

Summary
The coronavirus nucleocapsid (N) protein packages viral genomic RNA into a ribonucleoprotein complex. Interactions between N proteins and RNA are thus crucial for the assembly of infectious virus particles. The 45 kDa recombinant nucleocapsid N protein of coronavirus infectious bronchitis virus (IBV) is highly sensitive to proteolysis. We obtained a stable fragment of 14.7 kDa spanning its N-terminal residues 29–160 (IBV-N29-160). Like the N-terminal RNA binding domain (SARS-N45-181) of the severe acute respiratory syndrome virus (SARS-CoV) N protein, the crystal structure of the IBV-N29-160 fragment at 1.85 Å resolution reveals a protein core composed of a five-stranded antiparallel β sheet with a positively charged β hairpin extension and a hydrophobic platform that are probably involved in RNA binding. Crosslinking studies demonstrate the formation of dimers, tetramers, and higher multimers of IBV-N. A model for coronavirus shell formation is proposed in which dimerization of the C-terminal domain of IBV-N leads to oligomerization of the IBV-nucleocapsid protein and viral RNA condensation.
Introduction
Coronaviruses are large enveloped single-stranded RNA viruses of positive polarity which cause a wide spectrum of diseases affecting humans and animals (reviewed in Lai and Holmes, 2001). In 2003, the causative agent for the outbreak of atypical pneumonia with a high fatality rate was identified as the severe acute respiratory syndrome (SARS) coronavirus (SARS-CoV) (Peiris et al., 2003), and its genome was rapidly sequenced and characterized (Marra et al., 2003, Rota et al., 2003). The potential risks for public health posed by SARS-CoV and the current lack of specific antiviral agents or vaccines against this emerging pathogen have triggered a global research effort in order to characterize this family of viruses at the molecular level. Coronavirus infectious bronchitis virus (IBV) causes an acute and contagious disease in chickens, with a significant impact on the poultry industry worldwide.
In structural terms, coronavirus virions are roughly spherical, with an approximate diameter of 120 nm. Their detailed in vivo morphology is still a matter of debate but might be composed of three structural layers: a lipid envelope with three or four glycoproteins, a protein core, and a tubular or helicoidal nucleocapsid, as shown for the porcine transmissible gastroenteritis virus (TGEV) (Escors et al., 2001). Low-resolution electron micrographs have highlighted the crown-like structure that surrounds the coronavirus envelope (Sturman et al., 1980). These spikes contain the S protein, a class I fusion glycoprotein (Bosch et al., 2004, Lescar et al., 2001) which is also responsible for binding to the receptor (Lai and Holmes, 2001). Two integral membrane proteins, M (about 230 amino acids) and E (about 100 amino acids), are essential for the maturation of newly formed virions, and are sufficient for the formation of a closed viral particle (Vennema et al., 1996). The M protein is thought to possess three transmembrane segments and a large C-terminal endodomain that interacts with the nucleocapsid and possibly also with the RNA genome (Sturman et al., 1980, Kou and Masters, 2002, Narayanan et al., 2003). The nucleocapsid protein of IBV (IBV-N) is a phosphoprotein of 409 amino acids that is well-conserved across various IBV strains (Williams et al., 1992) and is also important for cell-mediated immunity. It forms a protective shell that packages the viral genomic RNA of 27.6 kb and is also thought to participate in viral RNA replication and transcription. Specific packaging of viral genetic material is usually performed via the recognition of a particular nucleotide sequence by a nucleocapsid protein. Such “packaging signals” have been identified at the 3′ end of the viral genomes of mouse hepatitis virus (MHV) (Fosmire et al., 1992) and bovine coronavirus (BCV) (Cologna and Hogue, 2000) and at the 5′ end of the TGEV genome (Escors et al., 2003), but not unambiguously for the IBV genome. In elegant structural studies performed in other viral families with RNA genomes, such as HIV (De Guzman et al., 1998) and the MS2 bacteriophage (Valegard et al., 1997), the packaging signals were seen to form a stem-loop structure that is recognized by the nucleocapsid protein. In the case of the IBV genome, this special RNA structure has not been determined with certainty, although previous studies demonstrated that the IBV-N protein interacts specifically with RNA sequences located at the 3′ noncoding region of the viral genome (Zhou et al., 1996). Both the N- and C-terminal domains of IBV-N, but not its middle region, bind to an oligoribonucleotide of 155 nucleotides, located at the 3′ end of the viral genome nontranslated region, but little is known about the details of this interaction and how it relates to virus assembly (Zhou and Collisson, 2000).
In an attempt to define how the viral genome is incorporated into newly formed viral particles and how this process is coupled with nucleocapsid assembly, we have undertaken functional and structural studies using the full-length N protein from IBV expressed in bacteria. The full-length recombinant IBV-N protein expressed in Escherichia coli is unstable. Through cleavage by E. coli proteases, a stable fragment of 14.7 kDa comprising its N-terminal residues 29–160 can be obtained. We report here the crystal structure of this N-terminal fragment refined at 1.85 Å resolution and compare it to the N-terminal RNA binding domain of the SARS-CoV N protein (SARS-N45-181), which was solved recently using NMR (Huang et al., 2004). We demonstrate the formation of multimers of the IBV-N protein in vitro and propose that dimers and possibly tetramers of IBV-N, which are stabilized predominantly via dimerization of their C-terminal domains, act as elementary building blocks for RNA genome condensation and nucleocapsid assembly.
Results and Discussion
Identification of a Stable Proteolytic Fragment of IBV-N
The full-length IBV-N protein comprising 409 residues was expressed in E. coli in a soluble form and purified as described in the Experimental Procedures. Crystallization trials with the full-length protein produced crystals that grew from a precipitate after about 3 months. Analysis of dissolved crystals using SDS-PAGE reveals that they contain a fragment of the full-length protein of about 14.7 kDa ( Figure 1). A domain of similar size could be obtained by incubating the IBV-N protein at room temperature for the same period (Figure 1). Thus, this polypeptide fragment presumably derives from slow proteolysis of IBV-N by traces of E. coli proteases present in the crystallization solution. In order to identify its nature, this proteolytically stable fragment was subjected to mass spectrometry, which revealed a mass of 14,692 Da. N-terminal amino acid sequencing identified residues Ser-Ser-Gly-Asn-Ala-Ser-Trp, which are located at positions 29–35 of the IBV-N amino acid sequence. Given that Ser-29 is the first amino acid of the fragment, the closest mapping onto the sequence gives Leu-160 as the C-terminal residue (calculated mass 14,691 Da). The IBV-N29-160 protein shares 37% amino acid sequence identity with the N-terminal RNA binding domain of a comparable domain from the SARS-CoV N protein (SARS-N45-181), whose structure was reported recently ( Figure 2).
Figure 1.

Structural Domains of the IBV-N Protein
(A) Schematic representation of the IBV-N protein depicting its various domains and clustering of positive charges, as inferred from the present and other studies.
(B) SDS-PAGE analysis of the full-length recombinant IBV-N protein of 44.9 kDa (lane 1, arrow) and the N-terminal proteolytically stable fragment of 14.7 kDa spanning residues 29–160 of the sequence which was crystallized (lane 2). The recombinant IBV-N29-160 is shown in lane 3.
(C) Typical plate-shaped crystals of the recombinant IBV-N29-160 protein.
Figure 2.

Overall Fold of the IBV-N Protein
(A and B) Comparison of the folds adopted by IBV-N29-160 ([A]; shown as a stereoview, top) and the N-terminal domain of the SARS-CoV nucleocapsid protein (B) (Huang et al., 2004). The two proteins are displayed in the same orientation. Secondary structure elements and some residue numbers are indicated.
(C) Topology diagram of the IBV-N29-160 protein. Its N- and C-terminal ends are labeled.
Structure Determination and Quality of the Model
Overexpression of the recombinant N-terminal IBV-N29-160 fragment readily gave crystals diffracting beyond 2.0 Å (Figure 1). Attempts to solve the structure by molecular replacement using the averaged NMR structure of a SARS-CoV nucleocapsid N-terminal domain deposited in the Protein Data Bank (PDB) (Huang et al., 2004) were unsuccessful, even though the two structures turned out to adopt a related fold (Figure 2). The IBV-N29-160 protein is devoid of methionine and cysteine residues. Thus, in order to assist structure determination using the multiwavelength anomalous dispersion (MAD) method, Ile-62, Leu-104, and Val-116 were mutated to methionine. These hydrophobic amino acid residues have been shown to introduce little perturbation in the native protein structure when substituted by methionine residues (Gassner and Matthews, 1999). In addition, the presumably exposed residue Lys-85 (as suggested by an amino acid sequence alignment with the SARS-CoV N protein) was mutated to Cys in order to introduce a potential binding site for mercury compounds. This mutated fragment of IBV-N29-160 was used for structure determination using the MAD method with crystals containing the selenomethionyl protein. Data collection, phasing, and refinement statistics are summarized in Table 1, Table 2 for the selenomethionine-derivatized crystal (SeMet) and for the native protein crystal. Overall, the path of the main chain is unambiguously defined in clear electron density for the two IBV-N29-160 molecules present in the asymmetric unit in each crystal form. A total of 134 protein residues per molecule (two extra residues at the N terminus derive from the cloning procedure) were included in the final models, which have excellent stereochemical parameters as well as 182 and 188 well-defined water molecules, respectively (Table 2). Electron density is absent for the Lys-81 side chain which is exposed to the solvent.
Table 1.
Crystallographic Data Collection and Phasing Statistics
| Data Set |
Native |
SeMet |
||
|---|---|---|---|---|
| IBV-N: 29–160 |
IBV-N: 29–160 (Three Residues Mutated to Met) |
|||
| Peak | Inflection | Remote | ||
| Wavelength | 1.5418 | 0.97943 | 0.97956 | 0.98729 |
| Cell parameters (Å, °), P1 | a = 35.48 | a = 34.77 | ||
| b = 35.72 | b = 35.37 | |||
| c = 56.11 | c = 55.95 | |||
| α = 99.05 | α = 100.51 | |||
| β = 93.93 | β = 95.48 | |||
| γ = 109.53 | γ = 110.16 | |||
| Resolution (Å) | 20–1.85 | 20–1.95 | ||
| Total number of reflections | 75,798 | 76,265 | 64,999 | 72,832 |
| No. of unique reflection | 20,031 | 20,083 | 17,032 | 19,684 |
| Completeness (%)a | 92.4 (88.8) | 96.6 (95.0) | 96.5 (95.2) | 95.6 (87.8) |
| Multiplicityb | 3.8 (3.7) | 3.8 (3.7) | 3.8 (3.6) | 3.7 (3.5) |
| Rmergec | 0.064 (0.625) | 0.05 (0.118) | 0.05 (0.131) | 0.06 (0.177) |
| I/σ(I) | 7.4 (1.1) | 8.6 (3.7) | 8.3 (4.4) | 9.1 (6.0) |
| Solvent content (%) | 43.3 | 40.6 | ||
| No. of Se sites | — | 6 | ||
| Phasing powerd | — | 0.7/0.6 | 0.6/0.4 | 0.2/1.1 |
| f′/ f″e | — | −8.1/5.7 | −10.5/3.3 | −4.3/0.5 |
| Figure of meritf 20–2.5 Å | — | 0.61/0.793 | ||
The numbers in parentheses refers to the last (highest) resolution shell.
For the SeMet crystal, Friedel pairs are treated as different reflections.
Rmerge = ΣhΣi|Ihi − <Ih>|/Σh,i Ihi, where Ihi is the ith observation of the reflection h, while <Ih> is its mean intensity.
Anomalous phasing power/dispersive phasing power, where anomalous phasing power is |λiFh| − |λiF−h|/anomalous lack of closure and dispersive phasing power is |λiFh| − |λjFh|/dispersive lack of closure.
Values of f′ and f″ where estimated from a scan of the absorption edge using the program CHOOCH (Evans and Pettifer, 2001).
Figures of merit are given before and after real space density modification, respectively.
Table 2.
Refinement Statistics
| Native | SeMet | |
|---|---|---|
| Resolution range (Å) | 19.92–1.85 | 20.0–1.95 |
| Intensity cutoff (F/σ(F)) | none | none |
| No. of reflections: completeness (%) | 100.0 | 96.1 |
| Used for refinement | 18,921 | 16,077 |
| Used for Rfree calculation | 1,026 | 881 |
| No. of nonhydrogen atoms | ||
| Protein | 2130 | 2128 |
| Water molecules | 188 | 182 |
| R factor (%)a | 22.96 | 22.73 |
| Rfree (%)b | 27.03 | 27.59 |
| Rms deviations from ideality | ||
| Bond lengths (Å) | 0.007 | 0.008 |
| Bond angles (°) | 1.05 | 1.14 |
| Ramachandran Plot | ||
| Residues in most favored regions (%) | 88.8 | 90.3 |
| Residues in additional allowed regions (%) | 10.2 | 9.2 |
| Residues in generously allowed regions (%) | 1.0 | 0.5 |
| Overall G factorc | 0.10 | 0.04 |
| PDB accession code | 2BXX | 2BTL |
R factor = Σ ‖Fobs| − |Fcalc‖/Σ |Fobs|.
Rfree was calculated with 5% of reflections excluded from the whole refinement procedure.
G factor is the overall measure of structure quality from PROCHECK (Laskowski et al., 1993).
Overall Structure
The two monomers present in the asymmetric unit can be superimposed with a root mean square (rms) deviation of 0.5 Å for their main chain atoms. The IBV-N29-160 monomer has approximate overall dimensions of 35 Å × 35 Å × 30 Å and consists of a core formed by a five-stranded antiparallel β sheet with the topology β4-β2-β3-β1-β5, which faces a smaller antiparallel sheet composed of only two strands, β1′-β4′, which are absent in the SARS-N protein (Figure 2). A long flexible hairpin loop β2′-β3′, which is inserted between the β2 and β3 strands, protrudes largely from the protein core. This extension is mobile, as shown by higher than average temperature factors, and contains several basic residues which are conserved across various coronavirus N protein sequences ( Figure 3). Extended loops spanning up to 30 residues connect the various secondary structure elements, presumably introducing flexibility to the overall architecture. This potential adaptability to various structural contexts might be important for assembly and disassembly of the nucleocapsid during the virus life cycle. The overall fold is similar to the SARS-N45-181 protein (Figure 2) with a few structural differences, such as the presence of a short 310 helix connecting strands β1′ and β2. Overall, a three-dimensional structural alignment between the SARS-CoV and IBV nucleocapsid N-terminal domains using the program DALI (Holm and Sander, 1993) shows that a total of 124 equivalent Cα atoms can be superimposed, with an rms deviation of 3.0 Å. The Z score is 10.4, confirming the global similarity of the two folds. The rather large difference between the SARS-CoV and IBV nucleocapsid N-terminal domain structures accounts for the failure of molecular replacement procedures to solve the latter structure using the former as a model. The important structural differences we observe between the SARS-CoV and IBV nucleocapsid N-terminal domain structures may stem from an inherent mobility of the coronavirus nucleocapsid structure or from a large uncertainty of the atomic positions determined by NMR, or both. A search through the PDB did not return any other protein with a statistically significant Z score, emphasizing the uniqueness of this fold as noted by Huang et al. (2004).
Figure 3.

Structure-Based Alignment of Coronavirus Nucleocapsid Amino Acid Sequences Corresponding to the Proteolytically Stable N-Terminal Fragment
Secondary structure elements are labeled above the sequence for IBV-N29-160 and below for the SARS-CoV N-terminal fragment (Huang et al., 2004). Sequences of IBV (infectious bronchitis virus, strain Beaudette, NP_040838); H-CoV (human coronavirus, strain HKU1, YP_173242); MHV (murine hepatitis virus, strain 1, AAA46439); TGEV (porcine transmissible gastroenteritis virus, strain RM4, AAG30228); and SARS (SARS-CoV, 1SSK_A) were obtained from GenBank. Conserved residues are shaded.
Dimer Formation
In our crystal structure, the two monomers assemble into a butterfly-shaped dimer related by a 180° rotation, burying in this interaction an accessible surface area of 560 Å2. The transformation is not a pure rotation, as a residual translation is needed to bring the two monomers into coincidence. The relatively small surface area suggests a rather weak binding affinity, an observation in agreement with the fact that, using size exclusion chromatography, the recombinant IBV-N29-160 protein predominantly elutes as a monomer (see below). This is also consistent with our findings of a different dimeric interface adopted by the same recombinant IBV-N29-160 protein in a nonrelated crystal form (with space group C2) that diffracts only to medium resolution.
Nucleic Acid Binding
In order to package the viral genome of 27.6 kb, the IBV-N protein must provide extended surfaces to bind the viral RNA genome both specifically and nonspecifically (without a requirement for a special base sequence). N- and C-terminal regions of IBV-N encompassing residues 1–171 and 268–407, respectively, interact with noncoding regions of the viral genomic RNA located at its 3′ end (Zhou and Collisson, 2000). As the fragment 1–91 does not bind RNA, residues between 91 and 171 were proposed to either make direct contacts with RNA or be necessary for the integrity of the protein structure (Zhou and Collisson, 2000). Because the segment 92–95 includes strictly conserved hydrophobic residues which are buried in the protein core in our structure, we propose that the fragment 1–91 studied by Zhou and Collisson (2000) was probably poorly folded and thus nonactive. We tested nucleic acid binding by IBV-N29-160 and found that the recombinant fragment was able to bind an oligoribonucleotide from the 3′ end of the viral genome ( Figure 4). This result is in agreement with studies by Huang et al. (2004), who used NMR to demonstrate that SARS-CoV N45-181 could bind a 32-mer oligoribonucleotide located at the 3′ end of the SARS-CoV genome. Interestingly, this oligoribonucleotide has a highly conserved sequence across various coronaviruses including IBV, and adopts a unique tertiary structure (Robertson et al., 2005). A surface representation of electrostatic charges of the IBV-N29-160 protein shown in Figure 5 reveals a striking segregation in the charge distribution on the protein surface. The β2′-β3′ hairpin forms a basic patch at the thumb, whereas the base is acidic (Figure 5). These two charged patches are separated by a neutral and rather hydrophobic platform contributed by residues projecting from strands β4-β2-β3 that form a palm-like structure. An alignment of nucleocapsid protein amino acid sequences from various coronaviruses highlights the conservation of several residues exposed at the protein surface, suggesting that some might play a role in nucleic acid recognition (Figure 3, Figure 5). The topology of the protein and its charge distribution suggest a mode of RNA binding in which its phosphate groups would project toward the basic β2′-β3′ hairpin, possibly making electrostatic interactions with the conserved positively charged Arg-76 and Lys-78 residues, while the sugar and base moieties would contact the hydrophobic platform. In this model, the exposed hydrophobic residues Tyr-92 and Tyr-94 (strand β3) could form stacking interactions with the bases, as was observed, for instance, in complexes between the vaccinia virus protein VP39 and mRNA (Hu et al., 1999) or between the matrix protein VP40 from Ebola virus and a triribonucleotide (Gomis-Ruth et al., 2003). As suggested by Huang et al. (2004), additional favorable interactions might be formed upon closure of the flexible β2′-β3′ hairpin onto the incoming RNA ligand.
Figure 4.

Analysis of the RNA Binding Activity of the Full-Length IBV-N Protein and IBV-N29-160 and IBV-N218-329 Fragments
The purified IBV-N (lanes 2 and 7), IBV-N29-160 (lanes 3 and 8), IBV-N218-329 (lanes 4 and 9), His-tagged IBV-N29-160 (lanes 5 and 10), and GST (negative control, lanes 6 and 11) were separated on a 15% SDS-PAGE gel. The proteins were either visualized by Coomassie brilliant blue staining (lanes 1–6) or transferred to Hybond C extra membrane (Amersham) and detected by Northwestern blot with a digoxin-labeled RNA probe corresponding to the IBV genome sequence from nucleotides 26,539–27,608 (lanes 7–11). Molecular masses of standard proteins are indicated.
Figure 5.

Proposed RNA Binding Site of IBV-N
(A) Surface representation of the IBV-N29-160 fragment with electrostatic potentials colored in blue (positive) and red (negative). Residues which are suggested to participate in RNA binding are labeled. The N- and C-terminal ends of the polypeptide chains are indicated.
(B) Close-up view of the proposed RNA binding site of the IBV-N29-160 fragment. The Cα trace of IBV-N29-160 is displayed. Side chains which are likely to participate in nucleic acid binding are shown as sticks.
In Vitro Oligomerization of the IBV Nucleocapsid Protein
Oligomerization of N protein has been studied in MHV (Robbins et al., 1986) and SARS-CoV (He et al., 2004). Trimers of N subunits linked by intermolecular disulfide bonds were identified in MHV. Using mutational analysis, the Ser/Arg-rich motif spanning residues 184–196 (immediately downstream from our crystallized fragment) was shown to be essential for the multimerization of the N protein from SARS-CoV (He et al., 2004). We analyzed the oligomerization states of the full-length IBV-N protein, IBV-N29-160, and IBV-N218-329 in solution. Crosslinking experiments were performed using glutaraldehyde, a short self-polymerizing reagent mostly reacting with the amino and amine groups of lysine and histidine, respectively (Buehler et al., 2005), and suberic acid bis N-hydroxy-succinamide ester (SAB), a reagent which only crosslinks lysine residues at larger distances. Concentrations of crosslinking agent higher than 0.1 mM led to the formation of dimers, tetramers (but not trimers), and larger oligomers of IBV-N, along with the disappearance of monomeric species ( Figure 6). By contrast, an approximately 20-fold higher concentration of crosslinking agent (2 mM glutaraldehyde or 1 mM SAB; see Figure 6) was required to obtain equal amounts of monomers and dimers of IBV-N29-160. This suggests that regions within the C-terminal domain of IBV-N make a predominant contribution to the multimerization of IBV-N. Secondary structure predictions and limited proteolysis studies of the IBV-N protein suggest the presence of a structured—possibly α-helical—C-terminal domain of about 12 kDa, which is connected to IBV-N29-160 by a Ser/Arg/Ala/Gly-rich loop of approximately 50 amino acid residues (Figure 1). We expressed such a stable recombinant C-terminal domain encompassing residues 218–329 of the N protein in a soluble form. This C-terminal domain can bind RNA (Figure 4), fold independently, and was recently crystallized (H.F., D.X.L., and J.L., unpublished data). Crosslinking experiments show that IBV-N218-329 forms dimers, trimers, tetramers, and higher oligomers for concentrations of crosslinking agent higher than 1 mM with a concomitant decrease in monomer species, thus confirming the important contribution of the C-terminal domain of IBV to the formation of IBV-N multimers (Figure 6). As an independent confirmation, we subjected the IBV-N29-160 and IBV-N218-329 domains to size exclusion chromatography ( Figure 7). Under these conditions, the C-terminal domain IBV-N218-329 elutes faster than the N-terminal domain as a sharp symmetric peak corresponding to a dimer. The N-terminal domain elutes at a position intermediate between a monomer and a dimer (with an estimated size corresponding to a protein of molecular weight 18.1 kDa). This pattern of migration could be due to the asymmetric shape of the IBV N-terminal domain or could be indicative of the presence of a mixture of monomer and dimer of the N-terminal domain in solution.
Figure 6.

Crosslinking Experiments
(A) Full-length IBV-N protein.
(B) IBV-N29-160 protein, which was crystallized.
(C) C-terminal fragment, IBV-N218-329.
The nature and concentrations of crosslinking agent are shown. Monomer, dimer, trimer, and tetramer species of the recombinant proteins are indicated.
Figure 7.
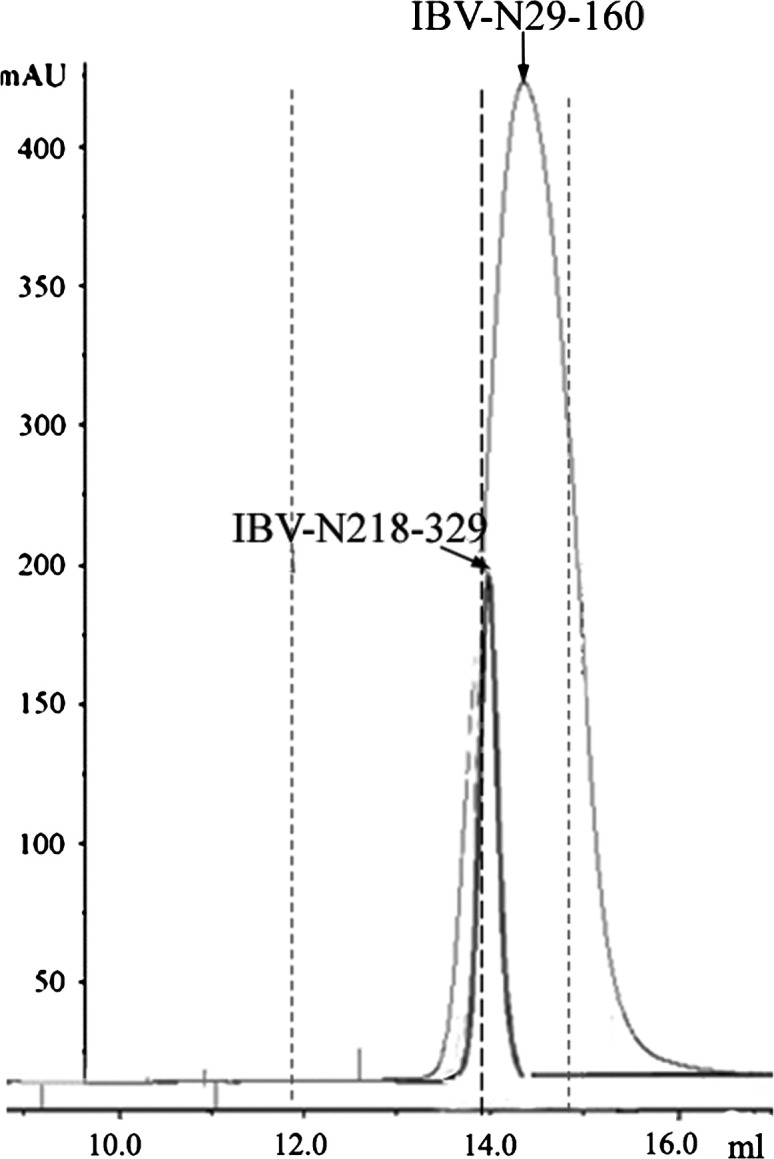
Size Exclusion Chromatography Elution Profiles of IBV-N29-160 and IBV-N218-329
The vertical axis shows absorbance at 280 nm. The horizontal axis indicates the elution volume in milliliters. Three thin vertical lines indicate the positions of molecular weight of protein standards (from left to right: ovalbumin, 43 kDa; chymotrypsinogen A, 25.0 kDa; and ribonuclease A, 13.7 kDa). The large difference in absorbance stems from the different individual molar absorbance coefficients at 280 nm of IBV-N29-160 (40,540 M−1cm−1) and IBV-N218-329 (4,080 M−1cm−1).
Implications for Coronavirus Nucleocapsid Assembly
Our data suggest that residues 218–329 at the C-terminal end of the IBV-N protein play a major role for its multimerization. This is consistent with results reported by Surjit et al. (2004), who studied SARS-CoV nucleocapsid dimerization using the yeast two-hybrid system, and points to conserved assembly properties between the SARS-CoV and IBV in spite of significant amino acid differences between their two nucleocapsid proteins. Can we ascribe a function to multimer formation by the N protein? One obvious explanation is that multimerization increases the protein surface area accessible for binding the viral genomic RNA, thus providing the elementary building block for nucleocapsid assembly. Indeed, several crystal structures of capsid proteins have revealed the presence of multimers that present continuous patches of basic residues at their surface: the capsid proteins of West Nile virus and Borna disease virus form tetrameric assemblies (Dokland et al., 2004, Rudolph et al., 2003) and the nucleocapsid protein of porcine respiratory syndrome virus, an arterivirus, forms dimers (Doan and Dokland, 2003). Unfortunately, because these structures were determined in the absence of an RNA ligand, it is difficult to evaluate to what extent multimer formation is coupled with nucleic acid recognition. In the Arteviridae, a viral family genomically related to the coronaviruses, the basic N-terminal half of the nucleocapsid protein is involved in RNA binding while its C-terminal domain forms a tight dimer (Doan and Dokland, 2003).
Further complexity for the study of coronavirus nucleocapsid assembly stems from its interaction with the M protein endodomain (Kou and Masters, 2002, Narayanan et al., 2000, Narayanan et al., 2003) and from the fact that several coronavirus proteins can interact with single-stranded RNA, including the nsp9 replicase protein from SARS-CoV (Egloff et al., 2004, Sutton et al., 2004). In the absence of a nucleic acid ligand, the N protein appears to be composed of two main globular domains loosely connected by Arg/Ser/Ala/Gly-rich loops that are highly sensitive to proteolysis. These connecting regions may undergo modifications (e.g., phosphorylation) that could influence the multimerization state of the protein and control its interaction with RNA. In a recent report, sumoylation of Lys-62 of SARS-CoV N protein expressed in mammalian cells was proposed to promote dimerization of the protein (Li et al., 2005). It is not known whether similar modifications of the IBV-N occur in virus-infected cells. Nevertheless, a working hypothesis for coronavirus nucleocapsid formation can be proposed ( Figure 8). In this model, viral genomic RNA binding by both the N- and C-terminal domains would lead to a clustering of N proteins. Dimerization of the C-terminal domains would trigger oligomerization of the N-terminal domains by increasing their local concentration above a certain threshold. In turn, this would trigger condensation of viral RNA. Interdomain flexibility we have defined in the linker regions could facilitate the necessary conformational changes during the transition to a more compact form of the ribonucleocapsid (Figure 8).
Figure 8.

Hypothetical Model for the Assembly of the IBV Ribonucleoprotein Complex
(A) Both the N- (cyan) and C-terminal (green) domains of the IBV-N protein can bind RNA (represented as a thin orange line). The basic patch in IBV-N29-160 is depicted by plus signs. Dimerization of the C-terminal domains (arrows) leads to a clustering of IBV-N proteins and to their oligomerization.
(B) The endodomain of the integral membrane protein M can provide further contacts to the ribonucleocapsid (see text). However, the precise coupling between RNA recognition and IBV-N multimerization remains uncertain.
Further studies are underway to elucidate the three-dimensional structure of the globular C-terminal domain of IBV-N, to define the interactions between the IBV-N protein and viral RNA, and to characterize the morphology of the ribonucleocapsid.
Experimental Procedures
Cloning and Expression
The gene encoding the IBV-N protein was amplified by PCR using the Pfu polymerase (Stratagene, Singapore) with the forward (5′-ATTATT CAT ATG GCA AGC GGT AAA GCA GC-3′) and reverse primer (5′-ATTATT CTC GAG TCA AAG TTC ATT CTC TCC TA-3′) and cloned into the pET 29b vector using T4 ligase (Research Biolabs, Singapore). The underlined sequences correspond to NdeI and XhoI sites, respectively. Proteins (lacking the His6 tag due to the insertion of a stop codon in the reverse primer) were expressed in E. coli BL21(DE3). The cells were grown at 37°C in Luria-Bertani medium containing 100 μg/ml ampicillin until the culture reached an OD600 of 0.7. Protein expression was induced by the addition of 1 mM isopropyl-β-D-thiogalactopyranoside for 3 hr at 30°C. Cells harvested and resuspended at 4°C in a buffer containing 20 mM Na3PO4 (pH 7.8) were lysed by sonication and the remaining insoluble material was removed by centrifugation at 20,000 × g for 20 min at 4°C. N- and C-terminal fragments of the IBV-N gene coding for residues 29–160 and 218–329, respectively, were cloned into pET-16b using the following primers: 5′-AATA CATATG TCT TCT GGA AAT GCA TCT TG-3′; 5′-AATA CTC GAG TCA CAG GGG AAT GAA GTC CC-3′ and 5′-A AATA CAT ATG AAG GCA GAT GAA ATG GC-3′; 5′-AA ATA CTC GAG TCA CGT TCC TAC ACC ATC GAC-3′. These two proteins (hereafter named IBV-N29-160 and IBV-N218-329, respectively) were expressed as described above for IBV-N, yielding truncated fragments having a His10 tag at their N terminus followed by a Factor Xa cleavage site. The His10 tags were cleaved during purification. Expression of the selenomethionylated protein IBV-N29-160 was carried out as described in Doublié (1997).
Analysis of the Proteolytically Stable Fragment Derived from IBV-N
Automated N-terminal amino acid sequence determination of the proteolytic fragment obtained by degradation of IBV-N was performed using an Applied Biosystems (Singapore) Procise sequencer. The molecular mass of purified proteins was analyzed using a MALDI-TOF mass spectrometer (API 300 MS/MS; Applied Biosystems).
Protein Purification
The IBV-N protein precipitated with ammonium sulfate at 30% saturation, was centrifuged and resuspended in PBS, dialyzed against buffer A (20 mM HEPES, 1 mM EDTA, 1 mM DTT [pH 6.8]), and loaded onto a cation exchange chromatography column (Mono S HR 5/5; GE Biosciences, Singapore) preequilibrated with buffer A. Elution was carried out using an NaCl gradient of buffer B (20 mM HEPES, 1 mM EDTA, 1 mM DTT, 1 M NaCl [pH 6.8]). Fractions containing the protein—as shown by SDS-PAGE—were pooled and concentrated to 10–15 mg/ml by ultrafiltration using a Centriprep device (Millipore, Singapore) with a molecular weight cutoff of 10 kDa. Size exclusion chromatography (Superdex 75; Amersham) was carried out in a buffer containing 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM DTT, 0.1% NaN3. The protein was concentrated to 10 mg/ml as determined by the Bradford assay (Bio-Rad, Singapore), using BSA as a standard. The truncated recombinant IBV-N29-160 was resuspended in PBS and loaded onto an Ni-NTA column (Qiagen, Singapore) preequilibrated with 20 mM KH2PO4, 50 mM NaCl (pH 7.8). After washing with 20 mM KH2PO4, 1 M NaCl, 10 mM imidazole (pH 7.2), IBV-N29-160 was eluted using a buffer containing 20 mM KH2PO4, 0.5 M NaCl, 0.5 M imidazole (pH 6.0). The His10 tag was removed by proteolysis with Factor Xa in a buffer containing 100 mM NaCl, 2 mM CaCl2, 10 mM Tris (pH 8.0) using a substrate enzyme molar ratio of 50:1 for 4 hr at room temperature. The cleavage mixture was loaded onto a benzamidine column to eliminate Factor Xa, and the IBV-N29-160 protein recovered in the flow through was subjected to two final steps of purification as described above for the full-length IBV-N protein. Purification of the recombinant IBV-N218-329 was carried out using a similar protocol. Purification of the selenomethionine-substituted protein was performed using the same protocol as the native protein.
Crystallization of IBV-N29-160
Crystals of the recombinant IBV-N29-160 were grown at 18°C by vapor diffusion using the hanging drop method. Two microliters of the protein at a concentration of 10 mg/ml was mixed with an equal volume of the precipitating solution from the well (0.1 M sodium sulfate, 20% PEG 3350), yielding plate-shaped crystals growing to maximum dimensions of about 0.3 × 0.3 × 0.05 mm3 in about 2 weeks (Figure 1). Crystals of the selenomethionine protein were obtained under the same conditions.
Data Collection, Structure Determination, and Refinement
For data collection, crystals were soaked in a cryoprotecting solution (25% glycerol, 0.1 M sodium sulfate, 20% PEG 3350 [pH 6.5]) before being mounted and cooled to 100 K in a nitrogen gas stream (Oxford Cryosystems, Oxford, UK). Diffraction intensities at three wavelengths (Table 1) were recorded from a selenomethionine (SeMet)-derivatized IBV-N29-160 crystal on beamline NW12 at the Photon Factory (Tsukuba, Japan) on an ADSC charge-coupled device (CCD) detector (ADSC Corporation, Poway, CA) using an attenuated beam of dimensions 0.1 × 0.1 mm2 (Table 1). Integration, scaling, and merging of the intensities were carried out using programs from the CCP4 (1994). The six selenium atoms present within the two molecules of the asymmetric unit were located using the program SOLVE (Terwilliger, 2003). An initial electron density map was calculated and modified using the program RESOLVE (Terwilliger, 2003), using these selenium atom positions to locate the noncrystallographic symmetry (ncs) axis relating the two molecules in the asymmetric unit, and model building was first carried out in this map using the program O (Jones et al., 1991). For subsequent cycles, electron density maps were calculated using partial model phases combined with experimental MAD phases with the program REFMAC5 from the CCP4 (1994), which was used for the initial refinement of the structure, that included ncs restraints. A few cycles of refinement using molecular dynamics with a slow cooling protocol using a maximum likelihood target incorporating phase probability distribution encoded in the form of Hendrickson Lattman coefficients were subsequently carried out using the program CNS (Brunger et al., 1998), with ncs restraints. A data set for the native protein was collected on an R axis IV++ image plate detector using CuKα radiation from a Micromax-007 rotating anode (Rigaku/MSC, The Woodlands, TX) operating at 20 mA and 40 kV (Table 1). The SeMet model was placed in the native crystal form and adjustments to the model were carried out using difference Fourier maps calculated with REFMAC5, which was used for refinement. Superposition of structures and rms deviation calculations were carried out using the program LSQKAB from the CCP4 (1994). Figure 2, Figure 5 were produced with the program PyMOL (DeLano, 2002).
Crosslinking Experiments
The purified recombinant proteins IBV-N, IBV-N29-160, and IBV-N218-329 were incubated with either glutaraldehyde or SAB (Sigma-Aldrich, St. Louis, MO) for 2 hr at 20°C using a constant amount of protein (5 μg) with increasing amounts of the crosslinking agent. The samples were submitted to electrophoresis on an 8%–15% SDS-PAGE gel and stained with Coomassie blue.
Size Exclusion Chromatography
A Superdex 75 10/300 GL size exclusion chromatographic column (Amersham) mounted on an AKTA FPLC (GE Biosciences, Singapore) was used to analyze the homogeneity and apparent multimerization states of IBV-N29-160 and IBV-N218-329, respectively. Protein concentrations used were 10 mg/ml and the loaded sample volume was 0.1 ml. The buffer was 10 mM Tris-HCl, 0.2 M NaCl, 3 mM β-mercapto-ethanol (pH 7.5) and the flow rate was 0.5 ml/min. Standard protein markers (Amersham) used for calibration were ribonuclease A, 13.7 kDa, elution 14.88 ml; chymotrypsinogen A, 25.0 kDa, 13.81 ml; ovalbumin, 43.0 kDa, 11.81 ml; BSA, 67.0 kDa, 10.89 ml. Apparent size/molecular weights were deduced by plotting Kav versus log (MW) with Kav = (Ve − V0)/(Vt − V0), where Ve is the elution volume of the protein, Vt is the total column bed volume, and V0 is the void volume.
RNA Binding Assay
The full-length IBV-N protein and the IBV-N29-160 and IBV-N218-329 fragments were expressed in E. coli BL21 cells and purified as described above. The polyhistidine tags of the truncated proteins were removed by digestion with Factor Xa. The purified proteins were separated on 15% SDS-PAGE, transferred to Hybond C extra membrane (Amersham), and probed with digoxin-labeled RNA representing the negative sense of the IBV genome from nucleotides 25,873–27,608. The probe was made by in vitro transcription using SP6 polymerase in the presence of digoxin according to the manufacturer's instructions (Roche, Singapore).
Acknowledgments
We thank Jacques d'Alayer (Institut Pasteur) for performing N-terminal amino acid sequencing, and Terje Dokland and Soichi Wakatsuki for help and very useful discussions. Financial support via grants from NTU (SUG 14/02), the Singapore Biomedical Research Council (03/1/21/20/291 and 02/1/22/17/043), and the Singapore National Medical Research Council (NMRC/SRG/001/2003) to the Lescar laboratory is acknowledged as well as provision of excellent beam time by the Photon Factory (Japan).
Published: December 13, 2005
Contributor Information
Ding Xiang Liu, Email: dxliu@imcb.a-star.edu.sg.
Julien Lescar, Email: julien@ntu.edu.sg.
References
- Bosch B.J., Martina B.E., Van Der Zee R., Lepault J., Haijema B.J., Versluis C., Heck A.J., De Groot R., Osterhaus A.D., Rottier P.J. Severe acute respiratory syndrome coronavirus (SARS-CoV) infection inhibition using spike protein heptad repeat-derived peptides. Proc. Natl. Acad. Sci. USA. 2004;101:8455–8460. doi: 10.1073/pnas.0400576101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Buehler P.W., Boykins R.A., Jia Y., Norris S., Freedberg D.I., Alayash A.I. Structural and functional characterization of glutaraldehyde polymerized bovine hemoglobin and its isolated fractions. Anal. Chem. 2005;77:3466–3478. doi: 10.1021/ac050064y. [DOI] [PubMed] [Google Scholar]
- CCP4 (Collaborative Computational Project, Number 4) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Cologna R., Hogue B.G. Identification of a bovine coronavirus packaging signal. J. Virol. 2000;74:580–583. doi: 10.1128/jvi.74.1.580-583.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Guzman R.N., Wu Z.R., Stalling C.C., Pappalardo L., Borer P.N., Summers M.F. Structure of the HIV-1 nucleocapsid protein bound to the SL3 ψ-RNA recognition element. Science. 1998;279:384–388. doi: 10.1126/science.279.5349.384. [DOI] [PubMed] [Google Scholar]
- DeLano W.L. DeLano Scientific; San Carlos, CA: 2002. The PyMOL User's Manual. [Google Scholar]
- Doan D.N.P., Dokland T. Structure of the nucleocapsid protein of porcine reproductive and respiratory syndrome virus. Structure. 2003;11:1445–1451. doi: 10.1016/j.str.2003.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokland T., Walsh M., Mackenzie J.M., Khromykh A.A., Ee K.H., Wang S. West Nile virus core protein; tetramer structure and ribbon formation. Structure. 2004;12:1157–1163. doi: 10.1016/j.str.2004.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doublié S. Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 1997;276:523–530. [PubMed] [Google Scholar]
- Egloff M.-P., Ferron F., Campanacci V., Longhi S., Rancurel C., Dutartre H., Snijder E.J., Gorbalenya A.E., Cambillau C., Canard B. The severe acute respiratory syndrome coronavirus replicative protein nsp9 is a single stranded RNA-binding subunit unique in the RNA virus world. Proc. Natl. Acad. Sci. USA. 2004;101:3792–3796. doi: 10.1073/pnas.0307877101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escors D., Ortego J., Laude H., Enjuanes L. The membrane M protein carboxy terminus binds to transmissible gastroenteritis coronavirus core and contributes to core stability. J. Virol. 2001;75:1312–1324. doi: 10.1128/JVI.75.3.1312-1324.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escors D., Izeta A., Capiscol C., Enjuanes L. Transmissible gastroenteritis coronavirus packaging signal is located at the 5′ end of the virus genome. J. Virol. 2003;77:7890–7902. doi: 10.1128/JVI.77.14.7890-7902.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans G., Pettifer R.F. CHOOCH: a program for deriving anomalous-scattering factors from X-ray fluorescence spectra. J. Appl. Crystallogr. 2001;34:82–86. [Google Scholar]
- Fosmire J.A., Hwang K., Makino S. Identification and characterization of a coronavirus packaging signal. J. Virol. 1992;66:3522–3530. doi: 10.1128/jvi.66.6.3522-3530.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassner N.C., Matthews B.W. Use of differentially substituted selenomethionine proteins in X-ray structure determination. Acta Crystallogr. D Biol. Crystallogr. 1999;55:1967–1970. doi: 10.1107/s0907444999013347. [DOI] [PubMed] [Google Scholar]
- Gomis-Ruth F.X., Dessen A., Timmins J., Bracher A., Kolesnikowa L., Becker S., Klenk H.D., Weissenhorn W. The matrix protein VP40 from Ebola virus octamerizes into pore-like structures with specific RNA binding properties. Structure. 2003;11:423–433. doi: 10.1016/S0969-2126(03)00050-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He R., Dobie F., Ballantine M., Leeson A., Li Y., Bastien N., Cutts T., Andonov A., Cao J., Booth T.F. Analysis of multimerization of the SARS coronavirus nucleocapsid protein. Biochem. Biophys. Res. Commun. 2004;316:476–483. doi: 10.1016/j.bbrc.2004.02.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L., Sander C. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- Hu G., Gershon P.D., Hodel A.E., Quiocho F.A. mRNA cap recognition: dominant role of enhanced stacking interactions between methylated bases and protein aromatic side chains. Proc. Natl. Acad. Sci. USA. 1999;96:7149–7154. doi: 10.1073/pnas.96.13.7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q., Yu L., Petros A.M., Gunasekera A., Liu Z., Xu N., Hajduk P., Mack J., Fesik S.W., Olejniczak E.T. Structure of the N-terminal RNA-binding domain of the SARS CoV nucleocapsid protein. Biochemistry. 2004;43:6059–6063. doi: 10.1021/bi036155b. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zhou J.-Y., Cowan S.W., Kjeldgaard M. Improved methods for the building of protein models in electron density and the location of errors in these models. Acta Crystallogr. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Kou L., Masters P.S. Genetic evidence for a structural interaction between the carboxy termini of the membrane and nucleocapsid proteins of mouse hepatitis virus. J. Virol. 2002;76:4987–4999. doi: 10.1128/JVI.76.10.4987-4999.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.M.C., Holmes K.V. 4th Edition. Lippincott Raven; Philadelphia: 2001. Coronaviridae: The Viruses and Their Replication in Fundamental Virology. [Google Scholar]
- Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- Lescar J., Roussel A., Wien M.W., Navaza J., Fuller S.D., Wengler G., Wengler G., Rey F.A. The fusion glycoprotein shell of Semliki Forest virus: an icosahedral assembly primed for fusogenic activation at endosomal pH. Cell. 2001;105:137–148. doi: 10.1016/s0092-8674(01)00303-8. [DOI] [PubMed] [Google Scholar]
- Li F.Q., Xiao H., Tam J.P., Liu D.X. Sumoylation of the nucleocapsid protein of severe acute respiratory syndrome associated coronavirus. FEBS Lett. 2005;579:2387–2396. doi: 10.1016/j.febslet.2005.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y. The genome sequence of SARS-associated coronavirus. Science. 2003;300:1399–1403. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- Narayanan K., Maeda A., Maeda J., Makino S. Characterization of the coronavirus M protein and nucleocapsid in infected cells. J. Virol. 2000;74:8127–8134. doi: 10.1128/jvi.74.17.8127-8134.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan K., Chen C.J., Maeda J., Makino S. Nucleocapsid-independent specific viral RNA packing via viral envelope protein and viral RNA signal. J. Virol. 2003;77:2922–2927. doi: 10.1128/JVI.77.5.2922-2927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris J.S., Chu C.M., Cheng V.C., Chan K.S., Hung I.F., Poon L.L., Law K.I., Tang B.S., Hon T.Y., Chan C.S. Clinical progression and viral load in a community outbreak of coronavirus associated SARS pneumonia: a prospective study. Lancet. 2003;361:1767–1772. doi: 10.1016/S0140-6736(03)13412-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins S.G., Frana M.F., McGowan J.J., Boyle J.F., Holmes K.V. RNA-binding proteins of coronavirus MHV: detection of monomeric and multimeric N protein with an RNA overlay-protein blot assay. Virology. 1986;150:402–410. doi: 10.1016/0042-6822(86)90305-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson M.P., Igel H., Baertsch R., Haussler D., Ares M., Jr., Scott W.G. The structure of a rigorously conserved RNA element within the SARS virus genome. PLoS Biol. 2005;3:e5. doi: 10.1371/journal.pbio.0030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Peñaranda S., Bankamp B., Maher K., Chen M.-h. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1398. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- Rudolph M.G., Kraus I., Dickmanns A., Eickmann M., Garten W., Ficner R. Crystal structure of the borna disease virus nucleoprotein. Structure. 2003;11:1219–1226. doi: 10.1016/j.str.2003.08.011. [DOI] [PubMed] [Google Scholar]
- Sturman L.S., Holmes K.V., Behnke J. Isolation of coronavirus envelope glycoproteins and interaction with the viral nucleocapsid. J. Virol. 1980;33:449–462. doi: 10.1128/jvi.33.1.449-462.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surjit M., Liu B., Kumar P., Chow V.T.K., Lal S.K. The nucleocapsid protein of the SARS coronavirus is capable of self association through a C-terminal 209 amino acid interaction domain. Biochem. Biophys. Res. Commun. 2004;317:1030–1036. doi: 10.1016/j.bbrc.2004.03.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton G., Fry E., Carter L., Sainsbury S., Walter T., Nettleship J., Berrow N., Owens R., Gilbert R., Davidson A. The nsp9 replicase protein of SARS-coronavirus, structure and functional insights. Structure. 2004;12:341–353. doi: 10.1016/j.str.2004.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger T.C. SOLVE and RESOLVE: automated structure solution and density modification. Methods Enzymol. 2003;374:22–37. doi: 10.1016/S0076-6879(03)74002-6. [DOI] [PubMed] [Google Scholar]
- Valegard K., Murray J.B., Stonehouse N.J., van den Worm S., Stockley P.G., Liljas L. The three-dimensional structures of two complexes between recombinant MS2 capsids and RNA operator fragments reveal sequence-specific protein-RNA interactions. J. Mol. Biol. 1997;270:724–738. doi: 10.1006/jmbi.1997.1144. [DOI] [PubMed] [Google Scholar]
- Vennema H., Godeke G.J., Rossen J.W.A., Voorhout W.F., Horzinek M.C., Opstelten D.J., Rottier P.J.M. Nucleocapsid-independent assembly of coronavirus-like particles by co-expression of viral envelope protein gene. EMBO J. 1996;15:2020–2029. doi: 10.1002/j.1460-2075.1996.tb00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A.K., Wang L., Sneed L.W., Collisson E.W. Comparative analysis of the nucleocapsid genes of several strains of infectious bronchitis virus and other coronaviruses. Virus Res. 1992;25:213–222. doi: 10.1016/0168-1702(92)90135-V. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M., Collisson E.W. The amino and carboxyl domains of the infectious bronchitis virus nucleocapsid protein interact with 3′ genomic RNA. Virus Res. 2000;67:31–39. doi: 10.1016/S0168-1702(00)00126-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M., Williams A.K., Chung S.I., Wang L., Collisson E.W. The infectious bronchitis virus nucleocapsid protein binds RNA sequences in the 3′ terminus of the genome. Virology. 1996;217:191–199. doi: 10.1006/viro.1996.0106. [DOI] [PubMed] [Google Scholar]