

Abstract
In the diabetic retina, cellular changes in the retinal pigment epithelium (RPE) and neurons occur before vision loss or diabetic retinopathy can be identified clinically. The precise etiologies of retinal pathology are poorly defined, and it remains unclear if the onset and progression of cellular dysfunction differ between type 1 and type 2 diabetes. Three mouse models were used to compare the time course of RPE involvement in type 1 and type 2 diabetes. C57BL/6J mice injected with streptozotocin (STZ mice) modeled type 1 diabetes, whereas Leprdb/db mice on both BKS and B6.BKS background strains modeled type 2 diabetes. Electroretinogram (ERG)-based techniques were used to measure light-evoked responses of the RPE (direct current-coupled ERG, dc-ERG) and the neural retina (a-wave, b-wave). Following onset of hyperglycemia, a-wave and b-wave amplitudes of STZ mice declined progressively and by equivalent degrees. Components of the dc-ERG were also altered, with the largest reduction seen in the c-wave. Leprdb/db mice on the BKS strain (BKS.Lepr) displayed sustained hyperglycemia and a small increase in insulin, whereas Leprdb/db mice on the B6.BKS background (B6.BKS.Lepr) were transiently hyperglycemic and displayed severe hyperinsulinemia. BKS.Lepr mice exhibited sustained reductions in the dc-ERG c-wave, fast oscillation, and off response that were not attributable to reduced photoreceptor activity; B6.BKS.Lepr mice displayed transient reductions in the c-wave and fast oscillation that correlated with hyperglycemia and magnitude of photoreceptor activity. In summary, all mouse models displayed altered RPE function concomitant with the onset of hyperglycemia. These results suggest that RPE function is directly reduced by elevated blood glucose levels. That RPE dysfunction was reversible and mitigated in hyperinsulinemic B6.BKS.Lepr mice provides insight into the underlying mechanism.
Keywords: retinal pigment epithelium, electroretinogram, hyperglycemia
diabetic retinopathy (DR) is one of the most severe complications of diabetes and is the most common cause of blindness in working age adults (Klein et al. 1995). The prevalence of DR in patients with type 1 diabetes (T1D) is 40%, and that for patients with type 2 diabetes (T2D) is 20%. Within each cohort, prevalence increases with duration of diabetes. After 20 years of disease, 90% of T1D patients and 60% of T2D patients experience some level of clinical retinopathy (Klein 2007). Therapies, including laser photocoagulation and intraocular injection of VEGF inhibitors, have been successful at improving vision in patients with diabetic macular edema; however, these therapies do not reduce the risk of developing vision loss (Cheung et al. 2010; Robinson et al. 2012; Salam et al. 2011; Wilkinson-Berka and Miller 2008). Development of early detection procedures and identification of early indicators of dysfunction within the retina remain a significant challenge.
Patients with early stages of diabetes and no sign of DR have decreased light sensitivity and delayed dark adaptation recovery, indicative of outer retina dysfunction (Arden et al. 1998; Holopigian et al. 1997). Altered outer retinal neuron function has been documented in diabetic patients via electroretinography (ERG), even in the absence of clinical retinopathy (Bresnick and Palta 1987; Gardner et al. 2002; Juen and Kieselbach 1990; Parisi and Uccioli 2001; Yamamoto et al. 1996), and delays in multifocal electroretinography (mfERG) implicit times can predict the location of microaneurysm development (Bearse et al. 2006; Fortune et al. 1999; Harrison et al. 2011; Ng et al. 2008) and are found at very early times in adolescents with either T1D or T2D (Bronson-Castain et al. 2012).
ERGs measure the massed electrical response of the retina to light stimuli and demonstrate the function of multiple retinal cell types through analysis of the individual waveform components. ERGs recorded in response to strobe flashes allow for evaluation of the short-latency components generated by the neural retina (see Fig. 1D). The cornea-negative a-wave is generated by the light-induced decline in the dark current around the rod photoreceptor outer segments (Hood and Birch 1990; Lamb 1996; Penn and Hagins 1969). The positive polarity b-wave is generated by the activity of rod depolarizing bipolar cells and is dependent on output of the photoreceptor (Hood and Birch 1996; Kofuji et al. 2000; Robson and Frishman 1995). The higher frequency oscillatory potentials (OPs) superimposed on the b-wave reflect amacrine cell activity (Wachtmeister 1998). ERG protocols using longer duration stimuli and direct current amplification (dc-ERG) allow the slower responses of nonneuronal cell types to be examined. In the dc-ERG waveform (see Fig. 2E), the positive-polarity c-wave represents the summation of a [K+] decline-induced hyperpolarization of the RPE apical membrane and a negative Kir4.1-mediated slow PIII response of the Müller glia cells (Kofuji et al. 2000; Oakley and Green 1976; Schmidt and Steinberg 1971; Steinberg and Miller 1973; Steinberg et al. 1970; Witkovsky et al. 1975; Wu et al. 2004a). The c-wave is followed by the negative-polarity fast oscillation that reflects recovery of the subretinal [K+] and a [Cl−]-dependent hyperpolarization of the basal RPE membrane (Griff and Steinberg 1984; Linsenmeier and Steinberg 1982). The positive-polarity light peak is induced by depolarization of the basal membrane of the RPE due to increased Cl− permeability and is followed by the off response (Fujii et al. 1992; Gallemore and Steinberg 1989, 1993; Linsenmeier and Steinberg 1982; Wu et al. 2004b). None of the dc-ERG components are a direct response to light, but instead reflect the summed RPE/Müller cell responses generated subsequent to the response of rod photoreceptors. These components thus provide noninvasive measures of RPE function, although their interpretation must take into account the status of rod photoreceptor function (Samuels et al. 2010).
Fig. 1.

Type 1 diabetic (T1D) streptozotocin-injected (STZ) mice display reductions in strobe-flash electroretinograms (ERGs) beginning 2 wk following onset of hyperglycemia. A: schematic illustrating the STZ injection and ERG testing paradigms. B: systemic non-fasting glucose concentration. C: serum insulin levels at each time point of ERG testing. D: averaged ERG waveform tracings from 3 representative flash luminances. Control (CNTL) mice are represented by black bars and STZ-injected diabetic mice by gray bars. E: a-wave amplitudes obtained in response to a 1.4 log cd·s/m2 flash. F: b-wave amplitudes obtained in response to a 1.4 log cd·s/m2 flash. G: relative changes in a- and b-wave amplitudes observed in STZ mice. Data are plotted relative to the average of the age-matched CNTL mice. The diagonal line indicates an equivalent reduction in a- and b-wave amplitudes. Data points indicate the average ± SE. At each time point, n ≥ 3 for each group. *P < 0.05, CNTL vs. STZ by 2-way ANOVA with Tukey's post hoc analysis.
Fig. 2.

T1D STZ mice display reductions in retinal pigment epithelium (RPE) function by the direct current-coupled ERG (dc-ERG), which coincides with decreased photoreceptor function. Bar graphs show c-wave (A), fast oscillation (B), light peak (C), and off response amplitude (D) at the 3 time points tested. Control (CNTL) mice are represented by black bars and STZ-injected diabetic mice by gray bars. E: representative dc-ERG waveform traces at each time point. CNTL traces are shown in black and STZ traces in gray. The major dc-ERG components are labeled in the top trace. F: relative amplitude of the c-wave plotted as a function of the relative a-wave amplitude. G: relative amplitude of the fast oscillation plotted as a function of the relative a-wave amplitude. H: relative amplitude of the off response plotted as a function of the relative a-wave amplitude. Diamonds indicate the number of weeks following onset of hyperglycemia. Statistical analysis was performed separately at each time point between groups; n ≥ 3 for each group at each time point. *P < 0.05.
ERGs performed on diabetic patients demonstrate reductions in b-wave amplitude and increases in the implicit time of the high-frequency OPs, which are also reduced in amplitude (Shirao and Kawasaki 1998). These changes become more pronounced with disease progression, and the a-wave also becomes involved at later disease states (Bresnick et al. 1984; Holopigian et al. 1997; Pardue et al. 2014). Reports from patient studies and analysis of animal models indicate that the RPE is an early site of dysfunction (Kirber et al. 1980; Klein et al. 1980; Krupin et al. 1982; Tso et al. 1980; Vinores et al. 1989). The electrooculogram (EOG) can be used to measure function of the RPE in patients, and the fast oscillation component of the EOG is reduced in diabetic patients both with and without DR (Schneck et al. 2008). After 6 mo of diabetes, T1D mice display reductions in amplitude of several components of the dc-ERG, including the c-wave, fast oscillation, and off response (Samuels et al. 2012). Importantly, reductions in ERG components generated by the RPE are noted as early as 2 wk post onset of diabetes (Pautler and Ennis 1980), earlier than the reduction in OPs or b-wave, which appear 4 wk post onset of diabetes (Aung et al. 2013; Kohzaki et al. 2008; Li et al. 2002; Pardue et al. 2014; Phipps et al. 2004, 2006; Shinoda et al. 2007). Compared with animal models of T1D, models of T2D have been less well studied, although reductions in a-wave, b-wave, and OP amplitude and increased OP latencies have been documented at 16 wk of age in Leprdb/db mice (Bogdanov et al. 2014; Simo and Hernandez 2014). This work does not, however, define the time course over which retinal and RPE dysfunction develops.
Systemic glucose concentration is the only known predictor for DR (1993; King et al. 1999). The purpose of the present study is to identify and differentiate the earliest effects of hyperglycemia on the function of the RPE and outer retina in mouse models of T1D and T2D. In this report, we demonstrate reductions in the light-evoked electrical responses of the RPE in a well-established mouse model of T1D (STZ injection) and two strains of Leprdb/db mutant mice that model T2D (Lai and Lo 2013). In all models, RPE abnormalities appeared concomitantly with hyperglycemia and before or at the same time that photoreceptor dysfunction commenced. Moreover, low doses of insulin appear to delay RPE dysfunction, but not neuronal dysfunction. Our data indicate that RPE dysfunction is an early hallmark of T1D and T2D and that it may be predictive for development of clinical retinopathy.
METHODS
Ethical Approval
Treatment of animals was in compliance with the Association for Research in Vision and Ophthalmology resolution on treatment of animals in research, and all animal procedures were approved by the Institutional Animal Care and Use Committees of the Louis Stokes Cleveland Veterans Affairs Medical Center and the Cleveland Clinic.
Mice
Mouse model of T1D.
C57BL/6J mice from The Jackson Laboratory (Bar Harbor, ME) were bred, and 4- to 8-wk-old littermates were randomly assigned to diabetic (STZ) or nondiabetic control (CNTL) groups. Diabetes was induced by three sequential daily intraperitoneal injections of a freshly prepared solution of STZ in 0.1 M citrate buffer (pH 4.4) at 30 mg/kg body wt. Insulin was given to STZ-injected mice by intraperitoneal injection every other day, beginning at the onset of hyperglycemia, to prevent ketosis without preventing hyperglycemia and glucosuria [0–0.2 units of neutral protamine Hagedorn (NPH) Humulin N; Eli Lilly, Indianapolis, IN]. This was typical for all STZ mice to maintain body weight and hydration. CNTL-injected mice received citrate buffer only and did not receive insulin.
Mouse models of T2D.
Leprdb/db breeders were purchased from The Jackson Laboratory. Mice homozygous for the Leprdb/db allele on the B6.BKS background (000697) are referred to as “B6.BKS.Lepr,” whereas Leprdb/db mice on the pure BKS background (000642) are referred to as “BKS.Lepr.” For both strains, Lepr+/db animals were bred to generate Leprdb/db diabetic mice and control Lepr+/+ or Lepr+/db littermates.
Mice were genotyped for the Leprdb allele by PCR amplification by one of two sets of primers, depending on the experiment. For “db one” (20 μM), AGAACGGACACTCTTTGAAGTCTC, and “db two” (20 μM), CATTCAAACCATATTTAGGTTTGTGT, PCR amplification was performed for 35 cycles at 94°C for 30 s, 52°C for 45 s, and 72°C for 45 s, followed by 2 min at 72°C. PCR products were digested with Rsa1 overnight at 37°C and separated on a 2% agarose gel. For “db forward” (10 μM), ACCAACTTCCCAACAGTCCA, and “db reverse” (10 μM), TGATGCCCTGAAAATCAAGC, PCR amplification was carried out for 35 cycles at 94°C for 30 s, 52°C for 30 s, and 72°C for 30 s, followed by 3 min at 72°C and then direct sequencing with BigDye Terminator (v1.1; Applied Biosystems, Grand Island, NY).
Body weight and non-fasting blood glucose were measured at least monthly and at the time of electroretinography using a One-Touch Ultra glucometer (Bio-Rad Laboratories, Hercules, CA), and plasma insulin concentrations were measured using the Ultra Sensitive rat insulin ELISA kit with mouse insulin standards (Crystal Chem, Downer's Grove, IL). No T2D mice received insulin.
Electroretinography
After overnight dark adaptation, mice were anesthetized (65 mg/kg pentobarbital sodium), the cornea was anesthetized (1% proparacaine hydrochloride), and the pupils were dilated (1% tropicamide, 2.5% phenylephrine hydrochloride, and 1% cyclopentolate). Mice were placed on a temperature-regulated heating pad throughout each recording session. Responses of the outer retina were recorded using an Espion E3 ColorDome full-field ganzfeld (Diagnosys, Lowell, MA) with a Ag-AgCl electrode referenced to a Ag-AgCl pellet electrode placed in the mouth of the mouse in response to strobe-flash stimuli presented in the dark. Ten steps of increasing flash luminance [−3.6 to 2.1 log candela (cd)·s/m2] were presented in order of increasing flash strength, and the number of successive trials averaged together decreased from 20 for low-level flashes to 2 for the highest flash stimuli. The duration of the interstimulus interval increased from 4 s for low-luminance flashes to 90 s for the highest stimuli. The amplitude of the a-wave was measured 6.6 ms after flash onset from the prestimulus baseline. The amplitude of the b-wave was measured from the a-wave amplitude at 6.6 ms to the peak of the b-wave.
Immediately following the dark-adapted strobe-flash stimuli, components of the dc-ERG, generated by the RPE, were recorded in response to a 5 cd/m2 stimulus presented for 7 min. The amplitude of the c-wave was measured from the prestimulus baseline to the peak of the c-wave. The amplitude of the fast oscillation was measured from the c-wave peak to the trough of the fast oscillation. The amplitude of the light peak was measured from the fast oscillation trough to the asymptotic value. The amplitude of the off response was measured from the light peak asymptote to the peak of the off response.
Immediately after the conclusion of the dc-ERG recording, a steady 20 cd/m2 adapting field was presented in the ganzfeld bowl. After 4 min of light adaptation, cone ERGs were recorded to strobe-flash stimuli (−1 to 2 log cd·s/m2) superimposed on the adapting field. The amplitude of the cone ERG was measured from the prestimulus baseline to the positive peak of the waveform.
Statistical Analysis
For all analyses, data were compiled as averages ± SE, and statistics were performed using two-way ANOVA with Tukey's post hoc analysis or the Student's paired t-test. To normalize the dc-ERG responses to that of the a-wave, the amplitude of each individual animal was compared with the averaged control response and calculated as the relative amplitude. Relative amplitudes were then averaged and compared with the control average for statistical analysis. At least three animals per genotype per time point were used for all experiments.
Scanning Laser Ophthalmoscopy and Spectral-Domain Optical Coherence Tomography
Mice were anesthetized with 65 mg/kg pentobarbital sodium. Mydriasis was induced by administration of 1 μl of 0.5% Mydrin-P (tropicamide-phenylephrine combination) drops (Santen Pharmaceutical, Osaka, Japan). The drops were gently massaged into the eye by manually blinking the eyelids. Eyes were then immediately covered with Systane Ultra artificial tears (Alcon Laboratories, Ft. Worth, TX). Mice were then placed in a warmed, humidified, oxygenated acrylic plastic sheet chamber for a minimum of 5 min to permit time for pupil dilation. Mice were then removed for imaging by scanning laser ophthalmoscopy (SLO) (model HRA2; Heidelberg Engineering, Vista, CA) and spectral domain optical coherence tomography (SDOCT) (model Envisu SDOIS; Bioptigen, Research Triangle Park, NC). The SLO imaging involved collection of different imaging modalities, including dark-field reflectance and autofluorescent images with both blue (488 nm) and infrared (795 and 830 nm) illumination wavelengths. With the use of a wide-field objective lens with a 55° field of view (FOV), retinal images were collected with the optic nerve centrally positioned. Additional views of the peripheral regions were obtained to further investigate the nasal, temporal, superior, and inferior quadrants. Eyes were occasionally rehydrated with balanced salt solution or Systane Ultra artificial ears (Alcon Laboratories) and mechanically massaged to simulate blinking as needed. After SLO imaging, the mouse was transferred to the Envisu SDOIS system (Bioptigen) for SDOCT imaging. The SDOCT volumetric scans (250 a-scans per b-scan, × 250 b-scans per volume) were obtained with the optic nerve centrally located within the FOV. With the use of a 50° objective lens, the SDOIS system afforded a retinal FOV of ∼1.5 mm, with an axial, in-depth resolution of ∼6 μm. After imaging, both eyes received bacitracin zinc and polymyxin B sulfate ophthalmic ointment (Bausch & Lomb, Tampa, FL) to prevent corneal dehydration. During recovery, mice were placed in a bottom-warmed (33–36°C), oxygenated (21–60%) acrylic plastic sheet chamber until they fully recovered from general anesthesia.
RESULTS
STZ Model of T1D
We modeled T1D in mice by injection of STZ, which kills pancreatic β-cells, leading to loss of insulin production (Rossini et al. 1977). With the use of this paradigm, mice become hyperglycemic within 3 days of STZ injection and remain hyperglycemic for the duration of their lives. We assessed outer retina function at 1, 2, and 4 wk after onset of hyperglycemia (Fig. 1A) and measured blood glucose levels to confirm that all STZ-injected mice were significantly hyperglycemic (Fig. 1B). STZ mice were treated with exogenous insulin at levels that do not normalize blood glucose (Fig. 1B) or serum insulin (Fig. 1C) but that do prevent ketosis and dehydration.
STZ mice displayed significant reductions in a- and b-wave amplitudes beginning at 2 wk of diabetes (Fig. 1, D–F). When the relative b-wave is plotted against the relative a-wave amplitude for each time point, it was evident that the amplitude reduction in these two parameters was equivalent, indicating that the decreased photoreceptor response accounts for the loss of bipolar cell activity (Fig. 1G).
Assessment of RPE function via the dc-ERG demonstrated that STZ induces similar reductions in each of the major components of this waveform (c-wave, fast oscillation, light peak, and off response) at 2 and 4 wk (Fig. 2, A–E). Both the c-wave and fast oscillation were significantly reduced by 40% and 35%, respectively, at 2 wk (Fig. 1, A and B), localizing dysfunction to both the apical and basal RPE membranes. At 4 wk, reductions were not as overt in these components but remained significant, and the off response was found to be significantly lower in the STZ mice, as well. The light peak is the most variable component of the dc-ERG because of the duration of the stimulus and the requirement to maintain stability for the entirety of the recording period. Although we observed a reduction in light peak amplitude at all times, these reductions were not significant.
We did not find any signs of retinopathy via histology or SDOCT, nor did we find any evidence of RPE damage by SLO at any time point of analysis (data not shown). It is well documented that few or no abnormalities occur in the retina/RPE at these early time points in either STZ-treated mice or rats (Enzsoly et al. 2014; Robinson et al. 2012), and our data agree with these findings. We compared the time course over which reductions to photoreceptor (a-wave) and RPE (dc-ERG components) appear by normalizing the amplitude of each dc-ERG component from each STZ-treated animal to the control average of the corresponding dc-ERG component and then assessing that relative value in relation to the relative a-wave amplitude for individual animals. Figure 2, F–H, displays the average relative amplitude of the c-wave (Fig. 2F), fast oscillation (Fig. 2G), and off response (Fig. 2H) as a function of the relative a-wave amplitude for the time points examined. At 2 wk of hyperglycemia, the c-wave was significantly affected to a greater extent than the reduction in photoreceptor activity (Fig. 2F, P < 0.05); the 2-wk data point falls significantly below the diagonal line. At 1 and 4 wk, the data points fall along or near the line, which indicates an equivalent reduction of a- and c-wave amplitudes. When we measured the fast oscillation (Fig. 2G) and off response (Fig. 2H), the data also fell close to the diagonal line, indicating that these amplitude reductions were equivalent to the reduced photoreceptor response underlying the a-wave. Given that RPE components are generated secondary to photoreceptor activity (Berman 1991; Samuels et al. 2010; Wu et al. 2004b), it is possible that the reduction in the dc-ERG components reflects photoreceptor dysfunction, with the exception of the 2-wk c-wave.
Lepr model of T2D
DR occurs in T2D patients with only a slightly lower incidence than that in T1D patients (Klein 2007). Therefore, we conducted parallel studies in genetic mouse models of T2D over a 6-mo time course, assessing mice at 4-wk intervals through 16 wk, with endpoint testing at 24 wk (Fig. 3A). Mice harboring a homozygous G-to-T transversion mutation in the leptin receptor (Leprdb/db) display abnormal splicing and termination of the gene and loss of leptin-mediated signal transduction (Barinaga 1996; Chen et al. 1996; Lee et al. 1996). These mice recapitulate a subset of T2D characteristics including overt obesity (Fig. 3B), hyperglycemia (Fig. 3C), and increased insulin production (Fig. 3D). The Leprdb/db phenotype is influenced by the background strain on which the mutation is expressed (Coleman and Hummel 1973; Leiter et al. 1987) (Fig. 3). Homozygous Leprdb/db mice on a pure C57BLKS/J background, BKS.Lepr, are significantly and chronically hyperglycemic by 8 wk of age (Fig. 3C, right). In contrast, the BKS.Leprdb/db mutant mice bred onto the C57BL/6J background, B6.BKS.Lepr, display overt hyperglycemia as early as 4 wk of age, but their blood glucose levels fall to normal concentrations by 12 wk of age (Fig. 3C, right). Furthermore, B6.BKS.Lepr mice display significant hyperinsulinemia compared with BKS.Lepr mice (Fig. 3D; note the difference in scales). These two distinct strains of the Leprdb/db mutants permit ERG analyses that test whether differences in obesity, insulin levels, and/or hyperglycemia affect the function of the outer retina and RPE of mice sharing the same Leprdb mutation.
Fig. 3.

Leprdb/db mice are obese and display different extents of hyperglycemia and hyperinsulinemia based on background strain. A: schematic illustrating the experimental timeline of ERG testing for both strains of Lepr mice. B: body weight of B6.BKS.Lepr (left) and BKS.Lepr (right) mice over the time course of testing. C: systemic non-fasting blood glucose concentration over the time course of testing. Arrows indicate times of hyperglycemia. D: serum insulin concentration of the time course of testing. For all graphs, results for Lepr+/+,db/+ mice are shown in black and for Leprdb/db mice in gray. Data points indicate average ± SE; n ≥ 3 for each group. Student's t-test was performed between genotypes at each time point. *P < 0.05.
BKS.Lepr mice.
The BKS.Lepr mice displayed significant reductions in a-wave and b-wave amplitudes at 24 and 16 wk of age, respectively (Fig. 4, A–C). Both parameters declined with age, with the b-wave falling severely at 24 wk. At all ages, the extent of b-wave reduction was more pronounced than that of the a-wave (Fig. 4D).
Fig. 4.

Type 2 diabetic (T2D) BKS.Leprdb/db mice display reductions in a- and b-wave amplitude. A: averaged ERG waveform tracings from 3 representative flash luminance at the ages examined. B and C: average a-wave and b-wave amplitudes at each age in response to a 1.4 log cd·s/m2 flash. Results for Lepr+/+,db/+ mice are shown in black and for Leprdb/db mice in gray. Boxes surrounding ages indicate times of hyperglycemia. D: amplitude of the b-wave plotted relative to that of the a-wave. All data are normalized by the response amplitude of control littermates. The diagonal line indicates an equivalent reduction in a- and b-wave amplitude. The WT point represents the average relative amplitude ± SE calculated for Lepr+/+,db/+ mice that was utilized to determine statistical significance of Leprdb/db data. For all graphs, results for Lepr+/+,db/+ mice are shown in black and for Leprdb/db mice in gray. Diamonds indicate the age of mice at each time point of testing. Data points indicate average ± SE of at least 3 mice for each group. *P < 0.05 by 2-way ANOVA with Tukey's post hoc analysis (B and C) and Student's t-test (D).
When RPE function was examined in BKS.Lepr mice, a small and nonsignificant reduction in amplitude of the c-wave was observed at 4 wk (Fig. 5, A and E), when glucose levels were only slightly elevated (Fig. 3C). This reduction became more pronounced and was significant at each of the older ages (Fig. 5, A, E, and F), which correspond to the chronic hyperglycemia noted in this model (gray boxes, Fig. 3C). A similar progressive reduction was noted for the fast oscillation and off response, beginning at 8 and 12 wk of age, respectively (Fig. 5, B and E). These reductions correlated with that of the ERG b-wave (Fig. 4C). The dc-ERG light peak also tended to be reduced in BKS.Lepr mice at 4 and 8 wk compared with that in control littermates. Because of variability of the light peak component, statistical significance was not met. At later ages, the light peak amplitudes of control BKS.Lepr mice were abnormally low compared with those of other control mouse lines (Fig. 5, C and E).
Fig. 5.
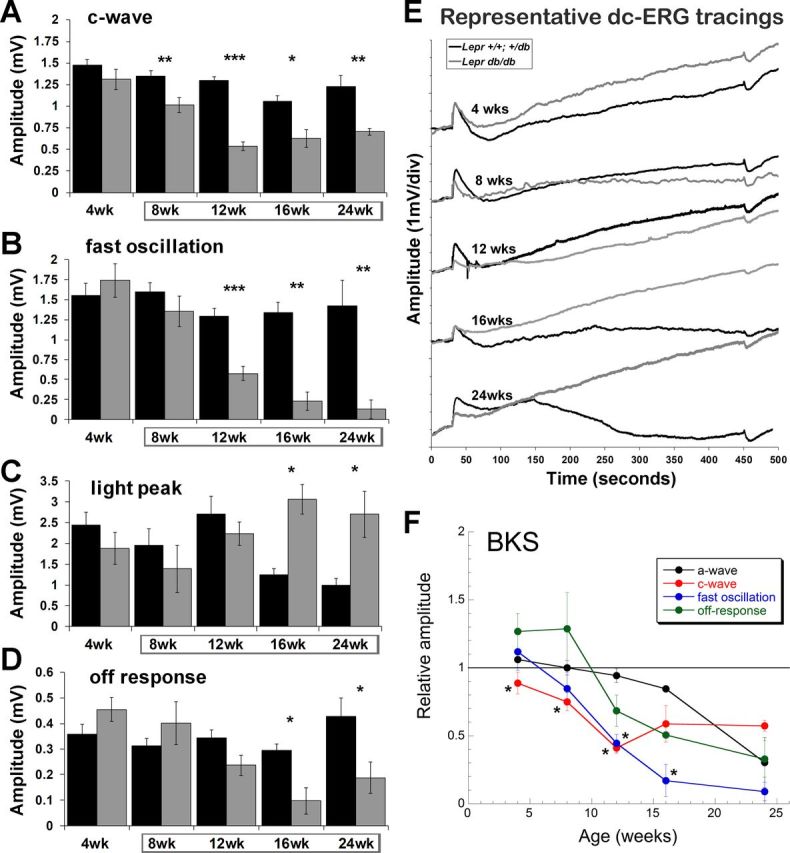
T2D BKS.Leprdb/db mice display reductions in RPE function by the dc-ERG that precede reductions in photoreceptor function. Bar graphs show c-wave (A), fast oscillation (B), light peak (C), and off response amplitude (D) of BKS.Lepr+/+,db/+ and BKS.Leprdb/db mice at the ages tested. Results for B6.BKS.Lepr+/+,db/+ mice are shown in black and for Leprdb/db mice in gray. Data points indicate average ± SE of at least 3 mice for each group. Boxes surrounding ages indicate times of hyperglycemia. E: representative dc-ERG waveform traces at each age. BKS.Lepr+/+,db/+ traces are shown in black and BKS.Leprdb/db traces in gray. F: relative amplitude of the a-wave, c-wave, fast oscillation, and off response as a function of age. Data are normalized to responses obtained from control BKS.Lepr+/+,db/+ littermates. Student's t-test was performed between genotypes at each time point. *P < 0.05; **P < 0.001; ***P < 0.0001.
To determine how dc-ERG defects and photoreceptor dysfunction correlated in BKS.Lepr mice, we plotted the normalized average amplitudes of the c-wave, fast oscillation, off response, and the a-wave as a function of age. If each dc-ERG component was affected to the same extent as the a-wave, the lines would superimpose. Instead, we found that the c-wave and fast oscillation were reduced to a greater extent than that of the a-wave at each time point through 12 and 16 wk, respectively (Fig. 5F). These data indicate that RPE dysfunction in BKS.Lepr mice cannot be attributed to a loss of photoreceptor function alone and must include an additional mechanism.
B6.BKS.Lepr mice.
B6.BKS.Lepr mice were also examined by ERG. B6.BKS.Lepr mice exhibited an initial reduction in b-wave amplitude that developed over a time frame similar to that seen in BKS.Lepr mice. Significant reductions were observed at 8 wk of age (Fig. 6, A and C) after 4 wk of hyperglycemia (Fig. 3C). At later ages, the b-wave reduction became progressively more pronounced (Fig. 6C). In comparison, statistically significant reductions in the a-wave were only found at 24 wk of age (Fig. 6, A and B). When compared, b-wave reductions of B6.BKS.Lepr mice exceeded those for the a-wave (Fig. 6D), similar to the pattern documented in BKS.Lepr mice (Fig. 4D) and distinct from that noted in the T1D model (Fig. 1G).
Fig. 6.

T2D B6.BKS.Leprdb/db mice display reductions in a- and b-wave amplitude. A: averaged ERG waveform tracings from 3 representative flash luminances over the time course of testing. B and C: average a-wave and b-wave amplitude at each age in response to a 1.4 log cd·s/m2 flash. Boxes surrounding ages indicate times of hyperglycemia. Results for B6.BKS.Lepr+/+,db/+ mice are shown in black and for Leprdb/db mice in gray. D: amplitude of the b-wave plotted relative to a-wave. Data are normalized to the responses obtained from control B6.BKS.Lepr+/+,db/+ mice at the same time point. The diagonal line indicates an equivalent reduction in a- and b-wave amplitudes. The WT point represents the average relative amplitude ± SE calculated for Lepr+/+,db/+ mice. Diamonds indicate the age of mice at each time point of testing. Data are averages ± SE; n ≥ 8 for each group. Two-way ANOVA with post hoc Tukey's test (B and C) and Student's t-test (D) was performed. *P < 0.05; **P < 0.001; ***P < 0.0001.
The time course of RPE involvement in B6.BKS.Lepr mice was also distinct from that in the other two models. Assessment of RPE function in these mice demonstrated insignificant but reproducible reductions to dc-ERG components (Fig. 7, A–D). When the amplitudes of each component were normalized to control levels and compared with the normalized a-wave amplitude, most reductions paralleled those of the a-wave (Fig. 7E; the lines superimpose). The sole exception was the reduction of the c-wave noted at 4 wk of age, the only age at which B6.BKS.Lepr mice are severely hyperglycemic (Fig. 3C). The return to normoglycemia following the initial hyperglycemia observed in this model could account for the recovery of the c-wave amplitude, but this recovery is specific to the RPE, because photoreceptor and bipolar cell function progressively decline at later ages where glucose levels are restored to normal (Fig. 6).
Fig. 7.
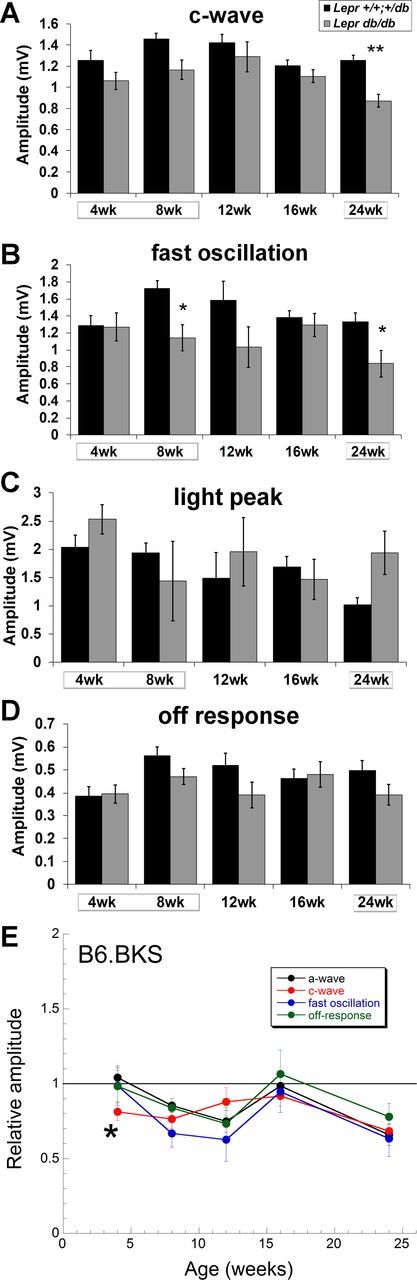
T2D B6.BKS.Leprdb/db mice display reductions in RPE function by the dc-ERG that are largely attributable to decreased photoreceptor activity. Bar graphs show c-wave (A), fast oscillation (B), light peak (C), and off response amplitude (D) of B6.BKS.Lepr+/+,db/+ and B6.BKS.Leprdb/db mice at the ages tested. Results for B6.BKS.Lepr+/+,db/+ mice are shown in black and for Leprdb/db mice in gray. Boxes surrounding ages indicate times of hyperglycemia. E: relative amplitude of the a-wave, c-wave, fast oscillation, and off response in B6.BKS.Leprdb/db mice as a function of age. Data are normalized to responses obtained from B6.BKS.Lepr+/+,db/+ mice; n ≥ 8 for each group. Student's t-test was performed between genotypes at each time point. *P < 0.05; **P < 0.001.
In both T2D mouse strains, reductions in the amplitude of either strobe-flash or dc-ERG components cannot be accounted for by cellular damage or degeneration, because no gross anatomic abnormalities were observed via SDOCT imaging (Fig. 8, A and B). Each retinal layer was distinctly identified, and no signs of retinal edema or microaneurysms were found. By SDOCT, an unhealthy retina is identifiable by the loss of specific banding demarking the retinal layers (Kim et al. 2008), which we found to be present in all mouse strains. The RPE also appeared to be normal as evidenced by SLO imaging. By using the AF modality of the SLO, changes to RPE are identifiable by the presence of hyperfluorescent foci and/or lesions (Luhmann et al. 2009), which were absent from both control and diabetic mice. These findings indicated that the electrophysiological changes do not reflect overt structural changes within the RPE or retina. In addition, we found no evidence of RPE leakage by fluorescein angiography in B6.BKS.Lepr or BKS.Lepr mice (data not shown). We did, however, observe some abnormal patterning on the red free dark reflectance modality of the SLO in B6.BKS.Lepr mice (Fig. 8B, arrowheads). The etiology of these spots is unknown; given the advanced age of the mice (50 wk), some abnormal patterning is not unexpected (Bell et al. 2012).
Fig. 8.

Leprdb/db mice do not show signs of RPE permeability or overt structural damage even at late ages. A: representative photomicrographs from scanning laser ophthalmoscopy (SLO) imaging and spectral domain optical coherence tomography (OCT) of BKS.Lepr+/+,db/+ (top) and BKS.Leprdb/db mice (bottom) at 16 wk of age. B: representative photomicrographs from SLO imaging and OCT of B6.BKS.Lepr+/+,db/+ (top) and B6.BKS.Leprdb/db mice (bottom) at 50 wk of age. No overt structural damage to the retina or RPE is observed in either strain. IRDF, infrared darkfield; RFDF, red free dark field; AF, autofluorescence. Arrowheads indicate abnormal patterning observed.
In BKS.Lepr mice, the decrease in amplitude of the dc-ERG components was not completely accounted for by a reduction in photoreceptor function (Fig. 5F). We therefore assessed the relationship between systemic glucose concentration and the amplitude of the a-wave, as well as each significantly affected dc-ERG component. In this model, the a-wave was not reduced until 24 wk (Fig. 4, A and B), although significant glucose elevations were seen at earlier ages (Fig. 9A, left; arrows denote the ages at which normal a-wave amplitudes are maintained at times of severe hyperglycemia, 8–16 wks). These data suggest that photoreceptor dysfunction occurs after a protracted period of hyperglycemia and that the initial hyperglycemic insult does not immediately induce a reduction to the a-wave. In comparison, the amplitude of the c-wave (Fig. 9B), fast oscillation (Fig. 9C), and off response (Fig. 9D) declined at the same ages that correlated with the onset and maintenance of hyperglycemia and fell below the blue line that demarks relative values equivalent to controls. This is a very different presentation from the B6.BKS.Lepr strain (Fig. 9, middle), where glucose levels are restored to the normal range by elevated serum insulin and RPE functional losses match those of photoreceptors (Figs. 7E and 9, A–D, middle). Note that if the B6.BKS.Lepr graphs in Fig. 9 were overlapped, all points would overlap except the c-wave at 4 wk, when B6.BKS.Lepr mice are severely hyperglycemic (Fig. 9B, middle; yellow arrowhead). This suggests that increased insulin delays RPE dysfunction by lowering serum glucose levels. Analysis of the a-wave, c-wave, fast oscillation, and off response relative to systemic blood glucose concentration in STZ animals compared with CNTL (Fig. 9, right) demonstrated that the a-wave is reduced at 2 wk of hyperglycemia, again illustrating the slight delay in a-wave defects. Each of the dc-ERG components was affected to a similar extent as the a-wave and superimpose with the a-wave symbols, with the exception of the 2-wk c-wave point (yellow arrowhead), which was more severely affected than the a-wave at that time point and correlated with the rise in blood glucose levels.
Fig. 9.

Reductions in RPE function of BKS.Leprdb/db mice are concomitant with increased glucose concentration. Averaged (±SE) relative a-wave (A), c-wave (B), fast oscillation (C), and off response amplitudes (D) of BKS.Leprdb/db (left), B6.BKS.Leprdb/db (middle), and STZ mice (right) plotted against average (±SE) blood glucose concentration. Amplitude measures are normalized to average control (either Lepr+/+;db/+ or CNTL) responses. Vertical blue lines highlight 250 mg/dl glucose, which is the cutoff for hyperglycemia. Horizontal blue lines highlight the average normalized control response. In BKS.Leprdb/db mice, arrows indicate a decrease in the amplitude of dc-ERG components at ages where a-wave amplitudes remain unaffected. A different pattern is seen in B6.BKS.Leprdb/db mice, however, where only the c-wave is selectively reduced at 4 wk, when mice are hyperglycemic (yellow arrowhead). In STZ mice, amplitude reductions are comparable for all ERG measures except the 2-wk c-wave (yellow arrowhead). Diamonds indicate the ages tested (BKS and B6.BKS) or the number of weeks following onset of hyperglycemia (STZ).
Within our testing paradigm, we also recorded light-adapted responses as the cone ERG. A six-step increasing luminance protocol was conducted. For each strain, equivalent findings were observed at each luminance, and data from the 1.4 log cd·s/m2 luminance are presented (Fig. 10, A–C). Whereas no reductions were observed in the T1D model at any time point (Fig. 10A), significant reductions in amplitude of the cone ERG were found in the B6.BKS.Lepr strain at 12 wk of age; these persisted and progressed through the 24-wk testing period (Fig. 10C). In comparison, smaller reductions were identified in the BKS.Lepr mice and were only significant at 24 wk (Fig. 10B). In STZ-treated rats, changes in cone morphology have been demonstrated at 12 wk (Enzsoly et al. 2014). This was noted to precede cone photoreceptor apoptosis, which has typically been found to peak at ∼6 mo (Barber et al. 1998; Park et al. 2003; Santiago et al. 2007) and correlates with our finding in BKS.Lepr mice. The earlier involvement of cone dysfunction that we observed in the B6.BKS.Lepr mice may indicate that cones or the cone pathway are impacted by high insulin.
Fig. 10.

Cone ERGs are reduced in T2D but not T1D mice. A: amplitude (right) and representative waveform tracings (left) of the cone ERG in CNTL and STZ mice in response to a 1.4 log cd·s/m2 flash at each time point examined. B: representative waveform tracings (top) and amplitude (bottom) of the cone ERG in BKS.Lepr+/+,db/+ and BKS.Leprdb/db mice in response to a 1.4 log cd·s/m2 flash at each age. C: representative waveform tracings (top) and amplitude (bottom) of the cone ERG in B6.BKS.Lepr+/+,db/+ and B6.BKS.Leprdb/db mice in response to a 1.4 log cd·s/m2 flash at each age. Note that B6.BKS.Leprdb/db mice develop reductions in the light-adapted response earlier than the BKS strain, and no changes are observed in the STZ mice at any of the time points analyzed. Results for CNTL and Lepr+/+,db/+ mice are shown in black and for STZ and Leprdb/db mice in gray. Bars indicate averages ± SE, and boxes surrounding ages indicate times of hyperglycemia; n ≥ 3 for each group. Student's t-test was performed between control and diabetic groups at each separate time point. *P < 0.05; **P < 0.001; ***P < 0.0001.
DISCUSSION
DR is a microvascular disorder, and the appearance of vision-impairing microaneurysms in the diabetic retina can be predicted by use of the mfERG (Bearse et al. 2006; Fortune et al. 1999; Harrison et al. 2011; Ng et al. 2008; Tyrberg et al. 2005); however, reductions in the amplitude and changes in the timing of the mfERG cannot be identified before development of vascular damage. We have utilized this idea that functional changes may predict development of pathology in the diabetic eye to investigate the role of RPE dysfunction as an early biomarker of DR.
This is the first study to systematically examine the onset of perturbations to RPE and outer retinal neuron function in mouse models of T1D and T2D. By evaluating the function of these cells in STZ and Leprdb/db mice, we demonstrate and establish that hyperglycemia leads to early defects within the outer retina/RPE despite differing etiologies. The functional defects documented in this work are not attributable to overt structural damage to the RPE or outer retina as evidenced by normal SLO fundus photographs and SDOCT scans (Fig. 8). Furthermore, in each strain, at least one component of the dc-ERG, the c-wave, is reduced by a greater magnitude than that of the a-wave. Because the light-evoked responses of the RPE are dependent on photoreceptor activity, this initial reduction is significant in demonstrating a specific and independent insult to the RPE concurrently with hyperglycemia. We further propose that insulin may prevent progression of RPE dysfunction, but not outer retinal neuron dysfunction, because of the different pattern of results obtained from BKS.Lepr and B6.BKS.Lepr mice.
Type 1 Diabetic Mice
STZ-injected T1D mice displayed reductions in RPE function (Fig. 2) and a-wave and b-wave amplitudes (Fig. 1, D–F) beginning at 2 wk after onset of hyperglycemia. We have previously shown in the PrphRd2/+ mouse model of photoreceptor degeneration (Samuels et al. 2010) that RPE function was preserved despite a significant loss in rod outer segment and outer nuclear layer thickness and the development of vacuoles and hypertrophy. This result indicates that the RPE function measured by the dc-ERG, although dependent on rod photoreceptor activity for its initiation, is not simply a surrogate measure of photoreceptor function. Here, dc-ERG changes exceed photoreceptor dysfunction, confirming a functional insult independent of the photoreceptor response.
Rodent models of diabetes have been widely used to understand the pathophysiological mechanisms of DR. For example, STZ-injected rats demonstrated early defects in psychophysical correlates of vision (visual acuity at 3 wk and contrast sensitivity at 9 wk post-STZ treatment) by the optokinetic tracking response (OKR) (Aung et al. 2014) and ERG OP changes at 4 wk (Hernandez et al. 2013; Layton et al. 2007; Shinoda et al. 2007; Zhang et al. 2011), as well as a- and b- wave changes (Aizu et al. 2002). Additional studies demonstrated decreases in visual acuity and OPs in STZ-injected mice at 3–4 wk (Aung et al. 2013) and in b-wave amplitude of alloxan-induced diabetic mice at similar time points (Johnsen-Soriano et al. 2008; Miranda et al. 2007). Initial evidence of neurodegeneration in the outer retina (photoreceptor inner segment swelling and vacuole presence, outer segment disorganization) and abnormal morphology of the RPE (decreased RPE thickness, reduced RPE65 staining) was identified at 12 wk following diabetes in two albino rat strains (Enzsoly et al. 2014). Akita mice, which mimic T1D by reduced insulin signaling as a result of a missense mutation in Ins2, display similar reductions in a- and b-wave amplitudes, although these deficits do not appear until 8 mo following onset of hyperglycemia (Barber et al. 2005; Gastinger et al. 2008; Han et al. 2013). Although it is not ideal to compare findings between rats and mice as phenotypes vary between strain, species and protocol for diabetes induction (Lai and Lo 2013), it is noteworthy that in all models of T1D, dysfunction and pathology attributable to the neural retina occur at times that follow the functional RPE defects identified in the present study. Notably, in contrast to our findings, the majority of the aforementioned studies do not report reductions in a-wave amplitude. Though the nature of this discrepancy is unknown, inconsistent reductions in a- and b-wave amplitude at early time points following hyperglycemia have been found (Aung et al. 2013). We hypothesize that the difference may reflect differences in anesthetic protocols for ERG testing. Most ERG recording paradigms utilize ketamine-xylazine mixtures for anesthesia, which are known to induce sustained hyperglycemia in mice (Brown et al. 2005). We utilized pentobarbital sodium, which does not impact blood glucose levels (Brown et al. 2005). Therefore, it is possible that ketamine-xylazine-induced increases in blood glucose concentrations of control animals masked differences in ERG component amplitudes between control and diabetic groups.
Type 2 Diabetic Mice
Investigations of DR and neurodegeneration in Leprdb/db mice have been performed for nearly 25 years (Cheung et al. 2005; Lai and Lo 2013; Midena et al. 1989; Robinson et al. 2012). Although structural abnormalities, including reductions in thickness of the retina and RPE basement membrane, have been identified (Clements et al. 1998; Tang et al. 2011), only one study conducted a functional assessment of the BKS.Lepr model (Bogdanov et al. 2014). These mice demonstrated reductions in a- and b-wave amplitudes, as well as other abnormalities, beginning at 16 wk of age (Bogdanov et al. 2014). Our studies of the BKS.Lepr strain, which has a similar time course of severe and sustained hyperglycemia, replicate these observations (Fig. 4). In the second T2D model studied here, B6.BKS.Lepr mice, a-wave reductions were noted at the same time point (Fig. 6A), while b-wave reductions were present earlier (Fig. 6B). Importantly, each of these defects occurs concurrently with or at time points that follow the identification of RPE defects and therefore may result as a secondary effect of the RPE dysfunction.
Our findings demonstrate that both strains of Leprdb/db mice exhibit altered RPE function evidenced by reduced dc-ERG waveform components. Significant reductions in the c-wave, fast oscillation and off response precede those of the a-wave, most notably in BKS.Lepr mice (Figs. 5 and 7). In this model, the fast oscillation was affected first (Figs. 5B, 7B, and 9C). The fast oscillation is generated by a restoration of subretinal [K+] and a [Cl−]-dependent hyperpolarization of the basal RPE membrane, so investigation of the channels underlying these currents and how they are affected by hyperglycemia will be important to examine in future studies. RPE dysfunction appears to be reversible, because the c-wave, fast oscillation, and off response in B6.BKS.Lepr mice return to control amplitudes (Fig. 7, A–D) when mice become normoglycemic (Fig. 3C). This return to normoglycemia in B6.BKS.Lepr mice is correlated with significantly elevated insulin levels (Fig. 3D). Although BKS.Lepr also display increased insulin levels, the elevation is only moderate. The Leprdb/db mutation is identical in BKS.Lepr and B6.BKS.Lepr mice; however, these strains differ in H2 haplotype, and it has been proposed that differences in metabolism of endogenous androgens and estrogen on the two backgrounds may account for their apparent differences in susceptibility and severity of diabetes (Coleman and Hummel 1973; Santiago et al. 2007). These differences make it difficult to conclude whether hyperinsulinemia is causative of RPE function stabilization. Importantly, no defects in RPE function are found before onset of hyperglycemia, and in B6.BKS.Lepr mice, when glucose concentrations are lowered because of hyperinsulinemia, RPE defects normalize to the reduction in the a-wave. Future studies to test the hypothesis that insulin reduces RPE defects could be performed by treating BKS.Lepr mice with exogenous insulin so that serum levels match those of B6.BKS.Lepr mice, and then determining if the amplitude of dc-ERG waveform components are normalized to the a-wave. In support of the possibility that insulin may protect the RPE is the recent finding by Enzsoly et al. (2014), who demonstrated that BKS.Lepr mice fed a restrictive diet to lower blood glucose levels did not display some of the ERG abnormalities found in their hyperglycemic counterparts. Additional support that elevations in glucose affect RPE function is provided by EOG data that demonstrated a reduction in fast oscillation amplitudes with acute elevations of d-glucose (Schneck et al. 2000) and data from mfERG studies that demonstrated an association of increased abnormal neuroretinal function with poor long-term glucose control (Lakhani et al. 2010; Laron et al. 2012).
b-Wave Reductions
Reductions in b-wave amplitude of diabetic mice and rats have been routinely reported by other groups (Barber et al. 1998; Hammes et al. 1995; Harrison et al. 2011; Lai and Lo 2013; Ng et al. 2008; Robinson et al. 2012; Zhang et al. 2008) and we demonstrate similar reductions here. In juxtaposition to our findings for dc-ERG components generated by the RPE, it appears that once a hyperglycemic insult has occurred, the reduction of the b-wave is immutable. Despite a return of normal glucose concentrations as observed in B6.BKS.Lepr mice, the b-wave continued to decline with age in both Lepr strains (Fig. 6B). It is important to bear in mind that the corneal-positive b- and c-waves are influenced by a cornea-negative ERG component, slow PIII, which is generated by a Kir4.1 conductance in Müller glia cells (Kofuji et al. 2000; Wu et al. 2004a). As a consequence, the b- and/or c-wave reductions could reflect an increase in slow PIII amplitude. We are using the Nyxnob mouse (Pardue et al. 1998), which lacks the ERG b-wave, to genetically isolate slow PIII and thus determine how the response properties of this component are impacted by diabetes.
Controlling Hyperglycemia
It is well established that chronic hyperglycemia is a major cause of DR and other microvascular complications associated with diabetes (Diabetes Control and Complications Trial Research Group 1993; King et al. 1999). Our findings support the notion that the RPE, responsible for transport of glucose across the blood-retinal barrier, is significantly impacted immediately following onset of hyperglycemia. Lu et al. (2013) have shown that modulation of the glucose transporter GLUT1 in the eye as a whole can modulate glucose concentrations within the retina and prevent diabetes-associated hallmarks of pathology. It is an intriguing possibility that specific reduction of GLUT1 within the RPE of diabetic mice could protect the retina from elevations in retinal glucose concentration. In addition, evaluation of RPE function in animal models of diabetes with the dc-ERG or in diabetic patients by the EOG following pharmacotherapy may be useful in measuring efficacy of treatments to slow and prevent diabetes-associated damage to the neural retina.
GRANTS
This work was supported by a Career Development Award from the Department of Veterans Affairs (to I. S. Samuels), the Foundation Fighting Blindness, and an unrestricted grant from Research to Prevent Blindness to the Cleveland Clinic Lerner College of Medicine of Case Western Reserve University.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
I.S.S. conception and design of research; I.S.S., B.A.B., A.J.P., and J.E.S. performed experiments; I.S.S., B.A.B., A.J.P., and J.E.S. analyzed data; I.S.S., B.A.B., and N.S.P. interpreted results of experiments; I.S.S. and N.S.P. prepared figures; I.S.S. drafted manuscript; I.S.S., B.A.B., and N.S.P. edited and revised manuscript; I.S.S., B.A.B., A.J.P., J.E.S., and N.S.P. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Gwen Sturgill-Short and Craig Beight for technical assistance and Dr. Brian Perkins for critical evaluation of the manuscript.
Present address of J. Saxon: Department of Outcomes Research, Anesthesiology Institute, Cleveland Clinic, Cleveland, OH 44195.
REFERENCES
- Aizu Y, Oyanagi K, Hu J, Nakagawa H. Degeneration of retinal neuronal processes and pigment epithelium in the early stage of the streptozotocin-diabetic rats. Neuropathology : 161–170, 2002. [DOI] [PubMed] [Google Scholar]
- Arden GB, Wolf JE, Tsang Y. Does dark adaptation exacerbate diabetic retinopathy? Evidence and a linking hypothesis. Vision Res : 1723–1729, 1998. [DOI] [PubMed] [Google Scholar]
- Aung MH, Kim MK, Olson DE, Thule PM, Pardue MT. Early visual deficits in streptozotocin-induced diabetic Long Evans rats. Invest Ophthalmol Vis Sci : 1370–1377, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aung MH, Park HN, Han MK, Obertone TS, Abey J, Aseem F, Thule PM, Iuvone PM, Pardue MT. Dopamine deficiency contributes to early visual dysfunction in a rodent model of type 1 diabetes. J Neurosci : 726–736, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber AJ, Antonetti DA, Kern TS, Reiter CE, Soans RS, Krady JK, Levison SW, Gardner TW, Bronson SK. The Ins2Akita mouse as a model of early retinal complications in diabetes. Invest Ophthalmol Vis Sci : 2210–2218, 2005. [DOI] [PubMed] [Google Scholar]
- Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest : 783–791, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barinaga M. Researchers nail down leptin receptor. Science : 913, 1996. [DOI] [PubMed] [Google Scholar]
- Bearse MA Jr, Adams AJ, Han Y, Schneck ME, Ng J, Bronson-Castain K, Barez S. A multifocal electroretinogram model predicting the development of diabetic retinopathy. Prog Retin Eye Res : 425–448, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell BA, Kaul C, Rayborn ME, Hollyfield JG. Baseline imaging reveals preexisting retinal abnormalities in mice. Adv Exp Med Biol : 459–469, 2012. [DOI] [PubMed] [Google Scholar]
- Berman ER. Biochemistry of the Eye. New York: Plenum, 1991. [Google Scholar]
- Bogdanov P, Hernández C, Corraliza L, Carvalho AR, Simó R. Effect of fenofibrate on retinal neurodegeneration in an experimental model of type 2 diabetes. Acta Diabetol. First published July 17, 2014; doi: 10.1007/s00592-014-0610-2. [DOI] [PubMed] [Google Scholar]
- Bresnick GH, Korth K, Groo A, Palta M. Electroretinographic oscillatory potentials predict progression of diabetic retinopathy. Preliminary report. Arch Ophthalmol : 1307–1311, 1984. [DOI] [PubMed] [Google Scholar]
- Bresnick GH, Palta M. Oscillatory potential amplitudes. Relation to severity of diabetic retinopathy. Arch Ophthalmol : 929–933, 1987. [DOI] [PubMed] [Google Scholar]
- Bronson-Castain KW, Bearse MA Jr, Neuville J, Jonasdottir S, King-Hooper B, Barez S, Schneck ME, Adams AJ. Early neural and vascular changes in the adolescent type 1 and type 2 diabetic retina. Retina : 92–102, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown ET, Umino Y, Loi T, Solessio E, Barlow R. Anesthesia can cause sustained hyperglycemia in C57/BL6J mice. Vis Neurosci : 615–618, 2005. [DOI] [PubMed] [Google Scholar]
- Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, Lakey ND, Culpepper J, Moore KJ, Breitbart RE, Duyk GM, Tepper RI, Morgenstern JP. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell : 491–495, 1996. [DOI] [PubMed] [Google Scholar]
- Cheung AK, Fung MK, Lo AC, Lam TT, So KF, Chung SS, Chung SK. Aldose reductase deficiency prevents diabetes-induced blood-retinal barrier breakdown, apoptosis, and glial reactivation in the retina of db/db mice. Diabetes : 3119–3125, 2005. [DOI] [PubMed] [Google Scholar]
- Cheung N, Mitchell P, Wong TY. Diabetic retinopathy. Lancet : 124–136, 2010. [DOI] [PubMed] [Google Scholar]
- Clements RS Jr, Robison WG Jr, Cohen MP. Anti-glycated albumin therapy ameliorates early retinal microvascular pathology in db/db mice. J Diabetes Complications : 28–33, 1998. [DOI] [PubMed] [Google Scholar]
- Coleman DL, Hummel KP. The influence of genetic background on the expression of the obese (Ob) gene in the mouse. Diabetologia : 287–293, 1973. [DOI] [PubMed] [Google Scholar]
- The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med : 977–986, 1993. [DOI] [PubMed] [Google Scholar]
- Enzsoly A, Szabo A, Kantor O, David C, Szalay P, Szabo K, Szel A, Nemeth J, Lukats A. Pathologic alterations of the outer retina in streptozotocin-induced diabetes. Invest Ophthalmol Vis Sci : 3686–3699, 2014. [DOI] [PubMed] [Google Scholar]
- Fortune B, Schneck ME, Adams AJ. Multifocal electroretinogram delays reveal local retinal dysfunction in early diabetic retinopathy. Invest Ophthalmol Vis Sci : 2638–2651, 1999. [PubMed] [Google Scholar]
- Fujii S, Gallemore RP, Hughes BA, Steinberg RH. Direct evidence for a basolateral membrane Cl− conductance in toad retinal pigment epithelium. Am J Physiol Cell Physiol : C374–C383, 1992. [DOI] [PubMed] [Google Scholar]
- Gallemore RP, Steinberg RH. Effects of DIDS on the chick retinal pigment epithelium. II. Mechanism of the light peak and other responses originating at the basal membrane. J Neurosci : 1977–1984, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallemore RP, Steinberg RH. Light-evoked modulation of basolateral membrane Cl− conductance in chick retinal pigment epithelium: the light peak and fast oscillation. J Neurophysiol : 1669–1680, 1993. [DOI] [PubMed] [Google Scholar]
- Gardner TW, Antonetti DA, Barber AJ, LaNoue KF, Levison SW. Diabetic retinopathy: more than meets the eye. Surv Ophthalmol , Suppl 2: S253–S262, 2002. [DOI] [PubMed] [Google Scholar]
- Gastinger MJ, Kunselman AR, Conboy EE, Bronson SK, Barber AJ. Dendrite remodeling and other abnormalities in the retinal ganglion cells of Ins2 Akita diabetic mice. Invest Ophthalmol Vis Sci : 2635–2642, 2008. [DOI] [PubMed] [Google Scholar]
- Griff ER, Steinberg RH. Changes in apical [K+] produce delayed basal membrane responses of the retinal pigment epithelium in the gecko. J Gen Physiol : 193–211, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes HP, Federoff HJ, Brownlee M. Nerve growth factor prevents both neuroretinal programmed cell death and capillary pathology in experimental diabetes. Mol Med : 527–534, 1995. [PMC free article] [PubMed] [Google Scholar]
- Han Z, Guo J, Conley SM, Naash MI. Retinal angiogenesis in the Ins2(Akita) mouse model of diabetic retinopathy. Invest Ophthalmol Vis Sci : 574–584, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison WW, Bearse MA Jr, Ng JS, Jewell NP, Barez S, Burger D, Schneck ME, Adams AJ. Multifocal electroretinograms predict onset of diabetic retinopathy in adult patients with diabetes. Invest Ophthalmol Vis Sci : 772–777, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez C, Garcia-Ramirez M, Corraliza L, Fernandez-Carneado J, Farrera-Sinfreu J, Ponsati B, Gonzalez-Rodriguez A, Valverde AM, Simo R. Topical administration of somatostatin prevents retinal neurodegeneration in experimental diabetes. Diabetes : 2569–2578, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holopigian K, Greenstein VC, Seiple W, Hood DC, Carr RE. Evidence for photoreceptor changes in patients with diabetic retinopathy. Invest Ophthalmol Vis Sci : 2355–2365, 1997. [PubMed] [Google Scholar]
- Hood DC, Birch DG. Beta wave of the scotopic (rod) electroretinogram as a measure of the activity of human on-bipolar cells. J Opt Soc Am A Opt Image Sci Vis : 623–633, 1996. [DOI] [PubMed] [Google Scholar]
- Hood DC, Birch DG. The A-wave of the human electroretinogram and rod receptor function. Invest Ophthalmol Vis Sci : 2070–2081, 1990. [PubMed] [Google Scholar]
- Johnsen-Soriano S, Garcia-Pous M, Arnal E, Sancho-Tello M, Garcia-Delpech S, Miranda M, Bosch-Morell F, Diaz-Llopis M, Navea A, Romero FJ. Early lipoic acid intake protects retina of diabetic mice. Free Radic Res : 613–617, 2008. [DOI] [PubMed] [Google Scholar]
- Juen S, Kieselbach GF. Electrophysiological changes in juvenile diabetics without retinopathy. Arch Ophthalmol : 372–375, 1990. [DOI] [PubMed] [Google Scholar]
- Kim KH, Puoris'haag M, Maguluri GN, Umino Y, Cusato K, Barlow RB, de Boer JF. Monitoring mouse retinal degeneration with high-resolution spectral-domain optical coherence tomography. J Vis : 17 11–11, 2008. [DOI] [PubMed] [Google Scholar]
- King P, Peacock I, Donnelly R. The UK prospective diabetes study (UKPDS): clinical and therapeutic implications for type 2 diabetes. Br J Clin Pharmacol : 643–648, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirber WM, Nichols CW, Grimes PA, Winegrad AI, Laties AM. A permeability defect of the retinal pigment epithelium. Occurrence in early streptozocin diabetes. Arch Ophthalmol : 725–728, 1980. [DOI] [PubMed] [Google Scholar]
- Klein BE. Overview of epidemiologic studies of diabetic retinopathy. Ophthalmic Epidemiol : 179–183, 2007. [DOI] [PubMed] [Google Scholar]
- Klein R, Klein BE, Wang Q, Jensen SC. Treatment and control of hypercholesterolemia and hypertension in persons with and without diabetes. Am J Prev Med : 329–335, 1995. [PubMed] [Google Scholar]
- Klein R, Wallow II, Ernest JT. Fluorophotometry. III. Streptozocin-treated rats and rats with pancreatectomy. Arch Ophthalmol : 2235–2237, 1980. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci : 5733–5740, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohzaki K, Vingrys AJ, Bui BV. Early inner retinal dysfunction in streptozotocin-induced diabetic rats. Invest Ophthalmol Vis Sci : 3595–3604, 2008. [DOI] [PubMed] [Google Scholar]
- Krupin T, Waltman SR, Szewczyk P, Koloms B, Farber M, Silverstein B, Becker B. Fluorometric studies on the blood-retinal barrier in experimental animals. Arch Ophthalmol : 631–634, 1982. [DOI] [PubMed] [Google Scholar]
- Lai AK, Lo AC. Animal models of diabetic retinopathy: summary and comparison. J Diabetes Res : 106594, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakhani E, Wright T, Abdolell M, Westall C. Multifocal ERG defects associated with insufficient long-term glycemic control in adolescents with type 1 diabetes. Invest Ophthalmol Vis Sci : 5297–5303, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb TD. Ida Mann Lecture. Transduction in human photoreceptors. Aust N Z J Ophthalmol : 105–110, 1996. [DOI] [PubMed] [Google Scholar]
- Laron M, Bearse MA Jr, Bronson-Castain K, Jonasdottir S, King-Hooper B, Barez S, Schneck ME, Adams AJ. Interocular symmetry of abnormal multifocal electroretinograms in adolescents with diabetes and no retinopathy. Invest Ophthalmol Vis Sci : 316–321, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layton CJ, Safa R, Osborne NN. Oscillatory potentials and the b-wave: partial masking and interdependence in dark adaptation and diabetes in the rat. Graefes Arch Clin Exp Ophthalmol : 1335–1345, 2007. [DOI] [PubMed] [Google Scholar]
- Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, Friedman JM. Abnormal splicing of the leptin receptor in diabetic mice. Nature : 632–635, 1996. [DOI] [PubMed] [Google Scholar]
- Leiter EH, Le PH, Coleman DL. Susceptibility to db gene and streptozotocin-induced diabetes in C57BL mice: control by gender-associated, MHC-unlinked traits. Immunogenetics : 6–13, 1987. [DOI] [PubMed] [Google Scholar]
- Li Q, Zemel E, Miller B, Perlman I. Early retinal damage in experimental diabetes: electroretinographical and morphological observations. Exp Eye Res : 615–625, 2002. [DOI] [PubMed] [Google Scholar]
- Linsenmeier RA, Steinberg RH. Origin and sensitivity of the light peak in the intact cat eye. J Physiol : 653–673, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Seidel CP, Iwase T, Stevens RK, Gong YY, Wang X, Hackett SF, Campochiaro PA. Suppression of GLUT1; a new strategy to prevent diabetic complications. J Cell Physiol : 251–257, 2013. [DOI] [PubMed] [Google Scholar]
- Luhmann UF, Robbie S, Munro PM, Barker SE, Duran Y, Luong V, Fitzke FW, Bainbridge JW, Ali RR, MacLaren RE. The drusenlike phenotype in aging Ccl2-knockout mice is caused by an accelerated accumulation of swollen autofluorescent subretinal macrophages. Invest Ophthalmol Vis Sci : 5934–5943, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midena E, Segato T, Radin S, di Giorgio G, Meneghini F, Piermarocchi S, Belloni AS. Studies on the retina of the diabetic db/db mouse. I. Endothelial cell-pericyte ratio. Ophthalmic Res : 106–111, 1989. [DOI] [PubMed] [Google Scholar]
- Miranda M, Muriach M, Almansa I, Arnal E, Messeguer A, Diaz-Llopis M, Romero FJ, Bosch-Morell F. CR-6 protects glutathione peroxidase activity in experimental diabetes. Free Radic Biol Med : 1494–1498, 2007. [DOI] [PubMed] [Google Scholar]
- Ng JS, Bearse MA Jr, Schneck ME, Barez S, Adams AJ. Local diabetic retinopathy prediction by multifocal ERG delays over 3 years. Invest Ophthalmol Vis Sci : 1622–1628, 2008. [DOI] [PubMed] [Google Scholar]
- Oakley B 2nd, Green DG. Correlation of light-induced changes in retinal extracellular potassium concentration with c-wave of the electroretinogram. J Neurophysiol : 1117–1133, 1976. [DOI] [PubMed] [Google Scholar]
- Pardue MT, Barnes CS, Kim MK, Aung MH, Amarnath R, Olson DE, Thule PM. Rodent hyperglycemia-induced inner retinal deficits are mirrored in human diabetes. Transl Vis Sci Technol : 6, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardue MT, McCall MA, LaVail MM, Gregg RG, Peachey NS. A naturally occurring mouse model of X-linked congenital stationary night blindness. Invest Ophthalmol Vis Sci : 2443–2449, 1998. [PubMed] [Google Scholar]
- Parisi V, Uccioli L. Visual electrophysiological responses in persons with type 1 diabetes. Diabetes Metab Res Rev : 12–18, 2001. [DOI] [PubMed] [Google Scholar]
- Park SH, Park JW, Park SJ, Kim KY, Chung JW, Chun MH, Oh SJ. Apoptotic death of photoreceptors in the streptozotocin-induced diabetic rat retina. Diabetologia : 1260–1268, 2003. [DOI] [PubMed] [Google Scholar]
- Pautler EL, Ennis SR. The effect of induced diabetes on the electroretinogram components of the pigmented rat. Invest Ophthalmol Vis Sci : 702–705, 1980. [PubMed] [Google Scholar]
- Penn RD, Hagins WA. Signal transmission along retinal rods and the origin of the electroretinographic a-wave. Nature : 201–204, 1969. [DOI] [PubMed] [Google Scholar]
- Phipps JA, Fletcher EL, Vingrys AJ. Paired-flash identification of rod and cone dysfunction in the diabetic rat. Invest Ophthalmol Vis Sci : 4592–4600, 2004. [DOI] [PubMed] [Google Scholar]
- Phipps JA, Yee P, Fletcher EL, Vingrys AJ. Rod photoreceptor dysfunction in diabetes: activation, deactivation, and dark adaptation. Invest Ophthalmol Vis Sci : 3187–3194, 2006. [DOI] [PubMed] [Google Scholar]
- Robinson R, Barathi VA, Chaurasia SS, Wong TY, Kern TS. Update on animal models of diabetic retinopathy: from molecular approaches to mice and higher mammals. Dis Model Mech : 444–456, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson JG, Frishman LJ. Response linearity and kinetics of the cat retina: the bipolar cell component of the dark-adapted electroretinogram. Vis Neurosci : 837–850, 1995. [DOI] [PubMed] [Google Scholar]
- Rossini AA, Like AA, Chick WL, Appel MC, Cahill GF Jr.. Studies of streptozotocin-induced insulitis and diabetes. Proc Natl Acad Sci USA : 2485–2489, 1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salam A, Mathew R, Sivaprasad S. Treatment of proliferative diabetic retinopathy with anti-VEGF agents. Acta Ophthalmol : 405–411, 2011. [DOI] [PubMed] [Google Scholar]
- Samuels IS, Lee CA, Petrash JM, Peachey NS, Kern TS. Exclusion of aldose reductase as a mediator of ERG deficits in a mouse model of diabetic eye disease. Vis Neurosci : 267–274, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels IS, Sturgill GM, Grossman GH, Rayborn ME, Hollyfield JG, Peachey NS. Light-evoked responses of the retinal pigment epithelium: changes accompanying photoreceptor loss in the mouse. J Neurophysiol : 391–402, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago AR, Cristovao AJ, Santos PF, Carvalho CM, Ambrosio AF. High glucose induces caspase-independent cell death in retinal neural cells. Neurobiol Dis : 464–472, 2007. [DOI] [PubMed] [Google Scholar]
- Schmidt R, Steinberg RH. Rod-dependent intracellular responses to light recorded from the pigment epithelium of the cat retina. J Physiol : 71–91, 1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneck ME, Fortune B, Adams AJ. The fast oscillation of the electrooculogram reveals sensitivity of the human outer retina/retinal pigment epithelium to glucose level. Vision Res : 3447–3453, 2000. [DOI] [PubMed] [Google Scholar]
- Schneck ME, Shupenko L, Adams AJ. The fast oscillation of the EOG in diabetes with and without mild retinopathy. Doc Ophthalmol : 231–236, 2008. [DOI] [PubMed] [Google Scholar]
- Shinoda K, Rejdak R, Schuettauf F, Blatsios G, Volker M, Tanimoto N, Olcay T, Gekeler F, Lehaci C, Naskar R, Zagorski Z, Zrenner E. Early electroretinographic features of streptozotocin-induced diabetic retinopathy. Clin Experiment Ophthalmol : 847–854, 2007. [DOI] [PubMed] [Google Scholar]
- Shirao Y, Kawasaki K. Electrical responses from diabetic retina. Prog Retin Eye Res : 59–76, 1998. [DOI] [PubMed] [Google Scholar]
- Simo R, Hernandez C. Neurodegeneration in the diabetic eye: new insights and therapeutic perspectives. Trends Endocrinol Metab : 23–33, 2014. [DOI] [PubMed] [Google Scholar]
- Steinberg RH, Miller S. Aspects of electrolyte transport in frog pigment epithelium. Exp Eye Res : 365–372, 1973. [DOI] [PubMed] [Google Scholar]
- Steinberg RH, Schmidt R, Brown KT. Intracellular responses to light from cat pigment epithelium: origin of the electroretinogram c-wave. Nature : 728–730, 1970. [DOI] [PubMed] [Google Scholar]
- Tang L, Zhang Y, Jiang Y, Willard L, Ortiz E, Wark L, Medeiros D, Lin D. Dietary wolfberry ameliorates retinal structure abnormalities in db/db mice at the early stage of diabetes. Exp Biol Med (Maywood) : 1051–1063, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tso MO, Cunha-Vaz JG, Shih CY, Jones CW. Clinicopathologic study of blood-retinal barrier in experimental diabetes mellitus. Arch Ophthalmol : 2032–2040, 1980. [DOI] [PubMed] [Google Scholar]
- Tyrberg M, Ponjavic V, Lovestam-Adrian M. Multifocal electroretinography (mfERG) in insulin dependent diabetics with and without clinically apparent retinopathy. Doc Ophthalmol : 137–143, 2005. [DOI] [PubMed] [Google Scholar]
- Vinores SA, Gadegbeku C, Campochiaro PA, Green WR. Immunohistochemical localization of blood-retinal barrier breakdown in human diabetics. Am J Pathol : 231–235, 1989. [PMC free article] [PubMed] [Google Scholar]
- Wachtmeister L. Oscillatory potentials in the retina: what do they reveal. Prog Retin Eye Res : 485–521, 1998. [DOI] [PubMed] [Google Scholar]
- Wilkinson-Berka JL, Miller AG. Update on the treatment of diabetic retinopathy. ScientificWorldJournal : 98–120, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkovsky P, Dudek FE, Ripps H. Slow PIII component of the carp electroretinogram. J Gen Physiol : 119–134, 1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Marmorstein AD, Kofuji P, Peachey Contribution of Kir4 NS.1. to the mouse electroretinogram. Mol Vis : 650–654, 2004a. [PMC free article] [PubMed] [Google Scholar]
- Wu J, Peachey NS, Marmorstein AD. Light-evoked responses of the mouse retinal pigment epithelium. J Neurophysiol : 1134–1142, 2004b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Kamiyama M, Nitta K, Yamada T, Hayasaka S. Selective reduction of the S cone electroretinogram in diabetes. Br J Ophthalmol : 973–975, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wu Y, Jin Y, Ji F, Sinclair SH, Luo Y, Xu G, Lu L, Dai W, Yanoff M, Li W, Xu GT. Intravitreal injection of erythropoietin protects both retinal vascular and neuronal cells in early diabetes. Invest Ophthalmol Vis Sci : 732–742, 2008. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhang J, Wang Q, Lei X, Chu Q, Xu GT, Ye W. Intravitreal injection of exendin-4 analog protects retinal cells in early diabetic rats. Invest Ophthalmol Vis Sci : 278–285, 2011. [DOI] [PubMed] [Google Scholar]