

Graphical abstract

Keywords: Respiratory syndrome; Coronavirus; 3CL protease; 1,2,3-triazole; Biological evaluation
Highlights
-
•
A novel series of fused 1,2,3-triazoles was accessed via an organocatalytic route.
-
•
Compounds 14d, 14n, 14q, 18f and 18i are active against coronavirus 229E.
-
•
Molecular modelling work flow was developed to identify key molecular interactions.
-
•
All active compounds established the key interactions based on molecular modelling.
Abstract
Synthesis and biological evaluation of a novel library of fused 1,2,3-triazole derivatives are described. The in-house developed multicomponent reaction based on commercially available starting materials was applied and broad biological screening against various viruses was performed, showing promising antiviral properties for compounds 14d, 14n, 14q, 18f and 18i against human coronavirus 229E. Further in silico studies identified the key molecular interactions between those compounds and the 3-chymotrypsin-like protease, which is essential to the intracellular replication of the virus, supporting the hypothesis that the protease is the target molecule of the potential antiviral derivatives.
Coronaviruses are single-stranded RNA viruses associated with mild to severe respiratory symptoms. Human coronaviruses (HCoV) strains HCoV-229E and HCoV-OC43 were first described in the 1960s as causes for respiratory tract infections in humans, including common cold and pneumonia.1 In 2002–2003, a new human coronavirus, named SARS-CoV, was identified as the etiological agent for the global outbreak of severe acute respiratory syndrome (SARS), which caused the death of over 800 individuals among 8000 cases worldwide, representing a fatality rate of almost 10%.2, 3, 4 Since then three additional coronaviruses have been recognized. Initially, HCoV-NL635 and HCoV-HKU1,6 were reported causing acute respiratory diseases of lower severity compared to the SARS-CoV and more recently, Middle East respiratory syndrome (MERS-CoV) causing lethal respiratory diseases.7, 8 To date, there are no approved antiviral drugs or vaccines available for the prevention and/or treatment of SARS-like viruses making the development of effective antiviral agents an imperative need.9
Coronaviruses express two proteases, a papain-like protease (PLpro) and a 3-chymotrypsin-like protease (3CLpro). The 3CLpro enzyme, also referred to as Main protease (Mpro), is essential to the intracellular viral replication, making it an attractive target for the development of novel inhibitors.10 Reports in the literature classify potential antiviral compounds into two main categories: i) the peptidomimetics (Fig. 1 A) and ii) small molecule-based inhibitors (Fig. 1B) presenting both activities in μM and nM range. Despite the satisfying initial results, the majority of those promising compounds did not proceed to clinical studies due to nonideal physicochemical properties.9
Fig. 1.

Representative A) peptidic13 and B) small molecule SARS-CoV 3CLpro inhibitors.14, 15, 16, 17, 18, 19
References on non-peptidic inhibitors accentuate the presence of the benzotriazole group. The importance of this benzotriazole motif relies on the formation of key interactions with the catalytic dyad, Cys145 and His41, of the 3CLpro active site.11 Considering our interest in the chemistry of 1,2,3-triazole bioactive molecules and their interesting binding mode, we wanted to prepare a novel library of fused 1,2,3-triazoles and subsequently determine their biological activity.
Here, we reported our preliminary results on the development of a novel series of fused 1,2,3-triazole compounds and their potential antiviral activity. For this purpose, we implemented the multicomponent reaction developed within our group, which resulted in a plethora of 1,2,3-triazole derivatives in a single step starting from readily available enolizable carbonyl compounds, primary amines and 4-nitrophenyl azide. In general, the method proceeds via an equilibrium of imine/enamine followed by [3 + 2] cycloaddition with the azide.12 This leads to a triazoline intermediate which after elimination of 4-nitroaniline results in the final fused 1,2,3-triazole analogues.
The synthesis, presented in Scheme 1 , began by following a general method to generate the oxopiperidine carboxylate intermediate which involves Michael addition of aniline onto ethyl acrylate followed by an intramolecular Dieckmann condensation.20 Subsequent nucleophilic substitution with benzyl bromide provided the starting material 11 in 65% overall yield. Once 11 was obtained, we proceeded for the fused triazole formation using a collection of primary amines and 4-nitrophenyl azide (PNA, 13).
Scheme 1.

Synthetic pathway towards derivatives 14a–r. Reagents and conditions:(a) AcOH, CuCl, 24 h, 110 °C, 55%, (b) NaH, Toluene, EtOH, 6 h, 100 °C, 90%, (c) BnBr, K2CO3, THF, 6 h, 70 °C, 65%,(d) Toluene, 18 h, 100 °C.
We commenced our investigations with benzylic amines bearing both electron-donating functional groups, (Table 1 14b, c) and electron-withdrawing groups, (Table 1 14d–j). Both families were obtained in moderate to good yields.
Table 1.
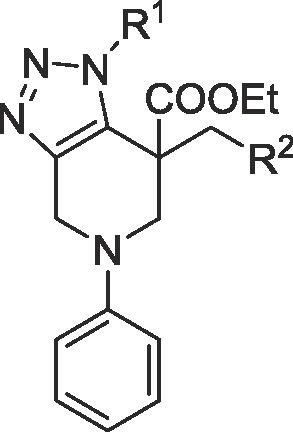
| Entry | Compound ID | R1 | R2 | Yield | EC50 (μM)a |
|---|---|---|---|---|---|
| 1 | 14a |  |
 |
80% | >100 |
| 2 | 14b |  |
85% | >100 | |
| 3 | 14c | 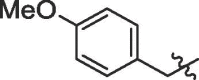 |
65% | >100 | |
| 4 | 14d | 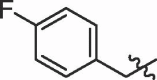 |
80% | 8.95 | |
| 5 | 14e | 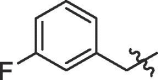 |
58% | >100 | |
| 6 | 14f | 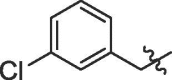 |
80% | >100 | |
| 7 | 14g | 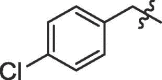 |
67% | >100 | |
| 8 | 14h | 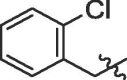 |
48% | >100 | |
| 9 | 14i | 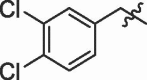 |
30% | >100 | |
| 10 | 14j | 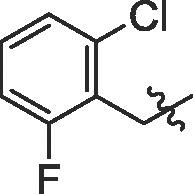 |
73% | >100 | |
| 11 | 14k |  |
55% | >100 | |
| 12 | 14l | 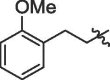 |
56% | >100 | |
| 13 | 14m | 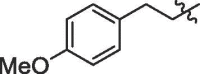 |
85% | >100 | |
| 14 | 14n | 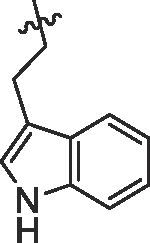 |
80% | 9.45 | |
| 15 | 14o | 57% | >100 | ||
| 16 | 14p | 51% | >100 | ||
| 17 | 14q | 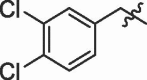 |
 |
50% | 9.45 |
| 18 | 14r |  |
 |
60% | >100 |
Concentration required to reduce virus-induced cytopathogenicity by 50%.
Further analysis using heterocyclic and aliphatic derivatives 14k–p proved the applicability of these substituents under the high temperature conditions used in this reaction (Table 1). Furthermore, we examined the influence of the introduction of a fluoro group on the benzyl bromide on the reactivity (Table 1, entries 17 and 18), which demonstrated that there is no effect on the outcome of the reaction.
A second series of fused 1,2,3-triazole analogues was prepared based on the previously described multicomponent method, starting from N-phenyl-4-piperidone material 17 which in turn was synthesized according to Buchwald-Hartwig Pd-catalyzed amination of different aryl bromides, presented in Scheme 2 .21 We initially examined the scope of this reaction with respect to primary amines. As we would expect based on the aforementioned results, we managed to synthesize an adequate number of examples in good to excellent yields (18a–d). Furthermore, the presence of electron withdrawing substituents on the aryl bromides was explored (18e–l). The results are summarized in Table 2 .
Scheme 2.

Reagents and conditions: (a) Pd(OAc)2, XPhos, NaO-t-Bu, Toluene/t-BuOH (5:1), 18 h, 120 °C, 65%, (b) aq. HCl (5 N), 3 h, 100 °C, 45%,(c) Toluene, 18 h, 100 °C.
Table 2.
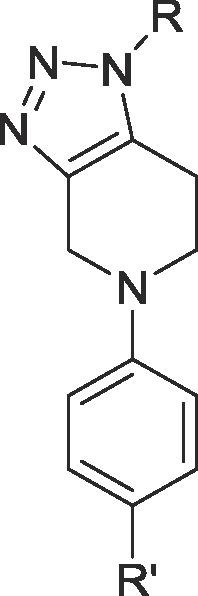
| Entry | Compound ID | R | R’ | Yield | EC50 (μM)a |
|---|---|---|---|---|---|
| 1 | 18a | 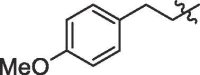 |
H | 85% | >100 |
| 2 | 18b | 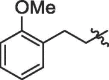 |
74% | >100 | |
| 3 | 18c | 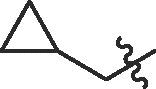 |
90% | >100 | |
| 4 | 18d | 48% | >100 | ||
| 5 | 18e | 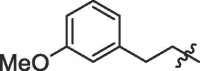 |
-CF3 | 90% | >100 |
| 6 | 18f | 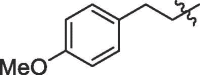 |
85% | 8.9 | |
| 7 | 18g | 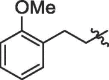 |
85% | >100 | |
| 8 | 18h | 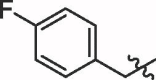 |
89% | >100 | |
| 9 | 18i | 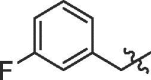 |
85% | 11.95 | |
| 10 | 18j | 89% | >100 | ||
| 11 | 18k | -F | 91% | >100 | |
| 12 | 18l | 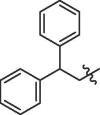 |
57% | >100 | |
Concentration required to reduce virus-induced cytopathogenicity by 50%.
The compounds were evaluated against a broad variety of viruses including HIV-1 (strain IIIB), HIV-2 (strain ROD) in MT-4 cells, herpes simplex virus type 1 (strain KOS), herpes simplex virus type 2 (strain G), herpes simplex virus type1 TK- (KOS) ACV res, vaccinia virus, adeno virus-2 and coronavirus (229E) in HEL cells and their inhibitory activity was compared to that of reference compounds as zidovudine, brivudine, cidofovir, acyclovir, ganciclovir, zalcitabine, alovudine and Urtica dioica agglutinin (UDA), respectively. All compounds were inactive towards all tested viruses, while 14d (EC50 = 8.95 µM), 14n (EC50 = 9.45 µM), 14q (EC50 = 9.45 µM), 18f (EC50 = 8.90 µM) and 18i (EC50 = 11.95 µM) showed moderate activity against human coronavirus (229E), but all approximately 50 fold lower than the activity observed with UDA (EC50 = 0.2 µM). No alterations of the normal cell morphology in confluent HEL cell cultures was observed at concentrations up to 100 µM (data not shown). The selectivity index (SI) (MCC/EC50 ratio) was >8 for all active compounds.
To determine the structure-activity relationship (SAR) for the synthesized library of fused 1,2,3-triazoles, it is of utmost importance to identify the key intermolecular contacts involved in the non-covalent interaction between 3CLpro and the structurally diverse inhibitors. With this aim, several molecular modeling techniques, including molecular docking, molecular dynamics, free energy of binding analyses and intermolecular interaction scanning, were applied in the search of such SAR knowledge. In a first stage, previously reported 3CLpro non-covalent inhibitors were modeled in order to define the key intermolecular interactions required for enzyme inhibition, while in a second stage the in silico analysis was extended to the family of fused 1,2,3-triazoles presented in this report.
The catalytic activity of 3CLpro has been well characterized, with extensive details regarding the structure of this enzyme and the corresponding catalytic site. In this respect, it is known that the active site is located within domains I and II, in which a catalytic dyad consisting of residues Cys145 and His41 is located.11 It has also been previously reported that the catalytic site of 3CLpro exhibits a stereoselective recognition of non-covalent inhibitors.18 In this context, our molecular modeling protocols initiated with the exploration of the binding mode, intermolecular interaction pattern and stereoselectivity of the non-covalent inhibitor of 3CLpro deposited in the Protein Databank under the code 3V3M. Both enantiomers of the bound ligand were docked within the catalytic site, and as can be seen in Fig S1a,b, the R enantiomer resulted in an identical interaction pattern to that observed in the experimentally obtained crystal. The ligand binding is stabilized by several hydrophobic and hydrogen bond (HB) interaction, of which those with Glu166, His163 and Gly143 are of particular relevance. Noteworthy, the S enantiomer was also able to establish several hydrophobic interactions, but was not able to establish HB with Glu166 and His163. Taking into account that it has been previously reported that only the R enantiomer is active,18 this finding suggests that the establishment of interactions with Glu166 and His163 constitutes a critical feature to inhibit the catalytic activity of the enzyme. To further study the persistence of these HB interactions, the intermolecular complexes were subjected to molecular dynamics (MD) analysis, with Table S1 presenting the persistence value (%) for each HB as calculated from the MD trajectories. As can be seen in Table S1, entry 1, the R enantiomer was able to maintain in relatively high frequencies the HB interactions with Glu166, His163 and Gly143, while the S enantiomer rearranged its binding to contact only Glu166 (60% persistence). Our findings not only validate the molecular modeling workflow we developed (i.e. the crystallographic binding pose was reproduced), but also strongly suggests that at least two stable electrostatic interactions within the active site are required for effective 3CLpro inhibition.21
To further study this hypothesis, a set of 12 previously reported 3CLpro inhibitors containing a fused 1,2,3 triazole ring and exhibiting a wide range of inhibitory activities (i.e. between 51 and 26000 nM, Tables S3 and S5), was subjected to our molecular modeling workflow. The lowest energy binding mode obtained for TS8 was in agreement with the reported crystallographic structure (Fig. S3d and pdb code 4MDS, respectively), further supporting an adequate parametrization and simulation conditions of the molecular modeling protocol. From inspection of the corresponding lowest energy docked poses for the whole set of compounds (Figs. S2–S4), we observed that all these inhibitors were indeed able to establish the two HB previously described for 3V3M, i.e. one with the backbone of Glu166 and another one with the side chain of His163. In these binding modes, the triazole ring is positioned in the proximity of the catalytic dyad. From the MD simulations and the quantification of the persistence of the above mentioned hydrogen bond interactions (Table S1, entries 3–14), we observed that all the compounds maintained the hydrogen bonds required for the inhibition of 3CLpro in high frequencies, and in particular that with Glu166. It is noteworthy that inhibitors that are enantiomerically pure and exhibiting submicromolar activities maintained high frequencies of HB with Glu166 and His163. In particular, TS-1, which is by far the most potent compound within the training set, exhibited also a single cluster of docked poses, suggesting not only an efficient pharmacodynamic interaction with 3CLpro, but also an adequate conformational preorganization that is compliant with the corresponding bioactive conformation.
Compounds 14a–r and 18a–l were subjected to the molecular modeling workflows in order to study whether 3CLpro may represent a plausible molecular target for their observed antiviral activity. Figs. S5 and S9 shows the lowest energy binding modes to 3CLpro found for compounds 14a–r. When the docked poses corresponding to the bioactive derivatives are observed (i.e. 14d and 14n, Figs. S5d, S8b, respectively), it can be seen that they establish the two HB interactions with Glu166 and His163, positioning also the triazole ring in the proximities of the catalytic dyad. As it was discussed in previous sections, this interaction pattern is required for the inhibition of 3CLpro catalytic activity, suggesting that this enzyme may be the antiviral target of these fused 1,2,3-triazoles. Further analysis by MD also showed that these interactions are maintained throughout the simulation, further supporting their bioactivities (Table S2, entries 4, 14 and 17). In contrast, the fused triazole derivatives that did not exhibited antiviral activities in infected cells, did not establish these two HB interactions in the lowest energy binding pose, or were not able to form them during the simulation trajectory when subjected to MD assays. Compounds 14c and 14m constitute two exceptions to this behavior, both of them bearing a methoxy substituent on the para position of the phenyl ring substituting the triazole central scaffold. These two compounds were able to establish and maintain the HB interaction pattern required for 3CLpro inhibition, (Table S2, entries 3 and 13) but did not exhibit antiviral activity. This fact may be due to a disfavorable entropic contribution upon binding to the 3CLpro catalytic site, since in order to maintain the interaction with His163, the rotation of the methoxy group is constrained within a dihedral angle of 10 Å. Clearly, this entropic cost is not present for the bioactive analogue bearing a fluorine atom in the para position (14d).
When derivatives 18a–l were analyzed (Figs. S10–S12), a similar behavior was observed, with the derivatives exhibiting antiviral activity (i.e. 18f and 18i) being able to establish two stable HB interactions within the catalytic site of 3CLpro (Figs. S11b and S12a). In particular, derivative 18f is anchored within the catalytic site through a stable interaction with His163, with two additional interactions being established with Glu166 and Thr25. Regarding the interaction of 18i, this compound establishes stable HB contacts with both Glu166 and His163, while further anchoring to residue Gln189. As a result, both compounds positioned the fused triazole scaffold in the vicinity of the catalytic dyad which is consistent with blocking the protease activity of the enzyme. Finally, when the interaction patterns of the inactive compounds within the series 18a–l were analyzed, we found that all of them failed to establish the two required HB interactions within the catalytic site of the enzyme.
In conclusion, we have succeeded in synthesizing a novel library of fused 1,2,3-triazoles using the in-house developed multicomponent reaction. The library was subjected to in vitro analysis using a broad variety of viruses to determine their antiviral properties and to in silico studies to determine the interactions with 3CLpro. Compounds 14d, 14n, 14q, 18f and 18i showed moderate activity against coronavirus 229E. A molecular modeling work flow was developed based on previously reported 3CLpro non-covalent inhibitors and helped the identification of key molecular interactions. Application of this model to our library, supports that the antiviral activity is mediated through the inhibition of 3CLpro. Additional studies on the structure-activity relationship will enable us to prepare new fused 1,2,3-triazole derivatives with enhanced antiviral properties.
Acknowledgments
This work was supported by Katholieke Universiteit Leuven (KU Leuven), grants C32/15/033 and ISPLA2/15/03. In addition, the authors gratefully acknowledge financial support from the Secretaria de Ciencia y Técnica of the Universidad Nacional de Córdoba (SECYT-UNC), the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), and the Agencia Nacional de Promoción Científica y Técnica (ANPCyT). The authors would also like to thank the GPGPU Computing Group from the Facultad de Matemática, Astronomía y Física (FAMAF), Universidad Nacional de Córdoba, Argentina, for providing access to computing resources. The authors also gratefully acknowledge the support of NVIDIA Corporation with the donation of the Titan Xp GPU used for this research. Mario A. Quevedo wishes to thank OpenEye Scientific Software and their Free Academic Licensing program for providing him with licenses to use the corresponding software packages.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmcl.2018.09.019.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Bradburne A.F., Bynoe M.L., Tyrrell D.A.J. Effects of a “New” human respiratory virus in volunteers. Br Med J. 1967;3:767–769. doi: 10.1136/bmj.3.5568.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drosten C., Günther S., Preiser W. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 3.Kuiken T., Fouchier R.A.M., Schutten M. Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet. 2003;362:263–270. doi: 10.1016/S0140-6736(03)13967-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Communicable Disease Surveillance and response; WHO. SARS case fatality ratio, incubation period. http://www.who.int/csr/sars/archive/2003_05_07a/en/. Published 7 May 2003.
- 5.Van Der Hoek L., Pyrc K., Jebbink M.F. Identification of a new human coronavirus. Nat Med. 2004;10:368–373. doi: 10.1038/nm1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woo P.C.Y., Lau S.K.P., Chu C. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol. 2005;79:884–895. doi: 10.1128/JVI.79.2.884-895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zaki A.M., Van Boheemen S., Bestebroer T.M., Osterhaus A.D.M.E., Fouchier R.A.M. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 8.Chan J.F.W., Lau S.K.P., To K.K.W., Cheng V.C.C., Woo P.C.Y., Yuen K.Y. Middle East Respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS-like disease. Clin Microbiol Rev. 2015;28:465–522. doi: 10.1128/CMR.00102-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zumla A., Chan J.F.W., Azhar E.I., Hui D.S.C., Yuen K.Y. Coronaviruses-drug discovery and therapeutic options. Nat Rev Drug Discov. 2016;15:327–347. doi: 10.1038/nrd.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S.H. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J Med Chem. 2016;59:6595–6628. doi: 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CL pro) structure : basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 12.Thomas J., Jana S., John J., Liekens S., Dehaen W. A general metal-free route towards the synthesis of 1,2,3-triazoles from readily available primary amines and ketones. Chem Commun. 2016;52:2885–2888. doi: 10.1039/c5cc08347h. [DOI] [PubMed] [Google Scholar]
- 13.Yang S., Chen S.J., Hsu M.F., Wu J.D., Tseng C.T.K., Liu Y.F., Chen H.C., Kuo C.W., Wu C.S., Chang L.W., Chen W.C., Liao S.Y., Chang T.Y., Hung H.H., Shr H.L., Liu C.Y., Huang Y.A., Chang L.Y., Hsu J.C., Peters C.J., Wang A.H.J., Hsu M.C. Synthesis, crystal structure, structure-activity relationships, and antiviral activity of a potent SARS coronavirus 3CL protease inhibitor. J Med Chem. 2006;49:4971–4980. doi: 10.1021/jm0603926. [DOI] [PubMed] [Google Scholar]
- 14.Chen L.R., Wang Y.C., Lin Y.W. Synthesis and evaluation of isatin derivatives as effective SARS coronavirus 3CL protease inhibitors. Bioorg Med Chem Lett. 2005;15:3058–3062. doi: 10.1016/j.bmcl.2005.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu C.Y., King K.Y., Kuo C.J. Stable benzotriazole esters as mechanism-based inactivators of the severe acute respiratory syndrome 3CL protease. Chem Biol. 2006;13:261–268. doi: 10.1016/j.chembiol.2005.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh A.K., Gong G., Grum-Tokars V. Design, synthesis and antiviral efficacy of a series of potent chloropyridyl ester-derived SARS-CoV 3CLpro inhibitors. Bioorg Med Chem Lett. 2008;18:5684–5688. doi: 10.1016/j.bmcl.2008.08.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J., Huitema C., Niu C. Aryl methylene ketones and fluorinated methylene ketones as reversible inhibitors for severe acute respiratory syndrome (SARS) 3Clike proteinase. Bioorg Chem. 2008;36:229–240. doi: 10.1016/j.bioorg.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacobs J., Grum-Tokars V., Zhou Y. Discovery, synthesis, and structure-based optimization of a series of N -(tert -Butyl)-2-(N -arylamido)-2(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL. J Med Chem. 2013;56:534–546. doi: 10.1021/jm301580n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turlington M., Chun A., Tomar S. Discovery of N(benzo[1,2,3]triazol-1-yl)-N-(benzyl)acetamido)phenyl) carboxamides as severe acute respiratory syndrome coronavirus (SARS-CoV) 3CLpro inhibitors: identification of ML300 and noncovalent nanomolar inhibitors with an induced-fit binding. Bioorg Med Chem Lett. 2013;23:6172–6177. doi: 10.1016/j.bmcl.2013.08.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gallagher M.J., Mann F.G. The structure and properties of certain polycyclic indolo- and quinolinoderivatives. Part XV. Derivatives of 1-phenyl-4-piperidone and its phosphorus and arsenic analogues. J Chem Soc. 1962;23:5110. [Google Scholar]
- 21.Schön U., Messinger J., Buckendahl M., Prabhu M.S., Konda A. An improved synthesis of N-aryl and Nheteroaryl substituted piperidones. Tetrahedron Lett. 2007;48:2519–2525. doi: 10.1016/j.tetlet.2007.02.053. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.