

Abstract
Viruses fully emerge by gaining the ability to sustainably infect new host populations. When the hosts are humans, emerging viruses can present major public health issues, as exemplified by the AIDS pandemic. Therefore, heuristic approaches to identify nascent diseases before they become pandemic would be valuable. Unfortunately, the current patient-based and epidemiological approaches are ill-suited in this regard because they are largely responsive and not predictive. Alternative approaches based on virus evolutionary ecology might have greater potential to predict virus emergence. However, given the difficulties encountered when studying metazoan viruses in this context, the development of new model systems is greatly desirable. Here, I highlight studies that show that bacteriophages are appropriate model organisms for virus emergence research because of the ease in which important population parameters can be manipulated. Ideally this research will permit identifying major factors determining the persistence or extinction of emerging viruses. If such viruses could be recognized in advance, patient-based and epidemiological strategies could be better mobilized to deal with them.
New approaches to virus emergence
Emerging viruses (EVs) are those that have entered new populations of hosts [1]. Recent emergence events, such as Severe Acute Respiratory Syndrome (SARS), Human Immunodeficiency Virus (HIV) and most recently Influenza A H1N1 (swine flu), have captured the popular imagination for their perceived (or actual) threat to humanity. Given that direct-contact transmitted HIV has caused around 60 million infections and 30 million deaths [2], the fear of a highly pathogenic airborne pathogen is justified. Because of the potential for a devastating pandemic, EVs have been identified as an existential threat to civilization [3].
Typically, EVs are considered using patient-based or epidemiological approaches [4]. The former emphasizes the diagnosis and treatment of pathogenic diseases and has led to the management and eradication of many formerly devastating diseases. The latter focuses on the behavioral, environmental and genetic influences on population-level patterns in disease manifestation. Although these approaches have been, in many respects, tremendously successful, one shortcoming is that they are largely responsive, rather than predictive, and thus cannot distinguish potential epidemics from sporadic, spillover diseases [5].
A new approach, called evolutionary epidemiology, is based on virus evolutionary ecology and has greater potential to generate predictive models of virus emergence 4, 6. This approach assumes that disease processes are the products of fundamental biological variables. An explosion of theoretical studies has emphasized such parameters as mutation rate, gene flow, contact rate, duration of infection and host population dynamics 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17. Many of these theoretical studies point to virus basic reproductive ratio, R 0, as the single most important epidemiological parameter. R 0 is defined as the mean number of secondary cases a typical single infected case will cause in a population 18, 19. Theoretically, if virus R 0>1, then the virus will tend to spread epidemically, and if not, the virus will disappear (Box 1 ).
Box 1. Epidemiology and population biology of infectious diseases.
In population biology, the change a population of size, N, is expressed similarly by the differential equation,
| (1) |
where B=birth, I=immigration, D=death and E=emigration rates, respectively. These terms are usually subsumed into one parameter, the per capita growth rate r, such that
| (2) |
In biology, r is significant because it is a measure of absolute fitness if construed for a particular genotype. In ecological theory, a source population is defined as one where r > 0, whereas a sink shows r<0. As defined, a source exhibits positive population growth and will continue to grow insofar as resources are available. By contrast, a sink shows negative population growth and will decline to eventual extinction unless rescued by either migration or adaptation.
Classic epidemiological models divide populations into compartments representing susceptible (S), infectious (I) and removed (R, i.e. recovered or dead) individuals (Figure I ). The compartments are dynamic in that individuals are entering and egressing each compartment over time. The change in the number of susceptible individuals over time depends on host birth rate and recovered individuals losing immunity. Passage from susceptible to infectious compartments depends on β, the rate constant of infectious transfer of viruses. Individuals are removed from the infectious compartment either by dying or recovering and gaining immunity. The removal rate is expressed by the variable γ. A set of three differential equations describes these transitions,
| (3) |
| (4) |
| (5) |
The most important epidemiological parameter is the basic reproductive number, R 0 [18], which is given by the equation
| (6) |
If R 0 > 1, then the pathogen spreads epidemically; if R 0 < 1, then the pathogen will die out.
Figure I.

Typical compartmental model of epidemiological theory.
Epidemiological theory overlaps population biology because it is here that R 0 can be connected to source/sink theory as follows,
| (7) |
This overlap permits application of the broader tenets of evolutionary ecological theory to infectious diseases, and, more specifically, can guide the investigation of disease emergence.
Theoretical modeling, however, has not been matched by concomitant experimental activity. The conventional wisdom is that, if we want to understand emergence, viruses should be studied in vivo or in mammalian cell culture models. However, from an evolutionary epidemiological standpoint, this approach is often impractical. In general, mammalian cell culture is somewhat trickier and more expensive than bacterial culture (even with regard to the relatively simple culture of HeLa) [20]. Cultured cells grow slowly and are prone to degeneration [21], contamination 22, 23, contact inhibition and differentiation [20] confounding results. Other factors include the fact that most primary cells have a limited lifespan (hence the requirement for immortalized cell lines) and are less amenable to reverse genetics. Multihost cell systems only compound these issues. Finally, cell culture removes cells from the whole organism context, thus negating one of its supposed advantages.
Therefore, an evolutionary epidemiological approach requires the development of new, simpler models to study the population and evolutionary dynamics of viral emergence. Among the possible model systems, bacteriophages and their bacterial hosts seem an excellent choice as they permit easy manipulation of conditions such as population sizes, environmental characteristics, host types, spatial structures and transmission rates (Table 1 ). Here, I review bacteriophage research that has contributed to our understanding of virus emergence. In the following sections, I discuss the implications of mutation, virulence, adaptation, sex, gene flow and spatial structure for viral emergence. A Glossary is included to define terms that might be unfamiliar to some readers.
Table 1.
Model bacteriophages used in studies of evolutionary ecology
| Name | ICTVdB numbera | Genome type and size (nt) | Typical laboratory host | Useful properties |
|---|---|---|---|---|
| T4 | 02.043.0.01.001. | dsDNA (169,000) | Escherichia coli | Best understood model for modern functional genomics and proteomics 85, 86, 87. Possesses eukaryote-like introns, high-speed DNA copying and DNA repair mechanisms. |
| λ | 02.066.0.01.001. | dsDNA (48,502) | Escherichia coli | Canonical temperate phage that has been the workhorse of molecular biology. Exceptionally well characterized lysis–lysogenic switch 87, 88, 89, 90. |
| PRD1 | 00.068.0.01.001. | dsDNA (14,927) | Salmonella typhimurium | Lipid-containing phage highly similar to adenovirus; broad host range, but plasmid-dependent [87]. Valuable tool for membrane studies [87]. |
| Mu | 02.043.0.04.001. | dsDNA (37,611) | Escherichia coli | Transposable, non-inducible temperate phage with many unusual and unique features [87]. |
| ΦX174 | 00.042.0.01.001. | ssDNA (5386) | Escherichia coli | First DNA genome sequenced; uses “antibiotic-like” proteins to lyse cells [87]. |
| M13 | 00.035.0.01.001. | ssDNA (6407) | Escherichia coli | Filamentous phage capable of secretion rather than cell lysis. Allows host to survive infection [87]. |
| Φ6 | 00.021.0.01.001. | dsRNA (13,379) | Pseudomonas syringae pv phaseolicola | One of few segmented, lipid-containing, dsRNA phage. Shows striking similarity to the Reoviridae. Well developed reverse genetic capability [87]. |
| MS2 | 00.037.0.01.001. | +ssRNA (3569) | Escherichia coli | Exceptionally small, ssRNA phage that infects male enterobacteria (i.e. F+, F′ or Hfr cells) by attaching to pilus [87]. |
ICTVdB: the Universal Virus Database of the International Committee on Taxonomy of Viruses (http://www.ncbi.nlm.nih.gov/ICTVdb/).
Mutation, the first step to emergence
In order to emerge, viruses must have the ability to productively infect new host types. For any virus, the probability of host range expansion is a function of several parameters: (i) its population size; (ii) growth rate; (iii) mutation rate; (iv) sequence space of available mutations that result in host range expansion; and (v) number of mutational steps required to access these genotypes in that sequence space. To understand this process, a systematic exploration of the sequence space of EVs might be required. For any particular virus–host association, how many mutations permit emergence, and what are the fitness consequences of those mutations on native and novel host types? It might be that some viruses are inherently more likely to emerge. By looking at mutations and their fitness consequences, we might be able to move beyond coarse-grained predictions (e.g. most EVs are RNA viruses [24]) to consider the correlates of emergence at the molecular level.
Research on bacteriophages can contribute to this effort by providing a theoretical foundation on which to base research on human pathogens. For example, Ferris et al. isolated phage Φ6 mutants that were able to grow on a previously non-permissive host, Pseudomonas glycinea, in addition to their typical laboratory host, Pseudomonas phaseolicola [25]. DNA sequencing revealed that, out of 40 isolates, 39 possessed a mutation in the gene coding for capsid spike protein P3, which is implicated in host attachment. Of these 39 isolates, 16 were unique substitutions. Using a statistical procedure based on the coupon–collection problem of probability theory [26], Ferris et al. estimate that 55 out of 4380, or 1.3%, of the possible nonsynonymous mutations in the P3 protein permit phage Φ6 to infect P. glycinea [25]. This specific prediction can be tested by collecting and sequencing a large pool of host range mutants. Currently, the model takes into account differential mutation probabilities (e.g. transitions more likely than transversions) but refinement is probably required. For example, the model can be further constrained by considering codon bias.
What mutation giveth, mutation also taketh away. An EV infecting a new host type can be likened to a single founder event in a new habitat. Founder effects commonly include reductions in fitness and genetic diversity [27]. The primary means of recovering genetic diversity is by mutation, but it has been shown that the majority of new mutations are deleterious [28]. Thus, the effects of emergence under these conditions could be analogous to a mutation–accumulation experiment. Here, organisms are serially passaged with severe bottlenecking on each passage such that ability of natural selection to remove non-lethal deleterious mutations is abolished. Bottlenecking inherent in the virus transmission process is probably exacerbated for EVs where within-host reproduction is low [29]. Such bottlenecking has been demonstrated to significantly reduce fitness with the phage MS2 [30]. Here, 20 passages with bottlenecking were sufficient to reduce phage fitness by almost two orders of magnitude. Given that each round of bottlenecking led to a fitness loss (up to 16% in some cases), within-host reproduction is expected to be reduced, thus probably increasing the bottleneck for future transmission events. As such, populations stuck in such demographic traps can rapidly become unsustainable.
In addition to mutation, viral genetic diversity can be regenerated through recombination or reassortment, processes that rely on viral coinfection of hosts [31]. Initial population sizes for EVs are likely to be small, thus coinfection could conceivably be rare. Moreover, founder effects increase the odds that virions inhabiting the same cell are genetically identical or nearly identical, thus negating the potential advantages of sex for viral evolution 31, 32. The interaction of small population sizes, lack of effective sex and high mutation rates might synergize to make EVs prone to Muller's ratchet and mutational meltdown (see Glossary) [33].
Virulence, a tradeoff between within-host growth and between-host transmission
In evolutionary biology, virulence is defined as the parasite-induced loss of host fitness 34, 35. Theoretical models suggest that parasites will evolve an optimal level of virulence based on a fitness tradeoff between within-host growth and between-host transmission 34, 35. Excessive virulence will lead to host death before transmission is effected and therefore leads to reductions in virus fitness. Empirical data from bacteriophage support this point. In phage Φ6 infections of P. phaseolicola, a negative correlation was observed between initial viral inoculum and virus growth rate [36]. The presumed mechanism for this result was a “tragedy of the commons” phenomenon where the renewable resource (bacterial hosts) was killed off before they had an opportunity to reproduce, thus reducing system-wide phage productivity. By contrast, insufficient virulence results in transmission failure and/or susceptibility to within-host competition from other genotypes.
Evidence for a tradeoff between horizontal and vertical transmission comes from studies of the filamentous bacteriophage f1. This phage establishes persistent infections of Escherichia coli cells where progeny are extruded from infected cells. Host cells suffer reduced growth, but are not killed, thus phage can also reproduce via vertical transmission. Messenger et al. experimentally manipulated the rates of horizontal and vertical transmission [37]. In the vertical transmission treatment, secreted phages were prevented from infecting new hosts. In the horizontal treatment, persistently infected cells were removed from lysates containing secreted phage. Following 24 days of treatment, phage and bacterial reproductive rates were assayed. Results indicated that natural selection favored increased virus virulence in horizontal treatment and decreased virulence in vertical treatment, corresponding to the expectations of the tradeoff hypothesis.
With regard to emerging human viruses, no studies have quantitatively explored the fitness effects of mutations resulting in host range expansion. Because of their tractability, phage studies have contributed significantly in this regard and have shown that fitness consequences of host range expansion mutations can be broad 25, 38. In human viruses, some circumstantial evidence points to frequent mismatches between virus growth and transmission. An anecdotal view seems to point to EVs frequently showing excessive virulence. For example, Ebola virus has been relatively unsuccessful perhaps because of its excessive virulence [39]. Other emerging pathogens, such as those causing avian influenza and SARS, were also highly lethal, and perhaps for this reason, did not spread widely. Perhaps intuitively this makes sense because the new hosts are not yet adapted to the emerging pathogen, but this argument cuts both ways. Pathogens are not yet evolved to new hosts. Moreover, this view is obviously skewed in that relatively benign EVs might not have achieved high visibility precisely because of their lack of virulence. Interestingly, studies show that most host range mutations in phages reduce virus growth rates 25, 40. The usual supposition is that virulence and lytic phage growth are directly correlated, although this might not be strictly true for other organisms [41]. Nevertheless, we can speculate that the inability of a virus to productively replicate in a new host might reduce its ability to reduce the fitness of a host. As such, we might expect that the majority of EVs could actually be relatively non-virulent. This conjecture is supported by mathematical modeling suggesting that less virulent pathogens are more likely to emerge [8]. Without further research, however, we cannot do more than speculate about the average virulence of EVs and their propensities to fully emerge.
Adaptation, the key to emergence
A key tenet of evolutionary theory is that most organisms are optimized for their present circumstances 42, 43 and are thus maladapted for other habitats. This tradeoff resulting from the use of multiple niches is well documented 44, 45. Likewise, a newly emerged virus usually experiences a reduction in fitness on its original host 38, 46 but has not yet had an opportunity to adapt to its new host. In fact, circumstantial evidence suggests that the basic reproductive rate, R 0 is <1 for most EVs (Box 1) 47, 48. This relative lack of local adaptation, and other factors such as demographic stochasticity, mutation accumulation and competition, might make it difficult for EVs to spread widely beyond their initial appearance. Thus, most emergence events probably are temporally transient [5]. An important consideration is that, because of fitness tradeoffs, most EVs are poorly adapted to novel hosts relative to native hosts. As such, EVs can initially show R 0<1, but to fully emerge, their R 0 must surpass 1.
In population theory, populations where the per capita growth rate, r, is <0 are termed sinks 49, 50. This parameter is fundamentally related to R 0, a relationship that is more thoroughly discussed in Box 1. Populations in sinks can only be sustained through migration or adaptation 50, 51, 52. The fundamentals of virus adaptation in simple habitats are well understood; fitness initially increases rapidly before decelerating over time 53, 54, 55, 56. A parsimonious explanation for this pattern of adaptation is that viruses rapidly ascend fitness peaks in habitats where they are initially unfit. The initial burst in fitness can occur in a matter of generations and high viral mutation rates, large population sizes and short generation times are probably responsible.
Given the opportunity, it seems that viruses could escape mutationally and demographically driven extinction easily; thus, the relative lack of emerging epidemic diseases could conceivably be construed as a mystery. Of course, this supposition assumes no prior knowledge of the population dynamics of viral emergence. In fact, it is this disconnect between our broad knowledge of evolutionary processes in simple habitats and a relative lack of knowledge of evolution in complex habitats that is most responsible for the lack of a predictive model of viral emergence. The key to understanding virus emergence will be moving virus research beyond the confines of simple habitats.
One way simple laboratory microcosms differ from the conditions faced by EVs in nature is that virus emergence usually occurs in habitats where multiple hosts are available [57]. Unfortunately, few experimental studies have explored how adaptation occurs in multihost habitats. If a fitness tradeoff encourages host specialization, how do viruses “decide” which host to focus on? This question is addressed in studies of phage T7 infecting E. coli where Heineman et al. apply optimal foraging theory to phage host range evolution [58]. Their results showed that T7 rapidly evolved to discriminate among different E. coli strains when one host strain was engineered to kill infecting phages but the other remained productive. In further experiments, the T7 study showed that host ratio and quality were important factors in determining whether host specialization was favored among phages. Host specialization was favored when better quality hosts were common and when there were large differences in host quality.
In another study, Guyader and Burch developed a mathematical model to describe the consequences of host specialization for bacteriophages [59]. Their model suggests that host range should be negatively correlated with host abundance. That is, when hosts are abundant, it pays for phage to specialize on a single host type. Guyader and Burch confirm the predictions of their model using phages ΦX174 and G4, a generalist and a specialist, respectively. When the density of E. coli C and Salmonella typhimurium was high, the specialist G4 (infecting only E. coli C) was selectively favored. When host densities were low, ΦX174, a phage able to infect both host types, was competitively superior. However, neither phage was able to evolve the alternate strategy when experimentally evolved under conditions favoring that strategy. When G4 was evolved at low host densities, it did not gain the ability to infect S. typhimurium, nor did ΦX174 evolve a reduced host range when passaged at high host densities. However, it is important to note that host range reduction will only be favored if a fitness tradeoff exists, which might not occur in laboratory settings.
Viral sex: boon or bane?
Sex is commonly defined as the combination of genetic material from parents to produce genetically unique offspring. Among DNA and RNA viruses, sex occurs by recombination. Here, a strand of genetic material is broken and swapped with similar (in homologous recombination) or dissimilar (in heterologous recombination) genetic material from a different source. Some viruses can undergo an additional process termed reassortment owing to the segmented nature of their genomes. In reassortment, hybrid progeny are created when two or more viruses infect the same cell. In the cell, replicated segments are packaged into capsids regardless of the parent of origin, thus progeny can contain segments from both parents (Figure 1 ). Because recombination among segmented viruses is rare relative to reassortment, reassortment is hypothesized to have evolved to provide segmented viruses with a means to have sex [60].
Figure 1.
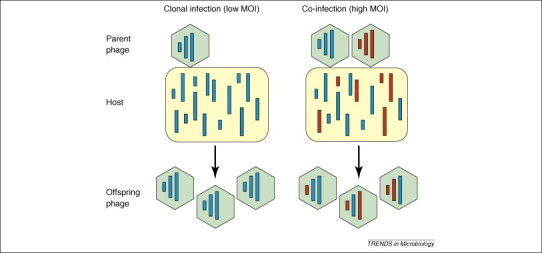
Reassortment of segmented RNA virus infections occurs at high multiplicities of infection (MOI). High MOIs enable coinfection of two or more virus genotypes. Assembled progeny can contain genetic material from both parents. By contrast, coinfection of genetically different phage usually does not occur at low MOI.
Sex is relevant to the study of emerging infectious diseases because it is presumed to facilitate adaptation in new environments or combat mutational load 61, 62. Experimental studies have begun to explore the adaptive significance of sex among viruses. Early results indicate that sex might counter Muller's ratchet, the accumulation of deleterious mutations 31, 32, but fails to assist adaptation in novel habitats 31, 32. Turner and Chao showed that the conditions where reassortment occurs (i.e. high multiplicities of infection) also permit the formation of cheaters (e.g. defective interfering particles) [63]. Defective interfering particles are viruses missing part of their genomes that rely on helper viruses to provide the necessary gene functions that they are missing.
Sex has also been assumed to increase genetic diversity. However, genetic sequencing has determined that sexually evolved populations of Φ6 appear to be no more diverse than asexually evolved populations (Dennehy et al., unpublished observations). Perhaps sexually evolved populations quickly accessed high fitness genotypes, thus reducing standing genetic variation [64]. Alternatively, or perhaps additionally, clonal interference inflated diversity in asexual populations. Clonal interference results from competition between beneficial alleles in different clonal lineages [65]. Clonal interference limits the effectiveness of selection, thus clonal populations might harbor more genetic diversity. However, this effect probably depends on the strength of selection. Pepin and Wichman have shown that clonal interference was a factor in phage ΦX174 evolution in a harsh environment but not in a benign environment [66].
Together, studies of sex among bacteriophages indicate that segmentation in RNA viruses probably did not evolve to provide the benefits of sex. Instead it is probable that segmentation aids genome packaging [67]. This hypothesis can be tested by evolving phage with experimentally manipulated segment lengths. Segment length chimeras can be constructed by repackaging restricted and ligated phage transcripts produced from plasmid-embedded sequences [67].
Gene flow and the source–sink perspective of viral emergence
Emerging genotypes must contend with several difficulties during establishment in novel host communities, such as competition, demographic stochasticity and temporal/spatial heterogeneity. One aspect of emergence that has received little attention is the effect of gene flow on adaptation. The effect of genotypes migrating from native hosts (sources) on populations evolving on novel hosts (sinks) is unknown. Conventional wisdom emphasizes the inhibitory effect of migration on niche evolution 68, 69, 70. Any beneficial mutations arising within a sink are presumed to be swamped by influxes of original or other genotypes and are lost to drift or other population processes.
The new source–sink theory by Holt et al. suggests that local adaptation within a sink can occur in spite of, or indeed because of, migration. These authors predict that the probability (or rate) of adaptation to sink habitats increases with the rate of migration from source populations [51]. The accelerating effect of migration is predicted to be particularly important in cases where local fitness is positively density-dependent. Here, migration increases resident genotype productivity by inflating local population density [52]. That is, if population growth of the pathogen is positively density-dependent, then migration can actually facilitate adaptation by enhancing local fitness of the pathogen. This phenomenon makes it easier for alleles of modest effect to be captured by natural selection, transforming the sink into a locally adapted population that can persist without migration [52].
Another theoretical consideration is that initial demographic state of the sink has important implications for the likelihood of sink escape and viral emergence. All else being equal, the lower the initial fitness of the EVs is in the sink, the more difficult full emergence might be [71]. If most novel beneficial mutations are limited in their impact on fitness, then it might take considerable time for enough beneficial mutations to accumulate to permit sink escape. The longer r<0 (Box 1), the greater the probability of extinction. However, there is some evidence of increased numbers of mutations of large effect when fitness is low [72]. The consequence of large effect beneficial mutations is that they might have multiple pleiotropic effects across the phenotype of an organism, which means it might be hard to predict precisely how EVs will differ from their ancestral progenitors. Virus fitness can also vary with host condition. If host immune systems are compromised in some way, then the initial r of the pathogen is higher, so initial adaptation might be quicker, and also the descendent emergent strain might be more similar overall in its properties to the ancestral strain.
Observations of correlations between migration rate and adaptation would lend empirical support for the theory proposed by Holt et al. Several studies report such observations. For example, Morgan et al. demonstrated such a correlation using Pseudomonas fluorescens and a DNA phage SBW25Φ2. In the absence of migration, SBW25Φ2 tended to be locally maladapted, whereas in the presence of migration populations showed evidence of adaptation 73, 74. Brockhurst et al. reported that bacterial resistance to phage was highest at intermediate levels of migration [75]. Similarly, Forde et al. showed that gene flow across a heterogeneous landscape increased the fitness of phage evolving in chemostats [76].
Naturally the default expectation is that migrant populations evolving on the same host will provide to sink populations more appropriate alleles for adaptation on novel hosts than will immigrants from populations evolving on different host types. However preliminary data indicate that this conjecture is not necessarily true. Experiments evaluating the effect of the source of immigrants on local adaptation suggested that source of migrants did not much matter as long as genetic variability was generated (Dennehy et al., unpublished observations). Future experiments manipulating migration rate and source might shed more light on how gene flow affects adaptation of EVs.
Space, the final frontier
Viral emergence implies spatial heterogeneity as a shift from one host type to another also entails a radical shift in habitat. Moreover, within- and between-host viral dynamics also requires a spatial perspective and lends itself theoretically to metapopulation approaches. Phage research has significantly contributed to our understanding of the spatial dimensions of viral dynamics on several fronts. One approach seeks to understand virus infection of a tissue by modeling phage spread on a two-dimensional bacterial lawn 77, 78, 79. The models emphasize the tradeoff between phage latent period and burst size, suggesting that these parameters are optimized based on resource availability. Ultimately, wave front analysis of plaque growth can update our understanding of virus invasion of tissue.
On a different spatial scale, another study used phage T4 to investigate the effect of migration between populations in a spatially structured habitat (a 96-well plate) [80]. Spatially restricted migration led to the evolution of reproductive “restraint”, whereas unrestricted migration selected for “rapacious” phage. In the latter treatment, phage that excessively exploited their habitat fell prey to the “tragedy of the commons” by eliminating their resource (i.e. lysing bacteria before they had opportunity to reproduce), thus slower phage growth was selected for. By contrast, the former treatment reduced the chance of population extinction by providing phage greater opportunity for fresh resources. These results are generally supported by theory suggesting that increased population viscosity favors cooperation [81].
Spatial structure can also increase biological diversity as evidenced by studies manipulating spatial structure and host density [82]. Here, differences in the relative fitness of two Φ6 strains narrowed as host density increased, allowing longer persistence of the weaker strain. Similar results come from studies of ΦX174 and α3 growing on E. coli. Longer incubation times provided greater spatiotemporal refuge for the weaker phage strain. It is clear that spatial structure has an outsized impact on virus ecology and will continue to be a fruitful field of research for some time.
Concluding remarks and future directions
EVs are major public health threats and previous theoretical efforts to generate predictive models of nascent threats have not been entirely successful. New approaches emphasizing virus evolutionary ecology promise to add more weapons to our arsenal [4]. However, such approaches necessitate new model systems. Most human or metazoan viruses are difficult to adapt to ecological or evolutionary experiments, even in vitro. One interesting approach uses insects, such as Galleria mellonella and Drosophila melanogaster, as models for current and emerging diseases [83]. Bacteriophages and bacterial cultures offer even more flexibility for manipulating genomes, populations and host types.
I do not argue that research on bacteriophages and their bacterial hosts can replace other areas of virus evolutionary ecology research, but rather supplement it. In other words, the approach taken here is “necessary, but not sufficient” for a broad understanding of the evolution of disease emergence. Phage/bacteria systems are limited in their ability to capture the complexities of within-host growth typical of metazoan systems. Specifically, the interplay between various metazoan cell types and tissues and metazoan viruses cannot be captured in prokaryotic systems. Coevolution concomitant with viral within-host population dynamics and host immune response are important in humans for instance, but can be more safely ignored in bacteria because of the ability to store bacteria in frozen stasis. Such coevolutionary processes could provide important constraints to virus evolutionary ecology, impacting their between-host dynamics. Another important limitation is that it is difficult to precisely simulate with bacteria the degrees of mixing that occur between different host species of metazoan viruses. For instance, avian influenza could result from very limited contact between avian and human species such that a single or few human hosts (super spreaders) can have a disproportionate influence on viral emergence [84]. Bacterial populations in laboratory settings, by contrast, are relatively homogeneous.
Although the specifics of metazoan virus infection can be qualitatively different from bacteriophage infections, the general evolutionary and ecological principles are very similar. Population characteristics, such as host density, host frequency, contact rate, spatial/temporal heterogeneity and community structure, should figure prominently in the persistence or extinction of new virus genotypes. These parameters are much more amenable to manipulation in bacteriophage systems. The hope is that bacteriophage research can identify important themes and parameters for viral persistence and identify areas worthy of significant effort for more sophisticated model systems. This effort is not without additional benefit. Bacteria are also a major source of infectious disease. Most studies overlook the impact of bacteriophages on bacterial population and evolutionary dynamics. Ongoing evolutionary dynamics among bacterial strains and their phages could be an important ‘hidden player’ in many infectious disease systems that are as yet poorly understood.
Acknowledgements
I thank Yoon Choi, Siobain Duffy, Nicholas Friedenberg, Rick Heineman, Robert Holt and Paul Turner for their insightful comments on the ideas presented herein. Editor Cesar Sanchez and three anonymous reviewers provided superb suggestions that considerably improved this paper. This work was supported by National Science Foundation Grant # DEB-0804039 and funding from the Professional Staff Congress of the City University of New York and the Research Foundation of The City University of New York.
Glossary
- Coupon-collection problem
problem in probability theory that refers to “collect all coupons and win” contests and asks how many samples from a population of coupons are required to collect one of each type.
- Demographic stochasticity
fluctuations in population size owing to random demographic events.
- Fitness tradeoff
situation where an increase in fitness in one context is opposed by a decrease in fitness owing to concomitant changes in another context.
- Gene flow
transfer of alleles from one population to another by interbreeding or migration.
- Horizontal and vertical transmission
pathogens can spread either horizontally from one host to another or vertically from parent to offspring.
- Muller's ratchet
a characteristic of an asexual population that prevents any lineage in the population from attaining a mutation load lower than that already existing in its least loaded lineage.
- Mutation accumulation experiments
experiments that serially passage organisms with extreme bottlenecks such that selection against deleterious mutations is effectively eliminated.
- Mutational load
reduction in population fitness owing to the accumulation of deleterious mutations.
- Mutational meltdown
process by which a population accumulates deleterious mutations, which leads to loss of fitness and decline of the population size, which could lead to further accumulation of deleterious mutations owing to inbreeding depression.
- Niche evolution
population adaptation to a sink habitat such that it can persist in the absence of migration.
- Optimal foraging theory
idea that natural selection favors organisms that maximize net energy gain per unit feeding time while pursuing sustenance.
- Sequence space
all permutations of a DNA, RNA or amino acid sequence of a given length.
References
- 1.Woolhouse M.E.J. Emerging pathogens: the epidemiology and evolution of species jumps. Trends Ecol. Evol. 2005;20:238–244. doi: 10.1016/j.tree.2005.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fauci A.S. 25 years of HIV. Nature. 2008;453:289–290. doi: 10.1038/453289a. [DOI] [PubMed] [Google Scholar]
- 3.Guerrant R.L., Blackwood B.L. Threats to global health and survival: the growing crises of tropical infectious diseases – Our “unfinished agenda”. Clin. Infect. Dis. 1999;28:966–986. doi: 10.1086/514765. [DOI] [PubMed] [Google Scholar]
- 4.Smith K.F. Ecological theory to enhance infectious disease control and public health policy. Front. Ecol. Environ. 2005;3:29–37. doi: 10.1890/1540-9295(2005)003[0029:ETTEID]2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Power A.G., Mitchell C.E. Pathogen spillover in disease epidemics. Am. Nat. 2004;164:S79–S89. doi: 10.1086/424610. [DOI] [PubMed] [Google Scholar]
- 6.Restif O. Evolutionary epidemiology 20 years on: challenges and prospects. Infect. Genet. Evol. 2009;9:108–123. doi: 10.1016/j.meegid.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 7.Andre J.B., Day T. The effect of disease life history on the evolutionary emergence of novel pathogens. Proc. R. Soc. B-Biol. Sci. 2005;272:1949–1956. doi: 10.1098/rspb.2005.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andre J.B., Hochberg M.E. Virulence evolution in emerging infectious diseases. Evolution. 2005;59:1406–1412. [PubMed] [Google Scholar]
- 9.Cross P.C. Utility of R-0 as a predictor of disease invasion in structured populations. J. R. Soc. Interface. 2007;4:315–324. doi: 10.1098/rsif.2006.0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Day T. The evolutionary emergence of pandemic influenza. Proc. R. Soc. B-Biol. Sci. 2006;273:2945–2953. doi: 10.1098/rspb.2006.3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Day T., Gandon S. Applying population-genetic models in theoretical evolutionary epidemiology. Ecol. Lett. 2007;10:876–888. doi: 10.1111/j.1461-0248.2007.01091.x. [DOI] [PubMed] [Google Scholar]
- 12.Fenton A., Pedersen A.B. Community epidemiology framework for classifying disease threats. Emerg. Infect. Dis. 2005;11:1815–1821. doi: 10.3201/eid1112.050306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gandon S., Day T. Evolutionary epidemiology and the dynamics of adaptation. Evolution. 2009;63:826–838. doi: 10.1111/j.1558-5646.2009.00609.x. [DOI] [PubMed] [Google Scholar]
- 14.Getz W.M. Modeling the invasion and spread of contagious diseases in heterogeneous populations. Dis. Evol.: Models Conc. Data Anal. 2006;71:113–144. [Google Scholar]
- 15.Kajita E. Modelling an outbreak of an emerging pathogen. Nat. Rev. Microbiol. 2007;5:700–709. doi: 10.1038/nrmicro1660. [DOI] [PubMed] [Google Scholar]
- 16.Reluga T. Reservoir interactions and disease emergence. Theor. Popul. Biol. 2007;72:400–408. doi: 10.1016/j.tpb.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yates A. How do pathogen evolution and host heterogeneity interact in disease emergence? Proc. R. Soc. B-Biol. Sci. 2006;273:3075–3083. doi: 10.1098/rspb.2006.3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heesterbeek J.A.P. A brief history of R-0 and a recipe for its calculation. Acta Biotheor. 2002;50:189–204. doi: 10.1023/a:1016599411804. [DOI] [PubMed] [Google Scholar]
- 19.Heffernan J.M. Perspectives on the basic reproductive ratio. J. R. Soc. Interface. 2005;2:281–293. doi: 10.1098/rsif.2005.0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu W.S., Aunins J.G. Large-scale mammalian cell culture. Curr. Opin. Biotechnol. 1997;8:148–153. doi: 10.1016/s0958-1669(97)80093-6. [DOI] [PubMed] [Google Scholar]
- 21.Arden N., Betenbaugh M.J. Life and death in mammalian cell culture: strategies for apoptosis inhibition. Trends Biotechnol. 2004;22:174–180. doi: 10.1016/j.tibtech.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 22.Cabrera C.M. Identity tests: determination of cell line cross-contamination. Cytotechnology. 2006;51:45–50. doi: 10.1007/s10616-006-9013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drexler H.G. False human hematopoietic cell lines: cross-contaminations and misinterpretations. Leukemia. 1999;13:1601–1607. doi: 10.1038/sj.leu.2401510. [DOI] [PubMed] [Google Scholar]
- 24.Woolhouse M., Gaunt E. Ecological origins of novel human pathogens. Crit. Rev. Microbiol. 2007;33:231–242. doi: 10.1080/10408410701647560. [DOI] [PubMed] [Google Scholar]
- 25.Ferris M.T. High frequency of mutations that expand the host range of an RNA virus. Genetics. 2007;176:1013–1022. doi: 10.1534/genetics.106.064634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown M. Coupon collecting. Prob. Eng. Informat. Sci. 2008;22:221–229. [Google Scholar]
- 27.Nei M. Bottleneck effect and genetic variability in populations. Evolution. 1975;29:1–10. doi: 10.1111/j.1558-5646.1975.tb00807.x. [DOI] [PubMed] [Google Scholar]
- 28.Eyre-Walker A., Keightley P.D. The distribution of fitness effects of new mutations. Nat. Rev. Genet. 2007;8:610–618. doi: 10.1038/nrg2146. [DOI] [PubMed] [Google Scholar]
- 29.Moury B. Estimation of the number of virus particles transmitted by an insect vector. Proc. Natl. Acad. Sci. U. S. A. 2007;104:17891–17896. doi: 10.1073/pnas.0702739104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de la Pena M. Effect of deleterious mutation-accumulation on the fitness of RNA bacteriophage MS2. Evolution. 2000;54:686–691. doi: 10.1554/0014-3820(2000)054[0686:eodmao]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 31.Chao L. The advantage of sex in the RNA virus phi 6. Genetics. 1997;147:953–959. doi: 10.1093/genetics/147.3.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chao L. Muller's ratchet and the advantage of sex in the RNA virus phi6. Evolution. 1992;46:289–299. doi: 10.1111/j.1558-5646.1992.tb02038.x. [DOI] [PubMed] [Google Scholar]
- 33.Lynch M. Mutation accumulation and the extinction of small populations. Am. Nat. 1995;146:489–518. [Google Scholar]
- 34.Frank S.A. Models of parasite virulence. Q. Rev. Biol. 1996;71:37–78. doi: 10.1086/419267. [DOI] [PubMed] [Google Scholar]
- 35.Bull J.J. Perspective – virulence. Evolution. 1994;48:1423–1437. doi: 10.1111/j.1558-5646.1994.tb02185.x. [DOI] [PubMed] [Google Scholar]
- 36.Dennehy J.J. Viral ecology and the maintenance of novel host use. Am. Nat. 2006;167:429–439. doi: 10.1086/499381. [DOI] [PubMed] [Google Scholar]
- 37.Messenger S.L. Virulence evolution in a virus obeys a trade-off. Proc. R. Soc. Lond. Ser. B-Biol. Sci. 1999;266:397–404. doi: 10.1098/rspb.1999.0651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duffy S. Pleiotropic costs of niche expansion in the RNA bacteriophage Phi 6. Genetics. 2006;172:751–757. doi: 10.1534/genetics.105.051136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takada A., Kawaoka Y. The pathogenesis of Ebola hemorrhagic fever. Trends Microbiol. 2001;9:506–511. doi: 10.1016/s0966-842x(01)02201-6. [DOI] [PubMed] [Google Scholar]
- 40.Pepin K.M. Variable pleiotropic effects from mutations at the same locus hamper prediction of fitness from a fitness component. Genetics. 2006;172:2047–2056. doi: 10.1534/genetics.105.049817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ebert D., Bull J.J. Challenging the trade-off model for the evolution of virulence: is virulence management feasible? Trends Microbiol. 2003;11:15–20. doi: 10.1016/s0966-842x(02)00003-3. [DOI] [PubMed] [Google Scholar]
- 42.Bull J.J. Optimality models of phage life history and parallels in disease evolution. J. Theor. Biol. 2006;241:928–938. doi: 10.1016/j.jtbi.2006.01.027. [DOI] [PubMed] [Google Scholar]
- 43.Parker G.A., Smith J.M. Optimality theory in evolutionary biology. Nature. 1990;348:27–33. [Google Scholar]
- 44.Elena S.F., Sanjuan R. Virus evolution: insights from an experimental approach. Annu. Rev. Ecol. Evol. Syst. 2007;38:27–52. [Google Scholar]
- 45.Kassen R. The experimental evolution of specialists, generalists, and the maintenance of diversity. J. Evol. Biol. 2002;15:173–190. [Google Scholar]
- 46.Elena S.F. Restrictions to RNA virus adaptation: an experimental approach. Antonie Van Leeuwenhoek Int. J. Gen. Mol. Microbiol. 2002;81:135–142. doi: 10.1023/a:1020589929125. [DOI] [PubMed] [Google Scholar]
- 47.Chattopadhyay S. Haplotype diversity in “source-sink” dynamics of Escherichiacoli urovirulence. J. Mol. Evol. 2007;64:204–214. doi: 10.1007/s00239-006-0063-5. [DOI] [PubMed] [Google Scholar]
- 48.Sokurenko E.V. Opinion – source-sink dynamics of virulence evolution. Nat. Rev. Microbiol. 2006;4:548–555. doi: 10.1038/nrmicro1446. [DOI] [PubMed] [Google Scholar]
- 49.Holt R.D. Parasite establishment in host communities. Ecol. Lett. 2003;6:837–842. [Google Scholar]
- 50.Pulliam H.R. Sources, sinks and population regulation. Am. Nat. 1988;132:652–661. [Google Scholar]
- 51.Holt R.D. The phenomology of niche evolution via quantitive traits in a ‘black-hole’ sink. Proc. R. Soc. Lond. Ser. B-Biol. Sci. 2003;270:215–224. doi: 10.1098/rspb.2002.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Holt R.D. Allee effects, immigration, and the evolution of species’ niches. Am. Nat. 2004;163:253–262. doi: 10.1086/381408. [DOI] [PubMed] [Google Scholar]
- 53.Burch C.L., Chao L. Evolution by small steps and rugged landscapes in the RNA virus phi 6. Genetics. 1999;151:921–927. doi: 10.1093/genetics/151.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elena S.F. Evolutionary dynamics of fitness recovery from the debilitating effects of Muller's ratchet. Evolution. 1998;52:309–314. doi: 10.1111/j.1558-5646.1998.tb01633.x. [DOI] [PubMed] [Google Scholar]
- 55.Novella I.S. Exponential increases of RNA virus fitness during large population transmissions. Proc. Natl. Acad. Sci. U. S. A. 1995;92:5841–5844. doi: 10.1073/pnas.92.13.5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wichman H.A. Different trajectories of parallel evolution during viral adaptation. Science. 1999;285:422–424. doi: 10.1126/science.285.5426.422. [DOI] [PubMed] [Google Scholar]
- 57.Benmayor R. Host mixing and disease emergence. Curr. Biol. 2009;19:764–767. doi: 10.1016/j.cub.2009.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heineman R.H. Optimal foraging by bacteriophages through host avoidance. Am. Nat. 2008;171:E149–E157. doi: 10.1086/528962. [DOI] [PubMed] [Google Scholar]
- 59.Guyader S., Burch C.L. Optimal foraging predicts the ecology but not the evolution of host specialization in bacteriophages. PLoS ONE. 2008;3:e1946. doi: 10.1371/journal.pone.0001946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Turner P.E. Searching for the advantages of virus sex. Orig. Life Evol. Biosph. 2003;33:95–108. doi: 10.1023/a:1023973015054. [DOI] [PubMed] [Google Scholar]
- 61.West S.A. A pluralist approach to sex and recombination. J. Evol. Biol. 1999;12:1003–1012. [Google Scholar]
- 62.Michod R.E. Adaptive value of sex in microbial pathogens. Infect. Genet. Evol. 2008;8:267–285. doi: 10.1016/j.meegid.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 63.Turner P.E., Chao L. Sex and the evolution of intrahost competition in RNA virus phi 6. Genetics. 1998;150:523–532. doi: 10.1093/genetics/150.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Elena S.F. Intraclonal variation in RNA viruses: generation, maintenance and consequences. Biol. J. Linn. Soc. 2003;79:17–26. [Google Scholar]
- 65.Miralles R. Clonal interference and the evolution of RNA viruses. Science. 1999;285:1745–1747. doi: 10.1126/science.285.5434.1745. [DOI] [PubMed] [Google Scholar]
- 66.Pepin K.M., Wichman H.A. Experimental evolution and genome sequencing reveal variation in levels of clonal interference in large populations of bacteriophage phi X174. BMC Evol. Biol. 2008;8:85. doi: 10.1186/1471-2148-8-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Onodera S. Directed changes in the number of double-stranded RNA genomic segments in bacteriophage Phi 6. Proc. Natl. Acad. Sci. U. S. A. 1998;95:3920–3924. doi: 10.1073/pnas.95.7.3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kirkpatrick M., Barton N. Chromosome inversions, local adaptation and speciation. Genetics. 2006;173:419–434. doi: 10.1534/genetics.105.047985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ronce O., Kirkpatrick M. When sources become sinks: migrational meltdown in heterogeneous habitats. Evolution. 2001;55:1520–1531. doi: 10.1111/j.0014-3820.2001.tb00672.x. [DOI] [PubMed] [Google Scholar]
- 70.Kawecki T.J. Adaptation to marginal habitats: contrasting influence of the dispersal rate on the fate of alleles with small and large effects. Proc. R. Soc. Lond. Ser. B-Biol. Sci. 2000;267:1315–1320. doi: 10.1098/rspb.2000.1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Holt R.D., Gomulkiewicz R. The evolution of species’ niches: a population dynamic perspective. In: Othmer H.G., editor. Case Studies in Mathematical Modeling: Ecology, Physiology, and Cell Biology. Prentice Hall; 1997. pp. 25–50. [Google Scholar]
- 72.MacLean R.C., Buckling A. The distribution of fitness effects of beneficial mutations in Pseudomonasaeruginosa. PLoS Genet. 2009;5:7. doi: 10.1371/journal.pgen.1000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morgan A.D. Differential impact of simultaneous migration on coevolving hosts and parasites. BMC Evol. Biol. 2007;7:1. doi: 10.1186/1471-2148-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morgan A.D. The effect of migration on local adaptation in a coevolving host-parasite system. Nature. 2005;437:253–256. doi: 10.1038/nature03913. [DOI] [PubMed] [Google Scholar]
- 75.Brockhurst M.A. The impact of migration from parasite-free patches on antagonistic host-parasite coevolution. Evolution. 2007;61:1238–1243. doi: 10.1111/j.1558-5646.2007.00087.x. [DOI] [PubMed] [Google Scholar]
- 76.Forde S.E. Adaptation varies through space and time in a coevolving host-parasitoid interaction. Nature. 2004;431:841–844. doi: 10.1038/nature02906. [DOI] [PubMed] [Google Scholar]
- 77.Abedon S.T., Yin J. Bacteriophage plaques: theory and analysis. Methods Mol. Biol. 2009;501:161–174. doi: 10.1007/978-1-60327-164-6_17. [DOI] [PubMed] [Google Scholar]
- 78.Abedon S.T., Culler R.R. Bacteriophage evolution given spatial constraint. J. Theor. Biol. 2007;248:111–119. doi: 10.1016/j.jtbi.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 79.Abedon S.T., Culler R.R. Optimizing bacteriophage plaque fecundity. J. Theor. Biol. 2007;249:582–592. doi: 10.1016/j.jtbi.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 80.Kerr B. Local migration promotes competitive restraint in a host-pathogen ‘tragedy of the commons’. Nature. 2006;442:75–78. doi: 10.1038/nature04864. [DOI] [PubMed] [Google Scholar]
- 81.Lion S., van Baalen M. Self-structuring in spatial evolutionary ecology. Ecol. Lett. 2008;11:277–295. doi: 10.1111/j.1461-0248.2007.01132.x. [DOI] [PubMed] [Google Scholar]
- 82.Dennehy J.J. Host density impacts relative fitness of bacteriophage phi 6 genotypes in structured habitats. Evolution. 2007;61:2516–2527. doi: 10.1111/j.1558-5646.2007.00205.x. [DOI] [PubMed] [Google Scholar]
- 83.Scully L.R., Bidochka M.J. Developing insect models for the study of current and emerging human pathogens. FEMS Microbiol. Lett. 2006;263:1–9. doi: 10.1111/j.1574-6968.2006.00388.x. [DOI] [PubMed] [Google Scholar]
- 84.Kilpatrick A.M. Host heterogeneity dominates West Nile virus transmission. Proc. R. Soc. B-Biol. Sci. 2006;273:2327–2333. doi: 10.1098/rspb.2006.3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mathews C.K. American Society for Microbiology; 1983. Bacteriophage T4. [Google Scholar]
- 86.Karam J.D., Drake J.W. American Society for Microbiology; 1994. Molecular Biology of Bacteriophage T4. [Google Scholar]
- 87.Calendar R. Oxford University Press; 2006. The Bacteriophages. [Google Scholar]
- 88.Hershey A.D. Cold Spring Harbor Laboratory; 1971. The Bacteriophage Lambda. [Google Scholar]
- 89.Hendrix R.W. Cold Spring Harbor Laboratory; 1983. Lambda II. [Google Scholar]
- 90.Ptashne M. Cold Spring Harbor Laboratory Press; 2004. A Genetic Switch: Phage Lambda Revisited. [Google Scholar]