

Abstract
The mucosal surfaces of the lungs are a major portal of entry for virus infections and there are urgent needs for new vaccines that promote effective pulmonary immunity. However, we have only a rudimentary understanding of the requirements for effective cellular immunity in the respiratory tract. Recent studies have revealed that specialized cellular immune responses and lymphoid tissues are involved in the protection of distinct anatomical microenvironments of the respiratory tract, such as the large airways of the nose and the alveolar airspaces. This review discusses some of the anatomical features of anti-viral immunity in the respiratory tract including the role of local lymphoid tissues and the relationship between effector and memory T cells in the airways, the lung parenchyma, and lymphoid organs.
Keywords: NALT, BALT, Lung, Virus, Lymphocyte, Memory
1. Introduction
Respiratory virus infections are a major cause of morbidity and mortality throughout the world. Influenza virus infections alone result in the deaths of about 36,000 people per year in the United States and there is tremendous concern that highly lethal variants of this virus may emerge and cause a major pandemic [1], [2]. Moreover, the emergence of new respiratory pathogens, such as the coronavirus associated with Severe Acute Respiratory Syndrome, pose a continual threat [3]. Despite the medical significance of respiratory viral infections, satisfactory vaccines have not been developed. For example, in the case of influenza virus, the currently available vaccine elicits humoral immunity specific for viral coat proteins and must be reformulated yearly to be effective against new viral strains [4]. One possible approach to improve vaccine utility would be to develop supplementary vaccines that promote cellular immunity against relatively invariant viral proteins [5]. The broadly cross-reactive immunity generated by such vaccines would operate against different strains of virus and may be effective through multiple epidemics. Recent progress in our understanding of cellular immune responses in the lung will facilitate the development of such vaccines.
2. Immunity in the respiratory tract
Much of our understanding of immune responses to respiratory virus infections has been derived through experimental animal models. A particularly robust and well-characterized model is the infection of mice with mouse-adapted influenza virus [6], [7], [8]. When introduced through the nose, the virus establishes infection of lung epithelial cells and elicits powerful cellular and humoral immune responses in the lung. CD8+ cytotoxic T cells first appear in the lung airways on day 7 post-infection and play a key role in clearing virus [9]. Typically, this effector T-cell response peaks around day 10 or 11 [9]. Antibody is also generated in the response, but isotype switched antibody does not accumulate until day 7 and does not appear to play a critical role in the primary infection unless the viral titer is particularly high [10]. Following resolution of the infection, memory T cells persist in secondary lymphoid organs, such as the spleen and local draining lymph nodes, as well as a variety of peripheral sites, including the lung parenchyma and airways [11], [12]. These memory cells retain the capacity to mediate accelerated recall responses due to their semi-activated status and increased precursor frequencies relative to naïve T-cell populations [9], [12], [13], [14].
While the basic outline of T-cell immunity in the lung has been established, the specific details remain obscure. First, we have only a poor understanding of how primary and memory T-cell responses are initiated and regulated. Second, it remains unclear how T cells actually clear virus from the lungs, although both cytolytic activity and gamma-interferon have been shown to play a role [15], [16], [17]. Third, we have only a rudimentary understanding of the establishment, maintenance and recall of T-cell memory with respect to a mucosal infection. And fourth, it is unclear how the various lymphoid tissues that drain the upper and lower respiratory tract contribute to the initiation and regulation of primary immune responses as well as the maintenance and responsiveness of immune memory. Nonetheless, the field has made substantial progress over the last few years. The advent of new technologies for studying immune responses in vivo has allowed viral immunologists to begin to visualize key processes and dissect the underlying mechanisms. In addition, we have begun to appreciate the anatomical aspects of immune responses in the respiratory tract.
3. Primary T-cell responses
The infection of respiratory epithelial cells initiates a cascade of events that culminate in the activation of a cellular immune response in the lung. The initial infection induces the production of inflammatory mediators by epithelial cells, which alert the innate immune response to the infection [18]. In addition, dendritic cells (DCs) lining the upper respiratory tract also detect the presence of an infection via toll-like receptors (TLRs), which detect viral proteins [19] or products of viral replication, such as double stranded RNA [18], [19], [20], [21], [22]. The combination of inflammation and TLR signaling activates DCs, increases their expression of class I and class II Major Histocompatibility Complex (MHC) molecules and induces a wide array of costimulatory and adhesion molecules as well as inflammatory cytokines that are required for the induction of T-cell responses [23], [24]. Together, these changes in DC activity result in enhanced presentation of viral antigens to T cells [24]. Finally, the DCs acquire the ability to traffic to the lymph nodes, migrate into T-cell areas and interact directly with naïve T cells [25]. Relatively little is known about the trafficking of DCs under these conditions, although it appears that the majority of DC movement to the lymph nodes occurs within 48 h of infection [25], [26]. Once in the T-cell areas, mature DCs present antigen to naïve T cells, which then initiate a program of proliferation and maturation [27]. This results in a massive increase in the number of antigen-specific T cells and the production of large numbers of effector cells with the capacity to lyse infected epithelial cells and secrete anti-viral cytokines [28]. Finally, these effector cells acquire the capacity to traffic and subsequently move to the site of infection in the lung where they effectively terminate the infection [28]. The initial proliferative program of T-cells appears to take place exclusively in the lymphoid tissues and cells only leave these sites after acquiring appropriate homing signals and effector activities [28]. Thus, the accumulation of T cells in the lung airways results from the rapid output of cells from the lymph nodes (LN) rather than local proliferation. However, as discussed below, there is also the induction of de novo lymphoid tissues in the lung parenchyma, which may serve as sites for additional T-cell replication.
4. Role of different lymphoid sites
It has generally been assumed that local encapsulated LN play the central role in coordinating the T-cell response to infection [28]. However, emerging data suggest that other lymphoid tissues may also play a critical role. In particular, the Nasal-Associated Lymphoid Tissue (NALT) [29], [30] and the Bronchus-Associated Lymphoid Tissue (BALT) [31], [32], [33] have the potential to prime lymphocytes in response to respiratory infections. Unlike classical LN, these mucosal lymphoid tissues are not encapsulated and are not supplied by afferent lymphatics [34]. Instead, these lymphoid tissues are in direct contact with the mucosal epithelium and respond to antigens that cross the epithelial barrier either by active transport or by infection [34]. Because of their position at mucosal sites, these tissues are poised to respond rapidly to infections in the nose and upper respiratory tract (NALT) as well as infections in the lung and lower respiratory tract (BALT).
The NALT of mice is thought to be equivalent to Waldeyer’s ring in humans, which includes the tonsils and adenoids [30]. As shown in Fig. 1 , NALT is a classic example of a mucosal lymphoid organ, with a prominent central B-cell follicle underneath a dome epithelium containing M cells that transport antigen from the lumenal surface of the epithelium to the APCs directly underneath [35]. NALT functions as an inductive lymphoid organ for immune responses to antigens administered in the nose [36], [37], [38], [39]. This is most clearly demonstrated for B-cell responses to antigens administered with cholera toxin, as antigen-specific germinal center B cells develop first in the NALT and subsequently in the draining LNs [40]. NALT also supports the generation of anti-viral effector T cells in response to vaccination with peptides [38], [41], proteins [36], or virus [42], [43] although it is not clear from the kinetics of these responses if CD8 cells are initially primed in the NALT or if they are primed in other lymphoid organs and subsequently traffic back to the NALT. Although the magnitude of the T-cell response in NALT is small relative to that in LNs or spleen [43], it is likely that the cells primed in NALT have specialized homing properties that allow them to protect the upper airways and nasal mucosa. Thus, while the relative contribution of NALT to the overall respiratory immune responses is minor, the NALT may be the primary lymphoid organ that generates cells that protect the upper respiratory tract and nasal passages. Given that natural respiratory infections often begin in the nose and upper airways, the NALT is likely to be one of the first sites of antigen recognition. As such, the role of NALT in initiating immune responses and directing the generation of effector cells that specifically protect the upper respiratory tract deserves closer study.
Fig. 1.
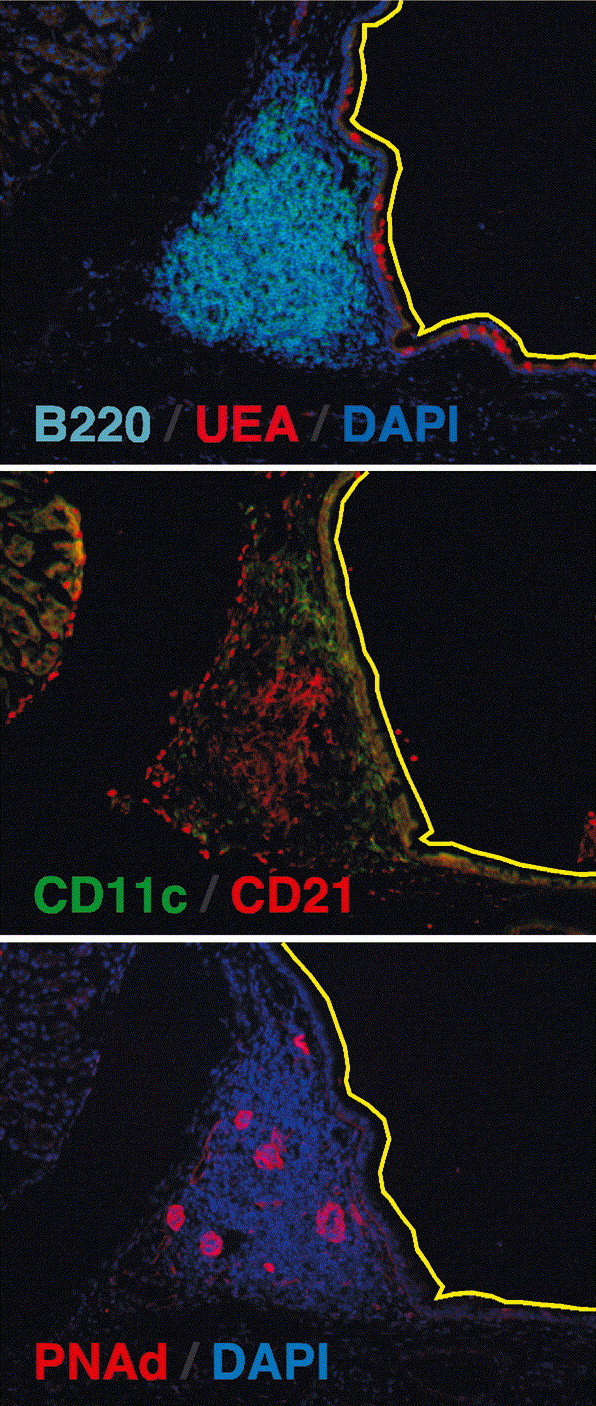
Structure of NALT. Serial frozen sections of NALT from the decalcified heads of C57BL/6 mice were probed with anti-B220 to identify B cell follicles and the lectin, Ulex europaeus agglutinin (UEA), to identify M cells in the dome epithelium (top panel), anti-CD11c to identify DCs and anti-CD21 to identify follicular DCs (middle panel), anti-PNAd to identify high endothelial venules expressing the peripheral LN addressin and DAPI to counterstain the NALT. The yellow line delineates the nasal epithelium and the black space in the upper right corner of each panel is the lumen of the nasal passage.
BALT was originally described as a submucosal lymphoid tissue found in the major bronchi of some species and is similar in structure to Peyer’s patches and NALT [44]. Like other lymphoid organs, classical BALT is formed independently of antigen by a pre-programmed pathway that unfolds during late embryonic development [45]. However, the presence of BALT in both murine and human lungs is controversial [46], [47]. Although there are numerous reports that organized lymphoid tissue and even germinal centers can be found in murine and human lungs [47], [48], [49], [50], [51], these lymphoid areas seem to appear as animals age or after infection or inflammation [47], [48], [49], [50], [51]. Furthermore, the inducible lymphoid areas that appear in the lung after infection do not necessarily fulfill the classical definition of BALT, as they are often found in perivascular, peribronchial and even interstitial areas in the lower airways of the lung and do not always occur under a dome epithelium. In addition, the size of the lymphoid areas in the lung also varies widely from small clusters of B, T, and DCs, to the well-developed follicular B-cell areas and inter-follicular T-cell areas shown in Fig. 2 . Furthermore, although the BALT-like areas observed after infection are clearly capable of supporting B- and T-cell proliferation (Fig. 2), it is not entirely clear whether these structures are merely collections of effector cells that were initially primed in other more conventional lymphoid tissues or whether these structures act like conventional lymphoid organs and are able to recruit and prime naïve lymphocytes.
Fig. 2.
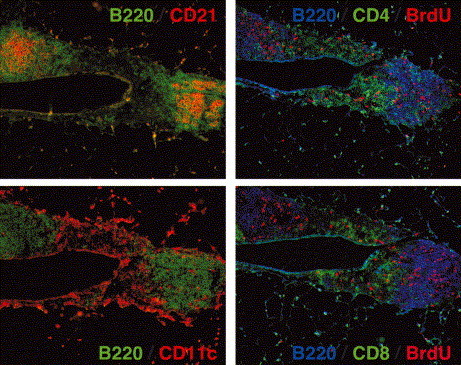
Structure of BALT. Mice were infected with influenza 14 days previously were pulsed with Bromo-deoxy-Uridine (BrdU) for 1 h prior to sacrifice. Serial frozen sections of lungs from influenza-infected mice were probed with anti-B220 to identify B cell follicles and anti-CD21 to identify follicular DCs (upper left panel), anti-B220 to identify B cell follicles and anti-CD11c to identify DCs in the interfollicular regions (lower left panel). Anti-BrdU antibodies were used to identify proliferating B220+ B cells and proliferating CD4+ T cells (upper right panel) as well as B220+ B cells and CD8+ T cells (lower right panel). Note that the two B cell follicles in these sections are separated by an interfollicular region that contains CD11c+ DCs and T cells. These lymphoid structures surround a major airway.
As discussed above, a central paradigm in immunology is that rare naïve lymphocytes recirculate exclusively through secondary lymphoid organs and are primed in these locations by DCs that were activated in peripheral non-lymphoid tissues [52]. Once the lymphocytes are primed and expanded in lymphoid tissues, they recirculate back to the infected tissue and exert their effector functions [52]. However, the ability of organized lymphoid tissues to form in the lung parenchyma after infection or inflammation blurs the distinction between lymphoid and non-lymphoid organs. Once BALT-like structures are formed in the lung, it is likely that the trafficking patterns of DCs and lymphocytes will be altered and that antigen will be handled differently. In fact, this may explain the observation that influenza-activated DCs only traffic to the LNs that drain the lung for the first day or two of infection and then cease to leave the lung [25]. Perhaps new BALT-like structures are rapidly formed in the lung after infection and trap migrating DCs before they leave. In addition, the BALT-like areas formed in response to a primary infection may play a much more important role in recruiting and priming lymphocytes in response to subsequent infections with the same or even heterologous viruses [53]. Consistent with this idea, DCs are reported to stay in the lungs of lymphotoxin-deficient mice after immunization and prime naïve CD4 cells in situ [54]. Since lymphotoxin-deficient mice lack peripheral LNs and often have BALT-like areas in their lungs [55], it is likely that BALT functioned as an inductive lymphoid organ in these experiments. Thus, the lung may function as a non-lymphoid target of infection and may also have the ability to function as a lymphoid organ and directly prime lymphocytes. However, the relative contribution of BALT to the priming and expansion of the overall T-cell response to respiratory viral infection remains unclear.
In addition to the role of NALT and BALT in lymphocyte priming and expansion, the environment of these mucosal lymphoid tissues may impart specialized properties on the effector cells that they generate. For example, B cell responses in Peyer’s patches are strongly biased towards the production of IgA [56] due to the production of TGFβ by T cells in the Peyer’s patch [57]. This response is specialized for the anatomical structure of the gut as IgA is efficiently transported across the mucosal epithelium of the intestine by the Polymeric Ig receptor [58], [59]. In contrast, B cell differentiation in NALT and BALT results in the production of both IgA and IgG [40]. This probably reflects differences in the structure of the gut and respiratory tract. While IgA is transported across the mucosal epithelium of the upper airways, where it serves to neutralize and clear viral infections, IgG is the primary Ig isotype responsible for the protection of the lower respiratory tract [60]. In fact, high affinity IgG, but not IgA, is of paramount importance for the neutralization of virus in the lung [60]. This is particularly true in the delicate alveolar airspaces, in which IgG entry is mediated by transudation rather than active transport. In fact, the IgG produced in IgA-deficient mice is sufficient to clear influenza in a primary infection and to neutralize the majority of virus in both the lungs and nasal passages upon secondary challenge [61]. Thus, the B cell responses in NALT and BALT are adapted to the requirements of the respiratory tract, rather than to classic mucosal immune responses as defined in the gut.
Mucosal immune responses also generate effector and memory T lymphocytes with unique homing properties [62]. For example, it has been shown that DCs from Peyer’s patches prime T cells that preferentially home back to the gut [63]. These T cells upregulate the integrin α4β7, which binds the mucosal addressin MAdCAM [64], as well as the chemokine receptor CCR9, which binds the gut-expressed chemokine CCL25 [65]. These homing molecules allow memory and effector cells primed in the gut to home back to the gut. Although the paradigm of a common mucosal immune system is often cited to suggest that cells primed at one mucosal site can home to any mucosal site [66], it appears that the well-defined homing properties of cells primed in the gut do not necessarily apply to the cells primed in the mucosal sites of the respiratory tract [67]. Similarly, the homing receptors and chemokines expressed in the gut do not necessarily match those expressed in the respiratory tract. For example, although the mucosal addressin, MAdCAM, is the primary homing molecule expressed in Peyer’s patches [62], NALT expresses both MAdCAM and the peripheral LN homing receptor PNAd [68]. Furthermore, PNAd is the primary addressin used for lymphocyte entry to the NALT [68]. In contrast, the HEVs of BALT express PNAd, but not MAdCAM, and lymphocyte entry into murine BALT is mediated by PNAd and VCAM [69]. This suggests that homing to NALT and BALT is more similar to homing to peripheral LNs than homing to Peyer’s patches. Future studies will have to determine whether there are NALT- or BALT-specific homing molecules that regulate migration to the mucosal tissues of the respiratory tract.
5. Memory T-cell subsets and the maintenance and recall of respiratory immunity
A signature feature of the adaptive immune response is the establishment of memory T cells capable of mediating accelerated and enhanced recall responses to secondary infection [70]. The rapid nature of the recall response is due to the persistence of increased frequencies of memory T cells in the airways, peripheral non-lymphoid tissues and secondary lymphoid organs [71], [72]. Long-term maintenance of these memory subsets is a dynamic process that involves continuous cell division and turnover [73]. However, the relationship between memory T cells found in the lung airways and the mucosal lining of the respiratory tract and the subsets of memory T cells that are found in the NALT, other secondary lymphoid organs, and BALT remains unclear.
Memory T cells are often divided into two general subsets based on their homing properties. Those memory T cells that maintain some of the homing and activation properties of effector cells (e.g., lack CD62L and CCR7 expression) are termed effector memory cells [71], [74], [75]. Like effector cells, effector memory cells are found throughout the body in non-lymphoid tissues [71], [76] and can rapidly respond to antigen stimulation with immediate effector functions such as cytotoxicity or cytokine production [74], [75]. Not all memory T cells are excluded from lymphoid tissues however, and other memory T cells, termed central memory cells, express CD62L and CCR7 expression, allowing them to recirculate through lymphoid organs [71], [74], [75], [76]. In contrast to effector memory cells, central memory cells do not have immediate effector functions [71], [74], [75], [76], but instead are thought to rapidly expand and differentiate into a second round of effector T cells in lymphoid organs. Although subsets of memory cells with different homing properties clearly exist after respiratory infection, it is not clear whether the ability of central memory cells to home to secondary lymphoid organs is important for the maintenance of memory T cells in the airways or for the maintenance of memory T cells as a whole.
6. Recall of memory T-cell subsets
While progress has been made in defining and characterizing distinct memory T-cell subsets, it is far from clear how each subset contributes to a recall response following secondary virus challenge in the lung. In particular, it is unclear whether the ability of central memory cells to home to secondary lymphoid organs is important for the expansion of memory T-cell populations upon secondary challenge. Although rapid secondary expansion was originally proposed to be a property of central memory cells and not effector memory cells, it is now clear that effector memory T cells can also rapidly proliferate and accumulate in the lungs of recipient mice upon adoptive transfer and challenge infection [72], [77], [78]. Thus, the functional differences between central and effector memory cells are not as clear-cut as originally described. Furthermore, the secondary expansion of memory cells in general is not dependent on the environment of secondary lymphoid organs, as adoptively transferred memory CD4 cells proliferate in response to antigen in mice that lack all peripheral lymphoid organs [79]. Thus, although memory T cells with different homing properties clearly persist in various lymphoid and non-lymphoid tissues after respiratory infection, it is not clear how the cells in each of these tissues are related, how they are maintained or how they contribute to secondary responses.
While the antigen-driven expansion of memory T cells in the secondary lymphoid organs has generally been considered the basis of a recall response, there is accumulating evidence that non-proliferating memory T cells are also critically involved [80], [81]. In this regard, recent data suggest that the recall response is comprised of several distinct phases that are temporally and anatomically separated and involve distinct subpopulations of memory T cells (Fig. 3 ) [81]. The first phase is mediated by memory T cells that are resident in the lung airways [11], [12], [14], [82], [83]. Importantly, these cells are able to respond to the first signs of infection when viral loads are very low. While unable to proliferate in response to infection due to the constraints of the airway environment, they can produce cytokines that may limit viral replication and spread in the epithelium [81], [84]. Interestingly, these cells are not able to mediate cytolytic activity, which has been attributed to the loss of CD11a, a unique feature of lung airway memory T cells [78], [85]. The second phase of the response is mediated by memory T cells that are rapidly recruited to the airways in the first few days of the response. These cells do not proliferate either prior to recruitment or at the site of infection but it is not clear whether these cells come from the lung parenchyma, local sites such as the NALT or BALT or even more distal sites via the circulation [80], [81]. In the mouse model, relatively large numbers of cells are recruited within four days of infection, but do not produce a sustained response and the response wanes by day 6 [81]. The third stage of the response is the antigen-driven expansion of memory T cells that occurs in the secondary lymphoid organs, including BALT, draining LNs and spleen [86]. These memory T cells proliferate for several days in the lymphoid organs and are only recruited to the lung airways after about day 5 of infection. In a normal recall response, these distinct phases are integrated to produce a sustained response to the infection in the lung.
Fig. 3.
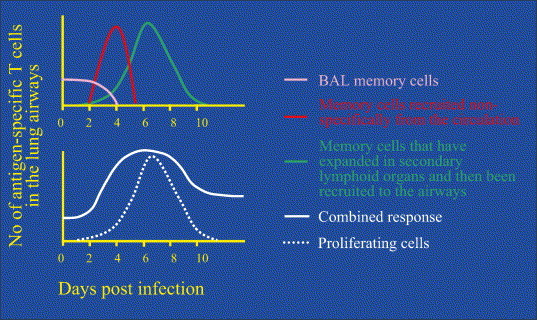
Recall responses to secondary virus infections occur in phases. The memory CD8+ T-cell response to secondary virus infections is comprised of three distinct phases (upper panel). The first phase of comprised of memory cells already resident in the lung airways (bronchoalveolar lavage cells, BAL) that respond immediately to infection. These cells do not proliferate and are largely eliminated by day 4 of the infection through inflammatory mechanisms. The second phase is the antigen non-specific recruitment of non-proliferating memory cells from the circulation. The numbers of cells peak around day 4 and then decline since this is a non-replicating population. Finally, proliferating effector cells generated by antigen-driven stimulation in the LN are recruited after day 4. The combined response is illustrated in the lower panel, which also indicates the fraction of the response that involves proliferating T cells. Note that the late phase of the combined response indicates that new memory cells persist in the lung airways following resolution of the infection.
The key feature of this model is that there are early memory T-cell responses to the virus that occur independently of the antigen-driven proliferation of memory T cells that occurs in lymphoid tissues. While these early T-cell responses are non-renewing (i.e., do not involve T-cell expansion), they are present at the site of infection when viral loads are relatively low. Furthermore, this early response appears to be a critical feature of effective T-cell immunity inasmuch as the strength of this response correlates with the efficacy of viral clearance. For example, it is well established that the relative efficacy of the recall response (in terms of controlling viral loads) declines rapidly over the first year [87], [88], [89]. Interestingly, this decline in efficacy correlates in part with a decline in the numbers of memory T cells in the lung airways suggesting that local T cells play a critical role in mediating protective immunity [12], [14]. This being the case, vaccines designed to promote protective immunity in the lung need to effectively generate memory T-cell populations that elicit these early phases of the T-cell response.
7. Concluding remarks
A major challenge for immunologists is deciphering the underlying mechanisms of immune protection at mucosal surfaces such as the lung epithelium. Over recent years, we have begun to develop a picture outlining the major processes involved. It has now emerged that these responses are highly orchestrated in anatomically distinct sites and regulated by a variety of factors. Furthermore, multiple lymphoid tissues appear to play critical roles, including de novo generated lymphoid sites in the lung parenchyma. Understanding the individual steps of an immune response in the lungs is essential if we are to develop vaccines that elicit effective immunity in the lung.
Acknowledgements
The authors are greatly indebted to Drs. Marcia Blackman and Frances Lund for critically reading the manuscript. This work was supported by the Trudeau Institute and NIH Grants U54AI057158, AI-055500, HL-69502, HL-63925, and HL-69409.
References
- 1.Nguyen-Van-Tam J.S., Hampson A.W. The epidemiology and clinical impact of pandemic influenza. Vaccine. 2003;21:1762–1768. doi: 10.1016/s0264-410x(03)00069-0. [DOI] [PubMed] [Google Scholar]
- 2.Horimoto T., Kawaoka Y. Pandemic threat posed by avian influenza A viruses. Clin Microbiol Rev. 2001;14:129–149. doi: 10.1128/CMR.14.1.129-149.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pearson H., Clarke T., Abbott A., Knight J., Cyranoski D. SARS: what have we learned? Nature. 2003;424:121–126. doi: 10.1038/424121a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gerdil C. The annual production cycle for influenza vaccine. Vaccine. 2003;21:1776–1779. doi: 10.1016/s0264-410x(03)00071-9. [DOI] [PubMed] [Google Scholar]
- 5.Woodland D.L., Hogan R.J., Zhong W. Cellular immunity and memory to respiratory virus infections. Immunol. Res. 2001;24:53–67. doi: 10.1385/IR:24:1:53. [DOI] [PubMed] [Google Scholar]
- 6.Renegar K.B. Influenza virus infections and immunity: a review of human and animal models. Lab. Anim. Sci. 1992;42:222–232. [PubMed] [Google Scholar]
- 7.Bender B.S., Small P.A. Influenza: pathogenesis and host defense. Semin. Respir. Infect. 1992;7:38–45. [PubMed] [Google Scholar]
- 8.Doherty P.C., Hou S., Tripp R.A. CD8+ T-cell memory to viruses. Curr Opin Immunol. 1994;6:545–552. doi: 10.1016/0952-7915(94)90139-2. [DOI] [PubMed] [Google Scholar]
- 9.Flynn K.J., Belz G.T., Altman J.D., Ahmed R., Woodland D.L., Doherty P.C. Virus-specific CD8+ T cells in primary and secondary influenza pneumonia. Immunity. 1998;8:683–691. doi: 10.1016/s1074-7613(00)80573-7. [DOI] [PubMed] [Google Scholar]
- 10.Graham M.B., Braciale T.J. Resistance to and recovery from lethal influenza virus infection in B lymphocyte-deficient mice. J. Exp. Med. 1997;186:2063–2068. doi: 10.1084/jem.186.12.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marshall D.R., Turner S.J., Belz G.T., Wingo S., Andreansky S., Sangster M.Y. Measuring the diaspora for virus-specific CD8+ T cells. Proc Natl Acad Sci USA. 2001;98:6313–6318. doi: 10.1073/pnas.101132698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hogan R.J., Zhong W., Usherwood E.J., Cookenham T., Roberts A.D., Woodland D.L. Protection from respiratory virus infections can be mediated by antigen-specific CD4+ T cells that persist in the lungs. J. Exp. Med. 2001;193:981–986. doi: 10.1084/jem.193.8.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cauley L.S., Hogan R.J., Woodland D.L. Memory T-cells in non-lymphoid tissues. Curr. Opin. Investig Drugs. 2002;3:33–36. [PubMed] [Google Scholar]
- 14.Hogan R.J., Usherwood E.J., Zhong W., Roberts A.A., Dutton R.W., Harmsen A.G. Activated antigen-specific CD8+ T cells persist in the lungs following recovery from respiratory virus infections. J. Immunol. 2001;166:1813–1822. doi: 10.4049/jimmunol.166.3.1813. [DOI] [PubMed] [Google Scholar]
- 15.Zhong W., Roberts A.D., Woodland D.L. Antibody-independent antiviral function of memory CD4+ T cells in vivo requires regulatory signals from CD8+ effector T cells. J. Immunol. 2001;167:1379–1386. doi: 10.4049/jimmunol.167.3.1379. [DOI] [PubMed] [Google Scholar]
- 16.Topham D.J., Cardin R.C., Christensen J.P., Brooks J.W., Belz G.T., Doherty P.C. Perforin and Fas in murine gammaherpesvirus-specific CD8+ T cell control and morbidity. J. Gen Virol. 2001;82:1971–1981. doi: 10.1099/0022-1317-82-8-1971. [DOI] [PubMed] [Google Scholar]
- 17.Topham D.J., Tripp R.A., Doherty P.C. CD8+ T cells clear influenza virus by perforin or Fas-dependent processes. J Immunol. 1997;159:5197–5200. [PubMed] [Google Scholar]
- 18.Diebold S.S., Montoya M., Unger H., Alexopoulou L., Roy P., Haswell L.E. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature. 2003;424:324–328. doi: 10.1038/nature01783. [DOI] [PubMed] [Google Scholar]
- 19.Kurt-Jones E.A., Popova L., Kwinn L., Haynes L.M., Jones L.P., Tripp R.A. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. 2000;1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 20.Akira S., Hemmi H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol. Lett. 2003;85:85–95. doi: 10.1016/s0165-2478(02)00228-6. [DOI] [PubMed] [Google Scholar]
- 21.Tripp R.A., Jones L.P., Haynes L.M., Zheng H., Murphy P.M., Anderson L.J. CX3C chemokine mimicry by respiratory syncytial virus G glycoprotein. Nat Immunol. 2001;2:732–738. doi: 10.1038/90675. [DOI] [PubMed] [Google Scholar]
- 22.Haynes L.M., Moore D.D., Kurt-Jones E.A., Finberg R.W., Anderson L.J., Tripp R.A. Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. J. Virol. 2001;75:10730–10737. doi: 10.1128/JVI.75.22.10730-10737.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moll H. Dendritic cells and host resistance to infection. Cell Microbiol. 2003;5:493–500. doi: 10.1046/j.1462-5822.2003.00291.x. [DOI] [PubMed] [Google Scholar]
- 24.Mellman I., Steinman R.M. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001;106:255–258. doi: 10.1016/s0092-8674(01)00449-4. [DOI] [PubMed] [Google Scholar]
- 25.Legge K.L., Braciale T.J. Accelerated migration of respiratory dendritic cells to the regional lymph nodes is limited to the early phase of pulmonary infection. Immunity. 2003;18:265–277. doi: 10.1016/s1074-7613(03)00023-2. [DOI] [PubMed] [Google Scholar]
- 26.Vermaelen K.Y., Carro-Muino I., Lambrecht B.N., Pauwels R.A. Specific migratory dendritic cells rapidly transport antigen from the airways to the thoracic lymph nodes. J. Exp. Med. 2001;193:51–60. doi: 10.1084/jem.193.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Norbury C.C., Malide D., Gibbs J.S., Bennink J.R., Yewdell J.W. Visualizing priming of virus-specific CD8+ T cells by infected dendritic cells in vivo. Nat. Immunol. 2002;3:265–271. doi: 10.1038/ni762. [DOI] [PubMed] [Google Scholar]
- 28.Roman E., Miller E., Harmsen A., Wiley J., Adrian U.H.V., Huston G. CD4 effector T cell subsets in the response to influenza: heterogeniety migration and function. J. Exp. Med. 2002;196:957–968. doi: 10.1084/jem.20021052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu H.Y., Nguyen H.H., Russell M.W. Nasal lymphoid tissue (NALT) as a mucosal immune inductive site. Scand. J. Immunol. 1997;46:506–513. doi: 10.1046/j.1365-3083.1997.d01-159.x. [DOI] [PubMed] [Google Scholar]
- 30.van der Ven I., Sminia T. The development and structure of mouse nasal-associated lymphoid tissue: an immuno- and enzyme-histochemical study. Regul. Immunol. 1993;5:69–75. [PubMed] [Google Scholar]
- 31.Bienenstock J., Johnston N., Perey D.Y. Bronchial lymphoid tissue. II. Functional characteristics. Lab Invest. 1973;28:693–698. [PubMed] [Google Scholar]
- 32.Bienenstock J., Johnston N., Perey D.Y. Bronchial lymphoid tissue. I. Morphologic characteristics. Lab Invest. 1973;28:686–692. [PubMed] [Google Scholar]
- 33.Sminia T., van der Brugge-Gamelkoorn G.J., Jeurissen S.H. Structure and function of bronchus-associated lymphoid tissue (BALT) Crit. Rev. Immunol. 1989;9:119–1150. [PubMed] [Google Scholar]
- 34.Bienenstock J., Johnston N. A morphologic study of rabbit bronchial lymphoid aggregates and lymphoepithelium. Lab Invest. 1976;35:343–348. [PubMed] [Google Scholar]
- 35.Tenner-Racz K., Racz P., Myrvik Q.N., Ockers J.R., Geister R. Uptake and transport of horseradish peroxidase by lymphoepithelium of the bronchus-associated lymphoid tissue in normal and bacillus Calmette-Guerin-immunized and challenged rabbits. Lab Invest. 1979;41:106–115. [PubMed] [Google Scholar]
- 36.Wu H.Y., Nikolova E.B., Beagley K.W., Russell M.W. Induction of antibody-secreting cells and T-helper and memory cells in murine nasal lymphoid tissue. Immunology. 1996;88:493–500. doi: 10.1046/j.1365-2567.1996.d01-690.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tamura S., Iwasaki T., Thompson A.H., Asanuma H., Chen Z., Suzuki Y. Antibody-forming cells in the nasal-associated lymphoid tissue during primary influenza virus infection. J Gen Virol. 1998;79:291–299. doi: 10.1099/0022-1317-79-2-291. [DOI] [PubMed] [Google Scholar]
- 38.Porgador A., Staats H.F., Itoh Y., Kelsall B.L. Intranasal immunization with cytotoxic T-lymphocyte epitope peptide and mucosal adjuvant cholera toxin: selective augmentation of peptide-presenting dendritic cells in nasal mucosa-associated lymphoid tissue. Infect Immun. 1998;66:5876–5881. doi: 10.1128/iai.66.12.5876-5881.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Asanuma H., Aizawa C., Kurata T., Tamura S. IgA antibody-forming cell responses in the nasal-associated lymphoid tissue of mice vaccinated by intranasal intravenous and/or subcutaneous administration. Vaccine. 1998;16:1257–1262. doi: 10.1016/s0264-410x(98)00048-6. [DOI] [PubMed] [Google Scholar]
- 40.Shimoda M., Nakamura T., Takahashi Y., Asanuma H., Tamura S., Kurata T. Isotype-specific selection of high affinity memory B cells in nasal-associated lymphoid tissue. J Exp Med. 2001;194:1597–1607. doi: 10.1084/jem.194.11.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Staats H.F., Bradney C.P., Gwinn W.M., Jackson S.S., Sempowski G.D., Liao H.X. Cytokine requirements for induction of systemic and mucosal CTL after nasal immunization. J. Immunol. 2001;167:5386–5394. doi: 10.4049/jimmunol.167.9.5386. [DOI] [PubMed] [Google Scholar]
- 42.Harmsen A., Kusser K., Hartson L., Tighe M., Sunshine M.J., Sedgwick J.D. Cutting edge: organogenesis of nasal-associated lymphoid Tissue (NALT) occurs independently of lymphotoxin-a (LTa) and retinoic acid receptor-related orphan receptor-g, but the organization of NALT in LTa dependent. J. Immunol. 2002;168:986–990. doi: 10.4049/jimmunol.168.3.986. [DOI] [PubMed] [Google Scholar]
- 43.Zuercher A.W., Coffin S.E., Thurnheer M.C., Fundova P., Cebra J.J. Nasal-associated lymphoid tissue is a mucosal inductive site for virus-specific humoral and cellular immune responses. J. Immunol. 2002;168:1796–1803. doi: 10.4049/jimmunol.168.4.1796. [DOI] [PubMed] [Google Scholar]
- 44.Plesch B.E., Gamelkoorn G.J., van de Ende M. Development of bronchus associated lymphoid tissue (BALT) in the rat, with special reference to T- and B-cells. Dev. Comp. Immunol. 1983;7:179–188. doi: 10.1016/0145-305x(83)90066-6. [DOI] [PubMed] [Google Scholar]
- 45.Delventhal S., Hensel A., Petzoldt K., Pabst R. Effects of microbial stimulation on the number, size and activity of bronchus-associated lymphoid tissue (BALT) structures in the pig. Int J Exp Pathol. 1992;73:351–357. [PMC free article] [PubMed] [Google Scholar]
- 46.Pabst R., Gehrke I. Is the bronchus-associated lymphoid tissue (BALT) an integral structure of the lung in normal mammals, including humans? Am J. Respir Cell Mol. Biol. 1990;3:131–135. doi: 10.1165/ajrcmb/3.2.131. [DOI] [PubMed] [Google Scholar]
- 47.Tschernig T., Pabst R. Bronchus-associated lymphoid tissue (BALT) is not present in the normal adult lung but in different diseases. Pathobiology. 2000;68:1–8. doi: 10.1159/000028109. [DOI] [PubMed] [Google Scholar]
- 48.Delventhal S., Brandis A., Ostertag H., Pabst R. Low incidence of bronchus-associated lymphoid tissue (BALT) in chronically inflamed human lungs. Virchows Arch. B Cell Pathol. Mol. Pathol. 1992;62:271–274. doi: 10.1007/BF02899692. [DOI] [PubMed] [Google Scholar]
- 49.Chvatchko Y., Kosco-Vilbois M.H., Herren S., Lefort J., Bonnefoy J.Y. Germinal center formation and local immunoglobulin E (IgE) production in the lung after an airway antigenic challenge. J. Exp. Med. 1996;184:2353–2360. doi: 10.1084/jem.184.6.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lund F.E., Partida-Sanchez S., Lee B.O., Kusser K.L., Hartson L., Hogan R.J. Lymphotoxin-alpha-deficient mice make delayed, but effective, T and B cell responses to influenza. J. Immunol. 2002;169:5236–5243. doi: 10.4049/jimmunol.169.9.5236. [DOI] [PubMed] [Google Scholar]
- 51.Brodie S.J., de la Rosa C., Howe J.G., Crouch J., Travis W.D., Diem K. Pediatric AIDS-associated lymphocytic interstitial pneumonia and pulmonary arterio-occlusive disease: role of VCAM-1/VLA-4 adhesion pathway and human herpesviruses. Am. J. Pathol. 1999;154:1453–1464. doi: 10.1016/S0002-9440(10)65400-4. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 52.Goodnow C.C. Chance encounters and organized rendezvous. Immunol Rev. 1997;156:5–10. doi: 10.1111/j.1600-065x.1997.tb00954.x. [DOI] [PubMed] [Google Scholar]
- 53.Chen H.D., Fraire A.E., Joris I., Brehm M.A., Welsh R.M., Selin L.K. Memory CD8+ T cells in heterologous antiviral immunity and immunopathology in the lung. Nat Immunol. 2001;2:1067–1076. doi: 10.1038/ni727. [DOI] [PubMed] [Google Scholar]
- 54.Constant S.L., Brogdon J.L., Piggott D.A., Herrick C.A., Visintin I., Ruddle N.H. Resident lung antigen-presenting cells have the capacity to promote Th2 T cell differentiation in situ. J Clin Invest. 2002;110:1441–1448. doi: 10.1172/JCI16109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Futterer A., Mink K., Luz A., Kosco-Vilbois M.H., Pfeffer K. The lymphotoxin beta receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity. 1998;9:59–70. doi: 10.1016/s1074-7613(00)80588-9. [DOI] [PubMed] [Google Scholar]
- 56.Butcher E.C., Rouse R.V., Coffman R.L., Nottenburg C.N., Hardy R.R., Weissman I.L. Surface phenotype of Peyer’s patch germinal center cells: implications for the role of germinal centers in B cell differentiation. J. Immunol. 1982;129:2698–2707. [PubMed] [Google Scholar]
- 57.Coffman R.L., Lebman D.A., Shrader B. Transforming growth factor beta specifically enhances IgA production by lipopolysaccharide-stimulated murine B lymphocytes. J. Exp. Med. 1989;170:1039–1044. doi: 10.1084/jem.170.3.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shimada S., Kawaguchi-Miyashita M., Kushiro A., Sato T., Nanno M., Sako T. Generation of polymeric immunoglobulin receptor-deficient mouse with marked reduction of secretory IgA. J. Immunol. 1999;163:5367–5373. [PubMed] [Google Scholar]
- 59.Johansen F.E., Pekna M., Norderhaug I.N., Haneberg B., Hietala M.A., Krajci P. Absence of epithelial immunoglobulin A transport with increased mucosal leakiness, in polymeric immunoglobulin receptor/secretory component-deficient mice. J. Exp. Med. 1999;190:915–922. doi: 10.1084/jem.190.7.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Palladino G., Mozdzanowska K., Washko G., Gerhard W. Virus-neutralizing antibodies of immunoglobulin G (IgG) but not of IgM or IgA isotypes can cure influenza virus pneumonia in SCID mice. J. Virol. 1995;69:2075–2081. doi: 10.1128/jvi.69.4.2075-2081.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mbawuike I.N., Pacheco S., Acuna C.L., Switzer K.C., Zhang Y., Harriman G.R. Mucosal immunity to influenza without IgA: an IgA knockout mouse model. J. Immunol. 1999;162:2530–2537. [PubMed] [Google Scholar]
- 62.Butcher E.C., Williams M., Youngman K., Rott L., Briskin M. Lymphocyte trafficking and regional immunity. Adv Immunol. 1999;72:209–253. doi: 10.1016/s0065-2776(08)60022-x. [DOI] [PubMed] [Google Scholar]
- 63.Mora J.R., Bono M.R., Manjunath N., Weninger W., Cavanagh L.L., Rosemblatt M. Selective imprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature. 2003;424:88–93. doi: 10.1038/nature01726. [DOI] [PubMed] [Google Scholar]
- 64.Berlin C., Berg E.L., Briskin M.J., Andrew D.P., Kilshaw P.J., Holzmann B. Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell. 1993;74:185–185. doi: 10.1016/0092-8674(93)90305-a. [DOI] [PubMed] [Google Scholar]
- 65.Svensson M., Marsal J., Ericsson A., Carramolino L., Broden T., Marquez G. CCL25 mediates the localization of recently activated CD8ab+ lymphocytes to the small-intestinal mucosa. J. Clin. Invest. 2002;110:1113–1121. doi: 10.1172/JCI15988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McDermott M.R., Bienenstock J. Evidence for a common mucosal immunologic system. I. Migration of B immunoblasts into intestinal respiratory and genital tissues. J Immunol. 1979;122:1892–1898. [PubMed] [Google Scholar]
- 67.Csencsits K.L., Walters N., Pascual D.W. Cutting edge: dichotomy of homing receptor dependence by mucosal effector B cells: Eα versus l-selectin. J. Immunol. 2001;167:2441–2445. doi: 10.4049/jimmunol.167.5.2441. [DOI] [PubMed] [Google Scholar]
- 68.Csencsits K.L., Jutila M.A., Pascual D.W. Nasal-associated lymphoid tissue: phenotypic and functional evidence for the primary role of peripheral node addressin in naive lymphocyte adhesion to high endothelial venules in a mucosal site. J. Immunol. 1999;163:1382–1389. [PubMed] [Google Scholar]
- 69.Xu B., Wagner N., Pham L.N., Magno V., Shan Z., Butcher E.C. Lymphocyte homing to bronchus-associated lymphoid tissue (BALT) is mediated by L-selectin/PNAd alpha4beta1 integrin/VCAM-1 and LFA-1 adhesion pathways. J. Exp. Med. 2003;197:1255–1267. doi: 10.1084/jem.20010685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dutton R.W., Bradley L.M., Swain S.L. T cell memory. Annu Rev Immunol. 1998;16:201–223. doi: 10.1146/annurev.immunol.16.1.201. [DOI] [PubMed] [Google Scholar]
- 71.Masopust D., Vezys V., Marzo A.L., Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 2001;291:2413–2417. doi: 10.1126/science.1058867. [DOI] [PubMed] [Google Scholar]
- 72.Seder R.A., Ahmed R. Similarities and differences in CD4+ and CD8+ effector and memory T cell generation. Nat Immunol. 2003;4:835–842. doi: 10.1038/ni969. [DOI] [PubMed] [Google Scholar]
- 73.Schluns K.S., Lefrancois L. Cytokine control of memory T-cell development and survival. Nat. Rev. Immunol. 2003;3:269–279. doi: 10.1038/nri1052. [DOI] [PubMed] [Google Scholar]
- 74.Harris N.L., Watt V., Ronchese F., Le Gros G. Differential T cell function and fate in lymph node and nonlymphoid tissues. J. Exp. Med. 2002;195:317–326. doi: 10.1084/jem.20011558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sallusto F., Lenig D., Forster R., Lipp M., Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 76.Reinhardt R.L., Khoruts A., Merica R., Zell T., Jenkins M.K. Visualizing the generation of memory CD4 T cells in the whole body. Nature. 2001;410:101–105. doi: 10.1038/35065111. [DOI] [PubMed] [Google Scholar]
- 77.Wherry E.J., Teichgraber V., Becker T.C., Masopust D., Kaech S.M., Antia R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol. 2003;3:3. doi: 10.1038/ni889. [DOI] [PubMed] [Google Scholar]
- 78.Ely K.H., Roberts A.D., Woodland D.L. Cutting edge: effector memory CD8+ T cells in the lung airways retain the potential to mediate recall responses. J. Immunol. 2003;171:3338–3342. doi: 10.4049/jimmunol.171.7.3338. [DOI] [PubMed] [Google Scholar]
- 79.Chalasani G., Dai Z., Konieczny B.T., Baddoura F.K., Lakkis F.G. Recall and propagation of allospecific memory T cells independent of secondary lymphoid organs. Proc Natl Acad Sci USA. 2002;99:6175–6180. doi: 10.1073/pnas.092596999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Topham D.J., Castrucci M.R., Wingo F.S., Belz G.T., Doherty P.C. The role of antigen in the localization of naïve, acutely activated, and memory CD8+ T cells to the lung during influenza pneumonia. J Immunol. 2001;167:6983–6990. doi: 10.4049/jimmunol.167.12.6983. [DOI] [PubMed] [Google Scholar]
- 81.Ely K.H., Cauley L.S., Roberts A.D., Brennan J.W., Cookenham T., Woodland D.L. Nonspecific recruitment of memory CD8+ T cells to the lung airways during respiratory virus infections. J Immunol. 2003;170:1423–1429. doi: 10.4049/jimmunol.170.3.1423. [DOI] [PubMed] [Google Scholar]
- 82.Wiley J.A., Hogan R.J., Woodland D.L., Harmsen A.G. Antigen-specific CD8+ T cells persist in the upper respiratory tract following influenza virus infection. J Immunol. 2001;167:3293–3299. doi: 10.4049/jimmunol.167.6.3293. [DOI] [PubMed] [Google Scholar]
- 83.Ostler T., Hussell T., Surh C.D., Openshaw P., Ehl S. Long-term persistence and reactivation of T cell memory in the lung of mice infected with respiratory syncytial virus. Eur J Immunol. 2001;31:2574–2582. doi: 10.1002/1521-4141(200109)31:9<2574::aid-immu2574>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 84.Hogan R.J., Cauley L.S., Ely K.H., Cookenham T., Roberts A.D., Brennan J.W. Long-term maintenance of virus-specific effector memory CD8+ T cells in the lung airways depends on proliferation. J. Immunol. 2002;169:4976–4981. doi: 10.4049/jimmunol.169.9.4976. [DOI] [PubMed] [Google Scholar]
- 85.Lefrancois L. Dual personality of memory T cells. Trends Immunol. 2002;23:226–228. doi: 10.1016/s1471-4906(02)02190-7. [DOI] [PubMed] [Google Scholar]
- 86.Cauley L., Cookenham T., Hogan R., Crowe S., Woodland D. Renewal of peripheral CD8 memory T cells during secondary viral infection of antibody sufficient mice. J Immunol. 2003;170:5597–5606. doi: 10.4049/jimmunol.170.11.5597. [DOI] [PubMed] [Google Scholar]
- 87.Sonoguchi T., Naito H., Hara M., Takeuchi Y., Fukumi H. Cross-subtype protection in humans during sequential, overlapping. J. Infect Dis. 1985;151:81–88. doi: 10.1093/infdis/151.1.81. [DOI] [PubMed] [Google Scholar]
- 88.Frank A.L., Taber L.H., Wells J.M. Individuals infected with two subtypes of influenza A virus in the same season. J. Infect Dis. 1983;147:120–124. doi: 10.1093/infdis/147.1.120. [DOI] [PubMed] [Google Scholar]
- 89.Woodland D., Ely K., Crowe S., Tighe M., Brennan J., Harmsen A. Antiviral memory T-cell responses in the lung. Microbes Infect. 2002;4:1091. doi: 10.1016/s1286-4579(02)01633-7. [DOI] [PubMed] [Google Scholar]