

Abstract
Previous estimates of a generic level phylogeny for the ubiquitous parasitoid wasp subfamily Microgastrinae (Hymenoptera) have been problematic due to short internal branches deep in the phylogeny. These short branches might be attributed to a rapid radiation among the taxa, the use of genes that are unsuitable for the levels of divergence being examined, or insufficient quantity of data. We added over 1200 nucleotides from four nuclear genes to a dataset derived from three genes to produce a dataset of over 3000 nucleotides per taxon. While the number of well-supported short branches in the phylogeny increased, we still did not obtain strong bootstrap support for every node. Parametric and nonparametric bootstrap simulations projected that an enormous, and likely unobtainable, amount of data would be required to get bootstrap support greater than 50% for every node. However, a marked increase in the number of well-supported nodes was seen when we conducted a Bayesian analysis of a combined dataset generated from morphological characters added to the seven gene dataset. Our results suggest that, in some cases, combining morphological and genetic characters may be the most practical way to increase support for short branches deep in a phylogeny.
Keywords: Bayesian, Braconidae, Hymenoptera, Morphology, Support
1. Introduction
Uncertainty in phylogenetic estimation at higher taxonomic levels is inevitable, due to the confounding effects of factors that may indicate alternative patterns. These factors include the convergence of morphological characters from similar ecological forces, and multiple substitutions in genetic data (“saturation”). Convergence and saturation often result in low bootstrap support values, poor Bremer decay indices or low Bayesian posterior probability values for some branches (Swofford et al., 1996). However, poor branch support can also be caused by failure to use a sufficient quantity of data (Fishbein et al., 2001), use of data that are inappropriate for the level of divergence that is being analysed (de Queiroz et al., 1995), or rapid evolutionary radiations among taxa (Fishbein et al., 2001). Often, it is difficult to know which factors are operating in any particular case.
Although phylogenies without strong support for all branches are sometimes well accepted, there are situations, such as the study of cophylogenetic relationships between hosts and associates when well-supported phylogenies are important. For example, reconciliation analysis (Page, 1995), the method most commonly used to examine cophylogenetic relationships (Brooks and McLennan, 2003), infers cophylogenetic history from the topology of the host and associate phylogenies and thus requires robust phylogenies to reconstruct the evolutionary history of the relationship between hosts and associates. Other situations requiring robust phylogenies include the forensic use of phylogenies to identify the source of infections such as human immunodeficiency virus (Korber et al., 2000, Rambaut et al., 2001, Worobey et al., 2004) and severe acute respiratory syndrome (SARS) (Guan et al., 2003).
One example of poor support possibly caused by several factors occurs in the phylogenies estimated for microgastrine wasps (Whitfield et al., 2002). Microgastrinae, a subfamily of Braconidae (Hymenoptera), is a speciose group with approximately 1400 described species in over 55 genera, and it has been estimated that there may actually be 5000 to 10,000 species worldwide (Whitfield, 1997b, Whitfield et al., 2002). Microgastrine wasps lay their eggs on lepidopteran larvae, and the wasp larvae develop while consuming the tissues of the lepidopteran larvae (Whitfield, 1997b, Whitfield et al., 2002). Many microgastrine wasp species have been transferred around the world to aid in the control of crop pests (Whitfield, 1997b, Whitfield et al., 2002). All microgastrine wasps have inherited an association with polydnaviruses, which are incorporated into the wasp genomes and help the wasp larvae evade lepidopteran immune systems (Whitfield and Asgari, 2003). It has therefore been of considerable coevolutionary interest to compare the phylogenetic histories of the wasps and those of the viruses. A robust phylogenetic framework is essential for producing a useful and informative classification for this large, economically and ecologically important insect group.
Previous work that estimated a phylogeny for the microgastrines from 2300 nucleotides from three genes (16S, 28S and COI) and 53 morphological characters found a tree with low bootstrap support for many branches (Mardulyn and Whitfield, 1999, Whitfield et al., 2002). The poorly supported branches in the microgastrine phylogenies are mainly short internal branches (Mardulyn and Whitfield, 1999, Whitfield et al., 2002). It was proposed that the short branches might have arisen from a rapid radiation as the microgastrines colonised new lepidopteran host species (Mardulyn and Whitfield, 1999), which themselves may have been diversifying in the early Tertiary (Grimaldi, 1999, Whitfield, 2002). Support for the rapid radiation of microgastrines was bolstered by the fact that the same branches were estimated to be short from multiple data sources. However, it was also acknowledged that the poorly supported short branches may have been due to insufficient data or the use of genes with rates of divergence that are inappropriate for the levels of divergence between the taxa (Mardulyn and Whitfield, 1999).
Here we present analyses of data from two mitochondrial and five nuclear genes, including the genetic data (16S, 28S and COI) and 53 morphological characters analysed by Whitfield et al. (2002). These analyses show that completely robustly supported phylogenies for Microgastrinae are unlikely to be estimated from genetic data alone. We use parametric and nonparametric bootstrapping of simulated datasets to estimate how much data would be required to resolve the phylogeny with every branch having nonparametric bootstrap values greater than 50%. The simulations show that unless an impractically large amount of molecular data is obtained, the use of morphological characters may be necessary to produce a completely robustly supported phylogeny that can be used to examine cophylogenetic relationships between microgastrine wasps and polydnaviruses.
2. Methods
Wasps were stored in 100% ethanol at 4 °C until genomic DNA could be extracted. Specimens were identified by JBW to genus, and to species where possible, using morphological characters and often also host data. Taxa from which sequences were obtained are listed in Table 1 . Because we had few sequences from Apanteles canarsiae we pooled sequences for A. canarsiae with A. galleriae and the resulting “chimera” is labelled Apanteles sp. in the phylogenies. Whole wasps were macerated using mini-mortar and pestles and the DNA extracted using Qiagen DNeasy tissue extraction kits. Polymerase chain reactions (PCR) were carried out with an Eppendorf Mastercycler thermocycler. PCR consisted of 2.5 μL of Hotmaster buffer (Eppendorf), 1.2 μL of dNTPs (8 mM), 2.5 μL of each primer (2.5 μM), 0.125 μL Hotmaster Taq (5 units/μL, Eppendorf), 0.8 μL of DNA and 15.375 μL water. PCRs consisted of an initial denaturing step of 94 °C for 2 min, followed by 35 cycles of 94 °C for 20 s, 20 s at the temperatures listed in Table 2 , 65 °C for 40 to 60 s depending on the size of the target region, and a final step of 65 °C for 5 min Primer sequences are listed in Table 2. A negative control was incorporated in each amplification round using water rather than DNA. PCR products were purified using Qiagen QIAquick kits. Sequencing was carried out on an ABI 3730 capillary sequencer.
Table 1.
Taxa sequenced and Genbank Accession numbers
Where accession numbers are absent, we failed to get sequences for that region of that taxon. Taxa in capitals are outgroups.
Table 2.
Primers used in this study
| Gene | Primer name | Sequence | Annealing temperature (°C) | Reference |
|---|---|---|---|---|
| 16S | 52–57 | |||
| Forward | 16S outer | CTTATTCAACATCGAGGTC | (Whitfield, 1997a) | |
| Reverse | 16SWb | CACCTGTTTATCAAAACAT | (Dowton and Austin, 1994) | |
| 28S | 55–62 | |||
| Forward | 28SF | CACCTGTTTATCAAAAACAT | (Mardulyn and Whitfield, 1999) | |
| Reverse | 28SR | TAGTTCACCATCTTTCGGGTCCC | (Mardulyn and Whitfield, 1999) | |
| Arginine kinase | 47–50 | |||
| Forward | F2 | GACAGCAARTCTCTGCTGAAGAA | (Kawakita et al., 2003) | |
| Forward | intF | GTNTCNACYCGTGRAGATGYGG | This study | |
| Reverse | R2 | GGTYTTGGCATCGTTGTGGTAGATAC | (Kawakita et al., 2003) | |
| Reverse | intR | AGRGTRTCRRCRTCDCCRAAGTC | This study | |
| COI | 50–53 | |||
| Forward | LCO1490 | GGTCAACAAATCATAAAGATATTGG | (Folmer et al., 1994) | |
| Reverse | HCO2198 | TAAACTTCAGGGTGACCAAAAAATCA | (Folmer et al., 1994) | |
| EF1a | 47–52 | |||
| Forward | EF1A1F | AGATGGGYAARGGTTCCTTCAA | (Belshaw and Quicke, 1997) | |
| Reverse | EF1A1R | AACATGTTGTCDCCGTGCCATCC | (Belshaw and Quicke, 1997) | |
| Rhodopsin | 47–55 | |||
| Forward | OpsFor2 | GGATGTASCTCCATTTGGTC | This study | |
| Reverse | Ops3′2 | AVHGATGCRACRTTCATTTTCT | This study | |
| Wingless | 47–53 | |||
| Forward | Wg1 | GARTGYAARTGYCAYGGYATGTCTGG | (Brower and DeSalle, 1998) | |
| Reverse | Wg2 | ACTICGCRCACCARTGGAATGTRCA | (Brower and DeSalle, 1998) |
2.1. Gene selection
The three genes, 16S, COI and 28S, originally used for this group were selected for their broad use among different groups of insects, ease of amplification across all taxa and because they provide resolution at several phylogenetic levels. 16S has been used to resolve intra-family relationships in Hymenoptera (Whitfield and Cameron, 1998). The nuclear gene 28S (including the D2 and D3 expansion loops) has provided a strong signal for intermediate and moderately deep levels in the phylogeny (Belshaw et al., 1998, Belshaw and Quicke, 1997, Cameron and Mardulyn, 2001, Cameron and Williams, 2003, Dowton and Austin, 1998, Mardulyn and Whitfield, 1999, Michel-Salzat and Whitfield, 2004, Whitfield, 2002, Whitfield et al., 2002, Wiegmann et al., 2003) and retains at least some signal at the species level. COI has been found to saturate quickly at the third position while remaining quite “conserved” at the first two positions due to a small number of sites free to vary (Mardulyn and Whitfield, 1999). Thus, it has proven highly useful at lower levels to detect species boundaries (Hebert et al., 2003a, Hebert et al., 2004, Hebert et al., 2003b) but has tended to fail at higher levels, especially in divergence time estimation studies (e.g., Whitfield, 2002).
We added sequences from four nuclear genes to the dataset of Whitfield et al. (2002). Arginine kinase has been used to resolve bee relationships at species and tribal level (Danforth et al., 2005, Kawakita et al., 2003). The nuclear gene EF1α has been used extensively to resolve lepidopteran relationships at intermediate phylogenetic levels (Cho et al., 1995, Friedlander et al., 1998, Mitchell et al., 1997, Mitchell et al., 2000, Mitchell et al., 2006, Wiegmann et al., 2000) and has been used recently in a number of studies on Hymenoptera (Cameron, 2003, Danforth et al., 2004, Danforth and Ji, 1998, Kawakita et al., 2003, Leys et al., 2002, Michel-Salzat et al., 2004). The gene occurs in at least two divergent copies in most Hymenoptera (originally reported by Danforth and Ji, 1998), but these copies are easily separated by PCR once taxon-specific primers are developed. We used primers that amplify the F2 copy in bumble bees (Kawakita et al., 2003).
Long wavelength rhodopsin (opsin) has been used to resolve relationships among bees (Mardulyn and Cameron, 1999), and is especially useful for intermediate levels of phylogeny (from species up to intergeneric and tribal levels—Cameron and Mardulyn, 2001, Cameron and Mardulyn, 2003, Cameron and Williams, 2003, Danforth et al., 2004, Kawakita et al., 2003, Michel-Salzat et al., 2004, Michel-Salzat and Whitfield, 2004), despite early reservations (Ascher et al., 2001). Wingless is less variable than many mtDNA genes, but more variable than most of the other nuclear protein-coding genes we sequenced in this study. Thus wingless tends to be useful at the generic level rather than at higher hierarchical levels (Brower and DeSalle, 1998).
2.2. Alignment
Sequences were aligned with Clustal X (Thompson et al., 1997). Alignment of COI, EF1α, and wingless sequences was straightforward as there were few insertions or deletions. The alignment of arginine kinase and opsin sequences was straightforward once an intron in each gene had been removed. There were several variable length-regions of 16S and 28S where it was difficult to assign homology. Regions of 16S and 28S that could not be aligned unambiguously were omitted from the analysis.
2.3. Testing for incongruence
We tested for incongruence between genes using the incongruence length difference test (Farris et al., 1994, Farris et al., 1995) implemented in PAUP*4.0b10 (Swofford, 2002) as the partition homogeneity test. We conducted 100 replicates and compared genes in both a pairwise manner and each gene to the rest of the combined sequence data with that gene excluded. Parsimony uninformative characters were removed before each test. We also tested the data for stationarity (equal nucleotide proportions between taxa) using the χ 2 test in PAUP*.
2.4. Phylogeny estimation
We used PAUP* to conduct maximum parsimony (MP), maximum likelihood (ML) and LogDet phylogenetic analyses. LogDet is a distance based method that is less affected than MP when taxa differ in their base frequencies (Lockhart et al., 1994). The Akaike Information Criterion as implemented in ModelTest 3.06 (Posada, 2000, Posada and Crandall, 1998) was used to select the model and estimate model parameters (GTR + gamma + proportion of invariable sites (Rodríguez et al., 1990, Tavaré, 1986, Yang et al., 1994); base frequencies A = 0.3166, C = 0.1589, G = 0.1819; rate matrix AC = 1.7463, AG = 11.0123, AT = 8.9781, CG = 2.1939, CT = 14.8349; γ = 0.6963; proportion of invariable sites = 0.3614) from all seven genes combined for the ML analysis.
MrBayes 3.1 (Huelsenbeck and Ronquist, 2001, Ronquist and Huelsenbeck, 2003) was used to generate Bayesian estimates of microgastrine phylogeny. We used a mixed model approach with eight partitions corresponding to the morphological characters and the seven gene regions. The models used for each of the seven genes were GTR (Tavaré, 1986) plus a proportion invariable sites plus gamma (Rodríguez et al., 1990, Yang et al., 1994). MrBayes estimated the model parameters from the data using one cold and three heated Markov chains. The Monte Carlo Markov chain length was 2,000,000 generations and we sampled the chain every 100 generations. We discarded the first 5000 samples as burnin and thus estimated our phylogeny and posterior probabilities from a consensus of the last 15,000 sampled trees.
2.5. Assessing the effect of branch length on bootstrap support
To compare the branch lengths of branches with bootstrap support greater than 50% to branches with less than 50% support, we reduced our data set to the 27 taxa for which we had data for all seven genes and estimated the phylogeny for the 27 taxa under the MP criterion. The MP analysis found three most parsimonious trees. We then loaded one of the three most parsimonious trees found from the MP analysis into PAUP* as a constraint tree and used MP to estimate the branch length for the constraint tree for individual genes. Branches for the constraint tree were categorised as either bootstrap support >50% or <50% and branch lengths for the branches for each gene were recorded and compared using a Student’s t test in Systat 9 (SPSS, 1998).
2.6. Assessing the amount of data needed
Pseudoreplicate datasets one and a half, two, three, four, five and 10 times the size of our dataset were constructed from the aligned data by altering the number of characters re-sampled in the nonparametric bootstrap command of PAUP*. These pseudoreplicate datasets were then analysed under the MP criterion and bootstrapped to estimate the amount of data that would be required to estimate phylogenies with all nodes having bootstrap support greater than 50%. Pseudoreplicate-data sets one and a half, two, three, four, five and 10 times the size of our dataset were also generated using a parametric approach with Seq-Gen (Rambaut and Grassly, 1997) from the ML equation calculated from the original data by Modeltest 3.06. Nonparametric bootstrap values were then obtained with PAUP* from the MP trees estimated from the data sets produced by Seq-Gen. This approach assumes that the data added will have similar properties to the data already obtained. This seems a valid assumption given that the genes we sequenced cover a range of evolutionary rates.
2.7. Assessing the effect of number of taxa
To assess the effect of altering the number of taxa, we randomly deleted taxa from our actual dataset to give datasets containing 15, 20, 25, 30, 35 and 40 taxa. We then obtained nonparametric bootstrap values for the branches from 100 replicates using MP.
3. Results
We added 1248 nucleotides to the previously published dataset and analysed a total of 3031 nucleotides (including gaps). We used primers that bound to a more conserved region of COI and thus reduced the previously published COI sequences (Mardulyn and Whitfield, 1999) from 1235 nucleotides to 419 nucleotides that were homologous with our sequences. Levels of variation between species and genera for the seven genes differed with 28S, arginine kinase, EF1α, opsin and wingless diverging more slowly than 16S and COI, which quickly saturated at the generic level (Fig. 1 ).
Fig. 1.
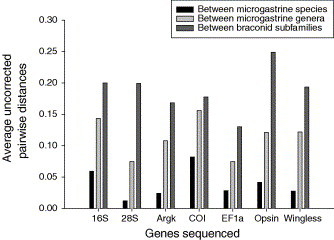
Average uncorrected pairwise distances between microgastrine species and genera, and braconid subfamilies for the seven genes sequenced. Arginine kinase (Argk), elongation factor 1 alpha (EF1α), opsin and wingless are the genes added to the original dataset of Mardulyn and Whitfield, 1999, Whitfield, 2002.
The ILD test found 17 of the 21 pairwise comparisons of the genes were significantly incongruent (P ⩽ 0.01). The four comparisons that were not significantly incongruent were 28S to arginine kinase, EF1α and wingless, and EF1α to 16S. Six of the seven comparisons of individual genes to the rest of the combined molecular data (with each individual gene excluded) revealed significant differences (P = 0.01). The exception was arginine kinase, which was not significantly heterogeneous with the combined data (P = 0.6).
The χ 2 test of sequence stationarity in PAUP* found there was significant heterogeneity in nucleotide proportions. A nonsignificant result was obtained when the third positions of codons in protein coding genes were excluded.
A maximum parsimony analysis of all seven genes for all taxa found three equally parsimonious trees of length 6861; consistency index, excluding uninformative characters = 0.30; retention index = 0.44 from 3031 characters of which 1494 were constant and 1207 were parsimony informative. The strict consensus tree of the three most parsimonious trees is shown in Fig. 2 .
Fig. 2.
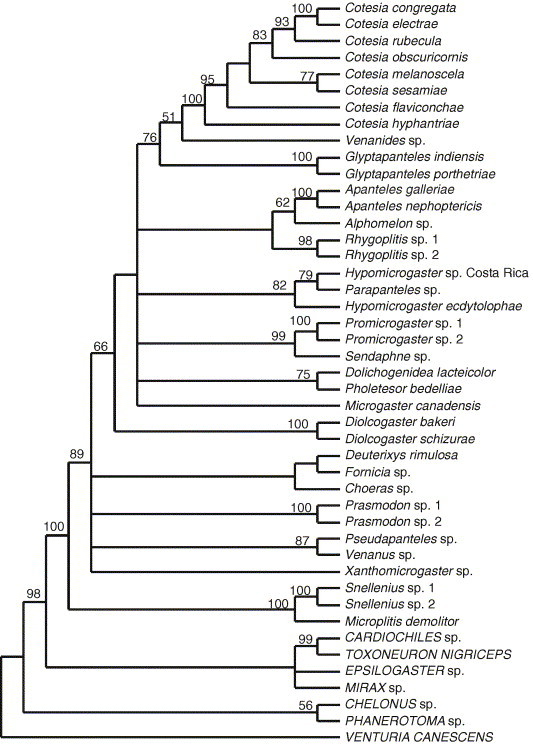
Strict consensus of the three most parsimonious trees (tree length 6861; consistency index, excluding uninformative characters, 0.30; retention index 0.44) obtained from a MP analysis of 3031 nucleotides from seven genes. Numbers above the branches are percentage bootstrap support values >50 (from 100 replicates).
A MP analysis found a single most parsimonious tree when third positions were excluded (result not shown) that was broadly congruent with the MP tree estimated from all positions (Fig. 2). Excluding third positions did not appreciably alter bootstrap support values obtained with MP, as 15 branches still had bootstrap support of less than 50%. Deleting third positions altered relationships found by MP within more recently diverged clades, but most deep and mid level relationships were not altered. The exception was that Choeras was placed as sister to Sendaphne and Promicrogaster, rather than with Fornicia and Deuterixys, when third positions were excluded, a more reasonable result based on morphology.
Both MP and Bayesian methods (Fig. 2, Fig. 3 ) supported Microgastrinae as a monophyletic group. Both methods found broadly similar relationships. However, the placement of Fornicia differed greatly depending on the method of analysis. Maximum parsimony placed Fornicia as a sister taxon to Deuterixys rimulosa whereas the Bayesian analysis placed Fornicia in a clade with Hypomicrogaster and Parapanteles. We were unable to get sequences from three genes for Fornicia, and it is possible that missing data are causing the two methods to differ in their placement of Fornicia.
Fig. 3.
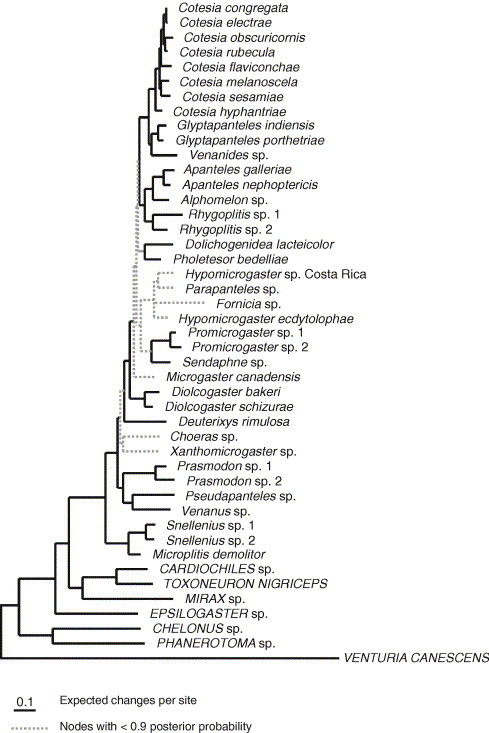
Majority rule consensus tree from 15,000 trees estimated from seven genes using MrBayes 3.1. Broken lines represent branches with posterior probabilities of less than 0.9.
The numbers of branches with high or low levels of support differed slightly for the phylogenies estimated using MP and Bayes. Maximum parsimony had 14 branches with bootstrap support of less than 50%, whereas the Bayesian tree had 10 branches with posterior probability values of less than 0.9. There was no obvious trend for branches to have higher Bayesian posterior probabilities than MP bootstrap support values. Some branches were supported with posterior probabilities higher than 0.9 but had low bootstrap support, while some branches that had posterior probabilities much less than 0.9 received high bootstrap support values. For example, grouping Hypomicrogaster ecdytolophae and Parapanteles sp. as sisters received a bootstrap value of 79% but had a posterior probability of 0.56. It must of course be kept in mind that in these comparisons, both the branch support measure and the optimality criterion for tree estimation differ.
The LogDet analysis found a single tree (result not shown) that was broadly congruent with both the MP and Bayesian trees. Nine nodes had bootstrap support of less than 50% (similar to the other methods). Nodes in the LogDet tree that differed from the MP and Bayesian trees did not have high levels of bootstrap support.
3.1. The effect of branch length on bootstrap support
Branches with bootstrap support of <50% in phylogenies obtained from the individual genes were significantly shorter than branches with bootstrap support >50% (meanbootstrap <50% = 5.5, SE = 0.7; meanbootstrap >50% = 10.5, SE = 0.7; Student’s t = 5.21, df = 127.3, P < 0.001) in the phylogeny (Fig. 4 ) estimated for the 27 taxa for which we had sequences for all seven genes.
Fig. 4.
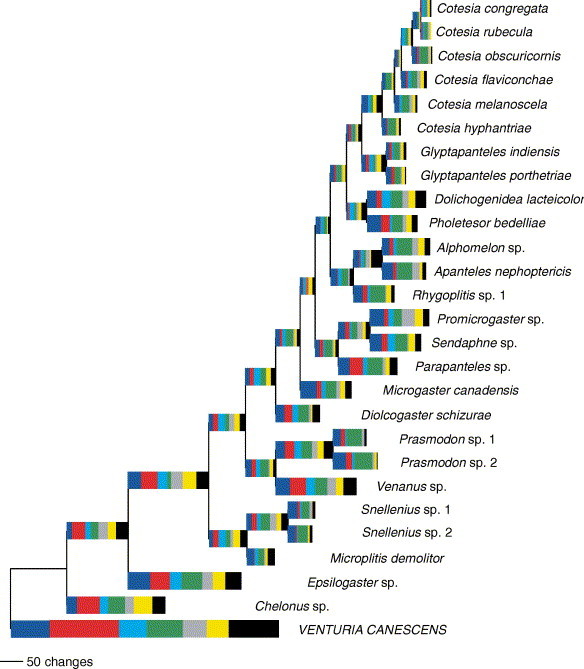
Maximum parsimony tree estimated for the taxa for which we have sequences for all seven genes. The horizontal length of each colour indicates the number of parsimony informative changes for each gene on the branch (blue = 16S, red = 28S, light-blue = arginine kinase, green = COI, grey = EF1α, yellow = opsin, black = wingless).
3.2. Simulations
The nonparametric approach using PAUP* to resample more characters from the original dataset found that the number of branches in the phylogeny with less than 50% bootstrap support reduced reasonably quickly until the dataset contained approximately 12,000 nucleotides (Fig. 5 ). After 12,000 nucleotides the rate of reduction of poorly supported branches decreased, and even with ten times more data than we have obtained five nodes still had bootstrap support of less than 50%. The parametric approach using Seq-Gen found that all branches of the phylogeny would have greater than 50% bootstrap support at around 12,000 nucleotides (Fig. 5). However, the nonparametric approach is more likely to be realistic, simulating normal “messy” data.
Fig. 5.
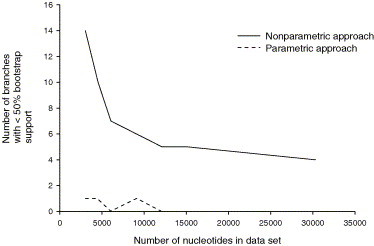
The number of branches with less than 50% bootstrap support obtained by increasing the number of characters resampled in the bootstrap function of PAUP* (nonparametric approach) or by using Seq-Gen to generate data from the likelihood equation calculated from the original data (parametric approach).
3.2.1. Number of taxa
Reducing the number of taxa in the dataset had little effect on the proportion of branches in the phylogeny with less than 50% bootstrap support (Fig. 6 ). Between 50% and 71% of the branches had bootstrap support of less than 50% depending of the number of taxa in the data set.
Fig. 6.
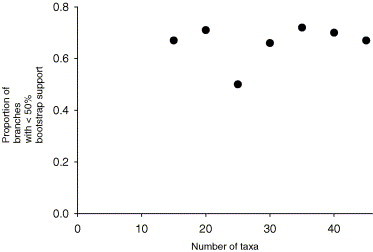
The proportion of branches with less than 50% bootstrap support for a phylogeny estimated using MP from the actual dataset of 45 taxa and with various numbers of taxa deleted randomly.
3.2.2. Addition of morphological characters
The Bayesian analysis of 53 morphological and 3031 molecular characters reduced the number of nodes with posterior probabilities of less than 0.9 from nine branches to three (Fig. 7 ). The phylogeny estimated from the Bayesian analysis of the seven genes alone differed from the phylogeny estimated from seven genes and 53 morphological characters in only two places. Fornicia moved from being part of a clade with Hypomicrogaster and Parapanteles (Fig. 3) in the phylogeny estimated from the genetic data alone, to being a sister to Venanides, Glyptapanteles and Cotesia (Fig. 7). Dolichogenidea and Pholetesor moved to being a sister to Hypomicrogaster, Promicrogaster, Parapanteles and Sendaphne in the combined genetic and morphological dataset. The placements of Fornicia and Pholetesor both had posterior probabilities of less than 0.9 in the phylogeny estimated from genes alone and only the branch placing Fornicia improved markedly in support (from 0.63 to 0.98).
Fig. 7.
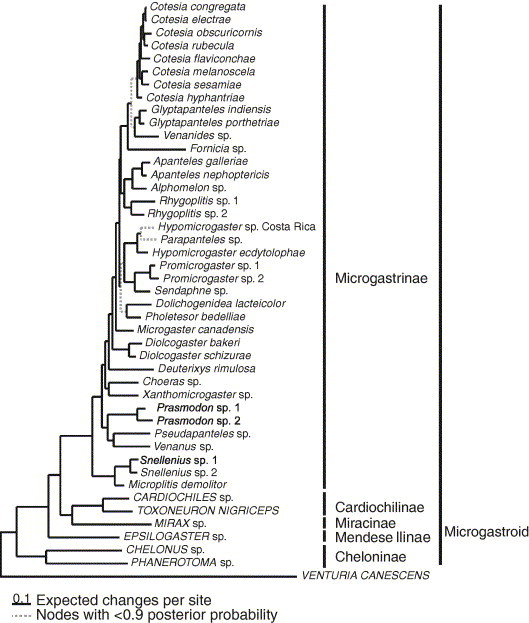
Phylogeny estimated from seven genes and 53 morphological characters using Bayesian mixed models (see text for details). Broken lines represent nodes with Bayesian posterior probabilities of less than 0.9.
4. Discussion
We identified genes that diverge more slowly than those already sequenced for Microgastrinae and their addition resulted in a more robust phylogeny for Microgastrinae, as assessed by higher nonparametric bootstrap proportions and posterior probabilities. However, despite substantially increasing the size of the dataset, we still did not obtain a completely robustly supported phylogeny using several methods of phylogeny estimation. Indeed, our nonparametric bootstrap simulations suggest we are unlikely to get a completely supported phylogeny from DNA alone. Although bootstrap support is not a direct measure of phylogenetic accuracy, most authors at least implicitly interpret the figures as rough measures of statistical support for a node (Buckley and Cunningham, 2002).
Support for the branches in our phylogeny could not be increased markedly by the method of analysis alone. The LogDet method of analysis is less affected by nonstationary data than is MP (Lockhart et al., 1994). However, using the LogDet transformation also did not produce a totally robustly supported tree. Excluding character sets did not produce a totally supported tree. For example, when a MP analysis of all data was compared to a MP analysis of the data with the third codon positions excluded, similar numbers of nodes had bootstrap support greater than 50%.
A marked improvement in support for our phylogeny was seen however when we added morphological characters and used a mixed model Bayesian analysis. Several other studies on diverse groups such as weevils (Marvaldi et al., 2002), molluscs (Collin, 2003) and feather mites (Dabert et al., 2001) have noted improvements in resolution and statistical support when analyses are conducted on combined morphological and DNA data. Dabert et al. (2001) also noted that molecular data alone tended to produce trees with better resolution and support at the terminal tips, and poor resolution and poor support deeper in the phylogeny, whereas the opposite (i.e., better resolution and support deeper in the phylogeny and poor resolution and support at the tips) occurred for phylogenies estimated from morphological data alone.
Short branches deep in a phylogeny are notoriously difficult to resolve. These branches will invariably have poor support as they are short due to a paucity of shared derived characters (Mardulyn and Whitfield, 1999) and we found that branches with less than 50% bootstrap support were significantly shorter than branches with greater than 50% support. In the case of the microgastrines, the short branches deeper in the phylogeny may be associated with the radiation of the Ditrysia (which contains 98% of present day lepidopteran species) that occurred from approximately 60 to 70 mya in the late-Cretaceous or early Cenozoic (Grimaldi, 1999). This radiation coincides approximately with the origin of the microgastrine group calculated by Whitfield (2002).
The ideal gene to resolve short deep branches should have a fast rate of divergence at the time of the radiation but then the gene’s rate of divergence needs to slow so that informative changes are not obscured by multiple substitutions at each site (Donoghue and Sanderson, 1992, Fishbein et al., 2001). It has been suggested that morphological characters may be more likely than nucleotide substitutions to undergo rapid changes followed by a slowing in the rate of change due to stabilising selection (Fishbein et al., 2001). Thus morphological characters may be a practical method to resolve short deep branches. Also, phenotypic variation is likely influenced by variation in many genes and morphological characters may be a cost effective way to indirectly increase the size of genetic datasets and improve levels of support for phylogenetic estimates.
4.1. Effect of methods
The increase in support for our phylogeny when morphological characters were added to the analysis was not due solely to changing from using bootstrap support of a MP analysis to using the posterior probabilities under a Bayesian approach. The Bayesian phylogeny estimated from the genetic data alone had lower levels of support than the Bayesian tree estimated from both genetic and morphological characters. It has been suggested that Bayesian posterior probabilities tend to be higher than MP bootstrap proportions for the same groups (Erixon et al., 2003). However, it has also been suggested that bootstrap values may be a conservative estimate of support for clades when support for clades is strong (Huelsenbeck and Rannala, 2004, Rannala and Yang, 1996) and it is likely that posterior probabilities give a better estimate of support than bootstrap proportions, especially when the most complex Bayesian models are used (Huelsenbeck and Rannala, 2004). We would have liked to compare bootstrap values of phylogenies generated by ML, rather than MP, to the posterior probabilities of the Bayesian trees but this was impractical due to computational constraints.
4.2. Simulations
The simulations using the bootstrap function in PAUP* to produce larger character sets from our data suggested that a phylogeny with every branch having more than 50% bootstrap support in a MP analysis was unlikely to be obtained without considerably more data. The approach using Seq-Gen was more optimistic, suggesting complete support was possible with around 12,000 nucleotides. However, the nonparametric method of simulating datasets produces data without gaps or missing data and thus produces a “perfect” dataset that is almost certainly unobtainable in reality. For example, we estimated a phylogeny with only one branch with less than 50% bootstrap support from a dataset simulated with Seq-Gen of the same size as our actual dataset. This compares to 14 branches with less than 50% bootstrap support in the phylogeny estimated from the actual data. The PAUP* based approach produces datasets that are more realistic as the simulated datasets have gaps in the sequences and missing data and is therefore more likely to give a better estimate of the data required to estimate a totally bootstrap supported phylogeny.
4.3. Incongruent phylogenies
4.3.1. ILD test
The P-values of <0.05 obtained for many of the ILD tests does not necessarily mean that conflict between individual genes has reduced bootstrap support for nodes in our phylogeny. There is significant disagreement as to what level of significance should be used to reject partition homogeneity. For example, Cunningham (1997) suggested a critical value of less than 0.01 should be used. There is also controversy over whether significant heterogeneity should preclude combining data derived from different genes for a phylogenetic analysis. Yoder et al. (2001) examined the effect of changing character weighting and/or data combinations on the phylogenetics of slow lorises and found that correct results were poorly supported and even incorrect results were obtained when character weighting and/or data combinations were altered to reduce incongruence (as assessed by the ILD). Likewise, Sullivan (1996) found that combining two heterogeneous datasets produced a better estimate of deer mouse and grasshopper mouse phylogenies than did either gene alone. Barker and Lutzoni (2002) also found from simulations that the ILD test was a relatively poor predictor of the effect of combining datasets on phylogenetic accuracy.
Dolphin et al. (2000) found that when rate differences between the two matrices being assessed reach a certain level, the ILD test could suggest significant heterogeneity despite the two matrices having similar underlying topologies. It seems likely that the marked differences in the divergence rates of the genes we analysed have generated the significant ILD test results. We suggest that we reduced the adverse effects of data heterogeneity by using complex evolutionary models for each partition of our data in a Bayesian analysis.
4.4. Wrong/Lack of data
Inappropriate gene choice has been suggested as a reason why it has been difficult to obtain a robust microgastrine phylogeny (Mardulyn and Whitfield, 1999). Several of the genes we used have been used in other studies of braconid phylogenetics (for example, Belshaw et al., 1998, Mardulyn and Whitfield, 1999, Michel-Salzat and Whitfield, 2004, Min et al., 2005, Whitfield et al., 2002), and in other studies of hymenopteran phylogenetics (Cameron and Mardulyn, 2001, Danforth et al., 2004, Kawakita et al., 2003, Sanchis et al., 2001, Weiblen, 2004). Given that our choice of genes covered deep, medium and shallow divergences and that these genes have been used successfully to estimate robust phylogenies for an enormous variety of taxa, we do not conclude that inappropriate gene choice has caused the poor support for some of the nodes. The contribution to the phylogenies from all the COI data is likely subject to long branch attraction (Felsenstein, 1978, Hendy and Penny, 1989) as this gene has the highest uncorrected pairwise divergences of the seven genes between microgastrine species and yet it has only the fifth highest levels of divergence between the braconid subfamilies. However, using model-based methods for estimating the phylogenies has probably lessened the effect of long branch attraction.
Missing data are also unlikely to have resulted in poor bootstrap support. A phylogeny estimated from those taxa for which we obtained sequences for all seven genes had six branches (of 51 total) with bootstrap support <50% in a MP analysis. The poorly supported branches were significantly shorter than branches with >50% bootstrap support. An examination of the contribution of individual genes to the branch length of the poorly supported branches found that there were few changes in all seven genes for these short branches, suggesting that a rapid radiation (i.e., truly short branches) has indeed occurred.
Insufficient data has also been suggested as a reason for poorly supported phylogenies. Rokas et al. (2003) suggested that 20 genes would be required to obtain mean bootstrap values of 95% with a 95% confidence interval for seven species of Saccharomyces yeasts. However, as few as eight genes would give mean bootstrap values of 95% with a 95% confidence interval if nonstationary genes (genes that have markedly shifted nucleotide frequencies for some taxa) were excluded from the Saccharomyces analysis (Collins et al., 2005). Deleting the third positions of codons from our data resulted in the genes becoming stationary. However, while deletion of third positions did not markedly alter the relationships estimated, it also did not markedly increase bootstrap support for our MP trees.
4.5. Effect of increasing taxa
There has been debate over whether it is better to add taxa or characters to an analysis to improve accuracy, given that resources are always finite. Rosenberg and Kumar (2001) suggested that longer sequences, rather than more taxa, will improve the accuracy of the phylogeny estimated. However, it was also argued that increasing the number of taxa equally reduces error in phylogenetic estimations (Pollock et al., 2002). The improvement in phylogenetic accuracy is in part determined by the length of sequences already obtained and by the levels of divergence between the taxa (Hillis et al., 2003). For example, if there are several long branches in the phylogeny, effort may be better expended on sequencing taxa that break up the long branches rather than adding more characters (Graybeal, 1998). In the microgastrine case, our simulations showed that neither adding taxa nor genetic data would increase bootstrap support for the short branches in the phylogeny.
4.6. Phylogenetic results
We found strong support (100% of bootstrap replicates from the MP analysis of the seven genes, and posterior probabilities of 0.99 for both the molecular data and the molecular data plus morphology) for monophyly of the microgastroid complex, sensu Wharton, 1993, Whitfield and Mason, 1994, Whitfield, 1997a, Dowton and Austin, 1998. Our finding of monophyly for Microgastrinae agrees with an earlier analysis of 16S data that also found the microgastroid complex to be monophyletic, although with equivocal bootstrap support (Dowton et al., 1998, Whitfield, 1997b). An analysis of portions of 16S and 28S rDNA also found strong bootstrap support for monophyly of the microgastroids (Dowton and Austin, 1998).
Our Bayesian analysis of the combined molecular and morphological characters found a relationship for the braconid subfamilies of (Cheloninae, (Mendesellinae, (Microgastrinae, (Cardiochilinae, Miracinae)))). Belshaw et al. (1998) found a similar relationship for the microgastrine subfamilies, excluding Mendesellinae, from an analysis of a portion of the 28S region. These results however, conflict with a MP analysis of portions of the 16S and 28S genes and 11 morphological characters (Dowton and Austin, 1998), and a Bayesian analysis of portions of 16S 18S and 28S regions and 96 morphological characters (Min et al., 2005), that found a relationship of (Adelinae + Cheloninae, (Miracinae, (Microgastrinae, Cardiochilinae))). We intend a more extensive examination of subfamily relationships within the microgastroid complex in the near future.
It is difficult to compare our estimate of relationships within Microgastrinae to other studies, as generally different or fewer microgastrine species were sampled in those studies (e.g., Belshaw et al., 1998, Dowton et al., 1998). We found Microplitis and Snellenius represent an early diverging lineage of microgastrines. A phylogeny estimated from portions of 16S and 28S also found Microplitis to be basal (Dowton and Austin, 1998) as did a phylogeny estimated from a portion of 28S (Mardulyn and Whitfield, 1999).
Acknowledgments
This study was primarily supported by National Science Foundation grant DEB 0316566 to J.B.W. It also includes work supported by two other grants to JBW: USDA NRI Grant 9501893 and NSF IBN 0344829. We wish to jointly thank those who contributed specimens for our analysis, especially Andy Austin, Martin Hauser, Mike Irwin, Dan Janzen, Angelica Penteado-Dias, and Mike Sharkey. We also wish to thank Andy Austin, Sydney Cameron, Bryan Danforth, Mark Dowton, and Heather Hines for helpful discussion concerning the genes used here, and John Huelsenbeck, Jack Sullivan and Dave Swofford for suggestions concerning the simulations. Two anonymous reviewers contributed helpful comments.
References
- Ascher J.S., Danforth B.N., Ji S. Phylogenetic utility of the major opsin in bees (Hymenoptera: Apoidea): a reassessment. Mol. Phylogenet. Evol. 2001;19:76–93. doi: 10.1006/mpev.2001.0911. [DOI] [PubMed] [Google Scholar]
- Barker F.K., Lutzoni F.M. The utility of the incongruence length difference test. Syst. Biol. 2002;51:625–637. doi: 10.1080/10635150290102302. [DOI] [PubMed] [Google Scholar]
- Belshaw R., Fitton M., Herniou H., Gimeno C., Quicke D.L.J. A phylogenetic reconstruction of the Ichneumonoidea (Hymenoptera) based on the D2 variable region of 28S ribosomal RNA. Syst. Entomol. 1998;23:109–123. [Google Scholar]
- Belshaw R., Quicke D.L.J. A molecular phylogeny of the Aphidiinae (Hymenoptera: Braconidae) Mol. Phylogenet. Evol. 1997;7:281–293. doi: 10.1006/mpev.1996.0400. [DOI] [PubMed] [Google Scholar]
- Brooks D.R., McLennan D.A. Extending phylogenetic studies of coevolution: secondary Brooks parsimony analysis, parasites, and the Great Apes. Cladistics. 2003;19:104–119. [Google Scholar]
- Brower A.V.Z., DeSalle R. Patterns of mitochondrial versus nuclear DNA sequence divergence among nymphalid butterflies: the utility of wingless as a source of characters for phylogenetic inference. Insect Mol. Biol. 1998;7:73–82. doi: 10.1046/j.1365-2583.1998.71052.x. [DOI] [PubMed] [Google Scholar]
- Buckley T.R., Cunningham C.W. The effect of nucleotide substitution model assumptions on estimates of nonparametric bootstrap support. Mol. Biol. Evol. 2002;19:394–405. doi: 10.1093/oxfordjournals.molbev.a004094. [DOI] [PubMed] [Google Scholar]
- Cameron, S.A., 2003. Data from the elongation factor-1alpha gene corroborate the phylogenetic pattern from other genes, revealing common ancestry of bumble bees and stingless bees (Hymenoptera: Apinae). In: Vandame, R., Rabanales, M.C.M.R., Díaz, M.C.M.A.G., Conesa, I.B.T.J.G., Guillén, M.C.D.S., (Eds.), III. Seminario Mesoamericano sobre Abejas sin Aguijón. El Colegio de la Frontera Sur, Tapachula, Chiapas, México, pp. 132–135.
- Cameron S.A., Mardulyn P. Multiple molecular data sets suggest independent origins of highly eusocial behavior in bees (Hymenoptera: Apinae) Syst. Biol. 2001;50:194–214. [PubMed] [Google Scholar]
- Cameron S.A., Mardulyn P. The major opsin gene is useful for inferring higher level phylogenetic relationships of the corbiculate bees. Mol. Phylogenet. Evol. 2003;28:610–613. doi: 10.1016/s1055-7903(03)00055-1. [DOI] [PubMed] [Google Scholar]
- Cameron S.A., Williams P.H. Phylogeny of bumble bees in the New World subgenus Fervidobombus (Hymenoptera: Apidae): congruence of molecular and morphological data. Mol. Phylogenet. Evol. 2003;28:552–563. doi: 10.1016/s1055-7903(03)00056-3. [DOI] [PubMed] [Google Scholar]
- Cho S., Mitchell A., Regier J.C., Mitter C., Poole R.W., Friedlander T.P., Zhao S. A highly conserved nuclear gene for low-level phylogenetics: elongation factor1-alpha recovers morphology-based tree for heliothine moths. Mol. Biol. Evol. 1995;12:650–656. doi: 10.1093/oxfordjournals.molbev.a040244. [DOI] [PubMed] [Google Scholar]
- Collin R. The utility of morphological characters in gastropod phylogenetics: an example from the Calyptraeidae. Biol. J. Linn. Soc. 2003;78:541–593. [Google Scholar]
- Collins T.M., Fedrigo O., Naylor G.J.P. Choosing the best genes for the job: the case for stationary genes in genome-scale phylogenies. Syst. Biol. 2005;54:493–500. doi: 10.1080/10635150590947339. [DOI] [PubMed] [Google Scholar]
- Cunningham C. Can three incongruence tests predict when data should be combined? Mol. Biol. Evol. 1997;14:733–740. doi: 10.1093/oxfordjournals.molbev.a025813. [DOI] [PubMed] [Google Scholar]
- Dabert J., Dabert M., Mironov S.V. Phylogeny of feather mite subfamily Avenzoariinae (Acari: Analgoidea: Avenzoariidae) inferred from combined analysis of molecular and morphological data. Mol. Phylogenet. Evol. 2001;20:124–135. doi: 10.1006/mpev.2001.0948. [DOI] [PubMed] [Google Scholar]
- Danforth B.N., Brady S.G., Sipes S.D., Pearson A. Single-copy nuclear genes recover Cretaceous-age divergences in bees. Syst. Biol. 2004;53:309–326. doi: 10.1080/10635150490423737. [DOI] [PubMed] [Google Scholar]
- Danforth B.N., Ji S. Elongation factor-1alpha occurs as two copies in bees: implications for phylogenetic analysis of EF-1alpha sequences in insects. Mol. Biol. Evol. 1998;15:225–235. doi: 10.1093/oxfordjournals.molbev.a025920. [DOI] [PubMed] [Google Scholar]
- Danforth B.N., Lin C.P., Fang J. How do insect nuclear ribosomal genes compare to protein-coding genes in phylogenetic utility and nucleotide substitution patterns? Syst. Entomol. 2005;30:549–562. [Google Scholar]
- de Queiroz A., Donoghue M.J., Junhyong K. Separate versus combined analysis of phylogenetic evidence. Annu. Rev. Ecol. Syst. 1995;26:657–681. [Google Scholar]
- Dolphin K., Belshaw R., Orme C.D.L., Quicke D.J. Noise and incongruence: interpreting results of the incongruence length difference test. Mol. Phylogenet. Evol. 2000;71:401–406. doi: 10.1006/mpev.2000.0845. [DOI] [PubMed] [Google Scholar]
- Donoghue M.J., Sanderson M.J. The suitability of molecular and morphological evidence in reconstructing plant phylogeny. In: Soltis P.S., Soltis D.E., Doyle J.J., editors. Molecular Systematics of Plants. Chapman Hall; New York: 1992. pp. 340–368. [Google Scholar]
- Dowton M., Austin A.D. Molecular phylogeny of the insect order Hymenoptera: Apocritan relationships. Proc. Natl. Acad. Sci. USA. 1994;91:9911–9915. doi: 10.1073/pnas.91.21.9911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowton M., Austin A.D. Phylogenetic relationships among the microgastroid wasps (Hymenoptera: Braconidae): combined analysis of 16S and 28S rDNA genes and morphological data. Mol. Phylogenet. Evol. 1998;10:354–366. doi: 10.1006/mpev.1998.0533. [DOI] [PubMed] [Google Scholar]
- Dowton M., Austin A.D., Antolin M.F. Evolutionary relationships among the Braconidae (Hymenoptera: Ichneumonoidea) inferred from partial 16S rDNA sequences. Insect Mol. Biol. 1998;7:129–150. doi: 10.1046/j.1365-2583.1998.72058.x. [DOI] [PubMed] [Google Scholar]
- Erixon P., Svennblad B., Britton T., Oxelman B. Reliability of bayesian posterior probabilities and bootstrap frequencies on phylogenetics. Syst. Biol. 2003;52:665–673. doi: 10.1080/10635150390235485. [DOI] [PubMed] [Google Scholar]
- Farris J.S., Källersjö M., Kluge A.G., Bult C. Testing the significance of congruence. Cladistics. 1994;10:315–320. [Google Scholar]
- Farris J.S., Källersjö M., Kluge A.G., Bult C. Constructing a significance test for incongruence. Syst. Biol. 1995;44:570–572. [Google Scholar]
- Felsenstein J. Cases in which parsimony or compatibility methods will be positively misleading. Syst. Zool. 1978;27:401–410. [Google Scholar]
- Fishbein M., Hibsch-Jetter C., Soltis D.E., Hufford L. Phylogeny of Saxifrageles (Angiosperms, Eudicots): analysis of a rapid ancient radiation. Syst. Biol. 2001;50:817–847. doi: 10.1080/106351501753462821. [DOI] [PubMed] [Google Scholar]
- Friedlander T.P., Horst K.R., Regier J.C., Mitter C., Peigler R.S., Fang Q.Q. Two nuclear genes yield concordant relationships within Attacini (Lepidoptera: Saturniidae) Mol. Phylogenet. Evol. 1998;9:131–140. doi: 10.1006/mpev.1997.9999. [DOI] [PubMed] [Google Scholar]
- Folmer O., Black M., Hoeh W., Lutz R., Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994;3:294–299. [PubMed] [Google Scholar]
- Graybeal A. Is it better to add taxa or characters to a difficult phylogenetic problem? Syst. Biol. 1998;47:9–17. doi: 10.1080/106351598260996. [DOI] [PubMed] [Google Scholar]
- Grimaldi D. The co-radiations of pollinating insects and angiosperms in the Cretaceous. Ann. Mo. Bot. Gard. 1999;86:373–406. [Google Scholar]
- Guan Y., Zheng B.J., He Y.Q., Liu X.L., Zhuang Z.X., Cheung C.L., Luo S.W., Li P.H., Zhang L.J., Guan Y.J., Butt K.M., Wong K.L., Chan K.W., Lim W., Shortridge K.F., Yuen K.Y., Peiris J.S.M., Poon L.L.M. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. 2003;302:276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- Hebert P.D.N., Cywinska A., Ball S.L., deWaard J.R. Biological identification through DNA barcodes. Proc. R. Soc. Lond. B. 2003;270:313–321. doi: 10.1098/rspb.2002.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert P.D.N., Penton E.H., Burns J.M., Janzen D.H., Hallwachs W. Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc. Natl. Acad. Sci. USA. 2004;101:14812–14817. doi: 10.1073/pnas.0406166101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert P.D.N., Ratnasingham N.S., deWaard J.R. Barcoding animal life: cyctochrome c oxidase subunit 1 divergences among closely related species. Proc. R. Soc. Lond. B. 2003;270:S96–S99. doi: 10.1098/rsbl.2003.0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendy M., Penny D. A framework for the quantitative study of evolutionary trees. Syst. Zool. 1989;38:297–309. [Google Scholar]
- Hillis D.M., Pollock D.D., McGuire J.A., Zwickl D.J. Is sparse taxon sampling a problem for phylogenetic inference? Syst. Biol. 2003;52:124–126. doi: 10.1080/10635150390132911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck J.P., Rannala B. Frequentist properties of bayesian posterior probabilities of phylogenetic trees under simple and complex substitution models. Syst. Biol. 2004;53:904–913. doi: 10.1080/10635150490522629. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck J.P., Ronquist F. MR BAYES: Bayesian inference of phylogeny. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- Kawakita A., Sota T., Ascher J.S., Ito M., Tanaka H., Kato M. Evolution and phylogenetic utility of alignment gaps within intron sequences of three nuclear genes in bumble bees (Bombus) Mol. Biol. Evol. 2003;20:87–92. doi: 10.1093/molbev/msg007. [DOI] [PubMed] [Google Scholar]
- Korber B., Muldoon M., Theiler J., Gao F., Gupta R., Lapedes A., Hahn B.H., Wolinsky S., Bhattacharya T. Timing the ancestor of the HIV-1 pandemic strain. Science. 2000;288:1789–1796. doi: 10.1126/science.288.5472.1789. [DOI] [PubMed] [Google Scholar]
- Leys R., Cooper S.J.B., Schwartz M.P. Molecular phylogeny and historical biogeography of the large carpenter bees, genus Xylocopa (Hymenoptera: Apidae) Biol. J. Linn. Soc. 2002;77:249–266. [Google Scholar]
- Lockhart P.J., Steel M.A., Hendy M.D., Penny D. Recovering evolutionary trees under a more realistic model of sequence evolution. Mol. Biol. Evol. 1994;11:605–612. doi: 10.1093/oxfordjournals.molbev.a040136. [DOI] [PubMed] [Google Scholar]
- Mardulyn P., Cameron S.A. The major opsin in bees (Insecta: Hymenoptera): A promising nuclear gene for higher level phylogenetics. Mol. Phylogenet. Evol. 1999;12:168–176. doi: 10.1006/mpev.1998.0606. [DOI] [PubMed] [Google Scholar]
- Mardulyn P., Whitfield J.B. Phylogenetic signal in COI, 16S, and 28S genes for inferring relationships among genera of Microgastrinae (Hymenoptera; Braconidae): evidence of a high diversification rate in this group of parasitoids. Mol. Phylogenet. Evol. 1999;12:282–294. doi: 10.1006/mpev.1999.0618. [DOI] [PubMed] [Google Scholar]
- Marvaldi A.E., Sequeira A.S., O’Brien C.W., Farrell B.D. Molecular and morphological phylogenetics of weevils (Coleoptera, Curculionoidea): do niche shifts accompany diversification? Syst. Biol. 2002;51:761–785. doi: 10.1080/10635150290102465. [DOI] [PubMed] [Google Scholar]
- Michel-Salzat A., Cameron S.A., Oliveira M.L. Phylogeny of the orchid bees (Hymenoptera: Apinae: Euglossini): DNA and morphology yield equivalent patterns. Mol. Phylogenet. Evol. 2004;32:309–323. doi: 10.1016/j.ympev.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Michel-Salzat A., Whitfield J.B. Preliminary evolutionary relationships within the parasitoid wasp genus Cotesia (Hymenoptera: Braconidae: Microgastrinae): combined analysis of four genes. Syst. Entomol. 2004;29:1–12. [Google Scholar]
- Min S., Xue-xin C., van Achterberg C. Phylogenetic relationships among the Braconidae (Hymenoptera: Ichneumonoidea) inferred from partial 16S rDNA, 28S rDNA D2, 18S rDNA gene sequences and morphological characters. Mol. Phylogenet. Evol. 2005;37:104–116. doi: 10.1016/j.ympev.2005.03.035. [DOI] [PubMed] [Google Scholar]
- Mitchell A., Cho S., Regier J.C., Mitter C., Poole R.W., Mathews M. Phylogenetic utility of elongation factor-1alpha in Noctuoidea (Insecta: Lepidoptera): the limits of synonymous substitution. Mol. Biol. Evol. 1997;14:381–390. doi: 10.1093/oxfordjournals.molbev.a025774. [DOI] [PubMed] [Google Scholar]
- Mitchell A., Mitter C., Regier J.C. More taxa or more characters revisited: combining data from nuclear protein-encoding genes for phylogenetic analyses of Noctuoidea (Insecta: Lepidoptera) Syst. Biol. 2000;49:202–224. [PubMed] [Google Scholar]
- Mitchell A., Mitter C., Regier J.C. Systematics and evolution of the cutworm moths (Lepidoptera: Noctuidae): evidence from two protein-coding nuclear genes. Syst. Entomol. 2006;31:21–46. [Google Scholar]
- Page, R.D.M., 1995. TreeMap, version 1.0b.
- Pollock D.D., Zwickl D.J., McGuire J.A., Hillis D.M. Increased taxon sampling is advantageous for phylogenetic inference. Syst. Biol. 2002;51:664–671. doi: 10.1080/10635150290102357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada, D., 2000. Testing Models of Evolution—Modeltest Version 3.06, version 3.06. Department of Zoology, Brigham Young University.
- Posada D., Crandall K.A. Modeltest: testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- Rambaut A., Grassly N. Seq-Gen: an application for the Monte Carlo simulation of DNA sequence evolution along phylogenetic trees. Comput. Appl. Biosci. 1997;13:235–238. doi: 10.1093/bioinformatics/13.3.235. [DOI] [PubMed] [Google Scholar]
- Rambaut A., Robertson D.L., Pybus O.G., Peeters M., Holmes E.C. Phylogeny and the origin of HIV-1. Nature. 2001:1047–1048. doi: 10.1038/35074179. [DOI] [PubMed] [Google Scholar]
- Rannala B., Yang Z. Probability distribution of molecular evolutionary trees: a new method of phylogenetic inference. J. Mol. Evol. 1996;43:304–311. doi: 10.1007/BF02338839. [DOI] [PubMed] [Google Scholar]
- Rodríguez F., Oliver J.F., Marín A., Medina J.R. The general stochastic model of nucleotide substitution. J. Theor. Biol. 1990;142:485–501. doi: 10.1016/s0022-5193(05)80104-3. [DOI] [PubMed] [Google Scholar]
- Rokas A., Williams B.L., King N., Carroll S.B. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature. 2003;425:798–804. doi: 10.1038/nature02053. [DOI] [PubMed] [Google Scholar]
- Ronquist F., Huelsenbeck J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Rosenberg M.S., Kumar S. Incomplete taxon sampling is not a problem for phylogenetic inference. Proc. Natl. Acad. Sci. USA. 2001;98:10751–10756. doi: 10.1073/pnas.191248498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchis A., Michelena J.M., Latorre A., Quicke D.L.J., Gardenfors U., Belshaw R. The phylogenetic analysis of variable-length sequence data: elongation factor-1alpha introns in European populations of the parasitoid wasp genus Pauesia (Hymenoptera: Braconidae: Aphidiinae) Mol. Biol. Evol. 2001;18:1117–1131. doi: 10.1093/oxfordjournals.molbev.a003882. [DOI] [PubMed] [Google Scholar]
- SPSS. 1998. Systat 9.01 Statistics. SPSS Inc.
- Sullivan J. Combining data with different distributions of among-site variation. Syst. Biol. 1996;45:375–380. [Google Scholar]
- Swofford D.L., Olsen G., Waddell P., Hillis D. Phylogenetic inference. In: Hillis D., Moritz C., Mable B., editors. Molecular Systematics. Sinauer Associates, Inc.; Sunderland: 1996. pp. 407–514. [Google Scholar]
- Swofford, D. L. 2002. PAUP* Phylogenetic analysis using parsimony (*and other methods), version 4. Sinauer and Associates.
- Tavaré L. Some probabilistic and statistical problems on the analysis of DNA sequences. Lect. Math. Life Sci. 1986;17:57–86. [Google Scholar]
- Thompson J.D., Gibson T.J., Plewniak F., Jeanmougin F., Higgins D.G. The Clustal X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research. 1997;24:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiblen G.D. Correlated evolution in fig pollination. Syst. Biol. 2004;53:128–139. doi: 10.1080/10635150490265012. [DOI] [PubMed] [Google Scholar]
- Wharton R.A. Bionomics of the Braconidae. Annual Reviews in Entomology. 1993;38:121–143. [Google Scholar]
- Whitfield J.B. Molecular and morphological data suggest a single origin of the polydnaviruses among braconid wasps. Naturwissenschaften. 1997;84:502–507. [Google Scholar]
- Whitfield, J.B., 1997b. Subfamily Microgastrinae. In: Wharton, R.A., Marsh, P.M., Sharkey, M.J. (Eds.), Identification Manual to the New World Genera of the Family Braconidae (Hymenoptera). International Society of Hymenopterists Special Publication 1, Washington, DC, pp. 333–364.
- Whitfield J.B. Estimating the age of the polydnavirus/braconid wasp symbiosis. Proc. Natl. Acad. Sci. USA. 2002;99:7508–7513. doi: 10.1073/pnas.112067199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield J.B., Asgari S. Virus or not? Phylogenetics of polydnaviruses and their wasp carriers. J. Insect Physiol. 2003;49:397–405. doi: 10.1016/s0022-1910(03)00057-x. [DOI] [PubMed] [Google Scholar]
- Whitfield J.B., Cameron S.A. Hierarchical analysis of variation in the 16S rRNA gene among Hymenoptera. Mol. Biol. Evol. 1998;15:1728–1743. doi: 10.1093/oxfordjournals.molbev.a025899. [DOI] [PubMed] [Google Scholar]
- Whitfield J.B., Mardulyn P., Austin A.D., Dowton M. Phylogenetic relationships among microgastrine braconid wasp genera based on data from the 16S, COI and 28S genes and morphology. Syst. Entomol. 2002;27:337–359. [Google Scholar]
- Whitfield J.B., Mason W.R.M. Mendesellinae, a new subfamily of braconid wasps (Hymenoptera, Braconidae) with a review of relationships within the microgastroid assemblage. Syst. Entomol. 1994;19:61–76. [Google Scholar]
- Wiegmann B.M., Mitter C., Regier J.C., Friedlander T.P., Wagner D.L., Nielsen E.S. Nuclear genes resolve Mesozoic-aged divergences in the insect order Lepidoptera. Mol. Phylogenet. Evol. 2000;15:242–259. doi: 10.1006/mpev.1999.0746. [DOI] [PubMed] [Google Scholar]
- Wiegmann B.M., Yeates D.K., Thorne J.L., Kishino H. Time flies, a new molecular time scale for brachyceran fly evolution without a clock. Syst. Biol. 2003;52:745–756. [PubMed] [Google Scholar]
- Worobey M., Santiago M.L., Keele B.F., Ndjango J.-B.N., Joy J.B., Labama B.L., Dhed’a B.D., Rambaut A., Sharp P.M., Shaw G.M., Hahn B.H. Contaminated polio vaccine theory refuted. Nature. 2004;428:820. doi: 10.1038/428820a. [DOI] [PubMed] [Google Scholar]
- Yang Z., Goldman N., Friday A. Comparison of models for nucleotide substitution used in maximum-likelihood phylogenetic estimation. Mol. Biol. Evol. 1994;11:316–324. doi: 10.1093/oxfordjournals.molbev.a040112. [DOI] [PubMed] [Google Scholar]
- Yoder A.D., Irwin J.A., Payseur B.A. Failure of the ILD to determine data combinability for slow loris phylogeny. Syst. Biol. 2001:50. [PubMed] [Google Scholar]