as described in Section 2 and commercially available purified aldolase (isoform A; Calbiochem) were used in these assays. Each of the proteins was incubated with unlabeled SHFV 3′(+)NCR RNA or with one of the following non-specific competitors, poly(I)–(C), yeast tRNA or WNV 3′(+)SL RNA prior to the addition of the 32P-labeled SHFV 3′(+)NCR RNA probe. The RPCs formed were resolved by 10% non-denaturing-PAGE and detected by autoradiography (Fig. 7A and D). In reactions containing higher His-PTB concentrations, multiple RPCs were observed which was likely due to the aggregation of the complexes. Two complexes were observed at all concentrations of aldolase tested and may represent dimers and tetramers of this protein. Partial competition was observed when a 10-fold molar excess of the specific competitor was used with either recombinant His-PTB or aldolase. Complete competition was observed with an 18-fold molar excess of the specific competitor with recombinant His-PTB and with a 56-fold molar excess of the specific competitor with aldolase. In contrast, no competition was observed when a 469-fold molar excess of poly(I)–poly(C), a 200-fold molar excess of yeast tRNA, or a 500-fold molar excess of WNV 3′(+)SL RNA was used. These data indicate that the interactions between the SHFV 3′(+)NCR RNA and both recombinant His-PTB and purified aldolase are specific.
Fig. 7.
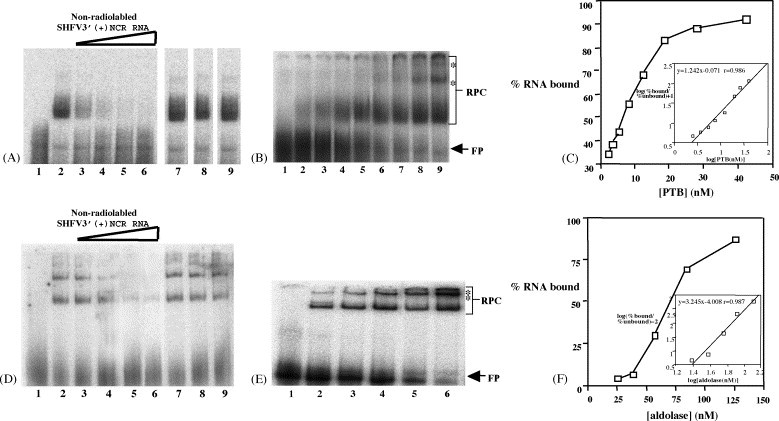
Analysis of the binding specificity of recombinant His-PTB and aldolase and quantification of their interactions with the SHFV 3′(+)NCR RNA. (A) Competition gel mobility shift assay using constant amounts of recombinant His-PTB and 32P-labeled SHFV 3′(+)NCR RNA and various amounts of different competitor RNAs. (Lane 1) free probe; (lane 2) no competitor RNA; lanes 3–6, increasing amounts of non-radiolabeled SHFV 3′(+)NCR RNA (10-, 18-, 56-, and 94-fold molar excess). (Lane 7) 469-fold molar excess of poly(I)–poly(C); (lane 8) 200-fold molar excess of yeast tRNA; (lane 9) 500-fold molar excess of WNV 3′(+)SL RNA. (B) Gel mobility shift assay. Increasing amounts of His-PTB were incubated with a constant amount of 32P-labeled SHFV 3′(+)NCR RNA. (Lane 1) free probe; (lanes 2–9) various concentrations (2.5, 3.7, 5.5, 8.2, 12.5, 18.7, 28, and 42.3 nM) of His-PTB. The locations of the RPC and free probe bands are indicated by arrows. Asterisks indicate the locations of the RPC aggregates that were detected with increasing protein concentrations. (C) Theoretical saturation binding curve. The relative amounts of RPC formation and free probe were measured using a FUJIFILM Bio Imaging Analyzer and Image Reader 1.8 and Image Gauge 3.0 software. The entire free probe region and all of the RPC band regions, as indicated by brackets, were included in the calculation. The percent of SHFV 3′(+)NCR RNA bound was plotted against the concentration of His-PTB. The data were transformed and used to calculate the relative equilibrium dissociation constant and stoichiometry of the interaction. (D) Competition gel mobility shift assay using constant amounts of aldolase and 32P-labeled SHFV 3′(+)NCR RNA and various amounts of different competitor RNAs. (Lane 1) free probe; (lane 2) no competitor RNA; (lanes 3–6) increasing amounts of non-radiolabeled SHFV 3′(+)NCR RNA (10-, 18-, 56-, and 94-fold molar excess). (Lane 7) 469-fold molar excess of poly(I)–poly(C); (lane 8) 200-fold molar excess of yeast tRNA; (lane 9) 500-fold molar excess of WNV 3′(+)SL RNA. (E) Gel mobility shift assay. Increasing amounts of aldolase were incubated with a constant amount of 32P-labeled SHFV 3′(+)NCR RNA. (Lane 1) free probe; (lanes 2–6) various concentrations (24.8, 37.3, 55.9, 83.9, and 125.8 nM) of aldolase. The locations of the RPC and free probe bands are indicated by arrows. Asterisks indicate the locations of the RPC aggregates that were detected with increasing protein concentrations. (F) Theoretical saturation binding curve (generated as described above).
3.10. Determination of the dissociation constants for the RNA–protein interactions
To determine whether the interactions between the SHFV 3′NCR RNA and PTB or aldolase were in the physiological range, the strength of each of these interactions was estimated using gel mobility shift assays. In these assays, a constant amount of 32P-labeled SHFV 3′(+)NCR RNA and increasing amounts of either purified recombinant His-PTB (Fig. 7B) or aldolase (Fig. 7E) were used and the amount of RPCs that formed was quantified. The minimum amount of protein needed to observe an RNA mobility shift was determined in preliminary experiments. The lowest concentration of aldolase required was 24.8 nM, while that for His-PTB was 2.5 nM. Because the His-PTB used was not purified to homogeneity, the concentrations for this protein were estimates. The relative amounts of the RPCs and free probe were measured and the values obtained from at least three experiments were averaged. A theoretical saturation binding curve was generated by plotting the percent of bound SHFV 3′(+)NCR RNA versus the concentration of either recombinant His-PTB (Fig. 7C) or aldolase (Fig. 7F). The entire free probe region and all of the RPC band regions were included in the calculations. The equilibrium dissociation constant (K d) values were calculated as previously described (Lu et al., 1992, Weeks and Crother, 1992) using the equation: log (% bound/% unbound) = n {log[PTB or aldolase (nM)]} – log K d. The relative K d for the His-PTB-SHFV 3′(+)NCR RNA interaction was estimated to be 7 nM, whereas the relative K d for the aldolase-viral RNA interaction was estimated to be 69 nM. The slope of the line in the inset graph for recombinant His-PTB was 1.24, indicating that approximately 1 His-PTB homodimer binds to one SHFV 3′(+)NCR RNA molecule. The slope of the line for aldolase was 3.25, indicating that approximately 3 aldolase homotetramers bind to one SHFV 3′(+)NCR RNA molecule.
4. Discussion
This is the first report identifying host cellular proteins that interact with an arterivirus 3′NCR RNA. PTB, which was identified as the 56 kDa MA104 protein, has multiple cellular functions. It can act as a negative or positive regulator of pre-mRNA splicing (Lou et al., 1999, Valcarcel and Gebauer, 1997) and is involved in regulating pre-mRNA polyadenylation (Lou et al., 1999, Moreira et al., 1998) and mRNA cytoplasmic localization (Cote et al., 1999). PTB belongs to the RNA recognition motif (RRM) family of RNA-binding proteins and each monomer of PTB contains four RRMs (RRM1–RRM4) (Perez et al., 1997). The three isoforms of PTB are expressed in all types of tissues (Patton et al., 1991).
PTB has previously been reported to regulate poliovirus (Hellen et al., 1993), hepatitis A virus (HAV) (Schultz et al., 1996), and human rhinovirus-2 (HRV) IRES-dependent translation (Hunt and Jackson, 1999). The binding of PTB to both the MHV 5′(+)NCR RNA and 3′(−)NCR RNA were shown to be important for virus replication (Li et al., 1999). PTB binding to the MHV 3′(−)NCR RNA was reported to induce a conformational change in this RNA (Huang and Lai, 1999). Further support for an involvement of PTB in MHV RNA replication was provided by the observation that transcription but not translation of MHV DI RNA was inhibited in cells over-expressing a truncated form of PTB (Choi et al., 2002). Three regions within the hepatitis C virus (HCV) genome RNA, namely the 5′NCR, a region in the capsid protein coding region, and the X region of the 3′NCR, were reported to bind to PTB (Ali and Siddiqui, 1997, Ito and Lai, 1999, Tsuchinhara et al., 1997). Zhang et al. (2004) showed that HCV replication was down-regulated by expression of siRNA against PTB. RNA interference studies unfortunately do not provide information about the roles that cellular co-factors play in viral replication cycles. Also, silencing of some cell genes can cause secondary effects on the cell that suppress virus replication indirectly.
The 42 kDa protein was identified as fructose 1,6 bisphosphate aldolase A, an abundant tetrameric cellular protein that is present throughout the cytoplasm where it binds to actin filaments of the cytoskeleton (O’Reilly and Clarke, 1993). The level of expression of the individual aldolase isoforms varies in different tissues and each isoform has distinct roles in the glycolytic pathway of the tissues in which it is expressed (Penhoet et al., 1966, Rottmann et al., 1987). Aldolase binds to the SHFV 3′(+)NCR RNA with a lower affinity (K d = 69 nM) than PTB. This is consistent with the observation that the PTB protein band in UV-induced cross-linking assays is more intense than the aldolase protein band even though the level of aldolase present in the cytoplasm is significantly higher than that of PTB.
There have been no previous reports of aldolase binding to a viral or a cellular RNA. However, another glycolytic enzyme, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), has been reported to bind to specific cellular and viral RNAs. GAPDH binds to tRNA in a sequence- and structure-specific manner in HeLa cell nuclear extracts where it is thought to participate in tRNA export (Singh et al., 1995). GAPDH was shown to have RNA helix-destabilizing activity and to bind to the HAV IRES (Schultz et al., 1996). It has also been reported to bind to the 3′(+)NCR and 3′(−)NCR RNAs of human parainfluenza virus type 3 (De et al., 1996). Both GAPDH and PTB were reported to bind to the HAV IRES and to compete for overlapping binding sites (Yi et al., 2000). The binding of GAPDH to the HAV IRES resulted in down regulation of translation while the binding of PTB stimulated translation.
Aldolase is also capable of organizing actin filaments into highly ordered three-dimensional structures (Clarke et al., 1985). It has been reported that a number of viruses manipulate the cellular actin cytoskeleton to facilitate various steps in their replication cycles (reviewed in Cudmore et al., 1997). The possibility of an involvement of the cytoskeleton in arterivirus replication has not yet been investigated.
The structural context of the binding sites on the SHFV 3′(+)NCR RNA appears to be important for interaction with both PTB and aldolase. Although the contributions of individual RNA sequence and structural elements to protein binding were not delineated in this study, some information about the relative locations of the protein binding sites in the viral 3′(+)NCR RNA was obtained. The limited mapping data for aldolase suggested that the binding site for this protein was located in the top portion of the SHFV 3′(+)NCR RNA SL.
The binding sites for PTB were mapped to the pyrimidine-rich top loop of the 3′SL and to an adjacent pyrimidine-containing bulge. Both of these sequences contained flanking guanines. A stretch of pyrimidines flanked by guanines was previously identified as a binding consensus sequence for PTB by SELEX (K d = ∼1–10 nM; Singh et al., 1995). The binding sites for PTB on the pre-mRNA sequences of α-tropomyosin and β-tropomyosin (K d = 1–5 nM) were mapped to a SL with a pyrimidine-rich terminal loop and a nearby single-stranded region containing a short stretch of pyrimidines (Singh et al., 1995). The K ds for the interactions between PTB and the SHFV 3′NCR RNA and between PTB and these two cellular pre-mRNAs were similar (∼K d = 7 nM). Interestingly, the mapped PTB binding site in the poliovirus IRES has a lower affinity (K d = 30–60 nM) and consists of a terminal loop containing a stretch of four pyrimidines without a nearby pyrimidine-rich bulge (Conte et al., 2000, Rust et al., 1999, Witherell et al., 1993). The information previously reported and the mapping data reported here suggest that two nearby binding sites presented within a structural context are required for a high affinity PTB–RNA interaction.
The different arterivirus 3′NCR RNAs vary in their lengths and sequences. The EAV and PRRSV 3′NCRs were shown to bind a 56 and 42 kDa MA104 cell protein in UV-induced cross-linking assays and specific competition was observed when they were used as competitors in a competition gel mobility shift assay with an SHFV 3′(+)NCR RNA probe. Although not identical, the predicted secondary structures of other arterivirus 3′NCR RNAs consist of a 3′SL with internal loop or bulge regions along the stem. In each case, pyrimidines are present in the top loop of the 3′SL and on the 3′ side of the stem in an internal loop or bulge located near the top of the stem. RNA structure probing analyses have not yet been reported for any of the other arterivirus 3′NCR RNAs.
Verheije et al. (2002) reported a tertiary (kissing) interaction between the top loops of a predicted SL located near the 5′ end of the PRRSV ORF7 (54 nts from the start codon) and a predicted SL structure within the PRRSV 3′(+)NCR. It is not yet known whether a similar tertiary interaction forms between the SL in the 3′NCR and an upstream SL structure in the SHFV genomic RNA. Possible roles of PTB and aldolase binding to the arterivirus 3′NCR RNA during the virus replication cycle are presently not known. The data reported here suggest that these two proteins could compete for an overlapping binding site and this competition may be functionally relevant.
Acknowledgements
We gratefully acknowledge the technical assistance of Stephanie Methven and Holly Starling. This work was supported by Public Health Service research grant AI 40688 from the National Institute of Allergy and Infectious Diseases and by grants from the Georgia State University Research Foundation. Support for T. Maines was provided by a Research Program Enhancement Award from the Georgia State University Office of Research and Sponsored Programs. The Beckman Research Institute facility was funded by the National Institutes of Health Cancer Center Support Grant CA33572.
References
- Ali N., Siddiqui A. The La antigen binds 5′ non-coding region of the hepatitis C virus RNA in the context of the initiator AUG codon and stimulates internal ribosome entry site-mediated translation. Proc. Natl. Acad. Sci. U.S.A. 1997;94:2249–2254. doi: 10.1073/pnas.94.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell J.L., Brinton M.A. BHK cell proteins that bind to the 3′ stem-loop structure of the West Nile virus genome RNA. J. Virol. 1995;69:5650–5658. doi: 10.1128/jvi.69.9.5650-5658.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell J.L., Brinton M.A. Translation elongation factor-1 alpha interacts with the 3′ stem-loop region of West Nile virus genomic RNA. J. Virol. 1997;71:6433–6444. doi: 10.1128/jvi.71.9.6433-6444.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blyn L.B., Swiderek K.M., Richards O., Stahl D.C., Semler B.L., Ehrenfeld E. Poly(rC) binding protein 2 binds to stem-loop IV of the poliovirus RNA 5′ non-coding region: identification by automated liquid chromatography-tandem mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 1996;93:11115–11120. doi: 10.1073/pnas.93.20.11115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blyn L.B., Towner J.S., Semler B.L., Ehrenfeld E. Requirement of poly(rC) binding protein 2 for translation of poliovirus DNA. J. Virol. 1997;71:6243–6246. doi: 10.1128/jvi.71.8.6243-6246.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinton M.A., Gavin E.I., Fernandez A.V. Genotypic variation among six isolates of lactate dehydrogenase-elevating virus. J. Gen. Virol. 1986;67:2673–2684. doi: 10.1099/0022-1317-67-12-2673. [DOI] [PubMed] [Google Scholar]
- Choi K.S., Huang P., Lai M.M.C. Polypyrimidine-tract binding protein affects transcription but not translation of mouse hepatitis virus RNA. Virology. 2002;303:58–68. doi: 10.1006/viro.2002.1675. [DOI] [PubMed] [Google Scholar]
- Clarke R.M., Morton D.J., Stephan P., Wiedemann J. In: Cell Motility: Mechanism and Regulation. Ishikawa H., Hatano S., Sato S., editors. University of Tokyo Press; Tokyo: 1985. pp. 235–250. [Google Scholar]
- Contag C.H., Chan S.P., Wietgrefe S.W., Plagemann P.G. Correlation between presence of lactate dehydrogenase-elevating virus RNA and antigens in motor neurons and paralysis in infected C58 mice. Vir. Res. 1986;6:195–209. doi: 10.1016/0168-1702(86)90069-9. [DOI] [PubMed] [Google Scholar]
- Conte M.R., Grune T., Ghuman J., Kelly G., Ladas A., Matthews S., Curry S. Structure of tandem RNA recognition motifs from polypyrimidine tract binding protein reveals novel features of the RRM fold. EMBO. 2000;19:3132–3141. doi: 10.1093/emboj/19.12.3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote C.A., Gautreau D., Denegre J.M., Dress T.L., Terry N.A., Mowry K.L. A Xenopus protein related to hnRNP I has a role in cytoplasmic RNA localization. Mol. Cell. 1999;4:431–437. doi: 10.1016/s1097-2765(00)80345-7. [DOI] [PubMed] [Google Scholar]
- Cudmore S., Reckmann I., Way M. Viral manipulations of the actin cytoskeleton. Trends Microbiol. 1997;4:141–148. doi: 10.1016/S0966-842X(97)01011-1. [DOI] [PubMed] [Google Scholar]
- Davis M.T., Lee T.D. Variable flow liquid chromatography-tandem mass spectrometry and the comprehensive analysis of complex protein digest mixtures. J. Am. Soc. Mass Spectrom. 1997;8:1059–1069. [Google Scholar]
- Davis M.T., Lee T.D. Rapid protein identification using a microscale electrospray LC/MS system on an ion trap mass spectrometer. J. Am. Soc. Mass Spectrom. 1998;9:194–201. doi: 10.1016/S1044-0305(97)00282-1. [DOI] [PubMed] [Google Scholar]
- De B.P., Gupta S., Zhao H., Drazba J.A., Banerjee A.K. Specific interaction in vitro and in vivo of glyceraldehyde-3-phosphate dehydrogenase and La protein with cis-acting RNAs of human parainfluenza virus type 3. J. Biol. Chem. 1996;271:24728–24735. doi: 10.1074/jbc.271.40.24728. [DOI] [PubMed] [Google Scholar]
- Ghetti A., Pinol-Roma S., Michael W.M., Morandi C., Dreyfuss G. HnRNP I, the polypyrimidine tract-binding protein: distinct nuclear localization and association with hnRNAs. Nucleic Acids Res. 1992;20:3671–3678. doi: 10.1093/nar/20.14.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil A., Sharp P.A., Jamison S.F., Garcia-Blanco M.A. Characterization of cDNAs encoding the polypyrimidine tract-binding protein. Genes Dev. 1991;5:1224–1236. doi: 10.1101/gad.5.7.1224. [DOI] [PubMed] [Google Scholar]
- Godeny E.K., Zeng L., Smith S.L., Brinton M.A. Molecular characterization of the 3′ terminus of the Simian hemorrhagic fever virus genome. J. Virol. 1995;69:2679–2683. doi: 10.1128/jvi.69.4.2679-2683.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel S.J., Hsue B., Dombrowski T.F., Masters P.S. Characterization of the RNA components of a putative molecular switch in the 3′ untranslated region of the murine coronavirus genome. J. Virol. 2004;78:669–682. doi: 10.1128/JVI.78.2.669-682.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B., Rong M., Lyakhov D., Gartenstein H., Diaz G., Castagna R., McAllister W.T., Durbin R.K. Rapid mutagenesis and purification of phage RNA polymerises. Protein Express. Purif. 1997;9:142–151. doi: 10.1006/prep.1996.0663. [DOI] [PubMed] [Google Scholar]
- Hellen C.U.T., Witherell G.W., Schmid M., Shin S.H., Pestova T.V., Gil A., Wimmer E. A cytoplasmic 57-kDa protein that is required for translation of picornavirus RNA by internal ribosomal entry is identical to the nuclear pyrimidine tract-binding protein. Proc. Natl. Acad. Sci. U.S.A. 1993;90:7642–7646. doi: 10.1073/pnas.90.16.7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsue B., Masters P.S. A bulged stem-loop strucute in the 3′ untranslated region of the genome of the coronavirus mouse hepatitis virus is essential for replication. J. Virol. 1997;71:7567–7578. doi: 10.1128/jvi.71.10.7567-7578.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsue B., Hartshorne T., Masters P.S. Characterization of an essential RNA secondary structure in the 3′ untranslated region of the murine coronavirus genome. J. Virol. 2000;74:6911–6921. doi: 10.1128/jvi.74.15.6911-6921.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P., Lai M.M.C. Polypyrimidine tract-binding protein binds to the complementary strand of the mouse hepatitis virus 3′ untranslated region, thereby altering RNA conformation. J. Virol. 1999;73:9110–9116. doi: 10.1128/jvi.73.11.9110-9116.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P., Lai M.M.C. Heterogeneous nuclear ribonucleoprotein A1 binds to the 3′ untranslated region and mediates potential 5′-3′ end cross talks of mouse hepatitis virus RNA. J. Virol. 2001;75:5009–5017. doi: 10.1128/JVI.75.11.5009-5017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt S.L., Jackson R.J. Polyprimidine-tract binding protein (PTB) is necessary, but not sufficient, for efficient internal initiation of translation of human rhinovirus-2 RNA. RNA. 1999;5:344–359. doi: 10.1017/s1355838299981414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang Y.K., Brinton M.A. A 68-nucleotide sequence within the 3′ non-coding region of Simian hemorrhagic fever virus negative-strand RNA binds to four MA104 cell proteins. J. Virol. 1998;72:4341–4351. doi: 10.1128/jvi.72.5.4341-4351.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inada T., Kikuchi H., Yamazaki S. Comparison of the ability of lactate dehydrogenase-elevating virus and its virion RNA to infect murine leukemia virus-infected or -uninfected cell lines. J. Virol. 1993;67:5698–5703. doi: 10.1128/jvi.67.9.5698-5703.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T., Lai M.M.C. An internal polypyrimidine-tract-binding protein-binding site in the hepatitis C virus RNA attenuates translation, which is relieved by the 3′-untranslated sequence. Virology. 1999;254:288–296. doi: 10.1006/viro.1998.9541. [DOI] [PubMed] [Google Scholar]
- Kim H.S., Kwang J., Yoon I.J., Foo H.S., Frey M.L. Enhanced replication of porcine reproductive and respiratory syndrome (PRRS) virus in a homogeneous subpopulation of MA104 cell line. Arch. Virol. 1993;133:477–483. doi: 10.1007/BF01313785. [DOI] [PubMed] [Google Scholar]
- Kreutz L.C. Cellular membrane factors are the major determinants of porcine reproductive and respiratory syndrome virus tropism. Virus Res. 1998;53:121–128. doi: 10.1016/s0168-1702(97)00134-2. [DOI] [PubMed] [Google Scholar]
- Li H.P., Huang P., Park S., Lai M.M.C. Polypyrimidine tract-binding protein binds to the leader RNA of mouse hepatitis virus and serves as a regulator of viral transcription. J. Virol. 1999;73:772–777. doi: 10.1128/jvi.73.1.772-777.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.J., Lai M.M.C. Deletion mapping of a mouse hepatitis virus defective interfering RNA reveals the requirement of an internal and discontiguous sequence for replication. J. Virol. 1993;67:6110–6118. doi: 10.1128/jvi.67.10.6110-6118.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.J., Liao C.L., Lai M.M.C. Identification of the cis-acting signal for minus-strand RNA synthesis of a murine coronavirus: implications for the role of minus-strand RNA in RNA replication and transcription. J. Virol. 1994;68:8131–8140. doi: 10.1128/jvi.68.12.8131-8140.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou H., Helfman D.M., Gagel R.F., Berget S.M. Polypyrimidine tract-binding protein positively regulates inclusion of an alternative 3′-terminal exon. Mol. Cell. 1999;19:78–85. doi: 10.1128/mcb.19.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C.D., Houghton J.E., Abdelal A.T. Characterization of the arginine repressor from Salmonella typhimurium and its interactions with the carAB operator. J. Mol. Biol. 1992;225:11–24. doi: 10.1016/0022-2836(92)91022-h. [DOI] [PubMed] [Google Scholar]
- Mathews D.H., Sabina J., Zuker M., Turner D.H. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- Meulenberg J.J.M., Bos-de Ruijter J.N.A., Wensvoort G., Moormann R.J.M. Infectious transcripts from cloned genome-length cDNA of porcine reproductive and respiratory syndrome virus. J. Virol. 1998;72:380–387. doi: 10.1128/jvi.72.1.380-387.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenkamp R., Rozier B.C., Greve S., Spaan W.J., Snijder E.J. Isolation and characterization of an arterivirus defective interfering RNA genome. J. Virol. 2000;74:3156–3165. doi: 10.1128/jvi.74.7.3156-3165.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira A., Takagaki Y., Brackenridge S., Wollerton M., Manley J.L., Proudfoot J.J. The upstream sequence element of the C2 complement poly(A) signal activates mRNA 3′ end formation by two distinct mechanisms. Genes Dev. 1998;12:2522–2534. doi: 10.1101/gad.12.16.2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nainan O.V., Margolis H.S., Robertson B.H., Balayan M., Brinton M.A. Sequence analysis of a new hepatitis A virus infecting cynomolgus macaques (Macaca fascicularis) J. Gen. Virol. 1991;72:1685–1689. doi: 10.1099/0022-1317-72-7-1685. [DOI] [PubMed] [Google Scholar]
- Nanda S.K., Leibowitz J.L. Mitochondrial aconitase binds to the 3′ untranslated region of the mouse hepatitis virus genome. J. Virol. 2001;75:3352–3362. doi: 10.1128/JVI.75.7.3352-3362.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly G., Clarke F. Identification of an actin binding region in aldolase. FEBS Lett. 1993;321:69–72. doi: 10.1016/0014-5793(93)80623-3. [DOI] [PubMed] [Google Scholar]
- Patton J.G., Mayer S.A., Tempst P., Nadal-Ginard B. Characterization and molecular cloning of polypyrimidine tract-binding protein: a component of a complex necessary for pre-mRNA splicing. Genes Dev. 1991;5:1237–1251. doi: 10.1101/gad.5.7.1237. [DOI] [PubMed] [Google Scholar]
- Penhoet E.E., Rajkumar T., Rutter W.J. Multiple forms of fructose diphosphate aldolase in mammalian tissues. Proc. Natl. Acad. Sci. U.S.A. 1966;56:1275–1282. doi: 10.1073/pnas.56.4.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez I., McAfee J.G., Patton J.G. Multiple RRMs contribute to RNA binding specificity and affinity for polypyrimidine tract binding protein. Biochemistry. 1997;36:11881–11890. doi: 10.1021/bi9711745. [DOI] [PubMed] [Google Scholar]
- Rottmann W.H., Deselms K.R., Niclas J., Camerato T., Holman P.S., Green C.J., Tolan D.R. The complete amino acid sequence of the human aldolase C isozyme derived from genomic clones. Biochimie. 1987;69:137–145. doi: 10.1016/0300-9084(87)90246-x. [DOI] [PubMed] [Google Scholar]
- Rust R.C., Ochs K., Meyer K., Beck E., Niepmann M. Interaction of eukaryotic initiation factor eIF4B with the internal ribosome entry site of foot-and-mouth disease virus is independent of the polypyrimidine tract-binding protein. J. Virol. 1999;73:6111–6113. doi: 10.1128/jvi.73.7.6111-6113.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagripanti J.L. Polyadenylic acid sequences in the genomic RNA of the togavirus of Simian hemorrhagic fever. Virology. 1985;145:350–355. doi: 10.1016/0042-6822(85)90171-0. [DOI] [PubMed] [Google Scholar]
- Sagripanti J.L., Zandomeni R.O., Weinmann R. The cap structure of Simian hemorrhagic fever virion RNA. Virology. 1986;151:146–150. doi: 10.1016/0042-6822(86)90113-3. [DOI] [PubMed] [Google Scholar]
- Sawicki S.G., Sawicki D.L. Coronaviruses use discontinuous extension for synthesis of subgenome-length negative strands. Adv. Exp. Med. Biol. 1995;380:499–506. doi: 10.1007/978-1-4615-1899-0_79. [DOI] [PubMed] [Google Scholar]
- Schultz D.E., Hardin C.C., Lemon S.M. Specific interaction of glyceraldehyde 3-phosphate dehydrogenase with the 5′-non-translated RNA of hepatitis A virus. J. Biol. Chem. 1996;271:14134–14142. doi: 10.1074/jbc.271.24.14134. [DOI] [PubMed] [Google Scholar]
- Singh R., Valcarcel J., Green M.R. Distinct binding specificities and functions of higher eukaryotic polypyrimidine tract-binding proteins. Science. 1995;268:1173–1176. doi: 10.1126/science.7761834. [DOI] [PubMed] [Google Scholar]
- Snijder E.J., Meulenberg J.J. Arteriviruses. In: Knipe P.M., Howley P.M., editors. Virology. fourth ed. Lippincott, Williams and Wilkins; Philadelphia, PA: 2001. pp. 1205–1220. [Google Scholar]
- Spagnolo J.F., Hogue B.G. Host protein interactions with the 3′ end of bovine coronavirus RNA and the requirement of the poly(A) tail for coronavirus defective genome replication. J. Virol. 2000;74:5053–5065. doi: 10.1128/jvi.74.11.5053-5065.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiderek K.M., Davis M.T., Lee T.D. The identification of peptide modifications derived from gel-separated proteins using electrospray triple quadrupole and ion trap analyses. Electrophoresis. 1998;19:989–997. doi: 10.1002/elps.1150190614. [DOI] [PubMed] [Google Scholar]
- Tolan D.R., Niclas J., Bruce B.D., Lebo R.V. Evolutionary implications of the human aldolase-A, -B, -C, and -pseudogene chromosome locations. Am. J. Hum. Genet. 1987;41:907–924. [PMC free article] [PubMed] [Google Scholar]
- Trousdale M.D., Trent D.W., Shelokov A. Simian hemorrhagic fever virus: a new togavirus. Proc. Soc. Exp. Biol. Med. 1975;150:707–711. doi: 10.3181/00379727-150-39111. [DOI] [PubMed] [Google Scholar]
- Tsuchinhara K., Tanaka T., Hijikata M., Kuge S., Toyoda H., Nomoto A., Yamamoto N., Shimotohno K. Specific interaction of poly-pyrimidine tract-binding protein with the extreme 3′-terminal structure of the hepatitis C virus genome, the 3′X. J. Virol. 1997;71:8868–8874. doi: 10.1128/jvi.71.9.6720-6726.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valcarcel J., Gebauer F. Post-transcriptional regulation: the dawn of PTB. Curr. Biol. 1997;7:R705–R708. doi: 10.1016/s0960-9822(06)00361-7. [DOI] [PubMed] [Google Scholar]
- van Berlo M.F., Horzinek M.C., van der Zeijst B.A. Equine arteritis virus-infected cells contain six polyadenylated virus-specific RNAs. Virology. 1982;118:345–352. doi: 10.1016/0042-6822(82)90354-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Marle G., Dobbe J.C., Gultyaev A.P., Luytjes W., Spaan W.J., Snijder E.J. Arterivirus discontinuous mRNA transcription is guided by base pairing between sense and antisense transcription-regulating sequences. Proc. Natl. Acad. Sci. U.S.A. 1999;96:12056–12061. doi: 10.1073/pnas.96.21.12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheije M.H., Olsthoorn R.C.L., Kroese M.V., Rottier P.J.M., Meulenberg J.J.M. Kissing interaction between 3′ non-coding and coding sequences is essential for porcine arterivirus RNA replication. J. Virol. 2002;76:1521–1526. doi: 10.1128/JVI.76.3.1521-1526.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks K.M., Crother D.M. RNA binding assays for Tat-derived peptides: implications for specificity. Biochemistry. 1992;31:10281–10287. doi: 10.1021/bi00157a015. [DOI] [PubMed] [Google Scholar]
- Wells S.E., Hillner P.E., Vale R.D., Sachs A.B. Circularization of mRNA by eukaryotic translation initiation factors. Mol. Cell. 1998;2:135–140. doi: 10.1016/s1097-2765(00)80122-7. [DOI] [PubMed] [Google Scholar]
- Williams G.D., Chang R.Y., Brian D.A. A phylogentetically conserved hairpin-type 3′ untranslated region pseudoknot functions in coronavirus RNA replication. J. Virol. 1999;73:8349–8355. doi: 10.1128/jvi.73.10.8349-8355.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witherell G.W., Gil A., Wimmer E. Interaction of polypyrimidine tract binding protein with the encephalomyocarditis virus mRNA internal ribosomal entry site. Biochemistry. 1993;32:8268–8275. doi: 10.1021/bi00083a030. [DOI] [PubMed] [Google Scholar]
- Yi M., Schultz D.E., Lemon S.M. Functional significance of the interaction of hepatitis A virus RNA with glyceraldehyde 3-phosphate dehydrogenase (GAPDH): opposing effects of GAPDH and polyprimidine tract binding protein on internal ribosome entry site function. J. Virol. 2000;74:6459–6468. doi: 10.1128/jvi.74.14.6459-6468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Yamada O., Sakamoto T., Yoshida H., Iwai T., Matsushita Y., Shimamura H., Araki H., Shimotohno K. Down-regulation of viral replication by adenoviral-mediated expression of siRNA against cellular cofactors for hepatitis C virus. Virology. 2004;320:135–143. doi: 10.1016/j.virol.2003.11.023. [DOI] [PubMed] [Google Scholar]
- Zuker M., Mathews D.H., Turner D.H. Algorithms and thermodynamics for RNA secondary structure prediction: a practical guide. In: Barciszewski J., Clark B.F.C., editors. RNA Biochemisty and Biotechnology. NATO ASI Series. Kluwer Academic Publishers; 1999. pp. 11–43. [Google Scholar]