

Abstract
Pharmacological ascorbate treatment (P-AscH−, high-dose, intravenous vitamin C) results in a transient short-term increase in the flux of hydrogen peroxide that is preferentially cytotoxic to cancer cells vs. normal cells. This study examines whether an increase in hydrogen peroxide is sustained post-treatment and potential mechanisms involved in this process. Cellular bioenergetic profiling following treatment with P-AscH− was examined in tumorigenic and non-tumorigenic cells. P-AscH− resulted in sustained increases in the rate of cellular oxygen consumption (OCR) and reactive oxygen species (ROS) in tumor cells with no changes in non-tumorigenic cells. Sources for this increase in ROS and OCR were DUOX 1 and 2, which are silenced in PDAC, but upregulated with P-AscH− treatment. An inducible catalase system, to test causality for the role of hydrogen peroxide, reversed the P-AscH−-induced increases in DUOX, while DUOX inhibition partially rescued P-AscH−-induced toxicity. In addition, DUOX was significantly downregulated in pancreatic cancer specimens compared to normal pancreas tissues. Together these results suggest that P-AscH−-induced toxicity may be enhanced by late metabolic shifts in tumor cells resulting in a feed-forward mechanism for generation of hydrogen peroxide and induction of metabolic stress through enhanced DUOX expression and rate of oxygen consumption.
Keywords: pancreatic cancer, pharmacological ascorbate, dual oxidases, catalase, hydrogen peroxide
INTRODUCTION
Pharmacological ascorbate (P-AscH−, high dose, intravenous vitamin C) has made a resurgence as an adjuvant therapy in cancer treatment. It is currently utilized in clinical trials as an adjuvant to chemotherapy and radiation for the treatment of pancreatic ductal adenocarcinoma (PDAC) and several other types of cancer (1–6). Anti-cancer effects of pharmacological ascorbate are achieved when levels reach supra-physiologic concentrations (10 – 20 mM) in the plasma through intravenous administration (7, 8). Most investigations have studied the immediate effects of P-AscH−, i.e. events occurring within the first 24 h after exposure (8–13). Principal findings are that P-AscH− generates hydrogen peroxide (H2O2), which ultimately leads to more cell death in tumor cells compared to normal cells.
We hypothesized that the initial production of H2O2 produced directly by the oxidation of P-AscH− may not be entirely responsible for the cytotoxic effects of P-AscH−. One possibility for later generation of H2O2 would be through the NADPH oxidase (NOX) family of enzymes, which is made up of 7 members (NOX1–5 and Dual Oxidase {DUOX 1 and 2}). These enzymes were first noted for their ability to produce reactive oxygen species (ROS) independent of detoxification processes. NOX family members have been associated with various forms of cancer; they are implicated in the development and progression of cancer. Despite these reports, data on the expression and distribution of these isoforms in cancer are far from conclusive. The function of NOXs in the development and progression of cancer are poorly understood. The NOX family of enzymes is characterized by the ability to reduce molecular oxygen to superoxide (O2.-) or hydrogen peroxide (H2O2) using NADPH as a source of reducing equivalents (14). While most NOX family members produce O2.-, NOX4 and DUOX1–2 produce H2O2. DUOX1 and 2 contain a peroxidase-like domain that contributes to the ability of these proteins to produce H2O2 and makes them distinct from other NOX family members (15). These plasma membrane-associated DUOX proteins can produce single to double-digit nanomolar concentrations of hydrogen peroxide (16–18). DUOX not only produces H2O2 but can also be activated by H2O2 (18). In addition to being highly expressed in the thyroid and lung these proteins are also expressed in other epithelial cell types including those of the kidney, testis, inner ear, heart, and pancreas (19–22). Studies in lung and hepatocellular cancers suggest that DUOXs may be silenced in cancer; however, their expression in pancreatic cancer has not been studied (21, 23, 24). Additionally, it is unknown if their expression can be induced by exogenous ROS (18).
Compared to normal pancreatic ductal epithelial cells, we found that both DUOX 1 and 2 are decreased in pancreatic cancer cell lines as well as tissue specimens from human pancreatic cancer. P-AscH− induces sustained oxidative stress that leads to cytotoxicity by enhancing DUOX expression and increased production of oxidants and oxygen consumption rate (OCR). Metabolic shifts in PDAC that result in a feed-forward mechanism of H2O2 generation and induction of metabolic stress via enhanced DUOX expression and increased OCR contributes to P-AscH−-induced toxicity. The DUOX enzymes may be potential targets for an adjuvant therapy.
METHODS
Cell Culture and Reagents-
Human pancreatic ductal adenocarcinoma cell lines MIA PaCa-2 and PANC-1 were cultured in DMEM (Gibco, 11965) supplemented with 10% FBS (Gibco, 26140) and 1% penicillin–streptomycin antibiotic (Gibco, 15140). The human patient-derived pancreatic ductal adenocarcinoma cell lines, 339 and 403, were cultured in DMEM/F-12 media (Gibco, 11320) supplemented with 10% FBS, insulin (Gibco, 12585), EGF (Gibco, PHG0311), hydrocortisone (Sigma, H0888), bovine pituitary extract (Gibco, 13028) and 1% penicillin–streptomycin antibiotic. The human non-small cell lung carcinoma cell line overexpressing catalase, H1299T-CAT, was generated as previously described (25, 26) and cultured in RPMI (Gibco, 11875) supplemented with 10% FBS. Non-tumorigenic HPV16-E6E7 immortalized cell line derived from normal pancreatic ductal epithelium (H6c7) with near normal genotype and phenotype of pancreatic duct epithelial cells is utilized for comparison. H6c7 cells were cultured in Serum Free Keratinocyte media (Gibco, 10724) supplemented with EGF (1 ng/mL), bovine pituitary extract (50 μg/mL), and 1% penicillin–streptomycin antibiotic. Human cell lines (MIA PaCa-2, PANC-1, H1299, and H6c7) were purchased directly from American Type Culture Collection and passaged for fewer than six months after receipt. No additional authentication was performed. The H1299T cells were characterized and verified by IDEXX-RADIL. Mycoplasma testing was performed routinely every 6 months. The patient-derived cell lines (339 and 403) were obtained from the Medical College of Wisconsin surgical oncology tissue bank (27, 28). HEK293 cells were a gift from Dr. William Nauseef at the University of Iowa. Regardless of varying cell type and media components, all cells were treated in fresh 10% DMEM media with ascorbate for 1 h at 37 °C. After the 1 h treatment, media was replaced i.e. ascorbate was removed, and cells were allowed to incubate for 48 h. Ascorbate came from a stock solution of 1 M (pH 7) made under argon and stored with a tight-fitting stopper at 4 °C. Ascorbate concentration was verified at 265 nm, ε = 14,500 M−11 cm−1 (29). To enhance rigor and reproducibility final concentrations were calculated in units of moles-per-cell to account for variation in media, cell density, and cellular metabolism (30, 31).
Measurement of pro-oxidants-
Bovine catalase (Sigma, C40) was utilized in the media at 100 μg/mL immediately prior to P-AscH− treatment (MIA PaCa-2, 1 mM/5 picomole cell−1; PANC-1, 2 mM/11 picomole cell−1) for 1 h and fresh media was replaced. Cells were allowed to proliferate for 48 h prior to the DCFH-DA (5-(and-6)-carboxy-2’,7’-dichlorodihydrofluorescein diacetate; Molecular Probes, C400) assay. Cells were incubated at 37 ° C, protected from light in 15 μM DCFH-DA for 15 min in HBSS). They were then harvested, re-suspended in 1x PBS and filtered before taking to Flow Cytometry. A CBD LSRII cytometer (BD Biosciences) was used to measure DCFH-DA oxidation at 504/529 nm. Data sets were analyzed using FlowJo Software.
Clonogenic Survival-
Approximately 2.5 × 105 cells were seeded 3 d prior to assay. Cells were treated with P-AscH− for 1 h and allowed to proliferate 48 h prior to being plated. Cells were trypsinized with TrypLE Express (Gibco, 12604) to form a single cell suspension and counted using a Countess II automated cell counter (Thermo Fisher) to determine the number of cells plated into each well. Cells formed colonies for 10 – 14 d before being fixed and stained for analysis. Colonies containing ≥ 50 cells were scored. Surviving fraction was defined as: SF = number of colonies counted/number of cells seeded. Each experimental condition was normalized to their own control to determine a normalized surviving fraction (9, 12, 13). Each condition was done in triplicate and experiments were performed at no less than n = 3.
Propidium Iodide (PI) Exclusion Assay-
48 h following P-AscH− treatment cells were trypsinized and centrifuged to form pellets. These pellets were then resuspended in ice cold PBS. PI was added at a concentration of 1 μg/mL and incubated for 5 min on ice prior to analysis. Flow cytometry was utilized to measure PI incorporation into these cells as a measure of the percentage of viable cells following P-AscH− treatment compared to control. The percentages of PI positive (non-viable) and PI negative (viable) cells were calculated using FlowJo software.
Cellular Bioenergetics-
To probe for potential changes in OCR (ΔOCR) upon exposure to P-AscH− a Clark electrode oxygen monitor (YSI Inc.) connected to an ESA Biostat multi-electrode system (ESA Products, Dionex Corp.) was employed. PANC-1, MIA PaCa-2, and H6C7 cells were treated with 2 mM, 1 mM, and 2 mM P-AscH− for 1 h. OCR was measured 48 h after P-AscH− exposure at 24 ° C. Changes in OCR after treatment with P-AscH− were determined by subtracting the untreated basal OCR from the increase following P-AscH− treatment to get ΔOCR after P-AscH−.
More detailed information on cellular bioenergetics was obtained using a Seahorse XF96 Extracellular Flux Analyzer (Seahorse Bioscience). The Cell Mito Stress Test medium was utilized supplemented with 2 mM GlutaMax, 1 mM sodium pyruvate, and 25 mM glucose. The assay was run according to manufacturer’s recommendations 48 h after the 1 h treatment with P-AscH−. Cells were plated immediately after P-AscH− treatment onto a Seahorse XF 96 analyzer plate; 2.5 × 104 or 3.0 × 104 for MIA PaCa-2 and PANC-1 cells, respectively. Actual number of cells per well were determined immediately after the Seahorse extra cellular flux experiment was completed. Results were normalized on a cell−1 s−1 basis (32). Cellular bioenergetics were examined using the Seahorse Cell Mito Stress Test. OCR measurements were performed in real time in control and P-AscH− treated PANC-1 cells at basal levels and following sequential addition of mitochondrial respiration inhibitors. Oligomycin, an inhibitor of ATP synthase, was used to distinguish between OCR due to synthesis of ATP (ATP-linked respiration) and OCR from other processes. Carbonyl cyanide-para-trifluoromethoxyphenyl-hydrazone (FCCP) treatment collapses the proton gradient and disrupts the mitochondrial membrane potential, which allows measurements of the maximal uncoupled respiration (Maximal Respiration). A combination treatment of rotenone, a complex I inhibitor, and antimycin A, a complex III inhibitor was used to shut down all mitochondrial OCR, which enabled us to distinguish between the mitochondrial (Basal Respiration) and non-mitochondrial (Non-mitochondrial OCR) form of respiration. The difference between maximal and basal respiration constitutes the Spare Capacity.
q-RT-PCR-
Total cellular RNA was isolated using Trizol reagent (Invitrogen, 15596026) in conjunction with the Direct-zol RNA Miniprep Kit (Zymo Research) according to the manufacture’s protocol. RNA was quantified using a ND1000 Nano-Drop spectrophotometer (Nanodrop). cDNA was synthesized using the high-capacity cDNA archive kit (Applied Biosystems). Quantitative real time PCR (q-RT-PCR) assays were performed using 2x Power SYBR green real-time master-mix (Applied Biosystems, 4368702) under the following set up: 95 ° C for 10 min, followed by 40 cycles of 95 ° C for 15 s and 60 ° C for 1 min (StepOne plus Sequence Detection System, Applied Biosystems). Primers were ordered from Integrated DNA Technology.
Primers sequences:
DUOX1 Forward-GGATGCTGAGATCCTTCATCGAGA.
DUOX1 Reverse- ACCTCCACCCCTTTGACACAGAG.
DUOX2 Forward- TGTGTATGAGTGGCTGCCCAGC.
DUOX2 Reverse- ACTGCTCAGAGGCCACCACAAA
Western Blotting- Protein samples were isolated and prepared in phosphosafe buffer (EDM Millipore, 71296) containing protease inhibitor cocktail (Sigma Aldrich). Protein content was measured using a Bradford Protein assay. Total protein (50 – 100 μg) was loaded on a 4 – 20% SDS-PAGE gradient gel. Protein was transferred to Immun-Blot PVDF membrane (Bio-Rad). Membranes were blocked in 5% bovine serum albumin in TBS-T. Primary Antibodies included: DUOX1 (Santa Cruz, H-9, 1:200), DUOX2 (Santa Cruz, E-8, 1:200), Tubulin (1:2000), actin (1:2000), and GAPDH (1:5000). Appropriate HRP-linked secondary antibodies were used at a concentration of 1:15,000–30,000. Blots were visualized using ECL 2 or Pico West substrate (Thermo Scientific, 34579) on x-ray film.
Catalase Activity Assay-
Catalase activity was determined using UV-Vis spectroscopy by following the rate of removal of hydrogen peroxide due to catalase (33, 34). Activity was normalized to the protein content of the sample as determined by the DC™ Protein Assay (Bio-Rad, 5000111) protein assay on a plate reader, which is based on the Lowry assay (35).
In Vivo Experiments-
The animal protocols were reviewed and approved by the Animal Care and Use Committee of The University of Iowa. Thirty-day-old athymic nude mice (Foxn1nu) were obtained from Envigo. Animals acclimated in the unit for 1 week before any manipulations were performed. For experiments growing xenografts with MIA PaCa-2 and H1299T-CAT cells (2 – 3 × 106), tumors were delivered subcutaneously into each flank region of nude mice with a 1-mL tuberculin syringe equipped with a 25-gauge needle. Tumors grew to approximately 5 mm in diameter before experimental treatment began and mice were treated twice daily with I.P. P-AscH− (4 g/kg) or saline (1 M) for 5 days. Mice were euthanized by CO2 asphyxiation and tumors were harvested and processed for experimental analyses. In separate groups of animals determining catalase levels utilizing the catalase activity assay in H1299T-CAT tumors, mice were injected with 106 H1299T-CAT cells. When tumors were palpable, mice were randomized into treatment groups and treated with control (1% sucrose) or doxycycline (2 mg/mL and 1% sucrose) in drinking water (changed every 2 – 3 d). On days 3, 7, and 10, two mice from each group were euthanized and tumors were collected in DETAPAC buffer and tested for catalase activity.
Immunofluorescent Staining-
The same protocol was used for ex vivo staining of both patient-derived normal pancreas and PDAC tumor samples and the matched patient-derived PDAC tumor and non-tumor adjacent pancreas samples. The matched patient samples were provided by the University of Iowa Gastrointestinal Molecular Epidemiologic Resource (GIMER). Tissues were paraformaldehyde fixed and imbedded in paraffin. Tissues were sectioned at 6 – 10 μm and placed on SuperFrost Plus slides and baked overnight at 42 – 45 ° C. Slides were de-paraffinized and were either subject to H&E staining or were further processed for immunohistochemistry. Immunofluorescent samples were subject to antigen unmasking protocol at 95 ° C for 15 min, cooled to room temperature, washed with double distilled water, and allowed to dry. Tissues were circled with a PAP pen and then samples were blocked with 5% normal goat serum prior to incubation with DUOX1 (1:10) and DUOX2 (1:10) primary antibodies in 1% normal goat serum at 4 ° C for 24 h. Slides were washed 3 times in PBS for 5 min. Goat anti-mouse secondary (1:400) conjugated to FITC in 1% normal goat serum at 4 °C overnight. Slides were washed 3 times with PBS for 5 min. Nuclear staining was performed using Topoisomerase-3 (1:1000) for 15 min and samples were washed once with PBS. Coverslips were mounted on slides with immunofluorescence mounting medium and visualized using a Zeiss Confocal Microscope 40x oil objective. Quantification was performed using only DUOX1 or DUOX2 staining images. ImageJ was used to quantify the fluorescence for each image and values were normalized to adjacent normal DUOX expression.
Statistical Methods-
Data are presented as the mean ± SEM. For statistical analyses of two groups, unpaired 2-tailed Student’s t-test were utilized. To study statistical differences between multiple comparisons, significance was determined using one-way ANOVA analysis with Tukey’s multiple-comparisons test. All analyses were performed in GraphPad Prism (GraphPad Software, Inc.).
RESULTS
Previous studies have demonstrated late ROS accumulation in radiation-induced oxidative stress, which results in toxicity to human cancer (pancreas, glioma, and head and neck) and normal cells (36–39). To determine if there were similar sustained effects of P-AscH− on the production of ROS, a time course (24 – 72 h) for the oxidation of DCFH-DA was undertaken in PANC-1 PDAC cells. Even though P-AscH− treatment was for only 1 h and the ascorbate removed, mean fluorescence intensity (MFI) for the oxidation of DCFH-DA was increased 48 h after exposure (Figure 1A). These studies were repeated at 48 h in both PANC-1 and MIA PaCa-2 cell lines as seen in Figure 1B–C with the addition of catalase to the media. Pretreatment with catalase completely reversed the increase in the MFI of ascorbate-induced oxidation of DCFH-DA at 48 h suggesting that sustained production of H2O2 occurs after short-term treatment with P-AscH− (Figure 1B–C).
Figure 1. P-AscH− increases ROS production 48 h after exposure, resulting in clonogenic cell death.
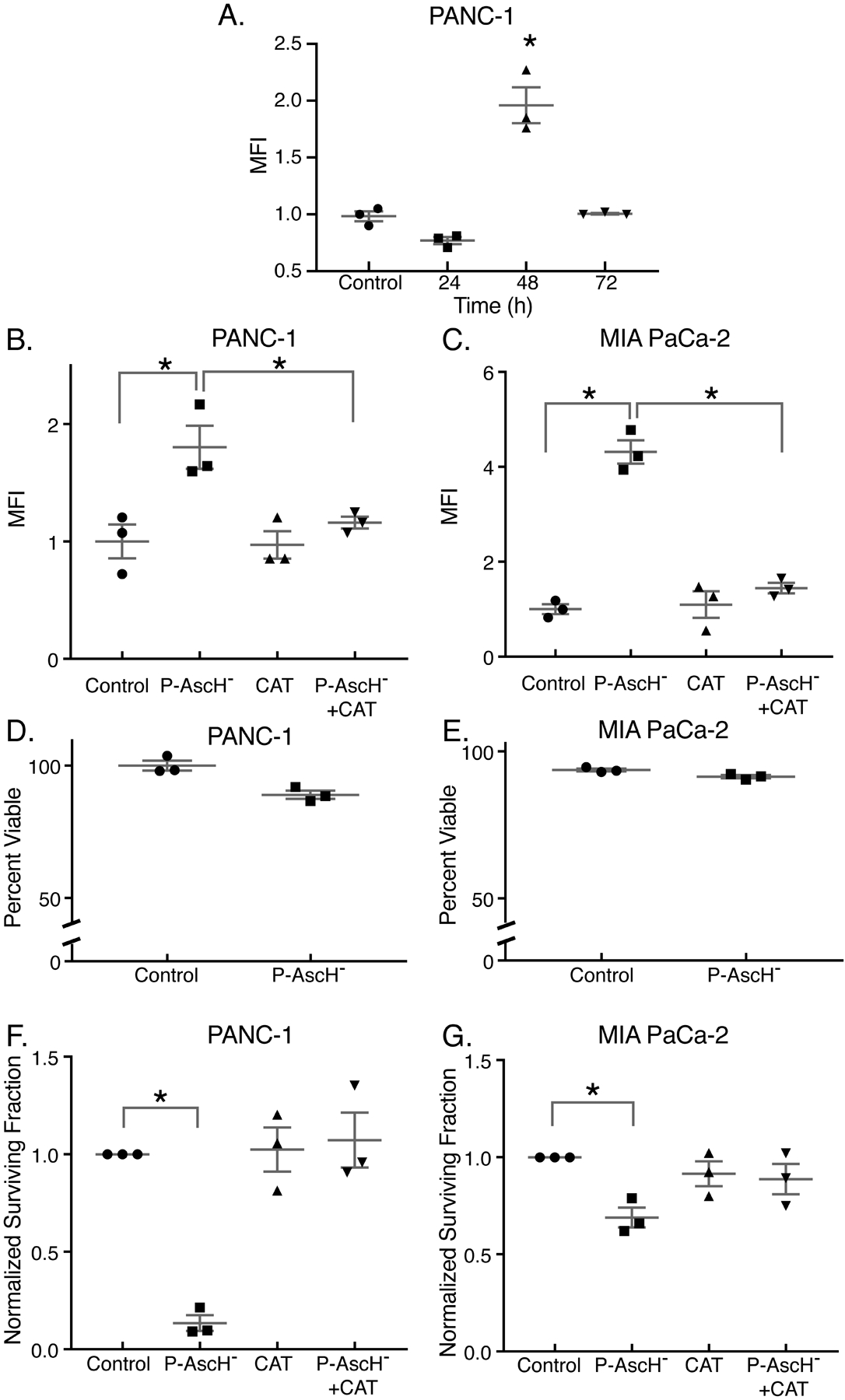
PDAC cell lines were treated with P-AscH− for 1 h for all experiments. Assays were performed 48 h after exposure, unless stated otherwise. Presented are means ± SEM for each set, n = 3; *p < 0.05 vs. control for all experiments. For panels A - C and F - G, ANOVA with Tukey’s multiple comparisons were used. For panels D - E, 2-tailed student’s t-test was used.
A. Time course (24 – 72 h) for the oxidation of DCFH-DA in PANC-1 cells treated with 2 mM ascorbate. Data is represented as mean fluorescent intensity (MFI) that is normalized to control.
B-C. PANC-1 and MIA PaCa-2 cells treated with P-AscH− (2 mM or 1 mM, respectively) exhibit increased DCFH-DA oxidation 48 h after treatment that is rescued by 100 μg/mL of bovine catalase in the medium.
D-E. PANC-1 and MIA PaCa-2 cells treated with 2 mM or 1 mM ascorbate respectively, exhibited no changes in viability using the propidium iodide (PI) exclusion assay (p > 0.05).
F-G. PANC-1 and MIA PaCa-2 cells treated with P-AscH− (2 mM or 1 mM, respectively) exhibit significant decreases in clonogenic survival.
To further investigate the role of P-AscH− in the toxicity to PDAC cells, MIA PaCa-2 and PANC-1 cells were treated during exponential growth for only 1 h with 1 mM and 2 mM P-AscH−, respectively, and then incubated for 48 h before experiments were performed. P-AscH− concentrations were chosen based on previous studies that demonstrated that some cell lines are more susceptible to P-AscH− based on the expression level of the H2O2 detoxifying enzyme, catalase (30). In both PANC-1 and MIA PaCa-2 cells there were no changes in the number of intact and viable cells at 48 h compared to controls (Figure 1D–E). However, despite these cells being intact and viable at 48 h, they displayed decreased doubling time and decreased clonogenic survival with P-AscH− treatment. In MIA PaCa-2, doubling time increased from 18.8 ± 0.6 h in controls to 23.4 h ± 1.9 with ascorbate treatment (5 picomole cell−1, Means ± SEM, P < 0.05 vs. control), and from 20.7 ± 1.0 to 33.1 ± 5.1 h in PANC-1 cells (16 picomole cell−1, Means ± SEM, P < 0.05 vs. control). This same trend was seen in clonogenic survival in both PANC-1 (Figure 1F) and MIA PaCa-2 cells (Figure 1G) compared to controls. This suggests that P-AscH− may initiate sustained oxidant formation after the initial insult, which may contribute to prolonged oxidative stress and cell death.
To investigate possible sources for this increased oxidant formation, as seen by the increase in the oxidation of DCFH-DA in Figures 1A–C, a Clark electrode was used to determine possible changes in the basal rate of oxygen consumption (OCR). Cellular OCR in MIA PaCa-2 and PANC-1 cells was significantly increased 48 h after exposure to P-AscH− while the basal OCR of the non-tumorigenic H6c7 pancreatic ductal epithelial cells showed little if any change (Figure 2A).
Figure 2. P-AscH− treatment results in increased basal oxygen consumption rate, ATP linked, Maximal Respiration, and Proton leak in PDAC cell lines.
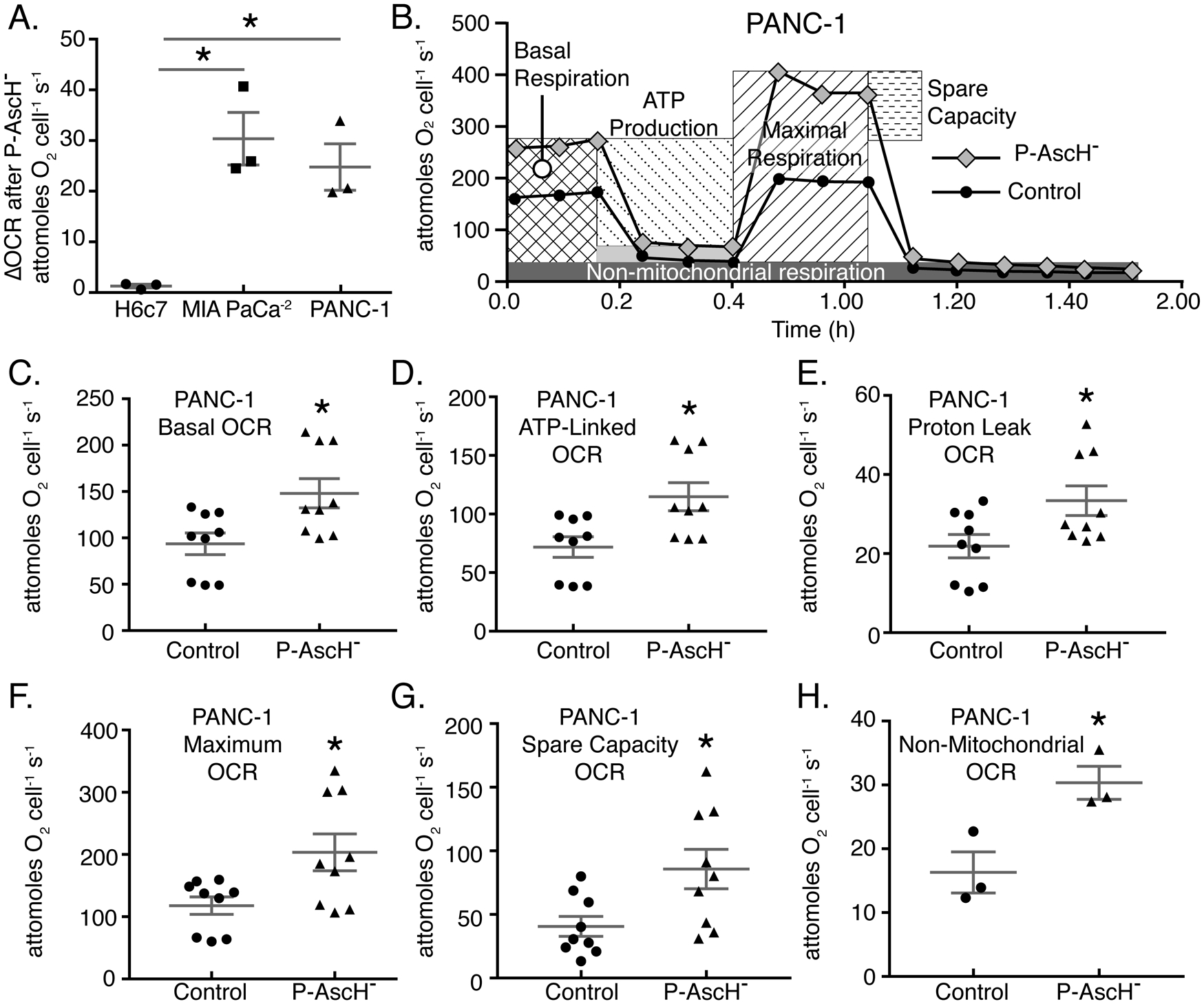
A. PANC-1, MIA PaCa-2, and H6c7 cells were treated with P-AscH− for 1 h and basal oxygen consumption rate (OCR) was measured 48 h after treatment using a Clarke Electrode. Changes in OCR after P-AscH− treatment were determined by subtracting the untreated basal OCR from the increase following P-AscH− treatment to get ΔOCR. PANC-1 cells treated with 2 mM P-AscH−, MIA PaCa-2 cells treated with 1 mM P-AscH− and H6c7 cells treated with 2 mM P-AscH− (Means ± SEM, n = 3, *p < 0.05 vs. control, ANOVA with Tukey’s multiple comparisons).
B. Example data from Seahorse XF96 instrumentation showing that PANC-1 cells treated with P-AscH− (2 mM) have alterations in the mitochondrial stress test curves 48 h after exposure.
C-H. PANC-1 cells demonstrate an increase in: basal respiration; D. ATP production; E. proton leak; F. maximal respiration; G. spare capacity and H. non-mitochondrial respiration 48 h after treatment (Means ± SEM, n = 9 C-G and n = 3 H, p < 0.05 vs. control, 2-tailed student’s t-test).
To gain greater understanding of the increase in OCR we observed in PDAC cell lines, a mitochondrial stress test was performed using a Seahorse XF96 analyzer to measure alterations in OCR as well as glycolytic flux following exposure to P-AscH−. Both PANC-1 (Figure 2B) and MIA PaCa-2 (Supplemental Figure 1) cells demonstrated significant alterations in their bioenergetic profiles, consistent with an increase in energy demand, as seen in the basal OCR at 48 h after P-AscH− treatment. The mitochondrial stress test determined overall changes in mitochondrial function. First, there is a significant increase in basal respiration (Figure 2C) in PANC-1 cells similar to the change in OCR observed in Figure 2A. In addition, there were also significant increases in ATP-linked OCR (Figure 2D), proton leak (Figure 2E), and maximal respiration (Figure 2F). These data suggest sustained alterations to mitochondrial function and sustained oxidative stress induced by P-AscH− treatment. Finally, we observed that there were increases in spare capacity (Figure 2G) as well as non-mitochondrial respiration (Figure 2H) in PANC-1 cells with similar results observed in MIA PaCa-2 cells (Supplemental Figure 1B–G). The increase in non-mitochondrial OCR (Figure 2G, supplemental Figure 1G) suggests that there may be an additional source of oxygen consumption in cells treated with P-AscH−.
One potential mechanism for the increase in sustained oxidative stress could be the production of H2O2 or O2.- from non-mitochondrial sources. NOX family members can generate increased fluxes of ROS, while DUOX expression can be induced by exogenous H2O2 (18). Since P-AscH− is a pro-drug for the delivery of H2O2 (11) in addition to the sustained increase in non-mitochondrial OCR, we hypothesized that there may be alterations in the expression of NOX family members following treatment with P-AscH−. First, PDAC cell lines were compared to non-tumorigenic pancreatic ductal cells by q-RT-PCR to determine the baseline mRNA expression of the DUOX genes. DUOX expression is significantly decreased in all PDAC cell lines tested in comparison to the pancreatic ductal epithelial cell line, H6c7 (Figure 3A–B). These results were also confirmed in western blot analysis that showed a dramatic reduction in DUOX1 and DUOX2 immunoreactive protein in all the PDAC cell lines as compared to the non-tumorigenic H6c7 cells, (Figure 3C–D). These results suggest that DUOX expression may be altered from PDAC to normal pancreatic ductal epithelium.
Figure 3. DUOX1 and DUOX2 mRNA expression is decreased in PDAC cell lines compared to ductal epithelium in vitro and is increased in a dose-dependent manner after exposure to P-AscH−.
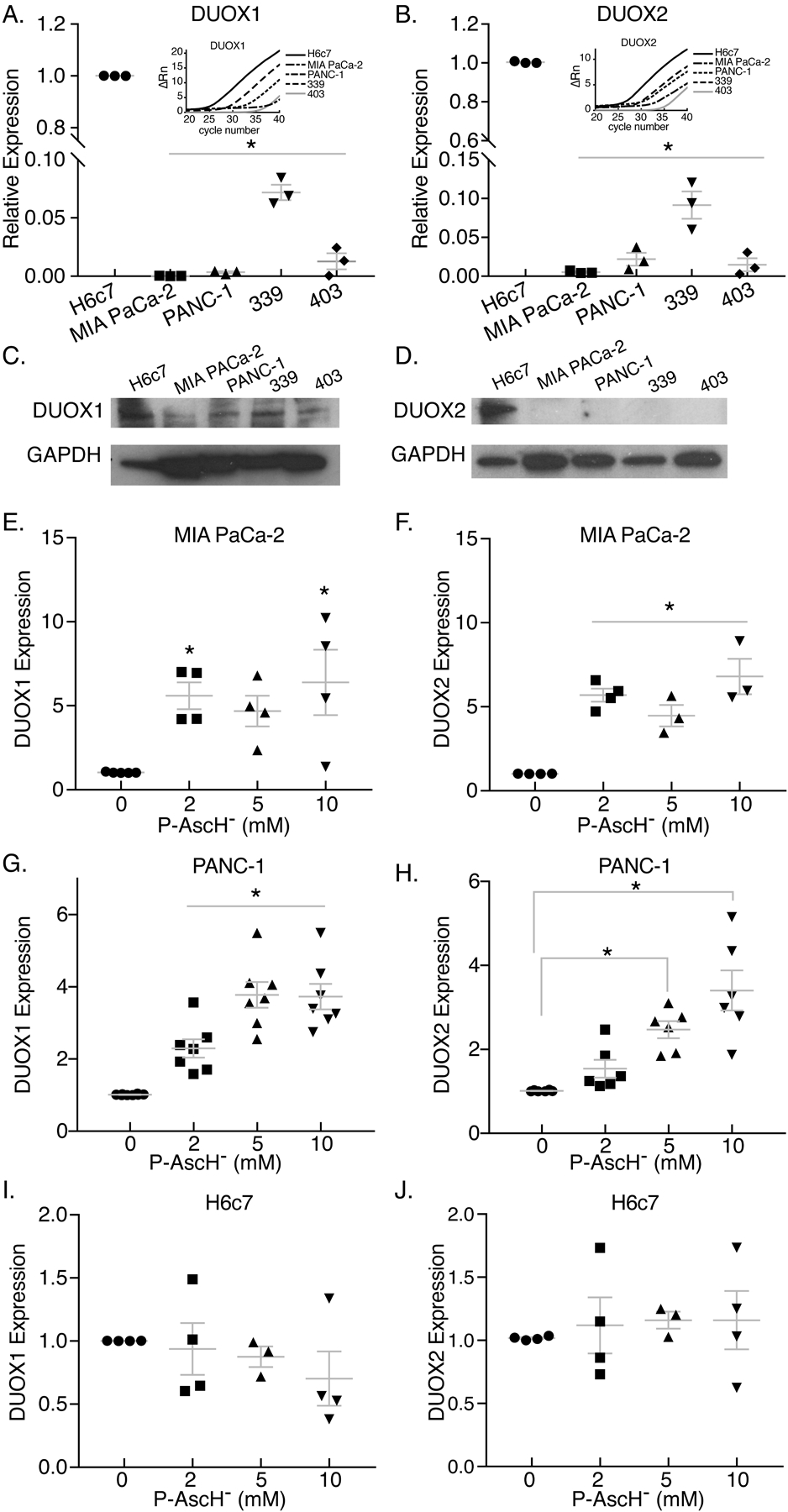
MIA PaCa-2, PANC-1 and H6c7 ductal epithelial cells were exposed to varying doses of P-AscH− for 1 h. mRNA expression was tested 24 h after exposure to P-AscH−. For figures A-B and E-J: Means ± SEM, ANOVA with Tukey’s multiple comparisons used.
A-B. DUOX1 and DUOX2 mRNA expression is decreased in PDAC cell lines MIA PaCa-2, PANC-1, 339, and 403 as compared to non-tumorigenic H6c7 cells (n = 3, *p < 0.05 vs. H6c7).
C-D. DUOX1 and DUOX2 immunoreactive protein is decreased in PDAC cell lines as compared to non-tumorigenic H6c7 cells. Representative blots shown.
E-F. DUOX1 and DUOX2 mRNA expression is increased in MIA PaCa-2 by increasing doses of P-AscH− (n = 3–5, *p < 0.05 vs. 0 mM).
G-H. DUOX1 and DUOX2 mRNA expression is increased in PANC-1 by increasing doses of P-AscH− (n = 6–7, *p < 0.05 vs. 0 mM).
I-J. DUOX1 and DUOX2 mRNA expression is unchanged in non-tumorigenic H6c7 cells by increasing doses of P-AscH− (n = 3–4, p > 0.05 vs. 0 mM).
Since expression of both DUOX1 and DUOX2 was decreased in all PDAC cell lines tested, we next sought to determine if their expression can be affected by treatment with P-AscH−. We observed that DUOX1 and DUOX2 mRNA is enhanced by treatment with P-AscH− in both dose- and time-dependent manners. We observed that DUOX expression was increased in a dose-dependent manner in MIA PaCa-2 cells (Figure 3E–F). Additionally, PANC-1 cells showed a similar trend with DUOX1 and DUOX2 mRNA being significantly increased at doses of 2 – 10 mM 24 h after treatment with P-AscH− (Figure 3G–H). In contrast, increasing doses of P-AscH− did not alter mRNA expression of DUOX1 and DUOX2 in the non-tumorigenic cell line H6c7 (Figure 3I–J). Temporal experiments indicate that P-AscH− induces DUOX1 and DUOX2 expression in MIA PaCa-2 (Figure 4A–B); western blotting (Figure 4C) demonstrates increases in protein expression 48 – 72 h after P-AscH− treatment. Similarly, P-AscH− increased DUOX in PANC-1 cells 24 – 72 h after treatment (Figure 4D–F). As mentioned above, DUOX1 expression can be induced by exogenous H2O2 (19). To determine if H2O2 generated by P-AscH− induced increases in DUOX, catalase was introduced to the cell culture medium and DUOX expression was determined 48 h after treatment with P-AscH−. Figures 4G–H demonstrate increases in DUOX expression that is reversed with catalase. Similar results were seen in the PANC-1 cell lines where the presence of catalase reversed P-AscH− induced increases in DUOX expression (Supplemental Figures 2A–B). This suggests that generation of H2O2 by P-AscH− leads to the observed increases in DUOX expression. In addition, the other NOX enzymes did not show a time-dependent increase in expression when PANC-1 cells were treated with P-AscH− (Supplemental Figure 3). As a control, we treated cells with ascorbate-2-phosphate, which does not oxidize and thus does not generate H2O2, but is taken up by cells increasing the level of intracellular ascorbate (40) Ascorbate-2-phosphate did not change expression of either DUOX 1 or 2 in either pancreatic cancer cell line (Supplemental Figure 4).
Figure 4. DUOX1 and DUOX2 expression is increased in a time dependent manner after exposure to P-AscH− in PDAC cells.
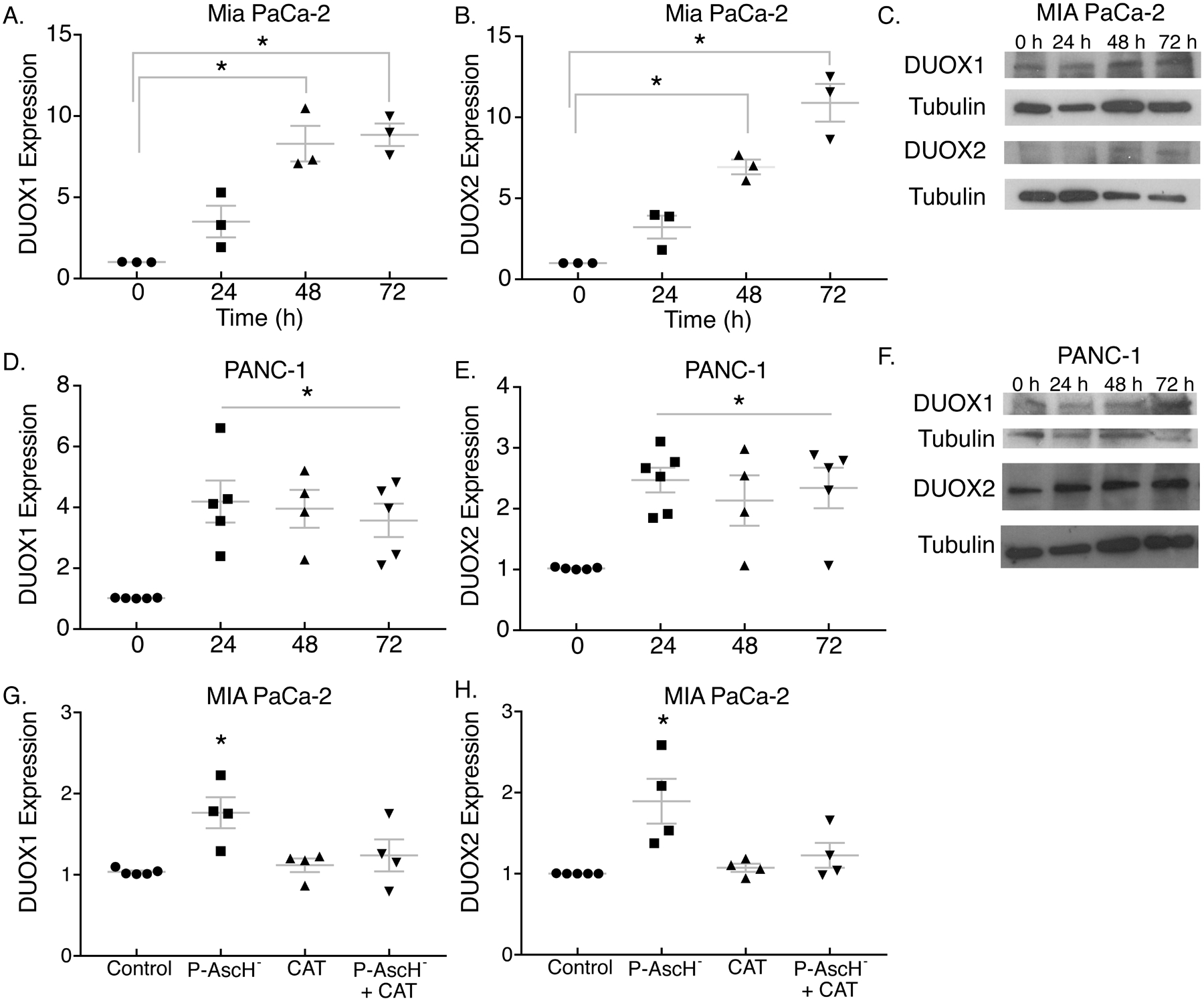
−MIA PaCa-2, PANC-1 and H6c7 ductal epithelial cells were exposed to P-AscH− and DUOX expression determined at 0–72 h. For figures A-B, D-E, and G-J: Means ± SEM, ANOVA with Tukey’s multiple comparisons used.
A-B. DUOX1 and DUOX2 mRNA expression is increased following P-AscH− (5 mM) in MIA PaCa-2 cells (n = 3, *p < 0.05 vs. 0 h).
C. DUOX1 and DUOX2 immunoreactive protein is increased in MIA PaCa-2 cells after P-AscH− (5 mM). Representative blots shown.
D-E. DUOX1 and DUOX2 mRNA expression is increased following P-AscH− (5 mM) in PANC-1 cells (n = 4 – 5, *p < 0.05 vs. 0 h).
F. DUOX1 and DUOX2 immunoreactive protein is increased in PANC-1 after P-AscH− (5 mM). Representative blots shown.
G-H. DUOX1 and DUOX2 mRNA expression is increased following P-AscH− (1 mM) in MIA PaCa-2 cells and is reversed by pretreatment with 100 μg/mL bovine catalase. (n = 4, *p < 0.05 vs. control).
To determine if the DUOX-induced increases in H2O2 contribute to P-AscH−-induced cytotoxicity, we used diphenyleneiodonium (DPI), an inhibitor of flavin-containing proteins, to inhibit the activity NOX proteins prior to treatment with P-AscH− in both MIA PaCa-2 and PANC-1 PDAC cell lines (Figure 5A–B). DPI treatment partially ameliorated the P-AscH−-induced decreases in clonogenic survival in both PANC-1 and MIA PaCa-2 cells. We repeated these studies and treated the cells with DPI, 48 h after treatment with P-AscH−. P-AscH− treatment decreased clonogenic survival to 57 ± 6%, while addition of DPI partially reversed this decrease to 78 ± 9% (Means ± SEM). To determine the role of DUOX-induced generation of H2O2 that occurs 48 h after P-AscH− treatment, we first incubated cells with catalase 24 after P-AscH− treatment and then left the catalase on for another 24 h. P-AscH− decreased clonogenic survival to 46 ± 2% which changed little to 65 ± 2% (Means ± SEM) with later treatment of catalase. Secondly, we used the doxycycline-inducible catalase (H1299T-CAT) cell line that has an induction of catalase 24 to 48 h after exposure to doxycycline. Figure 5C–D demonstrated that H1299T-CAT, showed a similar increase in DUOX1 and DUOX2 when treated with P-AscH− (5 mM). P-AscH− treatment (2 mM) also decreased clonogenic survival of the H1299T-CAT cell line (Figure 5E) and the decrease was partially reversed in cells exposed to doxycycline for 48 h. Because doxycycline has been shown to have antioxidant properties (41), we treated the parental H1299T cells with P-AscH−, doxycycline alone or P-AscH− + doxycycline (Supplemental Figure 5A) which demonstrated that doxycycline did not reverse P-AscH− induced decreases in clonogenic survival. Activity assays for catalase on the H1299T-CAT cells that were exposed to doxycycline displayed significant increases in intracellular catalase activity (Figure 5F). As seen by the RT-qPCR data in Figures 3A and B, the baseline levels of DUOX 1 and 2 are very low in all the PDAC cell lines examined. We hypothesized that inhibiting DUOX 1 or 2 would not have much of a detectable effect if these enzymes are nearly undetectable without the induction of expression shown with P-AscH−. Since both DUOX1 and 2 are induced by P-AscH−, we utilized the HEK293 cell line that stably overexpresses either DUOX 1 or 2 and then treated these cells with P-AscH− (1 – 4 mM). We hypothesized that increased of either DUOX1 or 2 expression leading to increased generation of H2O2 would add to P-AscH− induced decreases in clonogenic survival. Overexpression of DUOX 1 and 2 further decreased clonogenic survival when treated with P-AscH- (Figure 5G and H).
Figure 5. DUOX enhances P-AscH− induced cytotoxicity in PDAC cell lines.
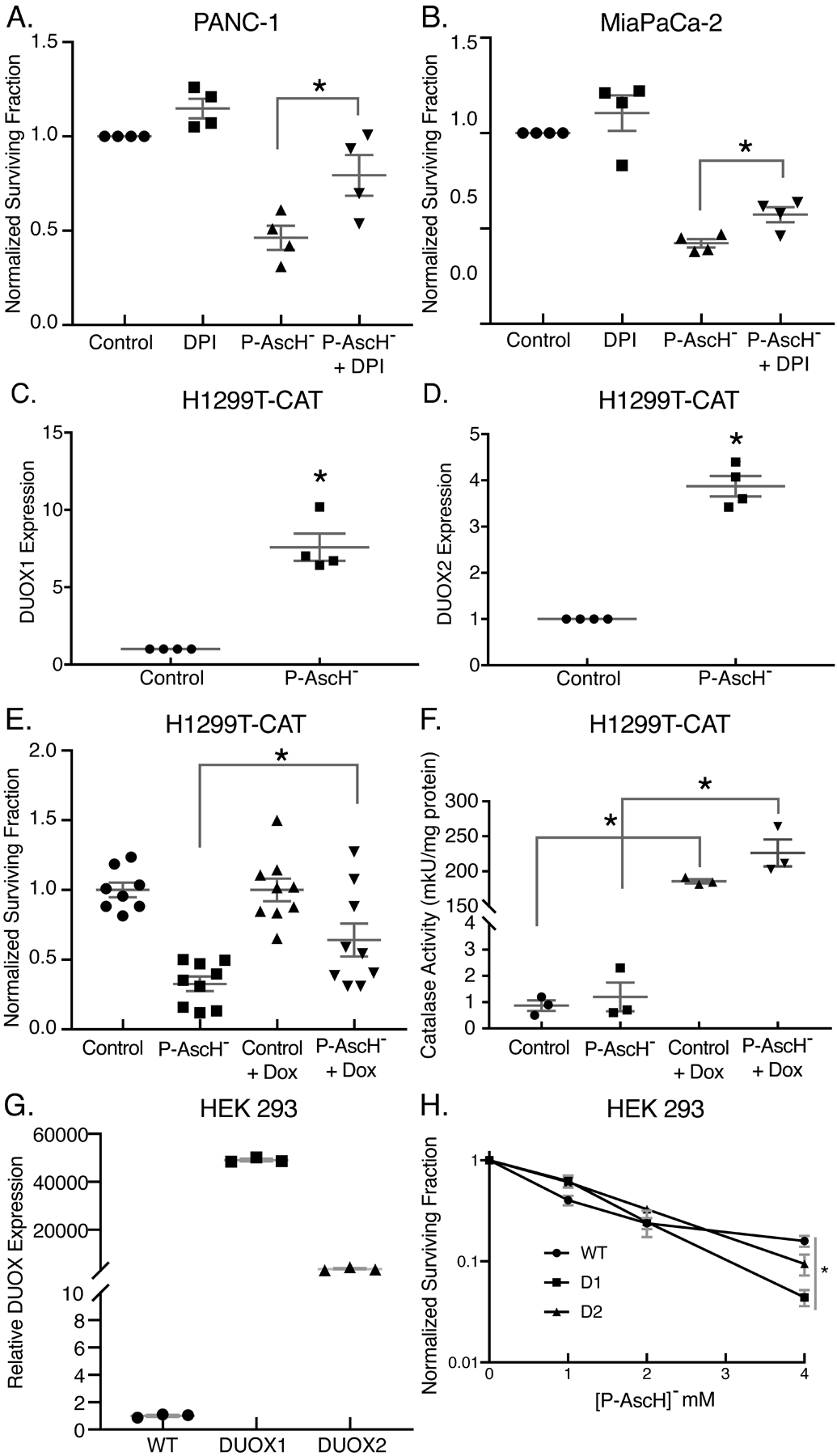
A-B. PANC-1 and MIA PaCa-2 cells were pretreated with DPI (5 μM) for 1 h prior to P-AscH− treatment (2 mM and 1 mM, respectively) and a clonogenic survival assay was performed 48 h later (Means ± SEM, n = 4, *p < 0.05 vs. P-AscH−, ANOVA with Tukey’s multiple comparisons).
C-D. DUOX1 and DUOX2 mRNA expression is increased in H1299T-CAT cells 48 h following P-AscH− treatment (5 mM) for 1 h (Means ± SEM, n = 4, *p < 0.05 vs. control, 2-tailed student’s t-test).
E. H1299T-CAT cells were treated with 2 mM P-AscH− for 1 h and 2 μg/mL doxycycline for 48 h before being plated in clonogenic survival assays. Over-expression of catalase demonstrated a partial reversal of P-AscH− toxicity (Means ± SEM, n = 9, *p < 0.05 vs. P-AscH−, ANOVA with Tukey’s multiple comparisons).
F. Catalase over-expressing H1299T-CAT were tested for catalase activity following 48 h treatment with 2 μg/mL doxycycline (Means ± SEM, n = 3, *p < 0.05 control vs. Control + Dox and P-AscH− vs. P-AscH− + Dox, ANOVA with Tukey’s multiple comparisons).
G. DUOX1 and DUOX2 over-expressing HEK 293 cells show increased DUOX1 and DUOX2 mRNA expression compared to control wild-type HEK 293 cells (Means ± SEM, n = 3, *p < 0.05 vs. WT, ANOVA with Tukey’s multiple comparisons).
H. WT, DUOX1, and DUOX2 over-expressing cells were treated with P-AscH− (0–4 mM) for 1 h and a clonogenic survival assay was performed (Means ± SEM, n = 3, *p < 0.05 vs. WT, ANOVA with Tukey’s multiple comparisons).
Next, we sought to determine if these results could be recapitulated in vivo. To this end, we used MIA PaCa-2 tumor xenografts in an athymic nude mouse model. Following formation of tumor xenografts to approximately 5 mm in diameter, mice were treated with either 1 M NaCl or P-AscH- 4 g/kg b.i.d. for 6 d. After treatment, mice were euthanized, tumors excised, and processed for western blots. DUOX1 and DUOX2 expression was significantly enhanced in vivo by P-AscH- treatment compared to controls (Figure 6A–B). To determine if the H2O2 produced during exposure to P-AscH− would lead to increased DUOX protein in vivo, H1299-CAT tumor xenografts were treated with twice daily P-AscH− 4 g/kg b.i.d. for 6 d. Mice received either sucrose (2%) or sucrose + doxycycline (2 mg/mL) in the drinking water. After treatment, mice were euthanized, and tumors excised for western blots to probe for immunoreactive DUOX1 and DUOX2 expression. Figure 6C demonstrates an increase in both DUOX1 and DUOX2 in the H1299T-CAT cell line consistent with what was seen in the MIA PaCa-2 cell line (Figure 6A). In mice that received doxycycline in the drinking water, catalase was significantly induced (Figure 6C) and there was little to no expression of DUOX1 or DUOX2 in tumor xenografts when compared to mice that were treated with P-AscH− alone. In a separate series of in vivo experiments, catalase was shown to be effectively overexpressed in tumors grown in mice receiving doxycycline and the induced catalase levels were maintained throughout the experimental time course and varying tumor volumes (Supplemental Figure 5B–D). These data are consistent with the in vitro results in Figures 4G–H demonstrating increases in DUOX expression that are reversed with catalase. Combined, these results suggest that treatment with P-AscH− results in non-mitochondrial responses, including enhanced DUOX1 and DUOX2 expression both in vitro and in vivo. These changes may contribute to cytotoxicity, potentially through a feed-forward mechanism of H2O2 production.
Figure 6. P-AscH− treatment increases DUOX1 and DUOX2 expression in vivo.
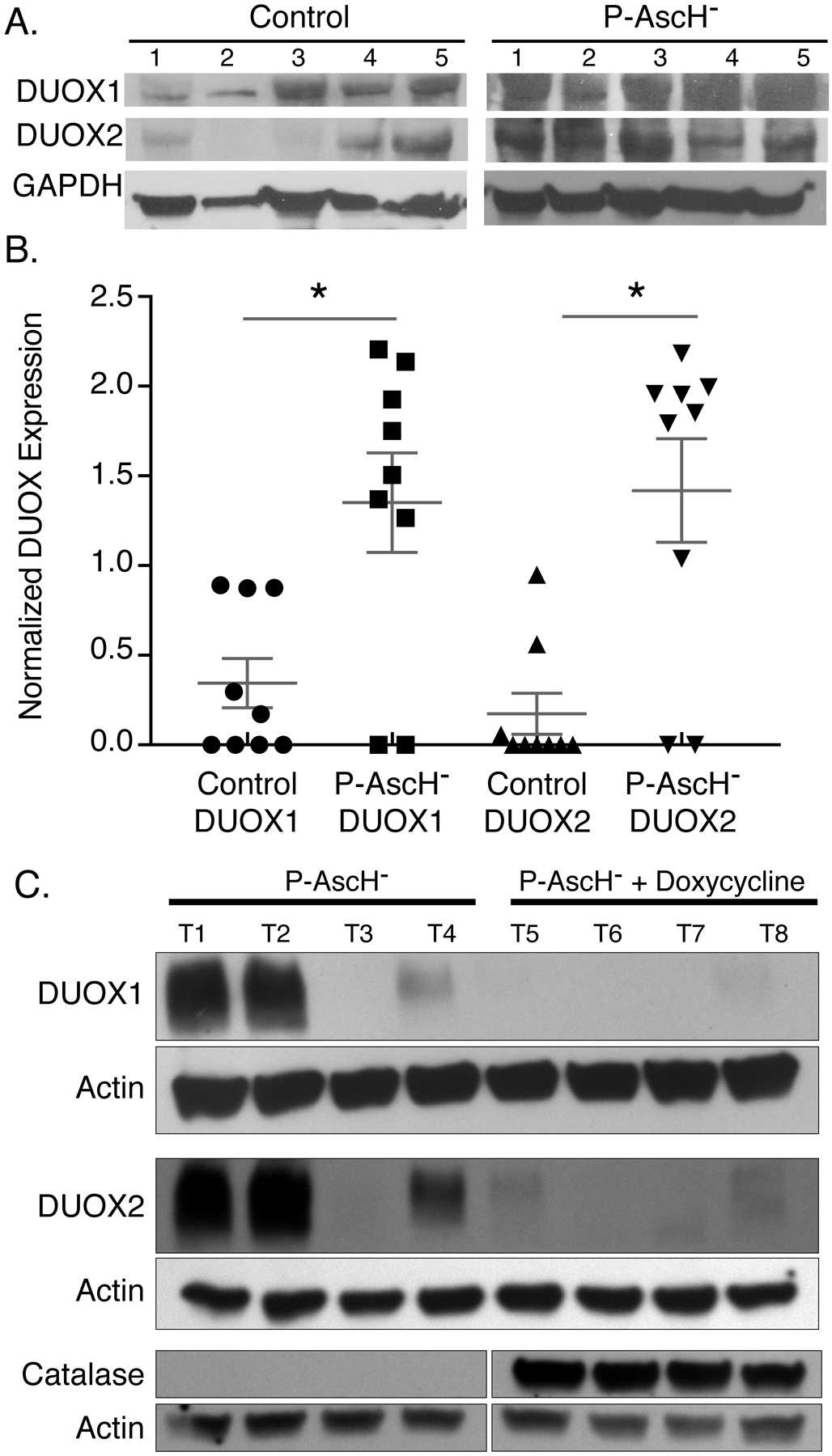
A. Athymic nude mice with heterotopic MIA PaCa-2 xenografts were treated for 5 days with 4 g/kg I.P. ascorbate b.i.d. Western blotting was performed to analyze protein levels of DUOXs in xenograft tumors that were excised from control and P-AscH− treated mice. DUOX1 and DUOX2 immunoreactive protein is increased in tumors of P-AscH− treated mice compared to saline treated mice (Representative blots shown).
B. Quantification of densitometric evaluation of western blots (Means ± SEM, n = 9, * p < 0.05 vs. controls, 2-tailed student’s t-test).
C. Athymic nude mice with heterotopic H1299T-CAT xenografts were treated for 5 d with 4 g/kg I.P. ascorbate b.i.d. All mice were given 1% sucrose in their drinking water ± doxycycline (2 mg/mL). Tumors were excised, and western blotting was performed to analyze protein levels of DUOXs and catalase in xenografts. DUOX1 and DUOX2 immunoreactive protein is increased in tumors of P-AscH− treated mice compared to mice in the P-AscH− + doxycycline group. Catalase immunoreactive protein is increased in tumors from the P-AscH− + doxycycline group compared to the P-AscH− group. (Representative blots shown, n = 4 mice per group).
DUOX expression can be induced by H2O2 but is silenced in several forms of cancer (23, 24). To determine the levels of DUOX expression in PDAC, patient-derived PDAC tumors and matched non-tumor adjacent samples were obtained for immunofluorescent analysis of DUOX1 and DUOX2. Hematoxylin and eosin staining demonstrated morphological differences between the normal adjacent tissue and the PDAC (Figure 7A). In addition, there were significant decreases in DUOX1 and DUOX2 expression in PDAC compared to normal adjacent pancreatic tissue (Figure 7B–C, respectively). To further confirm that DUOX expression is increased in normal pancreas, immunofluorescent staining on normal pancreas samples from non-tumor related pancreatic resections were compared to separate PDAC resections. Consistent with our previous observations, we observed that DUOX1 expression is greater in normal pancreas compared to PDAC (Supplemental Figure 6).
Figure 7. Resections for PDAC demonstrate differential expression of DUOX1 and DUOX2.
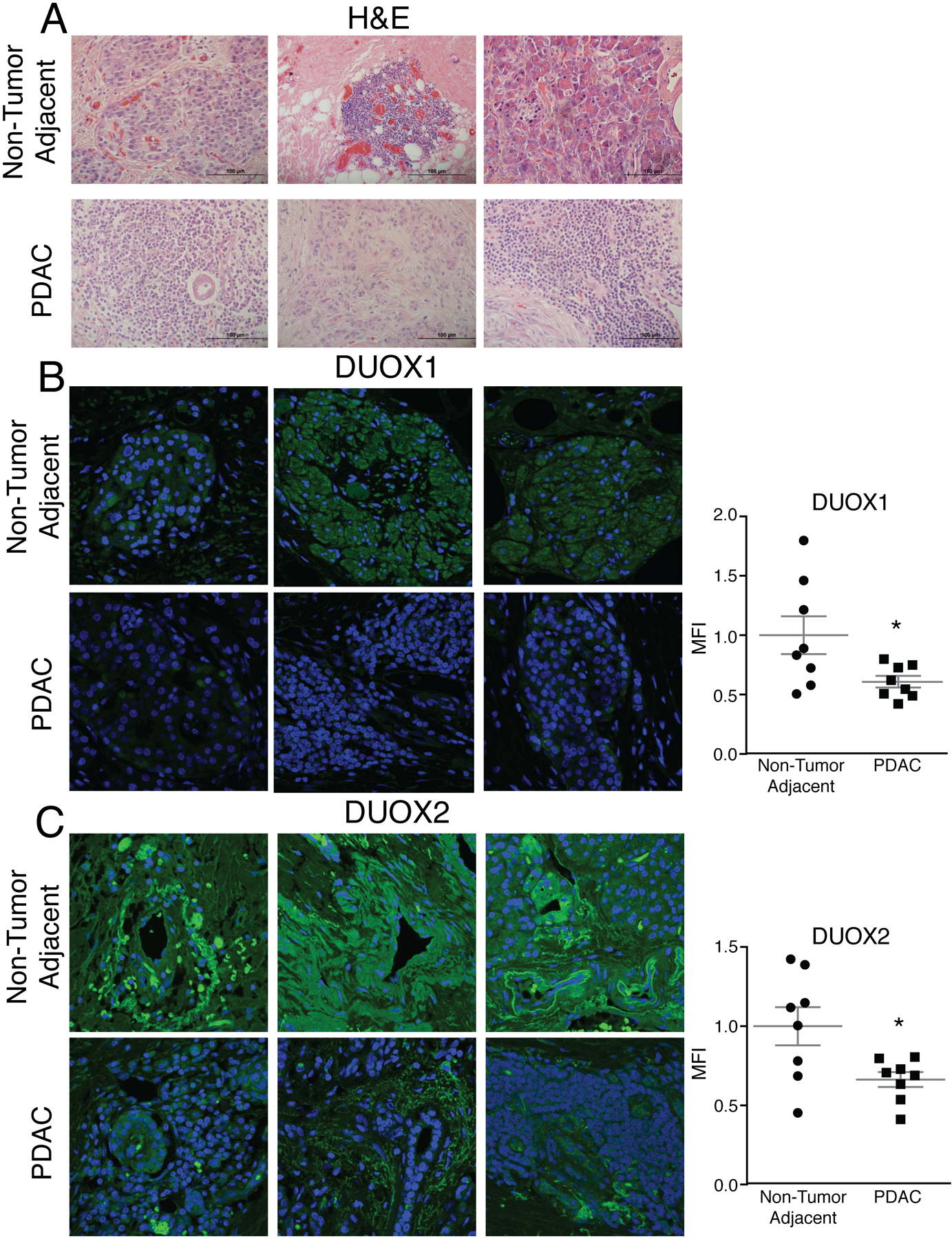
A. Hematoxylin and eosin staining performed on matched normal adjacent pancreas and PDAC from pancreaticoduodenectomy specimens demonstrated changes in morphology between normal adjacent and PDAC. Representative images shown.
B-C. DUOX1 and DUOX2 immunofluorescence staining was performed on non-tumor adjacent pancreas and PDAC in matched patient samples. Samples were visualized using a Zeiss Confocal Microscope 40x oil objective. Results show decreased DUOX1 and DUOX2 fluorescence in PDAC and increased DUOX1 and DUOX2 fluorescence in normal adjacent pancreas. Green staining is DUOX1/DUOX2 and blue staining is for nuclear Toppiosmerase-3. Representative images shown along with quantification demonstrating normalized mean fluorescence intensity (MFI) (Means ± SEM, n = 8, *p < 0.05 vs. adjacent normal, 2-tailed student’s t-test).
DISCUSSION
These results indicate that P-AscH− results in toxicity through mechanisms beyond those involving the oxidation of P-AscH− to produce H2O2 (9, 11). P-AscH−-treated cells are viable at 48 h, but are unable to form clones. We observed that these viable cells appear to have increased ROS generation, which may be due in part to a shift in metabolism from glycolysis to mitochondrial oxidative phosphorylation. ROS can be generated from exogenous sources including radiation, as well as endogenous sources including the mitochondrial electron transport system and oxygen-metabolizing enzymes such as Dual Oxidases 1 and 2. P-AscH− readily undergoes pH-dependent oxidation producing H2O2. In vitro, the mechanism for the toxicity of high-dose ascorbate centers on the generation of H2O2 by its oxidation (9–11). The oxidation of ascorbate, when at millimolar levels, will generate considerable amounts of H2O2, especially in the presence of catalytic metals (8, 42). Because cells rapidly and efficiently remove extracellular H2O2 via peroxiporins (30, 43), observed toxicities or cell killing induced by ascorbate is a function of the activities of intracellular antioxidant enzymes involved in the removal of H2O2, such as catalase, glutathione peroxidase, and the peroxiredoxins.
Numerous studies have demonstrated that P-AscH− induces selective oxidative stress and cytotoxicity in cancer cells vs. normal cells (10–13). Concomitant administration of the catalase inhibitor amino-1,2,4-triazole has been shown to enhance ascorbate toxicity to Ehrlich ascites cells in vitro (44). In these studies, the addition of various forms of both extracellular and intracellular catalase, which remove H2O2, completely reverse P-AscH−-induced cytotoxicity in a variety of cancer cell lines, while normal cells are resistant. Others have shown that adding glutathione and pyruvate directly to cell culture medium provides protection for the consequences of P-AscH− (45). However, greatly underappreciated is that these substances react directly with the H2O2 (30). Using conservative rate constants for the reaction of these substances with H2O2 (at pH 7.4 kGSH = 0.3 M−1 s−1; kpyruvate = 2 M−1 s−1) and a reasonable estimates for the cellular rate constant for the removal of extra cellular H2O2 (kcell = 3 × 10−12 s−1 L cell−1), millimolar levels of these substances will remove >95% of the H2O2 formed compared to the cells (<5%), thereby protecting the cells from extracellular H2O2 (30). These results provide evidence supporting a fundamental role for H2O2 in P-AscH−-induced toxicity. Although these reports support the hypothesis that H2O2 generated transiently upon exposure to P-AscH− could control cytotoxicity, the initial production of H2O2 may not be entirely responsible for the long-term biological effects of P-AscH− treatment. In our current study, we demonstrate that there is an increase in ROS measured by DCFH fluorescence, 48 h after treatment P-AscH− (Figure 1A), which may be attributed to induction of the DUOX enzymes as seen in Figures 3 and 4.
While most of the NOX family members produce superoxide, the DUOX members were originally discovered to produce H2O2 in the thyroid. This H2O2 production is required for thyroid hormone biosynthesis and since their initial discovery in the thyroid DUOX expression has been reported in other tissue types including the pancreas (19, 21) . This correlates with our present study where we demonstrated that DUOX 1 and 2 are expressed in normal pancreas and the expression is significantly diminished in PDAC (Figure 7, Supplementary Figure 6). In addition, DUOX production of H2O2 in the PDAC cells clearly contribute to P-AscH− toxicity since inhibiting the DUOX enzymes with diphenyleneiodonium, an inhibitor of flavin-containing proteins, partially reversed the P-AscH− induced decrease in clonogenic survival as seen in Figure 6A. Likewise, later induction of catalase using the doxycycline-inducible catalase vector also partially reversed the P-AscH− induced decrease in clonogenic survival (Figure 6E, F) and in orthotopic tumors as seen in Figure 6.
Looking at the bioenergetic data of Figure 2 quantitatively, there is an increase from approximately 100 to 150 amol cell−1 s−1, ≈50%, in basal OCR after exposure to P-AscH−. Although the fraction of mitochondrial OCR that leaks to form superoxide is unknown (46), if at most it is a generous 0.1%, then the rate of production of superoxide will be ≤≈ 0.001 × 150 amol cell−1 s−1 or 0.15 amol cell−1 s−1. Assuming superoxide dismutase captures most of this superoxide, then the rate of mitochondrial production of H2O2 will be ≤≈0.07 amol cell−1 s−1. Non-mitochondrial OCR increased from 17 to 30 amol cell−1 s−1, a change of 13 amol cell−1 s−1. This is about 9% of basal OCR (150 amol cell−1 s−1). Thus, this non-mitochondrial OCR from DUOX enzymes is consistent with the potential of DUOX enzymes to introduce an intracellular flux of H2O2.
NOX family members have been associated with various forms of cancer and they have been implicated in the development and progression of cancer. However, the expression and function of NOXs in the development and progression of cancer are not well understood. Studies in lung and hepatocellular cancer suggest that decreased DUOX expression results in a more aggressive phenotype and facilitates epithelial to mesenchymal transition (EMT) (21, 24). Restoration of DUOX1 results in growth arrest and inhibition of colony formation through G2/M cell cycle arrest and increased ROS generation in liver cancer (24). Reports in lung cancer indicate that DUOX silencing induces characteristic EMT alterations in cell phenotype while restoration of DUOX via overexpression causes cancer cells to revert to a more epithelial morphology and decrease ability to grow colonies (23). Combined, these studies suggest that the loss of DUOX leads to the initiation of EMT leading to invasive and metastatic disease. The function of DUOX expression in PDAC is unknown. Our current study indicates that DUOX is expressed in the pancreatic ductal epithelial cells, but expression is significantly decreased in PDAC cell lines as seen in Figure 3A–D. In addition, treatment with P-AscH− increased DUOX expression and led to inhibition of clonogenic survival, consistent with previous studies demonstrating a less aggressive phenotype with restoration of DUOX expression (23, 24). This suggests that loss of DUOX expression may give cancer cells an advantage, which allows them to evade apoptosis or other modes of cell death.
In the presence of catalytic metal ions, P-AscH− exerts a pro-oxidant effect (8). Ascorbate is an excellent one-electron reducing agent that can reduce ferric (Fe3+) to ferrous (Fe2+) iron, while being oxidized to ascorbate radical. Depending on coordination environment, Fe2+ can readily react with O2, reducing it to superoxide radical, which in turn dismutes to H2O2 and O2. This initial, large amount of H2O2 generated by P-AscH− is the main mediator of P-AscH−-induced toxicity in cancer cells as seen in Figures 1F, G (9–13). The current study demonstrates that late ROS accumulation enhances P-AscH− induced toxicity in PDAC cells and DUOXs among other members of redox enzymes have a role in this process. Pharmacological levels of ascorbate, which can only be achieved parenterally, create an environment of oxidative distress in PDAC. Furthermore, P-AscH− has been shown to be safe and tolerable in phase I clinical trials with suggestions of efficacy (1, 47, 48). Phase II trials are currently underway to further advance P-AscH− as a potential standard of care adjuvant treatment.
Supplementary Material
ACKNOWLEDGEMENTS
Supported by NIH grants P01CA217797, P30CA086862, CA184051, CA148062, CA169046, and CA086862. The authors would like to acknowledge use of the University of Iowa Central Microscopy Research Facility and the ESR Facility, the Holden Comprehensive Cancer Center and the Carver College of Medicine.
Supported by NIH grants P01CA217797, P30CA086862, CA184051, CA182804, CA148062, CA169046, GM007337, and F30CA213817
Footnotes
The authors declare no potential conflicts of interest.
REFERENCES
- 1.Welsh JL, Wagner BA, van’t Erve TJ, Zehr PS, Berg DJ, Halfdanarson TR, et al. Pharmacological ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic cancer (PACMAN): results from a phase I clinical trial. Cancer chemotherapy and pharmacology. 2013;71(3):765–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ma Y, Chapman J, Levine M, Polireddy K, Drisko J, Chen Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Science translational medicine. 2014;6(222):222ra18. [DOI] [PubMed] [Google Scholar]
- 3.Padayatty SJ, Sun H, Wang Y, Riordan HD, Hewitt SM, Katz A, et al. Vitamin C pharmacokinetics: implications for oral and intravenous use. Annals of internal medicine. 2004;140(7):533–7. [DOI] [PubMed] [Google Scholar]
- 4.Schoenfeld JD, Sibenaller ZA, Mapuskar KA, Wagner BA, Cramer-Morales KL, Furqan M, et al. O2(−) and H2O2-Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer cell. 2017;32(2):268. [DOI] [PubMed] [Google Scholar]
- 5.Polireddy K, Dong R, Reed G, Yu J, Chen P, Williamson S, et al. High Dose Parenteral Ascorbate Inhibited Pancreatic Cancer Growth and Metastasis: Mechanisms and a Phase I/IIa study. Scientific reports. 2017;7(1):17188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bodeker KL, Allen BG, Smith MC, Monga V, Sandhu S. First-in-human phase 1 clinical trial of pharmacological ascorbate combined with radiation and temozolomide for newly diagnosed glioblastoma. 2019. [DOI] [PMC free article] [PubMed]
- 7.New Reference Values for Vitamin C Intake. Annals of nutrition & metabolism. 2015;67(1):13–20. [DOI] [PubMed] [Google Scholar]
- 8.Du J, Cullen JJ, Buettner GR. Ascorbic acid: chemistry, biology and the treatment of cancer. Biochimica et biophysica acta. 2012;1826(2):443–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du J, Martin SM, Levine M, Wagner BA, Buettner GR, Wang SH, et al. Mechanisms of ascorbate-induced cytotoxicity in pancreatic cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16(2):509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Q, Espey MG, Krishna MC, Mitchell JB, Corpe CP, Buettner GR, et al. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver hydrogen peroxide to tissues. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(38):13604–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Q, Espey MG, Sun AY, Lee JH, Krishna MC, Shacter E, et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(21):8749–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rawal M, Schroeder SR, Wagner BA, Cushing CM, Welsh JL, Button AM, et al. Manganoporphyrins increase ascorbate-induced cytotoxicity by enhancing H2O2 generation. Cancer research. 2013;73(16):5232–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du J, Cieslak JA 3rd, Welsh JL, Sibenaller ZA, Allen BG, Wagner BA, et al. Pharmacological Ascorbate Radiosensitizes Pancreatic Cancer. Cancer research. 2015;75(16):3314–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Panday A, Sahoo MK, Osorio D, Batra S. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cellular & molecular immunology. 2015;12(1):5–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meitzler JL, Hinde S, Banfi B, Nauseef WM, Ortiz de Montellano PR. Conserved cysteine residues provide a protein-protein interaction surface in dual oxidase (DUOX) proteins. The Journal of biological chemistry. 2013;288(10):7147–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ameziane-El-Hassani R, Morand S, Boucher JL, Frapart YM, Apostolou D, Agnandji D, et al. Dual oxidase-2 has an intrinsic Ca2+-dependent H2O2-generating activity. The Journal of biological chemistry. 2005;280(34):30046–54. [DOI] [PubMed] [Google Scholar]
- 17.Dupuy C, Virion A, Ohayon R, Kaniewski J, Deme D, Pommier J. Mechanism of hydrogen peroxide formation catalyzed by NADPH oxidase in thyroid plasma membrane. The Journal of biological chemistry. 1991;266(6):3739–43. [PubMed] [Google Scholar]
- 18.Ameziane-El-Hassani R, Talbot M, de Souza Dos Santos MC, Al Ghuzlan A, Hartl D, Bidart JM, et al. NADPH oxidase DUOX1 promotes long-term persistence of oxidative stress after an exposure to irradiation. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(16):5051–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Deken X, Corvilain B, Dumont JE, Miot F. Roles of DUOX-mediated hydrogen peroxide in metabolism, host defense, and signaling. Antioxidants & redox signaling. 2014;20(17):2776–93. [DOI] [PubMed] [Google Scholar]
- 20.Harper RW, Xu C, Eiserich JP, Chen Y, Kao CY, Thai P, et al. Differential regulation of dual NADPH oxidases/peroxidases, Duox1 and Duox2, by Th1 and Th2 cytokines in respiratory tract epithelium. FEBS letters. 2005;579(21):4911–7. [DOI] [PubMed] [Google Scholar]
- 21.Luxen S, Belinsky SA, Knaus UG. Silencing of DUOX NADPH oxidases by promoter hypermethylation in lung cancer. Cancer research. 2008;68(4):1037–45. [DOI] [PubMed] [Google Scholar]
- 22.Edens WA, Sharling L, Cheng G, Shapira R, Kinkade JM, Lee T, et al. Tyrosine cross-linking of extracellular matrix is catalyzed by Duox, a multidomain oxidase/peroxidase with homology to the phagocyte oxidase subunit gp91phox. The Journal of cell biology. 2001;154(4):879–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Little AC, Sham D, Hristova M, Danyal K, Heppner DE, Bauer RA, et al. DUOX1 silencing in lung cancer promotes EMT, cancer stem cell characteristics and invasive properties. Oncogenesis. 2016;5(10):e261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ling Q, Shi W, Huang C, Zheng J, Cheng Q, Yu K, et al. Epigenetic silencing of dual oxidase 1 by promoter hypermethylation in human hepatocellular carcinoma. American journal of cancer research. 2014;4(5):508–17. [PMC free article] [PubMed] [Google Scholar]
- 25.Brandt KE, Falls KC, Schoenfeld JD, Rodman SN, Gu Z, Zhan F, et al. Augmentation of intracellular iron using iron sucrose enhances the toxicity of pharmacological ascorbate in colon cancer cells. Redox biology. 2018;14:82–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heer CD, Davis AB, Riffe DB, Wagner BA, Falls KC, Allen BG, et al. Superoxide Dismutase Mimetic GC4419 Enhances the Oxidation of Pharmacological Ascorbate and Its Anticancer Effects in an H2O2-Dependent Manner. Antioxidants 2018;7(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roy I, Zimmerman NP, Mackinnon AC, Tsai S, Evans DB, Dwinell MB. CXCL12 chemokine expression suppresses human pancreatic cancer growth and metastasis. PloS one. 2014;9(3):e90400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim MP, Evans DB, Wang H, Abbruzzese JL, Fleming JB, Gallick GE. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nature protocols. 2009;4(11):1670–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buettner GR. In the absence of catalytic metals ascorbate does not autoxidize at pH 7: ascorbate as a test for catalytic metals. Journal of biochemical and biophysical methods. 1988;16(1):27–40. [DOI] [PubMed] [Google Scholar]
- 30.Doskey CM, Buranasudja V, Wagner BA, Wilkes JG, Du J, Cullen JJ, et al. Tumor cells have decreased ability to metabolize H2O2: Implications for pharmacological ascorbate in cancer therapy. Redox Biol. 2016;10:274–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doskey CM, van ‘t Erve TJ, Wagner BA, Buettner GR. Moles of a Substance per Cell Is a Highly Informative Dosing Metric in Cell Culture. PloS one. 2015;10(7):e0132572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wagner BA, Venkataraman S, Buettner GR. The rate of oxygen utilization by cells. Free radical biology & medicine. 2011;51(3):700–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beers RF Jr., Sizer IW. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. The Journal of biological chemistry. 1952;195(1):133–40. [PubMed] [Google Scholar]
- 34.Aebi H Catalase in vitro. Methods in enzymology. 1984;105:121–6. [DOI] [PubMed] [Google Scholar]
- 35.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. The Journal of biological chemistry. 1951;193(1):265–75. [PubMed] [Google Scholar]
- 36.Gao Z, Sarsour EH, Kalen AL, Li L, Kumar MG, Goswami PC. Late ROS accumulation and radiosensitivity in SOD1-overexpressing human glioma cells. Free radical biology & medicine. 2008;45(11):1501–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fisher CJ, Goswami PC. Mitochondria-targeted antioxidant enzyme activity regulates radioresistance in human pancreatic cancer cells. Cancer biology & therapy. 2008;7(8):1271–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kalen AL, Sarsour EH, Venkataraman S, Goswami PC. Mn-superoxide dismutase overexpression enhances G2 accumulation and radioresistance in human oral squamous carcinoma cells. Antioxidants & redox signaling. 2006;8(7–8):1273–81. [DOI] [PubMed] [Google Scholar]
- 39.Eckers JC, Kalen AL, Xiao W, Sarsour EH, Goswami PC. Selenoprotein P inhibits radiation-induced late reactive oxygen species accumulation and normal cell injury. International journal of radiation oncology, biology, physics. 2013;87(3):619–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vislisel JM, Schafer FQ, Buettner GR. A simple and sensitive assay for ascorbate using a plate reader. Analytical biochemistry. 2007;365(1):31–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clemens DL, Duryee MJ, Sarmiento C, Chiou A, McGowan JD, Hunter CD, et al. Novel Antioxidant Properties of Doxycycline. Int J Mol Sci. 2018;19(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buettner GR, Jurkiewicz BA. Catalytic metals, ascorbate and free radicals: combinations to avoid. Radiation research. 1996;145(5):532–41. [PubMed] [Google Scholar]
- 43.Erudaitius D, Huang A, Kazmi S, Buettner GR, Rodgers VG. Peroxiporin Expression Is an Important Factor for Cancer Cell Susceptibility to Therapeutic H2O2: Implications for Pharmacological Ascorbate Therapy. 2017;12(1):e0170442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benade L, Howard T, Burk D. Synergistic killing of Ehrlich ascites carcinoma cells by ascorbate and 3-amino-1,2,4,-triazole. Oncology. 1969;23(1):33–43. [DOI] [PubMed] [Google Scholar]
- 45.Yun J, Mullarky E, Lu C, Bosch KN, Kavalier A, Rivera K, et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science (New York, NY). 2015;350(6266):1391–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murphy MP. How mitochondria produce reactive oxygen species. The Biochemical journal. 2009;417(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alexander MS, Wilkes JG, Schroeder SR, Buettner GR. Pharmacologic Ascorbate Reduces Radiation-Induced Normal Tissue Toxicity and Enhances Tumor Radiosensitization in Pancreatic Cancer. 2018;78(24):6838–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Monti DA, Mitchell E, Bazzan AJ, Littman S, Zabrecky G, Yeo CJ, et al. Phase I evaluation of intravenous ascorbic acid in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. PloS one. 2012;7(1):e29794. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.