

Abstract
Virus-specific memory T cell populations demonstrate plasticity in antigen recognition and in their ability to accommodate new memory T cell populations. The degeneracy of T cell antigen recognition and the flexibility of diverse antigen-specific repertoires allow the host to respond to a multitude of pathogens while accommodating these numerous large memory pools in a finite immune system. These cross-reactive memory T cells can be employed in immune responses and mediate protective immunity, but they can also induce life-threatening immunopathology or impede transplantation tolerance and graft survival. Here we discuss examples of altered viral pathogenesis occurring as a consequence of heterologous T cell immunity and propose models for the maintenance of a dynamic pool of memory cells.
Abbreviations: LCMV, lymphocytic choriomeningitis virus; PV, Pichinde virus; VV, vaccinia virus; RSV, respiratory syncytical virus; EBV, Epstein Barr virus; MCMV, murine cytomegalovirus; VSV, vesicular stomatitis virus; TdT, terminal deoxynucleotidyl transferase; NP, nucleoprotein; APC, antigen presenting cells; CFSE, 5-(and -6-)-carboxyfluorescein diacetate succinimidyl ester
Keywords: Memory T cell, Heterologous immunity, Cross-reactive, Allo-reactive, Memory attrition
1. Introduction
Naïve CD8 T cells expand and differentiate into cytokine-producing effector cells on encountering antigen under conditions of effective co-stimulation. After the peak of the immune response and clearance of the antigen, this programmed event is followed by a decrease of CD8 T cells by apoptosis, resulting in the generation of an antigen-specific CD8 memory T cell pool. The significance and characteristics of memory CD8 T cells in viral infections have been extensively described elsewhere [1], [2], [3], [4], [5], [6], [7]. Their main functions include providing protection on re-exposure to a pathogen and preventing the re-emergence of low-grade persistent viruses. They are able to achieve this, because their high frequency and elevated activation state leads to their rapid response to an antigenic challenge [8], [9], [10].
These antigen-specific memory T cells are accommodated into a finite immune system which already contains a large pool of pre-existing memory T cell populations. In fact, memory cells are part of a continually evolving interactive network, as immune responses to each new pathogen alter the frequencies, distributions and activities of memory T cells deposited from previous responses. This network is composed of a diverse repertoire of T cells, which compete with each other for niches in an ever-changing environment. This review will focus on how the immune system generates this dynamic network of T cell populations with a resilient plasticity to combat infections.
2. Degeneracy of T cell recognition
Although memory T cells are highly antigen specific, they maintain a diverse T cell receptor (TCR) repertoire [11], [12] and can be degenerate in the number of antigens that they can recognize. TCR diversity and degeneracy are potentially important features to prevent pathogen escape by mutation. The TCR of a CD8 T cell discriminates peptides of usually 8–10 amino acids that are embedded in MHC-I molecules [13]. Data from studies using crystal structures of peptide–MHC complexes suggest that only a few contact residues (often side chains of the amino acids) of the embedded peptide interact with the TCR (reviewed in [14], [15]). A TCR can tolerate certain amino acid substitutions in the peptide sequence and still become activated. For example, amino acid substitutions for a HLA B8-restricted Epstein Barr virus (EBV) peptide at positions 1, 2, and 8 were tolerated, while substitutions at positions 4, 6, and 7 were crucial for CTL recognition [16].
“Molecular mimicry”—where a different peptide retains sites that are necessary for interaction with the TCR [17]—is one of several paths to cross-reactive T cell responses. It is also possible that different regions of the same TCR can interact with two different targets [18], [19] and that a T cell can express two different TCR, due to incomplete allelic exclusion of the TCR alpha chain [20]. Taken together, these mechanisms make cross-reactivity very difficult to predict and, as we postulate, a fairly common event. Reports of pathogen-specific memory CD8 T cells recognizing cross-reactive epitopes on different proteins of the same pathogen, or proteins from closely related or totally unrelated pathogens are increasing and summarized in a recent review [4]. Mathematical calculations by Mason suggest that a single TCR should be able to react against 106 different nonamer peptides [21]. This feature may be valuable to the host, considering the large number of potential pathogenic antigens to which one is exposed over a lifetime. The host needs mainly to be concerned with cross-reactive T cells that are self-reactive, as occurs during conditions of autoimmunity, but a number of mechanisms serve to preclude that phenomenon. If T cells are as highly cross-reactive as estimated [21], one would expect that cross-reactivity could compensate for a situation with a limited TCR repertoire and still allow a normal immune response. Studies in mice deficient for the enzyme terminal deoxynucleotidyl transferase (TdT) have estimated that the size of their alpha/beta TCR repertoire is only 5–10% of that calculated for wild-type mice [22] due to an impaired TCR CDR3 diversification. When challenged with LCMV or Sendai virus, these mice demonstrated a surprisingly normal immune response and recovery from infection [23]. Since T cells from TdT-deficient mice are reported to be more ‘promiscuous’, i.e. cross-reactive, than those in wild-type mice [24], T cells with a highly cross-reactive profile may have compensated for the less diverse TCR repertoire. It is noteworthy that mice having very limited TCR diversity as a consequence of a transgenic TCR are also capable of responding to many antigens and resisting viral infections [25], [26]. T cell cross-reactivity might be even more pervasive and functionally relevant if the cross-reactive antigen is stimulating memory T cells, which are easier to activate than naïve cells and may be present in the host at a high frequency [27].
The first part of this review will discuss the topic of heterologous immunity [28] and the existing evidence that established memory T cell responses to a previously encountered pathogen can have a major impact on T cell immunodominance, protective immunity, and immunopathology during a subsequent infection with an unrelated pathogen (Fig. 1 ). These also may influence allo-specific T cell activity prior to and following transplantation (Fig. 2 ).
Fig. 1.
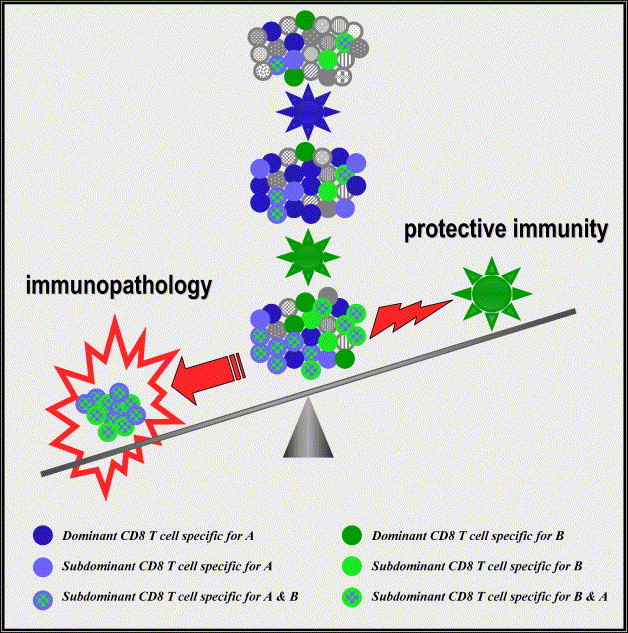
Consequence of modulation of the T cell repertoire during heterologous viral infections: The dots represent CD8 T cell populations that have different specificities. The naïve T cell repertoire encounters virus A (blue) and virus A-specific CD8 T cells expand and maintain into memory. If this memory T cell pool is exposed to an unrelated virus B (green), memory CD8 T cells that are cross-reactive will preferentially expand and dominate the response whereas non cross-reactive CD8 T cells specific for the first virus decrease in number. The now dominant cross-reactive CD8 T cells can participate in immunopathology and partial protective immunity.
Fig. 2.
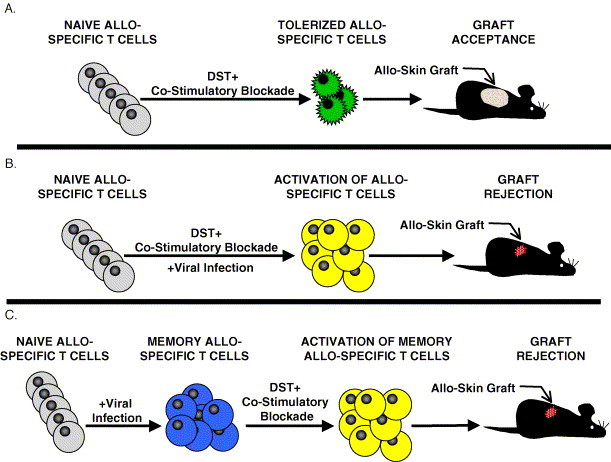
Viral infections interfere with the induction of tolerance against allo-antigens. (A) Naïve allo-specific T cells (colored grey) are deleted and become functionally anergic (colored green) after receiving a transfusion of donor allogeneic cells (donor-specific transfusion or DST) with a co-stimulatory blockade. Mice that have been tolerized to the allo-antigens can then accept allogeneic skin grafts. (B) A viral infection during the co-stimulatory blockade protocol will activate allo-specific T cells (colored yellow) and interfere with the establishment of tolerance, resulting in skin graft rejection. (C) Naïve mice infected with viruses generate memory allo-specific T cells (colored blue) that are refractory to the induction of tolerance using co-stimulatory blockade protocols and reject allogeneic skin grafts.
3. Cross-reactivity modulates the memory T cell repertoire
A CD8 T cell memory pool created after one virus infection demonstrates a distinct hierarchy of epitope-specific responses in a naïve host. While some viral epitopes are dominant and stimulate strong T cell responses, others are subdominant and stimulate weaker or barely detectable T cell responses [29]. Immunodominance is regulated by various parameters, including the efficiency of processing and presentation of the peptide, the affinity between peptide and the MHC-I, the availability of T cells with TCR that recognize the peptide–MHC complex, and the competition between T cells for domains on the antigen presenting cell [30]. When a memory CD8 T cell pool encounters a cross-reactive antigen, their high frequency and activation state gives them an advantage over naïve T cells, and can lead to a preferential expansion of the cross-reactive CD8 T cell population (Fig. 1). This cross-reactive expansion can alter the hierarchy of T cell responses. This was seen in sequential heterologous virus infections with two distantly related arenaviruses, lymphocytic choriomeningitis virus (LCMV) and Pichinde virus (PV). These viruses encode epitopes in the nucleoprotein (NP205) with 6 of 8 amino acids in common. This is normally a subdominant epitope for either virus in a naïve host. However, due to a selective expansion of NP205-specific cross-reactive memory CD8 T cells, this NP205-specific T cell response became dominant when LCMV-immune mice were infected with PV, or when PV-immune mice were infected with LCMV [31]. These data support the concept that expansions of cross-reactive T cell populations substantially contribute to the immune-hierarchies of T cell responses. This might help explain some of the variability in immunodominant hierarchies observed in human viral infections, where the host has been exposed to numerous infections throughout life. For instance, patients with identical haplotypes infected with HIV or hepatitis C show high variability in their T cell immune-hierarchies [32], [33]. Interestingly, cross-reactive CD8 T cell responses to other pathogens have been documented for both of these viruses [34], [35].
4. Heterologous immunity and the fine balance between protection and pathology during viral infections
Memory T cells cross-reactive with a heterologous virus can provide partial protective immunity and, in experimental models, can provide the difference between life and death in the infected individual [36], [37]. For example, LCMV-immune mice control PV infection, presumably due to the cross-reactivity of T cells specific for the subdominant NP205 epitope [31]. LCMV-immune mice also manifest strong protective immunity against infections with the large DNA poxvirus, vaccinia virus (VV) compared to naïve mice [36], [37]. In a respiratory model of infection this heterologous immunity prevented mortality to an otherwise lethal dose of VV [37]. Adoptive transfer studies demonstrated that CD4 and CD8 T cells from LCMV-immune mice were required to transfer protective immunity to naïve mice challenged with PV or VV [36]. Selective expansion of LCMV-specific memory CD8 T cells on VV infection suggested the possibility of cross-reactive CD8 T cell responses between these two viruses [37], [38]. In fact, we have identified VV-specific CD8 T cell epitopes in mice by searching for sequence similarity to a potentially cross-reactive LCMV epitope [4], (Selin and Cornberg, unpublished data).
Not unexpectedly, heterologous immunity is not as protective as homologous immunity, which elicits high affinity T cell and antibody responses against a previously encountered pathogen. A lower affinity cross-reactive T cell response might be more adept at stimulating immunopathology than conferring protective immunity. Due to the competition between cells that gives rise to immunodominance, low affinity cross-reactive memory cells might prevent the development of more effective high-affinity T cells responding to the normally immunodominant epitopes. This is reminiscent of the phenomenon of “original antigenic sin”, which was first described for B cell responses against influenza virus subtypes [39], but which has also been documented for CD8 T cells in viral infections. LCMV-immune mice infected with variant strains of LCMV that encode altered T cell epitopes generated wild-type LCMV-specific CD8 T cells that were less effective against the variant strain, leading to impaired viral clearance [40]. Another example of antigenic sin was observed in dengue virus infection. It has been shown that infection with a dengue virus serotype generated CD8 T cells with a higher affinity to a second and presumably previously encountered dengue virus serotype, suggesting that cross-reactive memory CD8 T cells had preferentially expanded over T cells more specific to the serotype causing infection [41]. This type of low affinity cross-reactive T cell may not be good for the host, as a more severe disease outcome, including hemorrhagic fever has been observed in subsequent infections with different dengue virus serotypes. These data suggest that cross-reactive T cells can make the difference in surviving a subsequent infection with an unrelated pathogen, but suboptimal clearance of the pathogen can also potentiate the ongoing immune response, leading to immunopathology (Fig. 1).
Another example of the fine balance that exists within heterologous immunity was observed during VV infection of LCMV-immune mice, where, once again, the price for partial protective T cell immunity was altered immunopathology. After an intraperitoneal inoculation, LCMV-immune mice challenged with VV developed necrosis of visceral fat, termed acute fatty necrosis or panniculitis [36]. This form of panniculitis is analogous to human erythema nodosum. In a respiratory infection model, reduced mortality of LCMV-immune mice infected with VV was accompanied by altered lung pathology. Their lungs were significantly infiltrated by LCMV-specific T cells, which contributed to obstruction of bronchioles by fibrin and inflammatory cells (bronchiolitis obliterans). In humans, erythema nodosum and bronchiolitis obliterans are of unknown etiology but can be seen in some viral and bacterial infections and are also associated with autoimmune diseases [42], [43]. The development of bronchiolitis obliterans in lung allografts is associated with transplant rejection [43].
Manifestations of heterologous immunity may therefore explain human disease pathogenesis variations thought previously to be due to genetic variations, the physiological condition of the patient, or the inoculation route and dose. The individual's history of infections may shape the T cell memory pool in ways that contribute to this variability. For example, the difference between a clinical and an asymptomatic acute EBV infection is the magnitude of the T-cell response, not the viral load [44]. Symptomatic disease is less likely in young children than in teenagers and young adults, who have a longer history of infections and presumably a more complex pool of memory cells than in young children [45]. In addition, autoimmunity has been associated with viral infections [46], and it is likely that an individual's history of virus infections and the unique composition of the cross-reactive memory T cell pool may either initiate or reactivate T cells with auto-immune potential.
An individual's history of infections and the numerous variations in infection sequences make heterologous immunity complicated and hard to predict. For example, it is not universal that LCMV protects against a subsequent infection, as immunity to LCMV enhanced respiratory syncytial virus (RSV) titers. Similarly, although immunity to influenza virus results in decreased titers of VV, it causes an enhancement in LCMV and murine cytomegalovirus (MCMV) titers on C57BL/6 mice [47].
Heterologous immunity does not just contribute to T cell numbers and population dynamics but can also alter T cell function and immune deviation. Immunity to previously encountered viruses can alter the cytokine response to subsequently encountered viruses. Mice immune to LCMV and challenged with VV make much higher levels of IFNγ and lower levels of IL-6 than naïve mice challenged with VV. Much of the enhanced IFNγ production comes directly from the LCMV epitope-specific T cells activated by the VV infection [37], [47]. A history of influenza infection protects mice from severe disease caused by infection with live RSV challenge after an RSV G-protein vaccination [48]. This study demonstrated that immunity to influenza shifted the usual TH2 response into a TH1 response and prevented severe eosinophilic infiltrates in the lung. Changes in TH1/TH2 responses due to infections or vaccinations early in childhood might explain why some children are more likely to develop allergies than others. Several studies reported beneficial effects of early infections in childhood on the development of asthma; this may be the consequence of an early skewing of the memory pool towards a TH1 phenotype that would inhibit TH2-based allergic responses (reviewed in [49]).
5. Cross-reactivity between virus-specific T cells and allo-antigens
The degenerate nature of antigen recognition by the TCR is exemplified by the ability of T cells to recognize non-self or allogeneic MHC molecules. Allo-specific T cells represent a substantial population of the naïve T cell repertoire, with between 0.1 and 10% of naïve T cells within an individual host being reactive with any unique allogeneic haplotype, as measured by limiting dilution analysis and quantitative measurements of cells responding to allo-antigens in vivo [50], [51], [52], [53]. This high frequency of allo-specific T cells allows a host to efficiently generate effector allo-specific T cells following exposure to allo-antigens in the form of blood transfusions, prior transplants, or pregnancy [54], [55]. Surprisingly, memory allo-specific T cells are detectable in patients that have never been obviously exposed to allo-antigens [56], suggesting that these T cells were activated by cross-reactive environmental antigens. Early studies in murine models demonstrated that CD8 T cells activated during an acute LCMV infection of H2b mice recognized both H2k and H2d allogeneic target cells in a standard cytotoxicity assay [57]. Cytotoxic, allo-specific CD8 T cells were also generated following infection with PV, VV, and MCMV [58]. Allo-specific CTL activity was also detected in humans infected with EBV during acute infectious mononucleosis [59], [60]. These results showed that allo-specific CD8 T cell responses are activated after viral infections, but did not address whether the allo-specific responses were activated by an antigen-dependent cross-reactive mechanism or by a non-specific bystander mechanism mediated by the massive production of cytokines after infection.
We now know that many virus-specific CD8 T cells generated in response to viral infections directly cross-react with allo-antigens [57], [61], [62], [63], [64], [65]. Short-term CD8 T cell clones derived from LCMV-infected mice were shown to lyse both virus-infected syngeneic targets and uninfected allogeneic target cells [64]. This cross-reactivity between LCMV-specific T cells and allo-antigens is consistent with earlier studies in which long-term murine CD8 T cell clones specific for either influenza virus or vesicular stomatitis virus (VSV), and human clones specific for EBV recognized allogeneic cells in cytotoxicity assays [61], [62], [66], [67]. In addition, allo-reactive T cells specific for H2Kk could be shown to lyse syngeneic H2b-target cells infected with influenza virus [68]. Cross-reactivity between virus-specific CD8 T cells and allo-antigens has been visualized directly from mice acutely infected with LCMV [63]. LCMV-specific CD8 T cells isolated from H2b mice produced IFNγ following a short in vitro stimulation with either H2d- or H2k-expressing cell lines. This cross-reactivity was broad-based, as a portion of T cells specific for each of the four LCMV-epitopes examined (GP33, NP205, GP276, and NP396) cross-reacted with H2d, yet it was distinctive, as different proportions of each epitope-specific population recognized H2d or H2k targets [63]. Together, these findings demonstrate the promiscuity of allo-antigen recognition in a variety of viral systems.
The studies described above show that cross-reactivity is an important mechanism for the activation of allo-specific CD8 T cells after a viral infection, but they do not exclude the possibility that allo-specific T cells would also be activated in a bystander manner through TCR-independent stimuli [69]. There is evidence to suggest that the cytokine-rich environment that develops after a viral infection is sufficient to activate T cells to homeostatically divide with no increase in overall number [58], [70]. However, during either a viral infection or an immune response against allogeneic cells, bystander activation of T cells as measured by the acquisition of cytotoxicity or by proliferation with an increase in number is not readily apparent [25], [52]. To evaluate the contribution of bystander activation to allo-specific responses generated after a viral infection, HY-TCR-transgenic (tg) mice were utilized [25]. TCR-tg HY-specific CD8 T cells have an extremely limited T cell repertoire, with 30–50% of the CD8 T cells expressing both transgenic α- and β-chains and the remaining cells expressing the transgenic β-chain with an endogenously expressed α-chain [71]. T cells derived from HY-TCR-tg mice respond equally well against both H2d- and H2k-expressing cells in mixed lymphocyte cultures, but an LCMV infection preferentially activated only H2k-specific CTL in those mice [25]. The stimulation of H2k-specific T cells and not H2d-specific T cells after the LCMV infections is in contrast to the activation of both populations in wild type mice and suggests that cross-reactivity and not a bystander effect drive the allo-specific responses. In addition, bystander-induced proliferation is not evident in an experimental model of a graft versus host response [52]. In this model, H2b-splenocytes were adoptively transferred into an allogeneic H2b/d host, which should be incapable of rejecting the donor cells. This allows the host-reactive T cells within the transferred population to proliferate extensively in the allogeneic environment, in a manner similar to a graft versus host reaction. However, co-adoptively transferred splenocytes that were completely syngeneic (H2b/d) with the host did not divide despite the vigorous anti-host response [52].
Regardless of the mechanisms driving the activation of allo-specific T cells after a viral infection, this phenomenon has potentially serious implications for the field of transplantation. Virus infections have long been known to precipitate the rejection of allografts, and the immunosuppressive drug treatment given to graft recipients makes the patient more susceptible to viral infections [72], [73]. One promising new strategy to increase the success of allogeneic transplantation and avoid the use of immunosuppressive drugs involves the specific suppression of allo-specific T cell responses by blocking co-stimulatory signaling during activation [74]. The activation of naïve T cells requires two signals: the first is engagement of the TCR by the appropriate MHC–peptide complex, and the second is the co-stimulation provided though pathways such as CD28–CD80/CD86 and CD40–CD40L(CD154) [75], [76]. TCR engagement in the absence of this second co-stimulatory signal will result in a non-functional or anergic population of antigen-specific T cells which may ultimately be lost by apoptosis [77]. Pretreatment of a transplant recipient with antibodies to co-stimulatory molecules such as CD40L and with an infusion of donor cells will specifically anergize T cells reactive to donor antigens and tolerize the recipient against donor tissues [78]. The mechanism mediating the induction of tolerance by co-stimulatory blockade appears to involve multiple pathways including the physical deletion of T cells, the functional anergization of T cells, and the immunoregulation of allo-specific T cells by regulatory T cells [79].
Co-stimulatory blockade effectively induces tolerance in the unperturbed T cell repertoire of a naïve host [78], [80], [81], [82], [83], but in “real-world” conditions a transplant recipient is continuously exposed to pathogens. Viral infections, in addition to precipitating graft rejection in traditionally treated transplant recipients [72], [73], may also impede the maintenance of tolerance against allo-antigens. In murine models, acute infections with viruses such as LCMV and PV shortly after transplantation (1–15 days post-transplant) and persistent LCMV infection interfered with the early maintenance of tolerance to allo-antigens and resulted in rapid rejection of donor skin grafts (Fig. 2) [69], [84], [85]. This virus-induced rejection was at least partially dependent on CD8 T cells, as depletion of CD8 cells from the recipient mice significantly delayed rejection. Interestingly, the viral interference in the induction of tolerance was transient, as recipient mice infected with LCMV 50 days post-transplant did not reject tissue grafts [84]. It is possible that multiple factors account for the inability of an LCMV infection at 50 days post-transplant to promote rejection, including the permanent deletion of the allo-specific T cells [86], the development of regulatory T cells that maintain peripheral tolerance to allo-antigens [87], or a combination of deletion and immunoregulation. The mechanism by which viral infections block this early maintenance of tolerance has not been completely elucidated, but the inability of poly(I:C), VV, or MCMV to interfere with the co-stimulatory blockade at this time point suggests that the production of cytokines alone is not sufficient [88].
Naïve T cells require co-stimulatory signaling to mount effective responses against an antigenic challenge, but memory T cells are less reliant on co-stimulation to become activated [89], [90]. The activation of memory T cells in the absence of co-stimulation suggests that the presence of allo-specific memory T cells may diminish the effectiveness of co-stimulatory blockade to induce tolerance against allogeneic tissues. Both CD8 and CD4 allo-specific memory T cells generated by exposure to allo-antigens are refractory to the induction of tolerance using the standard co-stimulatory blockades and efficiently reject allogeneic transplant [91], [92], [93]. Of interest is that prior exposure to allo-antigens is not the only mechanism for the generation of allo-specific memory T cells, as infection with viruses or bacteria also elicit allo-specific memory T cells [63], [64], [91], [94]. Allo-specific memory CD4 T cells generated by Leishmania major infection [94] and allo-specific memory CD8 T cells generated by LCMV infection [63] are refractory to the tolerance induced by co-stimulatory blockade and efficiently reject allogeneic skin grafts (Fig. 2). In addition, mice that have been sequentially infected with unrelated viruses have higher frequencies of allo-specific memory T cells and display heightened resistance to the co-stimulatory blockade [91].
A closer examination of allo-specific CD8 T cell populations in mice sequentially infected with heterologous viruses reveals that, while the frequency of T cells specific for a unique haplotype may increase after a secondary viral infection, there is a corresponding decline in the number T cells specific for alternative haplotypes, relative to the levels after the primary infection [63]. The selective alterations in the frequencies of T cells specific for distinct haplotypes after multiple viral infections suggests that T cell cross-reactivity contributes to these changes in repertoire and also implies that the activation of allo-specific T cells after an infection is an antigen-driven phenomenon. Whether the rejection of foreign transplants can be directly attributed to cross-reactive memory T cell responses remains to be resolved. One alternative explanation for the virus-induced rejection of transplants is that helper factors produced during a viral infection enhance the proliferation of non-cross-reactive allo-specific T cells that are stimulated by third party allo-antigens derived from the graft. Overall, these findings indicate that the real world environment presents a number of unique challenges for the use of co-stimulatory blockade protocols, and these issues will need to be solved to achieve success in human transplant patients.
6. Attrition and accommodation of virus-specific memory CD8 T cells
The peripheral CD8 T cell pool consists of naïve and memory subsets, often distinguished by the expression of CD44, and the homeostasis of the two compartments appears to be regulated independently of each other [95]. This independence, in fact, seems appropriate, as the purpose of these two compartments is quite distinct. While the homeostasis of the naïve T cell pool ensures continuous generation and maintenance of a diverse T cell repertoire, the priority of the memory CD8 T cell compartment is the preservation of a more focused antigen-experienced pool of memory cells specific to previously encountered pathogens.
Once generated, antigen-specific memory CD8 T cells are maintained in a remarkably stable manner [96], [97]. The stable nature of memory CD8 T cells is, in part, attributable to the self-renewal capacity of memory CD8 T cells, supported, in part, by a member of the γc cytokine family, IL-15 [98], [99]. In the absence of IL-15, virus-specific memory CD8 T cells decay slowly over time, due to the lack of “basal level homeostatic proliferation” [98].
The stability of memory CD8 T cells is disrupted by subsequent heterologous viral or bacterial infections [96], [100], [101], [102]. With the exception of cross-reactive CD8 T cells, which have been shown to be spared or to even increase in number, the majority of non-cross-reactive memory CD8 T cells reduce their frequencies and numbers upon successive heterologous virus or bacterial infections. This memory CD8 T cell attrition is not just limited to the secondary lymphoid tissues but is also applicable to T cell populations residing in non-lymphoid peripheral organs [103]. This attrition phenomenon intuitively provides solutions to dilemmas of the immune system as an information storage system. Instead of ever increasing the size of the secondary lymphoid tissues to harbor all the memory T cells generated from numerous previously encountered pathogens, the immune system sacrifices old memory T cells to accommodate new ones.
How and when this attrition of virus-specific memory CD8 T cell takes place has been the subject of several studies [103], [104], [105], [106]. We have proposed two hypothetical models, passive competition and active deletion, for this attrition. The passive competition model suggests that during heterologous virus infection, pre-existing memory CD8 T cells inevitably have to compete with newly generated pathogen-specific CD8 T cells for structural niches and/or survival factors such as cytokines, and eventually some memory CD8 T cells would lose this battle and die off. Virus-specific memory CD8 T cells also have to compete with T cells expanding homeostatically in response to lymphopenic environments, which are often induced during virus infections. The passive competition scenario may be exemplified in a study by Chapdelanie et al., in which an enhanced Mycobacterium bovis-specific CD8 T cell response, due to over-expression of IL-15, resulted in more profound attrition of pre-existing Listeria monocytogenes-specific CD8 T cells [104]. The active deletion model suggests that pre-existing memory CD8 T cells undergo a bystander apoptosis and get killed off by cytotoxic factor(s) released during the early phase of new virus infection. Thus far, our experimental data with mice suggest that the majority of virus-specific memory CD8 T cell attrition is a consequence of the active deletion model (Fig. 3 ) [103], [106].
Fig. 3.
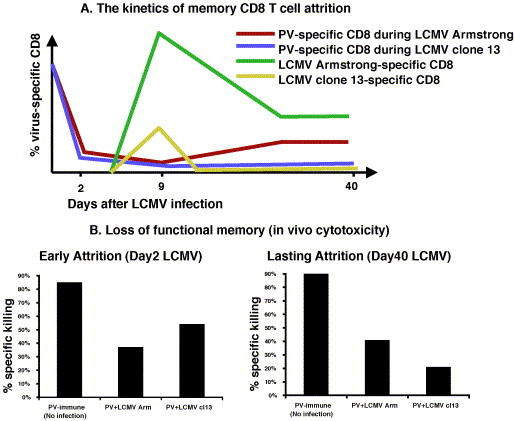
(A) The kinetics of non cross-reactive PV-specific memory CD8 T cell attrition demonstrates an ongoing loss of memory during heterologous persistent LCMV infection as compared to acute LCMV infection. PV-immune mice were infected with either acute LCMV Armstrong (red) or persistent clone13 (blue) infection. The frequency of dominant PV NP38-specific CD8 T cells in PBL was followed using IFNγ assay. The kinetics of attrition was overlaid with the kinetics of LCMV-specific CD8 T cell to show early depletion of PV-specific CD8 T cells. (B) Loss of functional PV-specific memory at the early and the late phase of LCMV infection as measured by an in vivo cytotoxicity assay. Using PV NP38-coated targets in PV-immune, PV-immune acutely infected with LCMV Armstrong or PV-immune mice infected with LCMV clone 13, which establishes a persistent infection, there is evidence for loss of functional memory both at the early phase (day 2) and the late phase (day 40) of LCMV infection with either LCMV strain [103].
Deletion of memory CD8 T cells occurs very early during acute virus infections [103], [105], [106] and is associated with generalized lymphopenia observed in many acute bacterial [107] and viral infections, including influenza [108], measles [109], Ebola [110], varicella zoster [111], E55+ murine leukemia [112], LCMV [103], [106] and SARS corona viruses [113]. Of note is that this lymphopenia preferentially affects some lymphocyte subsets more than others, and CD44hi memory CD8 T cells are particularly affected, more so than CD44lo (naïve) CD8 T cells [103], [105], [106]. This deletion of memory CD8 T cells occurs throughout the body and is mediated by apoptotic cell death, as many of the CD44hi CD8 T cells exhibit signs of apoptosis such as annexin-V reactivity and terminal deoxynucleotydyl transferase-mediated dUTP nick-end labelling (TUNEL) staining [106], [112]. This early lymphopenic phase parallels the peak of type I IFN [106], a signature cytokine produced by components of the innate immune system, including subsets of dendritic cells (DCs) activated via the Toll-like receptors [114]. It is unclear whether this lymphopenia is required for a subsequent optimal expansion of T cells specific to the pathogen. However, it is noteworthy that old mice, in contrast to young mice, were resistant to E55+ murine leukemia virus-induced lymphopenia, and the authors speculated that the lack of the lymphopenia was associated with subsequently generated lower CTL and antibody responses and suboptimal protective immunity [112]. The lymphopenia and memory CD8 T cell loss can be recapitulated by directly injecting mice with poly(I:C), a potent type I IFN inducer, and mice deficient in type I IFN receptors were resistant to the poly(I:C)-or LCMV-induced lymphopenia [106]. Type I IFN may not be the only factor, as downstream cell death pathways remain to be clarified. This apoptosis does not appear to require the conventional death pathways involving Fas, FasL, Bcl-2, Bcl-XL, perforin or IFNγ [106], [107].
During virus infection, the lymphopenic phase is soon followed by the proliferation of new virus-specific T cells and possibly some homeostatic proliferation of other cells to fill the available space, although the extent of homeostatic proliferation under those conditions is unknown [105]. The lymphopenia can be recapitulated without expansion of new antigen-specific T cells by inoculating the mice with poly(I:C) in the absence of an antigenic challenge [106]. Adoptive transfer studies have revealed that the poly(I:C) treatment reduced the number and the frequency (<70%) of CFSE-labeled virus-specific CD8 T cells as early as 18 h after treatment and prior to any signs of cell division [105]. This poly(I:C)-induced lymphopenia is immediately followed by an IL-15-mediated homeostatic division (recovery phase), and then that ensues a substantial but not complete recovery of memory CD8 T cells by 5 days after treatment [105]. In contrast, however, during a virus infection, which also poses an antigenic challenge, the initial loss of memory CD8 T cells is poorly restored [103]. The reduced number of pre-existing memory CD8 T cells that survived the initial lymphopenia now have to compete with proliferating T cells specific to the newly encountered pathogen (Fig. 3). Thus, some aspects of the competition model appear to be operating during the recovery phase and contribute to overall attrition.
The phenomenon of memory CD8 T cell attrition has been previously investigated almost exclusively in acute infections, where the pathogen gets cleared from host and the host immune system eventually returns to steady-state homeostasis. Some pathogens can evade or compromise host immune surveillance and establish persistent infections, which may disrupt the homeostasis of the host immune system by posing a continuous antigenic challenge [103], [115], [116]. Heightened levels of type I IFN, the key cytokine of attrition, can be continuously detected during persistent infections, and this continuous presence of type I IFN during chronic virus infection may intensify the level of attrition. We have examined the extent and the pattern of attrition of previously acquired memory CD8 T cells during acute or chronic virus infection by using the LCMV model system in the mouse [103]. The Armstrong strain of LCMV induces an acute sterilizing infection associated with a strong IFN and T cell responses and complete resolution of infection. In contrast, its highly disseminating clone 13 variant causes a long-term persistent infection in part by anergizing or deleting virus-specific T cells [117]. PV-, VSV-, or VV-immune mice were infected with either a moderate dose of LCMV-Armstrong to establish an acute infection or with a high dose of LCMV-clone 13, to establish a persistent infection. A far more profound level of attrition was detected among non cross-reactive virus-specific memory CD8 T cells during the persistent infection (Fig. 3). Furthermore, when CFSE-labeled memory CD8 T cells were transferred into persistently infected hosts, a continuous loss of donor memory CD8 T cells was detected, even long after the initial infection, and the loss was not due to the lack of IL-15-dependent basal level homeostatic proliferation [103]. Chronic virus infections are often associated with generalized immunosuppression, although the mechanisms are poorly understood [118], [119]. Chronic LCMV infection has been associated with aberrant antigen presenting cells (APCs) [116], [120], and with structural defects in the secondary lymphoid tissues [120]. In addition, our findings suggest that severe attrition of previously acquired memory CD8 T cells during chronic virus infection may also contribute to immunosuppression.
Repeated attrition of virus-specific memory CD8 T cells upon successive infections and vaccinations may eventually lead to impairment of protective immunity against the original virus. Supporting this notion, Smith et al. demonstrated compromised host protective immunity against tumor challenge due to reduced frequency of tumor-specific CD8 T cells caused by a heterologous bacterial infection [101]. Clearance of CSFE-labeled viral epitope-presenting target cells is also greatly impaired under these conditions as shown in in vivo cytotoxicity assays [103]. Furthermore, several reports have shown that the actual number and frequency of memory CD8 T cells are critical for protective immunity against viral or bacterial infections [121], [122].
Despite many demonstrations of infection-induced memory T cell attrition in the mouse system, it is presently not clear how important this is in human T cell homeostasis and protective immunity. Although virus-induced lymphopenia has been frequently reported in many human infections [109], [110], [111], [113], the link between the lymphopenia and memory T cell attrition in humans remains to be further clarified. Attrition in humans may not be as dramatic as in the mouse system, as the physically larger immune system may provide a buffering apparatus to the IFN-induced deletion. At the moment, there are no data to support or refute this idea. Cytokine-induced T cell attrition in humans may be more pronounced during persistent virus infections, including HIV. These issues remain to be further clarified and better understanding of these issues will eventually lead to more effective vaccine development.
7. Computer-generated modeling of passive versus active attrition
Some experimental data relating to active (cytokine secretion at the beginning of the response) and passive (competition for space and resources) mechanisms of T cell attrition and discussion above are shown in Fig. 3, but we have also asked whether computer-generated modeling can add insights into the significance of the active vs. passive attrition process. We have simulated “as if” situations by mathematical modeling, utilizing systems to consider the impact of both cross-reactive and non-cross-reactive T cells between an infectious agent which established the memory pool and a new infectious agent driving an active T cell response. We have used IMMSIM, a well-established model of the immune system, based on cellular automata and governed by probabilistic events. The program is available at http://www.immsin.org and can be downloaded for research and educational use. References to the model's past applications are found in [123], [124], [125], [126], [127].
The IMMSIM body consists of epithelial cells in a grid of 240 discrete “interaction sites” where 2500 cells of each type (TH1, TH2, Tc, B, macrophages) of the immune system are distributed, meet with each other and with antigens, and mount cellular and humoral responses whenever a virus infects the target epithelial cells. The specific interactions are governed by affinity and chance (via computer generated random numbers, RNs). Different RNs result in responses different in repertoire and events, simulating the variability within individual mice in vivo.
Each run begins with a virus inoculum at time step (TS) 1; the response builds a memory pool consisting of several clones. At TS 500, a second virus, different but having a cross-reactive epitope, is injected, and the changes in the memory cell clones can be followed (Fig. 4 ). To study repeated secondary responses starting with the same primary memory (a simulation of the in vivo adoptive transfer technique), we have used the same RN for the primary and allowed a different RN for each secondary infection.
Fig. 4.
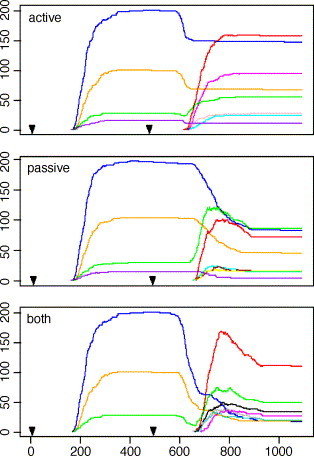
Mathematical modeling of memory T cell attrition by active, passive or both mechanisms: Tc memory clone dynamics during the primary and secondary response to cross-reacting viruses, under three models of attrition simulated by IMMSIM. Abscissa: time steps (TS) 0–1100. Ordinate: Tc memory clone cells numbers. Arrows at TS = 1 and TS = 500 indicate the time of inoculum of V1 (70 particles) and V2 (120 particles), respectively. V1 clearance is complete at TS ≅ 200 and V2 clearance at TS ≅ 700. Attrition is enacted in active by simulating IFN type 1 secretion by target epithelial cells at the time of the infection. The lymphokine diffuses locally and then causes the death of memory cells by contact, thus creating space for V2 specific cell growth. There is an early decrease in all memory clones; the cross-reacting ones show a typical “dip” before the secondary rise. Passive attrition is simulated by a drastic decrease of lifespan of memory cells when they reach a density threshold. It starts significantly later than active and its effect is felt for several hundreds TS, as it causes a characteristic decrease of the secondary clones after the secondary peak. The combination of both modes in both features early plunging of primary memory, a dip before the secondary growth and the after peak fall. The experiments shown also illustrate the significant findings of stimulation of new responding clones in active and both, and of favoring high affinity clones by active and both. This is epitomized by the relative behaviour of the green (low affinity for V1, medium affinity for V2) and the red (no reaction with V1, high affinity for V2) clones in the three attrition modes. By contrast note that clones blue and yellow have high affinity for V1 but do not bind V2, clones purple, lightblue and black do not bind V1 and have high affinity or medium affinity (black) for V2.
By enabling or disabling the attrition functions that we modeled in the IMMSIM code, responses with active only, passive only or both active and passive attrition are obtained. Fig. 4 shows a set of three experiments where CD8 memory clones develop in these three conditions. These runs are representative of a large number of repeated experiments, which have been compared and studied quantitatively. While a number of observations and facts about the runs are listed in the figure legend, the following findings were statistically tested and constitute the conclusions of this analysis.
After the challenge by the second virus, active and both active + passive attrition thwart the growth of cross-reactive clones, allowing an opportunity for new clones to start off. The number of the latter is significantly larger with both than with passive only (p < 0.01, n = 180, Student's t-test).
The pattern of dominance is influenced by the mode of attrition: extreme dominance of single clones is favored by active attrition, while co-dominance is more frequent with passive, which exerts a flattening effect. Furthermore, the dispersion measured among the highest three clones of each run of the secondary response is significantly wider in active and both than in passive (p < 0.01, n = 180, Student's t-test).
Affinity edge is important to establish clonal dominance, but in these experiments is only one of the factors; the success of the highest fit is favored by active and both but not by passive attrition (Fig. 4).
Experimental studies in mice, as discussed in the preceding section, have strongly implicated cytokine-dependent active attrition as being a major player in memory T cell loss. It is noteworthy that this mathematical model predicts that this active attrition thwarts the expansion of cross-reactive T cells. This active attrition may therefore serve to temper the immunodomination that could be imposed by low affinity but high frequency cross-reactive T cells on the development of more effective high affinity clones specific to well presented antigens.
8. Conclusion
The immune system has evolved such that highly diverse antigen-specific memory TCR repertoires to multiple pathogens over a lifetime can be accommodated within a confined space. With each new infection memory T cells are preferentially deleted by active cytokine-dependent mechanisms and possibly also by passive competition. However, memory T cells specific to previously encountered pathogens but cross-reactive with the newly encountered pathogen are preferentially maintained or expanded, such that the T cell repertoire specific to the previous pathogen becomes permanently altered. These stimulated cross-reactive memory T cells play a role in heterologous immunity by mediating effector function, by modulating the T cell immunodominance hierarchy, and by influencing the balance between protective immunity and immunopathology. Virus-specific T cells cross-reactive with allo-antigens can alter the memory allo-specific T cell pool and may modulate allograft survival and transplantation tolerance.
Acknowledgements
L.K.S., R.M.W. and F.C. are supported by the United States National Institutes of Health grants AI46578, AI49320, AR35506, AI054455 and DK32520. The contents of this article are solely the responsibility of the authors and do not represent the official views of NIH.
References
- 1.Kaech S.M., Ahmed R. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat Immunol. 2001;2:415–422. doi: 10.1038/87720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Stipdonk M.J., Lemmens E.E., Schoenberger S.P. Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat Immunol. 2001;2:423–429. doi: 10.1038/87730. [DOI] [PubMed] [Google Scholar]
- 3.Welsh R.M., Bahl K., Wang X.Z. Apoptosis and loss of virus-specific CD8(+) T-cell memory. Curr Opin Immunol. 2004;16:271–276. doi: 10.1016/j.coi.2004.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Welsh R.M., Selin L.K., Szomolanyi-Tsuda E. Immunological memory to viral infections. Annu Rev Immunol. 2004;22:711–743. doi: 10.1146/annurev.immunol.22.012703.104527. [DOI] [PubMed] [Google Scholar]
- 5.Ahmed R., Gray D. Immunological memory and protective immunity: understanding their relation. Science. 1996;272:54–60. doi: 10.1126/science.272.5258.54. [DOI] [PubMed] [Google Scholar]
- 6.Zinkernagel R.M. On differences between immunity and immunological memory. Curr Opin Immunol. 2002;14:523–536. doi: 10.1016/s0952-7915(02)00367-9. [DOI] [PubMed] [Google Scholar]
- 7.Dutton R.W., Bradley L.M., Swain S.L. T cell memory. Annu Rev Immunol. 1998;16:201–223. doi: 10.1146/annurev.immunol.16.1.201. [DOI] [PubMed] [Google Scholar]
- 8.Razvi E.S., Welsh R.M., McFarland H.I. In vivo state of antiviral CTL precursors. Characterization of a cycling cell population containing CTL precursors in immune mice. J Immunol. 1995;154:620–632. [PubMed] [Google Scholar]
- 9.Selin L.K., Welsh R.M. Cytolytically active memory CTL present in lymphocytic choriomeningitis virus-immune mice after clearance of virus infection. J Immunol. 1997;158:5366–5373. [PubMed] [Google Scholar]
- 10.Veiga-Fernandes H., Walter U., Bourgeois C., McLean A., Rocha B. Response of naive and memory CD8+ T cells to antigen stimulation in vivo. Nat Immunol. 2000;1:47–53. doi: 10.1038/76907. [DOI] [PubMed] [Google Scholar]
- 11.Pewe L.L., Netland J.M., Heard S.B., Perlman S. Very diverse CD8 T cell clonotypic responses after virus infections. J Immunol. 2004;172:3151–3156. doi: 10.4049/jimmunol.172.5.3151. [DOI] [PubMed] [Google Scholar]
- 12.Naumov Y.N., Naumova E.N., Hogan K.T., Selin L.K., Gorski J. A fractal clonotype distribution in the CD8+ memory T cell repertoire could optimize potential for immune responses. J Immunol. 2003;170:3994–4001. doi: 10.4049/jimmunol.170.8.3994. [DOI] [PubMed] [Google Scholar]
- 13.Falk K., Rotzschke O., Stevanovic S., Jung G., Rammensee H.G. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature. 1991;351:290–296. doi: 10.1038/351290a0. [DOI] [PubMed] [Google Scholar]
- 14.Bjorkman P.J. MHC restriction in three dimensions: a view of T cell receptor/ligand interactions. Cell. 1997;89:167–170. doi: 10.1016/s0092-8674(00)80195-6. [DOI] [PubMed] [Google Scholar]
- 15.Rudolph M.G., Wilson I.A. The specificity of TCR/pMHC interaction. Curr Opin Immunol. 2002;14:52–65. doi: 10.1016/s0952-7915(01)00298-9. [DOI] [PubMed] [Google Scholar]
- 16.Burrows S.R. Cross-reactive recognition of viral and self-peptides by a “public” T cell receptor expressed by cytotoxic T lymphocytes expanded in multiple unrelated individuals. Immunol Lett. 2004;93:7–9. doi: 10.1016/j.imlet.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 17.Barnett L.A., Fujinami R.S. Molecular mimicry: a mechanism for autoimmune injury. FASEB J. 1992;6:840–844. doi: 10.1096/fasebj.6.3.1740233. [DOI] [PubMed] [Google Scholar]
- 18.Daniel C., Horvath S., Allen P.M. A basis for alloreactivity: MHC helical residues broaden peptide recognition by the TCR. Immunity. 1998;8:543–552. doi: 10.1016/s1074-7613(00)80559-2. [DOI] [PubMed] [Google Scholar]
- 19.Speir J.A., Garcia K.C., Brunmark A., Degano M., Peterson P.A., Teyton L. Structural basis of 2C TCR allorecognition of H-2Ld peptide complexes. Immunity. 1998;8:553–562. doi: 10.1016/s1074-7613(00)80560-9. [DOI] [PubMed] [Google Scholar]
- 20.Alam S.M., Gascoigne N.R. Posttranslational regulation of TCR Valpha allelic exclusion during T cell differentiation. J Immunol. 1998;160:3883–3890. [PubMed] [Google Scholar]
- 21.Mason D. A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol Today. 1998;19:395–404. doi: 10.1016/s0167-5699(98)01299-7. [DOI] [PubMed] [Google Scholar]
- 22.Cabaniols J.P., Fazilleau N., Casrouge A., Kourilsky P., Kanellopoulos J.M. Most alpha/beta T cell receptor diversity is due to terminal deoxynucleotidyl transferase. J Exp Med. 2001;194:1385–1390. doi: 10.1084/jem.194.9.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilfillan S., Bachmann M., Trembleau S., Adorini L., Kalinke U., Zinkernagel R. Efficient immune responses in mice lacking N-region diversity. Eur J Immunol. 1995;25:3115–3122. doi: 10.1002/eji.1830251119. [DOI] [PubMed] [Google Scholar]
- 24.Gavin M.A., Bevan M.J. Increased peptide promiscuity provides a rationale for the lack of N regions in the neonatal T cell repertoire. Immunity. 1995;3:793–800. doi: 10.1016/1074-7613(95)90068-3. [DOI] [PubMed] [Google Scholar]
- 25.Zarozinski C.C., Welsh R.M. Minimal bystander activation of CD8 T cells during the virus-induced polyclonal T cell response. J Exp Med. 1997;185:1629–1639. doi: 10.1084/jem.185.9.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daly K., Nguyen P., Woodland D.L., Blackman M.A. Immunodominance of major histocompatibility complex class I-restricted influenza virus epitopes can be influenced by the T-cell receptor repertoire. J Virol. 1995;69:7416–7422. doi: 10.1128/jvi.69.12.7416-7422.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Selin L.K., Nahill S.R., Welsh R.M. Cross-reactivities in memory cytotoxic T lymphocyte recognition of heterologous viruses. J Exp Med. 1994;179:1933–1943. doi: 10.1084/jem.179.6.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Welsh R.M., Selin L.K. No one is naive: the significance of heterologous T-cell immunity. Nat Rev Immunol. 2002;2:417–426. doi: 10.1038/nri820. [DOI] [PubMed] [Google Scholar]
- 29.Chen W., Anton L.C., Bennink J.R., Yewdell J.W. Dissecting the multifactorial causes of immunodominance in class I-restricted T cell responses to viruses. Immunity. 2000;12:83–93. doi: 10.1016/s1074-7613(00)80161-2. [DOI] [PubMed] [Google Scholar]
- 30.Yewdell J.W., Bennink J.R. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu Rev Immunol. 1999;17:51–88. doi: 10.1146/annurev.immunol.17.1.51. [DOI] [PubMed] [Google Scholar]
- 31.Brehm M.A., Pinto A.K., Daniels K.A., Schneck J.P., Welsh R.M., Selin L.K. T cell immunodominance and maintenance of memory regulated by unexpectedly cross-reactive pathogens. Nat Immunol. 2002;3:627–634. doi: 10.1038/ni806. [DOI] [PubMed] [Google Scholar]
- 32.Betts M.R., Casazza J.P., Patterson B.A., Waldrop S., Trigona W., Fu T.M. Putative immunodominant human immunodeficiency virus-specific CD8(+) T-cell responses cannot be predicted by major histocompatibility complex class I haplotype. J Virol. 2000;74:9144–9151. doi: 10.1128/jvi.74.19.9144-9151.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koziel M.J., Walker B.D. Characteristics of the intrahepatic cytotoxic T lymphocyte response in chronic hepatitis C virus infection. Springer Semin Immunopathol. 1997;19:69–83. doi: 10.1007/BF00945026. [DOI] [PubMed] [Google Scholar]
- 34.Wedemeyer H., Mizukoshi E., Davis A.R., Bennink J.R., Rehermann B. Cross-reactivity between hepatitis C virus and Influenza A virus determinant-specific cytotoxic T cells. J Virol. 2001;75:11392–11400. doi: 10.1128/JVI.75.23.11392-11400.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hohn H., Kortsik C., Tully G., Nilges K., Necker A., Freitag K. Longitudinal analysis of Mycobacterium tuberculosis 19-kDa antigen-specific T cells in patients with pulmonary tuberculosis: association with disease activity and cross-reactivity to a peptide from HIVenv gp120. Eur J Immunol. 2003;33:1613–1623. doi: 10.1002/eji.200323480. [DOI] [PubMed] [Google Scholar]
- 36.Selin L.K., Varga S.M., Wong I.C., Welsh R.M. Protective heterologous antiviral immunity and enhanced immunopathogenesis mediated by memory T cell populations. J Exp Med. 1998;188:1705–1715. doi: 10.1084/jem.188.9.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen H.D., Fraire A.E., Joris I., Brehm M.A., Welsh R.M., Selin L.K. Memory CD8+ T cells in heterologous antiviral immunity and immunopathology in the lung. Nat Immunol. 2001;2:1067–1076. doi: 10.1038/ni727. [DOI] [PubMed] [Google Scholar]
- 38.Kim S.K., Brehm M.A., Welsh R.M., Selin L.K. Dynamics of memory T cell proliferation under conditions of heterologous immunity and bystander stimulation. J Immunol. 2002;169:90–98. doi: 10.4049/jimmunol.169.1.90. [DOI] [PubMed] [Google Scholar]
- 39.Fazekas de St G., Webster R.G. Disquisitions of original antigenic sin. I. Evidence in man. J Exp Med. 1966;124:331–345. doi: 10.1084/jem.124.3.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klenerman P., Zinkernagel R.M. Original antigenic sin impairs cytotoxic T lymphocyte responses to viruses bearing variant epitopes. Nature. 1998;394:482–485. doi: 10.1038/28860. [DOI] [PubMed] [Google Scholar]
- 41.Mongkolsapaya J., Dejnirattisai W., Xu X.N., Vasanawathana S., Tangthawornchaikul N., Chairunsri A. Original antigenic sin and apoptosis in the pathogenesis of dengue hemorrhagic fever. Nat Med. 2003;9:921–927. doi: 10.1038/nm887. [DOI] [PubMed] [Google Scholar]
- 42.Requena L., Requena C. Erythema nodosum. Dermatol Online J. 2002;8:4. [PubMed] [Google Scholar]
- 43.Schlesinger C., Meyer C.A., Veeraraghavan S., Koss M.N. Constrictive (obliterative) bronchiolitis: diagnosis, etiology, and a critical review of the literature. Ann Diagn Pathol. 1998;2:321–334. doi: 10.1016/s1092-9134(98)80026-9. [DOI] [PubMed] [Google Scholar]
- 44.Silins S.L., Sherritt M.A., Silleri J.M., Cross S.M., Elliott S.L., Bharadwaj M. Asymptomatic primary Epstein-Barr virus infection occurs in the absence of blood T-cell repertoire perturbations despite high levels of systemic viral load. Blood. 2001;98:3739–3744. doi: 10.1182/blood.v98.13.3739. [DOI] [PubMed] [Google Scholar]
- 45.Rickinson A.B., Kieff E. Epstein-Barr virus. In: Fields B.N., Knipe D.M., Howley P.M., editors. Vol. 2. Lippincott-Raven; Philadelphia: 1996. pp. 2397–2446. (Virology). [Google Scholar]
- 46.Zhao Z.S., Granucci F., Yeh L., Schaffer P.A., Cantor H. Molecular mimicry by herpes simplex virus-type 1: autoimmune disease after viral infection. Science. 1998;279:1344–1347. doi: 10.1126/science.279.5355.1344. [DOI] [PubMed] [Google Scholar]
- 47.Chen H.D., Fraire A.E., Joris I., Welsh R.M., Selin L.K. Specific history of heterologous virus infections determines anti-viral immunity and immunopathology in the lung. Am J Pathol. 2003;163:1341–1355. doi: 10.1016/S0002-9440(10)63493-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walzl G., Tafuro S., Moss P., Openshaw P.J., Hussell T. Influenza virus lung infection protects from respiratory syncytial virus-induced immunopathology. J Exp Med. 2000;192:1317–1326. doi: 10.1084/jem.192.9.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Douglass J.A., O’Hehir R.E. What determines asthma phenotype? Respiratory infections and asthma. Am J Respir Crit Care Med. 2000;161:211–214. doi: 10.1164/ajrccm.161.supplement_2.a1q4-13. [DOI] [PubMed] [Google Scholar]
- 50.Lindahl K.F., Wilson D.B. Histocompatibility antigen-activated cytotoxic T lymphocytes. II. Estimates of the frequency and specificity of precursors. J Exp Med. 1977;145:508–522. doi: 10.1084/jem.145.3.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ford W.L., Simmonds S.J., Atkins R.C. Early cellular events in a systemic graft vs. host reaction. II. Autoradiographic estimates of the frequency of donor lymphocytes which respond to each Ag-B-determined antigenic complex. J Exp Med. 1975;141:681–696. doi: 10.1084/jem.141.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suchin E.J., Langmuir P.B., Palmer E., Sayegh M.H., Wells A.D., Turka L.A. Quantifying the frequency of alloreactive T cells in vivo: new answers to an old question. J Immunol. 2001;166:973–981. doi: 10.4049/jimmunol.166.2.973. [DOI] [PubMed] [Google Scholar]
- 53.Heeger P.S. T-cell allorecognition and transplant rejection: a summary and update. Am J Transplant. 2003;3:525–533. doi: 10.1034/j.1600-6143.2003.00123.x. [DOI] [PubMed] [Google Scholar]
- 54.Bingaman A.W., Farber D.L. Memory T cells in transplantation: generation, function, and potential role in rejection. Am J Transplant. 2004;4:846–852. doi: 10.1111/j.1600-6143.2004.00453.x. [DOI] [PubMed] [Google Scholar]
- 55.Heeger P.S., Greenspan N.S., Kuhlenschmidt S., Dejelo C., Hricik D.E., Schulak J.A. Pretransplant frequency of donor-specific IFN-gamma-producing lymphocytes is a manifestation of immunologic memory and correlates with the risk of posttransplant rejection episodes. J Immunol. 1999;163:2267–2275. [PubMed] [Google Scholar]
- 56.Lombardi G., Sidhu S., Daly M., Batchelor J.R., Makgoba W., Lechler R.I. Are primary alloresponses truly primary? Int Immunol. 1990;2:9–13. doi: 10.1093/intimm/2.1.9. [DOI] [PubMed] [Google Scholar]
- 57.Yang H., Welsh R.M. Induction of alloreactive cytotoxic T cells by acute virus infection of mice. J Immunol. 1986;136:1186–1193. [PubMed] [Google Scholar]
- 58.Yang H.Y., Dundon P.L., Nahill S.R., Welsh R.M. Virus-induced polyclonal cytotoxic T lymphocyte stimulation. J Immunol. 1989;142:1710–1718. [PubMed] [Google Scholar]
- 59.Strang G., Rickinson A.B. Multiple HLA class I-dependent cytotoxicities constitute the “non-HLA-restricted” response in infectious mononucleosis. Eur J Immunol. 1987;17:1007–1013. doi: 10.1002/eji.1830170717. [DOI] [PubMed] [Google Scholar]
- 60.Tomkinson B.E., Maziarz R., Sullivan J.L. Characterization of the T cell-mediated cellular cytotoxicity during acute infectious mononucleosis. J Immunol. 1989;143:660–670. [PubMed] [Google Scholar]
- 61.Burrows S.R., Khanna R., Burrows J.M., Moss D.J. An alloresponse in humans is dominated by cytotoxic T lymphocytes (CTL) cross-reactive with a single Epstein-Barr virus CTL epitope: implications for graft-versus-host disease. J Exp Med. 1994;179:1155–1161. doi: 10.1084/jem.179.4.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Braciale T.J., Andrew M.E., Braciale V.L. Simultaneous expression of H-2-restricted and alloreactive recognition by a cloned line of influenza virus-specific cytotoxic T lymphocytes. J Exp Med. 1981;153:1371–1376. doi: 10.1084/jem.153.5.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brehm M.A., Markees T.G., Daniels K.A., Greiner D.L., Rossini A.A., Welsh R.M. Direct visualization of cross-reactive effector and memory allo-specific CD8 T cells generated in response to viral infections. J Immunol. 2003;170:4077–4086. doi: 10.4049/jimmunol.170.8.4077. [DOI] [PubMed] [Google Scholar]
- 64.Nahill S.R., Welsh R.M. High frequency of cross-reactive cytotoxic T lymphocytes elicited during the virus-induced polyclonal cytotoxic T lymphocyte response. J Exp Med. 1993;177:317–327. doi: 10.1084/jem.177.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jennings S.R. Cross-reactive recognition of mouse cells expressing the bm3 and bm11 mutations within H-2Kb by H-2Kb-restricted herpes simplex virus-specific cytotoxic T lymphocytes. J Immunol. 1985;135:3530–3536. [PubMed] [Google Scholar]
- 66.Sheil J.M., Bevan M.J., Lefrancois L. Characterization of dual-reactive H-2Kb-restricted anti-vesicular stomatitus virus and alloreactive cytotoxic T cells. J Immunol. 1987;138:3654–3660. [PubMed] [Google Scholar]
- 67.Burrows S.R., Silins S.L., Khanna R., Burrows J.M., Rischmueller M., McCluskey J. Cross-reactive memory T cells for Epstein-Barr virus augment the alloresponse to common human leukocyte antigens: degenerate recognition of major histocompatibility complex-bound peptide by T cells and its role in alloreactivity. Eur J Immunol. 1997;27:1726–1736. doi: 10.1002/eji.1830270720. [DOI] [PubMed] [Google Scholar]
- 68.Mullbacher A., Ashman R.B., Ada G.L. Alloreactive cytotoxic T lymphocytes lyse syngeneic influenza-infected tumour cell targets. Scand J Immunol. 1984;19:365–371. doi: 10.1111/j.1365-3083.1984.tb00943.x. [DOI] [PubMed] [Google Scholar]
- 69.Williams M.A., Tan J.T., Adams A.B., Durham M.M., Shirasugi N., Whitmire J.K. Characterization of virus-mediated inhibition of mixed chimerism and allospecific tolerance. J Immunol. 2001;167:4987–4995. doi: 10.4049/jimmunol.167.9.4987. [DOI] [PubMed] [Google Scholar]
- 70.Tough D.F., Borrow P., Sprent J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science. 1996;272:1947–1950. doi: 10.1126/science.272.5270.1947. [DOI] [PubMed] [Google Scholar]
- 71.Bluthmann H., Kisielow P., Uematsu Y., Malissen M., Krimpenfort P., Berns A. T-cell-specific deletion of T-cell receptor transgenes allows functional rearrangement of endogenous alpha- and beta-genes. Nature. 1988;334:156–159. doi: 10.1038/334156a0. [DOI] [PubMed] [Google Scholar]
- 72.Gaston J.S. Immune responses and allograft rejection. Lancet. 1987;1:860–861. doi: 10.1016/s0140-6736(87)91636-9. [DOI] [PubMed] [Google Scholar]
- 73.Jakel K.T., Loning T. Herpes virus infections, acute rejection, and transplant arteriosclerosis in human cardiac allografts. Transplant Proc. 1993;25:2029–2030. [PubMed] [Google Scholar]
- 74.Valujskikh A., Lakkis F.G. In remembrance of things past: memory T cells and transplant rejection. Immunol Rev. 2003;196:65–74. doi: 10.1046/j.1600-065x.2003.00087.x. [DOI] [PubMed] [Google Scholar]
- 75.Grewal I.S., Flavell R.A. CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol. 1998;16:111–135. doi: 10.1146/annurev.immunol.16.1.111. [DOI] [PubMed] [Google Scholar]
- 76.Lenschow D.J., Walunas T.L., Bluestone J.A. CD28/B7 system of T cell costimulation. Annu Rev Immunol. 1996;14:233–258. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 77.Schwartz R.H. T cell anergy. Annu Rev Immunol. 2003;21:305–334. doi: 10.1146/annurev.immunol.21.120601.141110. [DOI] [PubMed] [Google Scholar]
- 78.Parker D.C., Greiner D.L., Phillips N.E., Appel M.C., Steele A.W., Durie F.H. Survival of mouse pancreatic islet allografts in recipients treated with allogeneic small lymphocytes and antibody to CD40 ligand. Proc Natl Acad Sci USA. 1995;92:9560–9564. doi: 10.1073/pnas.92.21.9560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wood K.J., Sakaguchi S., Regulatory T cells in transplantation tolerance. Nat Rev Immunol. 2003;3:199–210. doi: 10.1038/nri1027. [DOI] [PubMed] [Google Scholar]
- 80.Markees T.G., Phillips N.E., Noelle R.J., Shultz L.D., Mordes J.P., Greiner D.L. Prolonged survival of mouse skin allografts in recipients treated with donor splenocytes and antibody to CD40 ligand. Transplantation. 1997;64:329–335. doi: 10.1097/00007890-199707270-00026. [DOI] [PubMed] [Google Scholar]
- 81.Larsen C.P., Elwood E.T., Alexander D.Z., Ritchie S.C., Hendrix R., Tucker-Burden C. Long-term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature. 1996;381:434–438. doi: 10.1038/381434a0. [DOI] [PubMed] [Google Scholar]
- 82.Kirk A.D., Harlan D.M., Armstrong N.N., Davis T.A., Dong Y., Gray G.S. CTLA4-Ig and anti-CD40 ligand prevent renal allograft rejection in primates. Proc Natl Acad Sci USA. 1997;94:8789–8794. doi: 10.1073/pnas.94.16.8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hancock W.W., Sayegh M.H., Zheng X.G., Peach R., Linsley P.S., Turka L.A. Costimulatory function and expression of CD40 ligand, CD80, and CD86 in vascularized murine cardiac allograft rejection. Proc Natl Acad Sci USA. 1996;93:13967–13972. doi: 10.1073/pnas.93.24.13967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Welsh R.M., Markees T.G., Woda B.A., Daniels K.A., Brehm M.A., Mordes J.P. Virus-induced abrogation of transplantation tolerance induced by donor-specific transfusion and anti-CD154 antibody. J Virol. 2000;74:2210–2218. doi: 10.1128/jvi.74.5.2210-2218.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Williams M.A., Onami T.M., Adams A.B., Durham M.M., Pearson T.C., Ahmed R. Cutting edge: persistent viral infection prevents tolerance induction and escapes immune control following CD28/CD40 blockade-based regimen. J Immunol. 2002;169:5387–5391. doi: 10.4049/jimmunol.169.10.5387. [DOI] [PubMed] [Google Scholar]
- 86.Turgeon N.A., Iwakoshi N.N., Phillips N.E., Meyers W.C., Welsh R.M., Greiner D.L. Viral infection abrogates CD8(+) T-cell deletion induced by costimulation blockade. J Surg Res. 2000;93:63–69. doi: 10.1006/jsre.2000.5962. [DOI] [PubMed] [Google Scholar]
- 87.van Maurik A., Herber M., Wood K.J., Jones N.D. Cutting edge: CD4+CD25+ alloantigen-specific immunoregulatory cells that can prevent CD8+ T cell-mediated graft rejection: implications for anti-CD154 immunotherapy. J Immunol. 2002;169:5401–5404. doi: 10.4049/jimmunol.169.10.5401. [DOI] [PubMed] [Google Scholar]
- 88.Welsh R.M., McNally J.M., Brehm M.A., Selin L.K. Consequences of cross-reactive and bystander CTL responses during viral infections. Virology. 2000;270:4–8. doi: 10.1006/viro.2000.0278. [DOI] [PubMed] [Google Scholar]
- 89.Flynn K., Mullbacher A. Memory alloreactive cytotoxic T cells do not require costimulation for activation in vitro. Immunol Cell Biol. 1996;74:413–420. doi: 10.1038/icb.1996.71. [DOI] [PubMed] [Google Scholar]
- 90.London C.A., Lodge M.P., Abbas A.K. Functional responses and costimulator dependence of memory CD4+ T cells. J Immunol. 2000;164:265–272. doi: 10.4049/jimmunol.164.1.265. [DOI] [PubMed] [Google Scholar]
- 91.Adams A.B., Williams M.A., Jones T.R., Shirasugi N., Durham M.M., Kaech S.M. Heterologous immunity provides a potent barrier to transplantation tolerance. J Clin Invest. 2003;111:1887–1895. doi: 10.1172/JCI17477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhai Y., Meng L., Gao F., Busuttil R.W., Kupiec-Weglinski J.W. Allograft rejection by primed/memory CD8+ T cells is CD154 blockade resistant: therapeutic implications for sensitized transplant recipients. J Immunol. 2002;169:4667–4673. doi: 10.4049/jimmunol.169.8.4667. [DOI] [PubMed] [Google Scholar]
- 93.Chen Y., Heeger P.S., Valujskikh A. In vivo helper functions of alloreactive memory CD4+ T cells remain intact despite donor-specific transfusion and anti-CD40 ligand therapy. J Immunol. 2004;172:5456–5466. doi: 10.4049/jimmunol.172.9.5456. [DOI] [PubMed] [Google Scholar]
- 94.Pantenburg B., Heinzel F., Das L., Heeger P.S., Valujskikh A. T cells primed by Leishmania major infection cross-react with alloantigens and alter the course of allograft rejection. J Immunol. 2002;169:3686–3693. doi: 10.4049/jimmunol.169.7.3686. [DOI] [PubMed] [Google Scholar]
- 95.Tanchot C., Rocha B. The organization of mature T-cell pools. Immunol Today. 1998;19:575–579. doi: 10.1016/s0167-5699(98)01344-9. [DOI] [PubMed] [Google Scholar]
- 96.Selin L.K., Vergilis K., Welsh R.M., Nahill S.R. Reduction of otherwise remarkably stable virus-specific cytotoxic T lymphocyte memory by heterologous viral infections. J Exp Med. 1996;183:2489–2499. doi: 10.1084/jem.183.6.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Murali-Krishna K., Altman J.D., Suresh M., Sourdive D.J., Zajac A.J., Miller J.D. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. 1998;8:177–187. doi: 10.1016/s1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- 98.Becker T.C., Wherry E.J., Boone D., Murali-Krishna K., Antia R., Ma A. Interleukin 15 is required for proliferative renewal of virus-specific memory CD8 T cells. J Exp Med. 2002;195:1541–1548. doi: 10.1084/jem.20020369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schluns K.S., Williams K., Ma A., Zheng X.X., Lefrancois L. Cutting edge: requirement for IL-15 in the generation of primary and memory antigen-specific CD8 T cells. J Immunol. 2002;168:4827–4831. doi: 10.4049/jimmunol.168.10.4827. [DOI] [PubMed] [Google Scholar]
- 100.Selin L.K., Lin M.Y., Kraemer K.A., Pardoll D.M., Schneck J.P., Varga S.M. Attrition of T cell memory: selective loss of LCMV epitope-specific memory CD8 T cells following infections with heterologous viruses. Immunity. 1999;11:733–742. doi: 10.1016/s1074-7613(00)80147-8. [DOI] [PubMed] [Google Scholar]
- 101.Smith D.K., Dudani R., Pedras-Vasconcelos J.A., Chapdelaine Y., van Faassen H., Sad S. Cross-reactive antigen is required to prevent erosion of established T cell memory and tumor immunity: a heterologous bacterial model of attrition. J Immunol. 2002;169:1197–1206. doi: 10.4049/jimmunol.169.3.1197. [DOI] [PubMed] [Google Scholar]
- 102.Liu H., Andreansky S., Diaz G., Turner S.J., Wodarz D., Doherty P.C. Quantitative analysis of long-term virus-specific CD8+-T-cell memory in mice challenged with unrelated pathogens. J Virol. 2003;77:7756–7763. doi: 10.1128/JVI.77.14.7756-7763.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kim S.K., Welsh R.M. Comprehensive early and lasting loss of memory CD8 T cells and functional memory during acute and persistent viral infections. J Immunol. 2004;172:3139–3150. doi: 10.4049/jimmunol.172.5.3139. [DOI] [PubMed] [Google Scholar]
- 104.Chapdelaine Y., Smith D.K., Pedras-Vasconcelos J.A., Krishnan L., Sad S. Increased CD8+ T cell memory to concurrent infection at the expense of increased erosion of pre-existing memory: the paradoxical role of IL-15. J Immunol. 2003;171:5454–5460. doi: 10.4049/jimmunol.171.10.5454. [DOI] [PubMed] [Google Scholar]
- 105.Peacock C.D., Kim S.K., Welsh R.M. Attrition of virus-specific memory CD8+ T cells during reconstitution of lymphopenic environments. J Immunol. 2003;171:655–663. doi: 10.4049/jimmunol.171.2.655. [DOI] [PubMed] [Google Scholar]
- 106.McNally J.M., Zarozinski C.C., Lin M.Y., Brehm M.A., Chen H.D., Welsh R.M. Attrition of bystander CD8 T cells during virus-induced T-cell and interferon responses. J Virol. 2001;75:5965–5976. doi: 10.1128/JVI.75.13.5965-5976.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jiang J., Lau L.L., Shen H. Selective depletion of nonspecific T cells during the early stage of immune responses to infection. J Immunol. 2003;171:4352–4358. doi: 10.4049/jimmunol.171.8.4352. [DOI] [PubMed] [Google Scholar]
- 108.Tumpey T.M., Lu X., Morken T., Zaki S.R., Katz J.M. Depletion of lymphocytes and diminished cytokine production in mice infected with a highly virulent influenza A (H5N1) virus isolated from humans. J Virol. 2000;74:6105–6116. doi: 10.1128/jvi.74.13.6105-6116.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Okada H., Kobune F., Sato T.A., Kohama T., Takeuchi Y., Abe T. Extensive lymphopenia due to apoptosis of uninfected lymphocytes in acute measles patients. Arch Virol. 2000;145:905–920. doi: 10.1007/s007050050683. [DOI] [PubMed] [Google Scholar]
- 110.Geisbert T.W., Hensley L.E., Gibb T.R., Steele K.E., Jaax N.K., Jahrling P.B. Apoptosis induced in vitro and in vivo during infection by Ebola and Marburg viruses. Lab Invest. 2000;80:171–186. doi: 10.1038/labinvest.3780021. [DOI] [PubMed] [Google Scholar]
- 111.Feldman S., Lott L. Varicella in children with cancer: impact of antiviral therapy and prophylaxis. Pediatrics. 1987;80:465–472. [PubMed] [Google Scholar]
- 112.Jiang J., Anaraki F., Blank K.J., Murasko D.M. Cutting edge: T cells from aged mice are resistant to depletion early during virus infection. J Immunol. 2003;171:3353–3357. doi: 10.4049/jimmunol.171.7.3353. [DOI] [PubMed] [Google Scholar]
- 113.Wong R.S., Wu A., To K.F., Lee N., Lam C.W., Wong C.K. Haematological manifestations in patients with severe acute respiratory syndrome: retrospective analysis. Br Med J. 2003;326:1358–1362. doi: 10.1136/bmj.326.7403.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Diebold S.S., Montoya M., Unger H., Alexopoulou L., Roy P., Haswell L.E. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature. 2003;424:324–328. doi: 10.1038/nature01783. [DOI] [PubMed] [Google Scholar]
- 115.Wherry E.J., Blattman J.N., Murali-Krishna K., van der Most R., Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Homann D., McGavern D.B., Oldstone M.B. Visualizing the viral burden: phenotypic and functional alterations of T cells and APCs during persistent infection. J Immunol. 2004;172:6239–6250. doi: 10.4049/jimmunol.172.10.6239. [DOI] [PubMed] [Google Scholar]
- 117.Zajac A.J., Blattman J.N., Murali-Krishna K., Sourdive D.J., Suresh M., Altman J.D. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998;188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Horohov D.W., Rouse B.T. Virus-induced immunosuppression. Vet Clin North Am Small Anim Pract. 1986;16:1097–1127. doi: 10.1016/s0195-5616(86)50131-5. [DOI] [PubMed] [Google Scholar]
- 119.Cohen J. What causes the immune system collapse seen in AIDS? Science. 1993;260:1256. doi: 10.1126/science.8098551. [DOI] [PubMed] [Google Scholar]
- 120.Borrow P., Evans C.F., Oldstone M.B. Virus-induced immunosuppression: immune system-mediated destruction of virus-infected dendritic cells results in generalized immune suppression. J Virol. 1995;69:1059–1070. doi: 10.1128/jvi.69.2.1059-1070.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Berger D.P., Homann D., Oldstone M.B. Defining parameters for successful immunocytotherapy of persistent viral infection. Virology. 2000;266:257–263. doi: 10.1006/viro.1999.0074. [DOI] [PubMed] [Google Scholar]
- 122.Badovinac V.P., Porter B.B., Harty J.T. Programmed contraction of CD8(+) T cells after infection. Nat Immunol. 2002;3:619–626. doi: 10.1038/ni804. [DOI] [PubMed] [Google Scholar]
- 123.Behn U., Celada F., Seiden P.E. Computer modeling in immunology. In: Lanzaveccia A., Malissen B., Sitia R., editors. Vol. 2. Academic Press; London: 2001. pp. 611–630. (Frontiers of life, part two). [Google Scholar]
- 124.Kohler B., Puzone R., Seiden P.E., Celada F. A systematic approach to vaccine complexity using an automaton model of the cellular and humoral immune system. I. Viral characteristics and polarized responses. Vaccine. 2000;19:862–876. doi: 10.1016/s0264-410x(00)00225-5. [DOI] [PubMed] [Google Scholar]
- 125.Celada F., Seiden P.E. Affinity maturation and hypermutation in a simulation of the humoral immune response. Eur J Immunol. 1996;26:1350–1358. doi: 10.1002/eji.1830260626. [DOI] [PubMed] [Google Scholar]
- 126.Celada F., Seiden P.E. A computer model of cellular interactions in the immune system. Immunol Today. 1992;13:56–62. doi: 10.1016/0167-5699(92)90135-T. [DOI] [PubMed] [Google Scholar]
- 127.Seiden P.E., Celada F. A model for simulating cognate recognition and response in the immune system. J. Theor. Biol. 1992;158:329–357. doi: 10.1016/s0022-5193(05)80737-4. [DOI] [PubMed] [Google Scholar]