

Abstract
While a lot is known about classical, anterograde neurotransmission, less is known about the mechanisms and molecules involved in retrograde neurotransmission. Our hypothesis is that N-acetylaspartylglutamate (NAAG), the most abundant dipeptide in the brain, may act as a retrograde transmitter in the brain. NAAG was predominantly localized in dendritic compartments of glutamatergic synapses in the intact hippocampus, where it was present in close proximity to synaptic-like vesicles. In acute hippocampal slices, NAAG was depleted from postsynaptic dendritic elements during neuronal stimulation induced by depolarizing concentrations of potassium or by exposure to glutamate receptor (GluR) agonists. The depletion was completely blocked by botulinum toxin B and strictly dependent on extracellular calcium, indicating exocytotic release. In contrast, there were low levels of NAAG and no effect by depolarization or GluR agonists in presynaptic glutamatergic terminals or GABAergic pre- and postsynaptic elements. Together these data suggest a possible role for NAAG as a retrograde signaling molecule at glutamatergic synapses via exocytotic release.
Keywords: dendrite, immunogold, neurotransmission, retrograde, synapse
Introduction
Retrograde neurotransmission is the release of a signaling molecule from the postsynaptic neuron that acts on receptors at the presynaptic nerve terminal, leading to alterations in synaptic transmission (Regehr et al. 2009). Thus, retrograde transmission provides postsynaptic neurons with the ability to control their own inputs. This mechanism has gained increasing focus recently, as a way of fine-tuning signaling between neurons. While a lot is known about how presynaptic terminals communicate to their postsynaptic counterparts, less is known about the mechanisms involved in retrograde neurotransmission.
The retrograde transmitters can be classified based on their mechanisms of release: 1) lipid derivatives or gasses, 2) neuropeptides, and 3) classic neurotransmitters. The most well-established retrograde transmitters, the endocannabinoids and nitric oxide, belong to the first group. Being lipophilic, they are not subjected to a vesicular release mechanism, and their release is regulated at the level of synthesis. Neuropeptides are released from dense-core vesicles as well as through nonvesicular mechanisms from postsynaptic elements, dendrites, and somata (Cheramy et al. 1981). Some classical neurotransmitters, in particular glutamate and gamma-aminobutyric acid (GABA), have been suggested to also act as retrograde transmitters (Zilberter 2000; Jenstad et al. 2009).
In anterograde neurotransmission, a rapid rise in intracellular calcium causes synaptic vesicles filled with neurotransmitters to fuse with the cell membrane and release their contents into the synaptic cleft; a release mechanism known as exocytosis. A number of proteins are involved in exocytosis, in particular the soluble N-ethylmaleimide-sensitive attachment protein receptor (SNARE) family. Although vesicles can dock at the active zone without the direct involvement of these proteins, the priming of vesicles is mediated by SNAREs. Some SNARE proteins have been identified postsynaptically (Kennedy et al. 2010; Lau et al. 2010; Suh et al. 2010; Jurado et al. 2013; Hussain and Davanger 2015), opening for the possibility that retrograde transmitters can be released by exocytosis.
N-Acetylaspartylglutamate (NAAG) is the most abundant dipeptide in the brain, where it is present at high concentrations (μM–mM) (Miyamoto et al. 1966; Koller et al. 1984; Guarda et al. 1988). Several lines of evidence suggest that NAAG acts as a neurotransmitter (Bergeron and Coyle 2012; Watanabe et al. 2018). Biochemical studies have shown that NAAG is released Ca2+-dependently from brain slices (Zollinger et al. 1994) and synaptosomes (Pittaluga et al. 1988). However, as synaptosomes contain both pre- and postsynaptic elements (Suresh and Dunaevsky 2015; Volgyi et al. 2018), these studies do not distinguish between pre- and postsynaptic release. Further supporting a neurotransmitter function, NAAG binds to N-methyl-D-aspartate (NMDA) receptors and group II metabotropic glutamate receptors (mGluRs) (Valivullah et al. 1994; Schaffhauser et al. 1998; Shave et al. 2001). In particular, NAAG activates mGluR3 (Wroblewska et al. 1997; Neale 2011), which are localized on presynaptic nerve terminals (Sanabria et al. 2004) and inhibit the release of glutamate (Di Iorio et al. 1996; Zhong et al. 2006). This suggests that NAAG may function as a modulating neurotransmitter. The remaining question is from which synaptic compartment NAAG is released. Immunocytochemistry places NAAG in both principal neurons and interneurons in the brain (Anderson et al. 1986; Frondoza et al. 1990; Moffett et al. 1993; Tsai et al. 1993), but apart from one study in the retina (Williamson and Neale 1988), the subcellular localization of NAAG in the central nervous system has not been studied. Therefore, it is not known which neuronal compartment contains the highest concentration of NAAG, which compartment is responsible for the release of NAAG in the brain, or the release mechanism, or whether NAAG exhibits retrograde signaling. To address these questions, we used high resolution postembedding immunogold cytochemistry to reveal the in vivo localization of NAAG in the hippocampus. In hippocampal brain slices, we studied the redistribution of NAAG in response to neuronal stimulation, as well as its sensitivity for botulinum toxin B and dependency on Ca2+.
Materials & Methods
Preparation and Incubation of In Vitro Hippocampal Slices
Acute hippocampal slices were obtained and treated largely as previously described (Holten et al. 2008). Adult Wistar rats were anesthetized with halothane or isoflurane (Abbott), decapitated, and the brains were put in ice-cold normal Krebs’ solution (in mM): 125.5 NaCl (Sigma-Aldrich, S7653), 2.5 KCl (Sigma-Aldrich, P9541), 1.0 NaH2PO4 (Sigma-Aldrich, S8282), 24 NaHCO3 (Sigma-Aldrich, S5761), 2.5 CaCl2 (Sigma-Aldrich, C5670), 2.0 MgSO4 (Sigma-Aldrich, M7506), and 15 mg glucose (Sigma-Aldrich, D9434). Hippocampi were dissected out, sliced (300 μm), and incubated for 45 min at 30 °C (after preincubations at 30 °C for 60 min) in oxygenated (95% O2, 5% CO2) normal (2.5 mM K+), or depolarizing (55 mM K+; Na+ reduced to 73 mM) Krebs’ solutions. Some of the slices were incubated at 2.5 mM K+ in the presence of a cocktail of GluR agonists 500 μM (S)-α-amino-3-hydroxy-5-methyl-4-isoxazoleprpionic acid (AMPA) (Tocris Bioscience, 83643-88-3), 100 μM cyclothiazide (Sigma Aldrich, 2259-96-3), 100 μM N-methyl-D-aspartic acid (NMDA) (Tocris Bioscience, 6384-92-5) and 100 μM (S)-3.5-dihydroxyphenylglycine (DHPG) (Tocris Bioscience, 162870-29-3). To reveal the mechanism of NAAG release, other slices were incubated with GluR agonists (in 2.5 mM K+) in the absence of calcium, or in the presence of botulinum toxin B (BoNT/B, 133 nM) (Sigma Aldrich, 93384-44-2). In the latter experiments, the slices were preincubated for 120 min with or without BoNT/B (133 nM) to increase the penetration of the toxin into the tissue before the incubation.
The slices were kept on a nylon mesh in a glass beaker with a continuous flow of O2 in the medium. After incubation, the slices were fixed for 1 h at room temperature (∼22 °C) in a mixture of 5% 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) (Thermo Fisher, 22981) and 1 mM N-hydroxysuccinimide (NHS) (Sigma-Aldrich, 130672) in 10 mM (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES) (Sigma-Aldrich, H7006) buffer (pH 7.4) or 30 min at room temperature in a mixture of 5% EDC and 1 mM NHS in 10 mM HEPES buffer, followed by 1 h at room temperature in a mixture of 5% EDC, 1 mM NHS, 1% formaldehyde (FA) (Sigma-Aldrich, 158 127), and 0.5% glutaraldehyde (GA) (AGAR scientific, R1010) or 2.5% GA and 1% FA in 0.1 M phosphate buffer (Sigma-Aldrich, S9638 and S7907) for 1 h at room temperature. The slices were kept overnight at 4 °C in the same fixative and then stored in the fixative diluted at 1:10.
In Vivo Perfusion Fixation
Adult Wistar rats were anesthetized by injection of pentobarbital (100 mg/kg, i.p.) and fixed by perfusion through the heart with 4% EDC plus 4% FA and 0.5% GA in 10 mM HEPES or 4% EDC, 1 mM NHS, and 1% dimethyl sulfoxide in 0.9% NaCl using a peristaltic pump.
Light Microscopic Immunocytochemistry
Slices used for staining of NAAG were allowed to sink in 30% sucrose (Sigma-Aldrich, S7903) before sectioning at 20 μm on a freezing microtome. Slices for glutamate staining were processed without further sectioning since BoNT/B is a large protein and therefore has a limited ability for diffusion into the slices, and hence only the surface of the slice could be used for analysis. Labeling with the NAAG antibodies (dilution 1:1000) was performed on free-floating sections. Labeling with the glutamate antibodies (Glu 607, dilution 1:60 000) was performed on free-floating slices, where complexes of GA-treated glutamine (Sigma-Aldrich, G3126), asparagine (Sigma-Aldrich, A0884), and aspartate (Sigma-Aldrich, A9256) (each at a concentration of 0.2 mM) were added to prevent any unspecific binding produced by the antibodies. The labeling was performed according to a three-layer biotinylated antibody-streptavidin-biotinylated peroxidase method (Hsu et al. 1981). High-resolution digital images of the NAAG staining were obtained using an automated Olympus BX52 microscope equipped with a 20× objective (Olympus Uplan Apo, NA 0.70), a motorized stage (LEP MAC5000, LUDL Electronic Products Ltd, Hawthorne, NY, USA), an Optronics MicroFire digital camera (Optronics Goleta, CA, USA), and Neurolucida 7.0 Virtual Slice software (MBF Bioscience, Inc, Williston, VT, USA). High-resolution digital images of the glutamate staining were obtained using an automated slide scanner system (Axioscan Z1, Carl Zeiss MircoImaging, Jena, Germany) with a 20×/0.8 plan apochromat objective and an Orca Flash 4.0 camera (Hamamatsu Photonics, Japan). Slides were scanned with extended depth of focus (EDF) for enhanced visualization of details from several focal planes.
Postembedding Electron Microscopic Immunocytochemistry
Slices from each experimental condition described above and specimens from perfusion-fixed animals were cryoprotected in glycerol (Sigma-Aldrich, G5516), quickly frozen in liquid propane, freeze-substituted with methanol, embedded in Lowicryl HM20 (polysciences), and further processed for immunogold labeling (Bergersen et al. 2008). Ultrathin sections (80–90 nm) were cut and mounted on nickel grids or formvar-coated one-hole grids. The slices were sectioned perpendicularly to the surface of the slice. Then the sections were processed in single labeling experiments with the NAAG antibodies (dilution 1:300, 1:500, or 1:600) or in double labeling experiments with NAAG (1:300) and GABA (1:100) or glutamine synthetase (GS) (BD transduction laboratories, #84458, 0.3 μg/mL) antibodies (Bergersen et al. 2008). In the double labeling experiments with GABA, the sections were first treated with the NAAG antibodies and then with the GABA antibodies to visualize NAAG in GABA terminals. Since both these primary antibodies were from rabbit, the sections were treated with FA vapor at 80 °C for 1 h between the NAAG labeling and the GABA labeling to prevent interference between the sequential labelings (Wang and Larsson 1985; Ottersen et al. 1992). A secondary antibody coupled to gold particles with a diameter of 10 nm was used in the first step (NAAG), and a secondary antibody with 15 nm gold particles was used in the second step (GABA). In the double labeling with GS, the sections were treated with the NAAG and GS antibodies at the same time.
Sera and Specificity Controls
The NAAG antiserum was raised in rabbits against NAAG bound to thyroglobulin by EDC (Cangro et al. 1987) and purified by adsorption on a series of agarose columns bearing sepharose-EDC-thyroglobulin and -BSA. Both the 990 GABA and the 607 glutamate antisera were raised in rabbits as described (Storm-Mathisen et al. 1983) by immunizations with GA–FA–BSA conjugates of GABA and glutamate, respectively. The GABA antiserum (Gundersen et al. 2004) and the glutamate antiserum (Ottersen et al. 1990; Gundersen et al. 1991) have been characterized previously and proven to be specific. As a precaution against cross-reactivities, soluble GA–FA complexes of L-glutamate (Sigma-Aldrich), glutamine (Sigma-Aldrich), and β-alanine (0.2 mM) (Sigma-Aldrich, A9920) were added to the diluted GABA antibodies, and soluble GA–FA complexes of aspartate, asparagine, and glutamine were added to the glutamate antibodies 3 h before applying it to the sections. The selectivity of the NAAG antibodies was tested before use, and selectivity controls were included in each individual experiment. As an intrinsic selectivity control for the light microscopic NAAG immunolabeling, conjugates of different acetylated molecules and carboxylates linked to brain macromolecules by EDC were spotted on cellulose nitrate–acetate filters, and these were processed together with the tissue sections. The NAAG antibodies selectively stained the NAAG spot (see Supplementary Fig. S1). Furthermore, the NAAG immunoreactivity of tissue sections and dot blots was blocked by adding 1.0 mM EDC-treated NAAG (Sigma-Aldrich A5930) to the NAAG antibodies (see Supplementary Fig. S1). As an in-experiment specificity test, some tissue sections were exposed to antibodies that had been pretreated with soluble NAAG EDC complexes for at least 3 h prior to immunolabeling. These preabsorption tests were performed along with both peroxidase (see Supplementary Fig. S1) and immunogold tissue labelings (Fig. 1 and Supplementary Fig. S2). The preabsorbed immunsera produced very low immunoreactivities in all experiments. As the immunogold method was performed on an ultrathin section after embedding in plastic-like resins, the possible problem of uneven antibody penetration between different subcellular structures is eliminated, as only molecules on the section surface are labeled.
Figure 1.
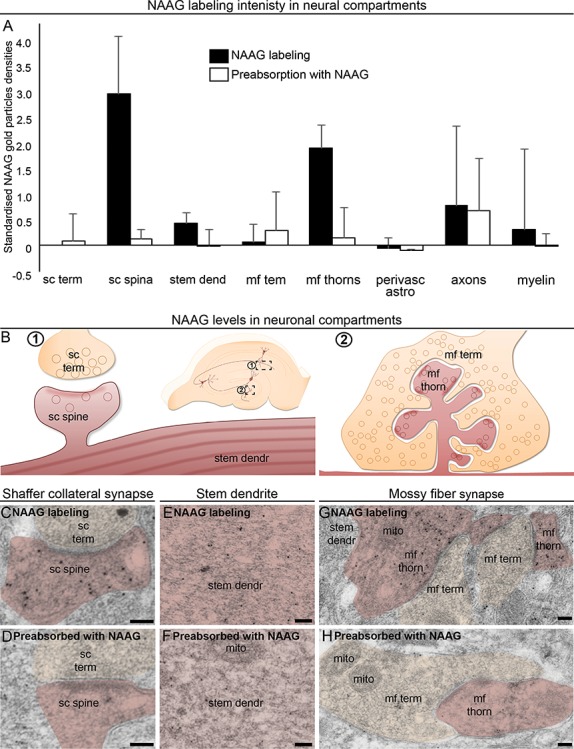
NAAG is predominantly located in postsynaptic dendritic spines/thorns. Subcellular structures from different cell types were evaluated in perfusion-fixed rat hippocampus by electron microscopy to investigate in which structures NAAG is found above background levels. This was performed in three different labeling experiments, with or without double labeling with GS (astrocyte marker). Therefore, the data were standardized to the Schaffer collateral terminals in each rat using z-transformation (A). The bars represent standardized mean and standard deviation. NAAG labeling was compared with the labeling in the same structures with NAAG antibodies preabsorbed with NAAG complexes. The latter gives a measure of the background, unspecific, labeling produced by the antibodies and was low in all compartments. Most of the nonsynaptic structures, however, were at the same low level, indicating low or nonexisting NAAG levels. The data were used qualitatively to determine which areas to focus on in the more detailed analysis, and hence no statistical analysis was performed. The NAAG staining appeared above background only in Shaffer collateral spines (B; C vs. D), stem dendrites (B; E vs. F), and mossy fiber thorns (B; G vs. H). Electron micrographs of perfusion-fixed tissue show that the density of gold particles signaling NAAG was much higher in dendritic spines (sc spine) forming excitatory synapses with Schaffer collateral terminals (sc term) (C) than in their presynaptic counterpart. The same was true for the dendritic thorns (mf thorn) compared to the mossy fiber terminals (mf term) forming excitatory synapses in dentate gyrus (E). Scale bars = 100 nm.
Quantitative Immunogold Analysis
The ultrathin sections were imaged in a FEI Tecnai 12 electron microscope. Electron micrographs were taken along random trajectories across the thickness of the slice in the stratum radiatum of CA1 and in the dentate gyrus at ×26.500–43.000 magnification. The BoNT/B-treated slices were cut perpendicular to the section surface so that the observations could be made at defined distances from the slice surface. BoNT/B is a relatively large protein, which restricts its diffusion into the tissue. The electron micrographs were taken within 30 μm from the surface of the slice. Here, the texture of the tissue was loose, permitting penetration of BoNT/B. In the CA1 hippocampus NAAG immunogold particles were quantified in terminals making asymmetrical synapses, in their postsynaptic spines, and in dendritic shafts. In the dentate gyrus NAAG immunoreactivity was quantified in mossy fiber terminals, as well as in their dendritic thorns, and in GABA positive nerve terminals and their postsynaptic compartments (granule cell somata and dendritic shafts). For slice experiments background NAAG labeling was calculated in sections labeled with NAAG antibodies that were preneutralized with soluble NAAG–EDC complexes for all tissue profiles included in the quantitative analysis and subtracted from the averages of the immunogold labeling produced by the NAAG antibodies. Quantification from perfusion-fixed tissue labeled either with NAAG antibodies or with NAAG antibodies preneutralized with soluble NAAG–EDC complexes is shown side-by-side in Figure 1 and Supplementary Figure S2. The areas of the various cellular and subcellular elements were recorded by ImageJ (http://rsb.info.nih.gov/ij/) and the coordinates were submitted to a program written in Python (http://www.python.org) for computation of particle densities (number of gold particles per μm2; (Larsson et al. 2005). The source code of ImageJ plugin and the Python program is available at https://old.liu.se/medfak/forskning/larsson-max/software?l=en. The association between gold particles and postsynaptic vesicle-like structures was measured as the distance from the center of the vesicle-like structure to center of the gold particle using ImageJ, compared to the distance from random points (generated by the ImageJ macro) to the center of the closest vesicle-like structure (Fig. 3D–G).
Figure 3.
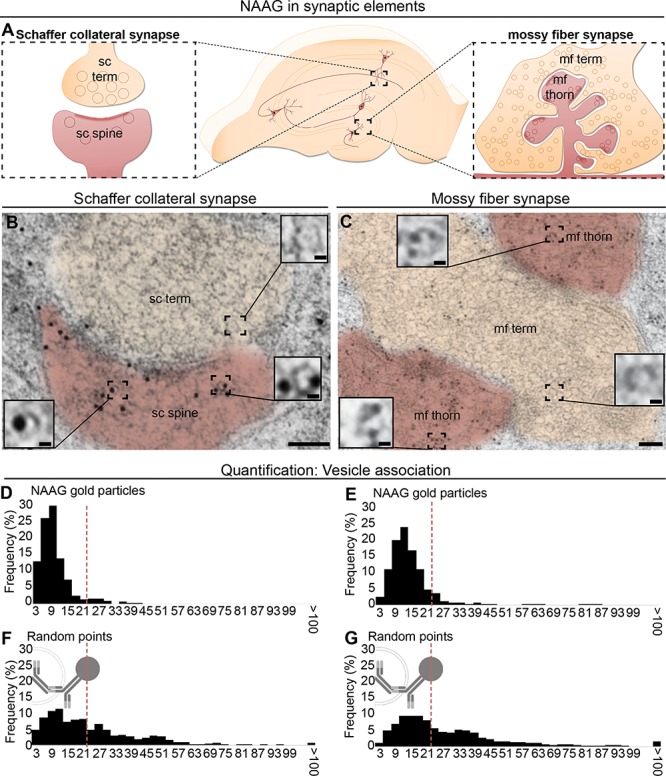
NAAG is located in vesicular structures in both postsynaptic dendritic spines in stratum radiatum (A, left) and in postsynaptic mossy fiber thorns in dentate gyrus (A, right). In the dendritic spines and thorns, gold particles representing NAAG were closely related to vesicular structures, which in size and shape resemble presynaptic vesicles (insets in B and C). Scale bars in (B and C): 100 nm; scale bar in insets: 10 nm. The distribution of distances of gold particles and the closest vesicle-like structures was quantified. In both postsynaptic compartments, the sc spines and the mf thorns, a large proportion of the NAAG particles were localized within 21 nm from the center of a vesicle (D and F). For comparison, for the randomly inserted points (E and G), only about half of the points were found within the same range from the vesicle (P = 9.1 × 10−43 for sc spines; and P = 8.9 × 10−50 for mf thorns; Pearson chi square test). 21 nm represents the theoretical distance between the center of the gold particle and the epitope (the radius of the gold particle [5 nm] + the length of the primary–secondary complex).
Statistics
The results were statistically evaluated (GraphPad) by the Students t-test when only comparing two groups, or with one-way ANOVA, Tukey’s post hoc test for multiple comparisons, to investigate differences in mean gold particle densities between individual experimental groups. In Figure 1, three rats, from three different staining experiments and with two different fixation regimes, have been standardized to the levels in Schaffer collateral terminals in each rat, using z-transformation. Statistical tests between perfusion-fixed brains labeled with NAAG antibodies, the same brains labeled with NAAG antibodies that had been preabsorbed with NAAG complexes, were not performed, as the latter is represented by two animals. The raw values (not standardized) are shown in Supplementary Figure S2. The distribution of distances between NAAG immunogold particles or random points and the closest vesicle-like structure was first converted to categorical data using the maximal theoretical distance of 21 nm as a cut-off. The difference in vesicle-association (portion of particles within 21 nm from the center of the vesicle) between NAAG immunogold particles and random points was analyzed by the Pearson’s chi-squared test (Microsoft Excel).
Results
NAAG is Located in Dendritic Spines/Thorns in the Brain
To unravel the subcellular compartment that contains the highest levels of NAAG, we performed peroxidase labeling. NAAG immunoreactivity was located in cell bodies of CA1 and CA3 pyramidal cells, granule cells, and cells in the dentate gyrus (see Supplementary Fig. S1A). It was evident that the NAAG staining appeared in parts of the dendritic tree of these cells, in addition to very small dots lining the dendritic trees (see Supplementary Fig. S1C,D), as expected for substances that have a synaptic localization. This synaptic-like labeling was completely absent in the hippocampal section labeled with the same antibodies preabsorbed with soluble NAAG–EDC complexes in excess (see Supplementary Fig. S1B,D,E).
Electron microscopy revealed high levels of NAAG in postsynaptic spine of the Schaffer collateral synapses in stratum radiatum, the post-synaptic thorns of the mossy fiber synapses in the dentate gyrus, and stem dendrites (Fig. 1). Other compartments, including presynaptic Schaffer collateral terminals and mossy fiber terminals, as well as perivascular astrocytes, myelin, and axons showed staining that was undistinguishable from the background (Fig. 1). Hence, we focused our further ultrastructural analysis on Schaffer collateral synapses in the hippocampal stratum radiatum and the mossy fiber synapses of the dentate gyrus.
The predominant localization of NAAG in postsynaptic spines/thorns opens up the possibility that NAAG is released as a retrograde transmitter from the postsynaptic compartment.
NAAG is Depleted From Dendritic Spines/Thorns During Neuronal Activation
As in the perfusion-fixed tissue, electron microscopic analysis from hippocampal slices incubated at physiological (2.5 mM) K+ concentrations revealed that the most prominent NAAG labeling was in postsynaptic dendritic spines of CA1 excitatory synapses (Fig. 2A,D) and dendritic thorns of mossy fiber synapses (Fig. 2E,H), while their corresponding nerve terminals (Schaffer collateral terminals and mossy fiber terminals, respectively) (Fig. 2A,D,E,H) and CA1 stem dendrites (Fig. 2I) displayed much weaker labeling (P < 0.01, One-way ANOVA, Tukey’s multiple comparisons test, GraphPad). To determine if NAAG could be released from spines/thorns, we exposed hippocampal slices to stimulation by high (55 mM) K+, or GluR agonists (100 μM DHPG, 100 μM NMDA, and 500 μM AMPA stabilized by 100 μM cyclotiazide). During K+ or GluR agonist-induced stimulation, immunogold labeling of NAAG was selectively depleted from the postsynaptic Schaffer collaterals (Fig. 2B,C vs. Fig. 2A, P < 0.01, one-way ANOVA, Tukey’s post hoc test for multiple comparisons, GraphPad) and mossy fiber thorns (Fig. 2F,G vs. Fig. 2E, P < 0.01, one-way ANOVA, Tukey’s multiple comparisons test, GraphPad). The stimulations did not affect the density of NAAG gold particle in the nerve terminals of these synapses (Fig. 2B,C,F,G, P > 0.05, one-way ANOVA, Tukey’s multiple comparisons test, GraphPad), or in dendritic shafts (Fig. 2I–K, P > 0.05, one-way ANOVA, Tukey’s multiple comparisons test, GraphPad).
Figure 2.
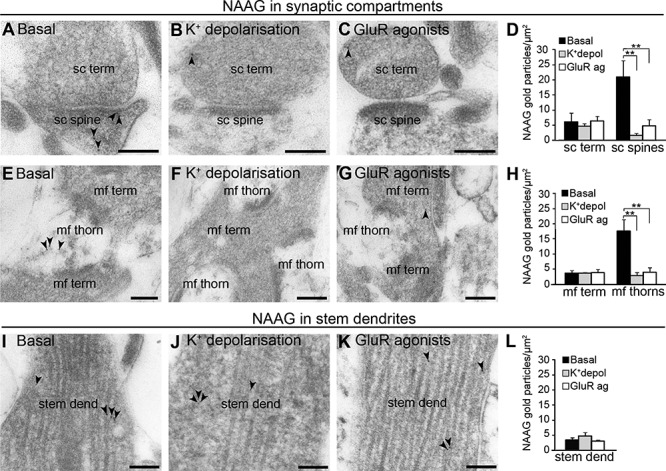
NAAG is depleted upon depolarization with high potassium or GluR agonists. Electron micrographs from slice experiments (A and E) confirmed what was found in the perfusion-fixed tissue: During basal conditions (2.5 mM K+) gold particles signaling NAAG were mainly present in dendritic spines (sc spine) and thorns (mf thorn) forming asymmetric synapses with Schaffer collateral terminals (sc term) or mossy fiber terminals (mf term). In response to membrane depolarization by K+ (55 mM; B, F and J) or by GluR agonists (C, G and K), NAAG immunogold particles were depleted from the postsynaptic sc spines (B; C compared to A) and mf thorns (F; G compared to E), without any change in the corresponding terminals or stem dendrites (J; K compared to I). Bar charts show the density of NAAG immunogold in Schaffer collateral presynaptic terminals (sc term) and postsynaptic spines (sc spine) (D) and stem dendrites (stem dend) (L) in the stratum radiatum, and in mossy fiber terminals (mf term) and thorns (mf thorn) of the dentate gyrus (H) of hippocampal slices incubated at 2.5 mM K+ (black bars), depolarizing conditions (55 mM K+; gray bars) or GluR agonists (white bars). The bars represent the average number of NAAG immunogold particles/μm2 ± SD. ** shows the difference from the same profile at basal conditions (P < 0.01, one-way ANOVA, Tukey’s multiple comparisons test, GraphPad). The total numbers of tissue profiles included in the quantifications were as follows (basal-K+-GluR): Schaffer collateral terminals and spines, 95-104-87; stem dendrites, 61-64-66, mossy fiber terminals, 62-69-61; and mossy fiber thorns, 174-156-180 (two Wistar rats, n = three slices in each condition).
NAAG is Located to Postsynaptic Vesicles
NAAG was localized at high densities in dendritic spines of the Schaffer collateral synapses (Fig. 3A,B) and thorns of the mossy fiber synapses (Fig. 3A,C). Interestingly, in the postsynaptic dendritic spines/thorns NAAG was associated with small, round, and clear structures (insets in Fig. 3B,C), resembling what we have previously observed for neurotransmitter labeling in synaptic vesicles in nerve terminals (Bergersen et al. 2008; Morland et al. 2013). The vesicle-association of NAAG gold particles was demonstrated statistically by comparing the proportion of NAAG gold particles that were located in close proximity to a vesicle, to an expected distribution (random points inserted in the same image). We demonstrate that 90.6% and 89.7% of the NAAG particles are localized within 21 nm from the center of the closest vesicle in Schaffer collateral spines and mossy fiber thorns, respectively (Fig. 3D,E). This proportion was significantly higher than what was found for random points, where 50.1% and 49.8% of the particles were located within 21 nm from the center of the closest vesicle for the two postsynaptic compartments, respectively (Fig. 1G,F), (P = 9.1 × 10−43 for Schaffer collateral spines and P = 8.9 × 10−50 for mossy fiber thorns; Pearson’s chisquared test) (Fig. 3G,F).
Characterization of Postsynaptic NAAG Depletion
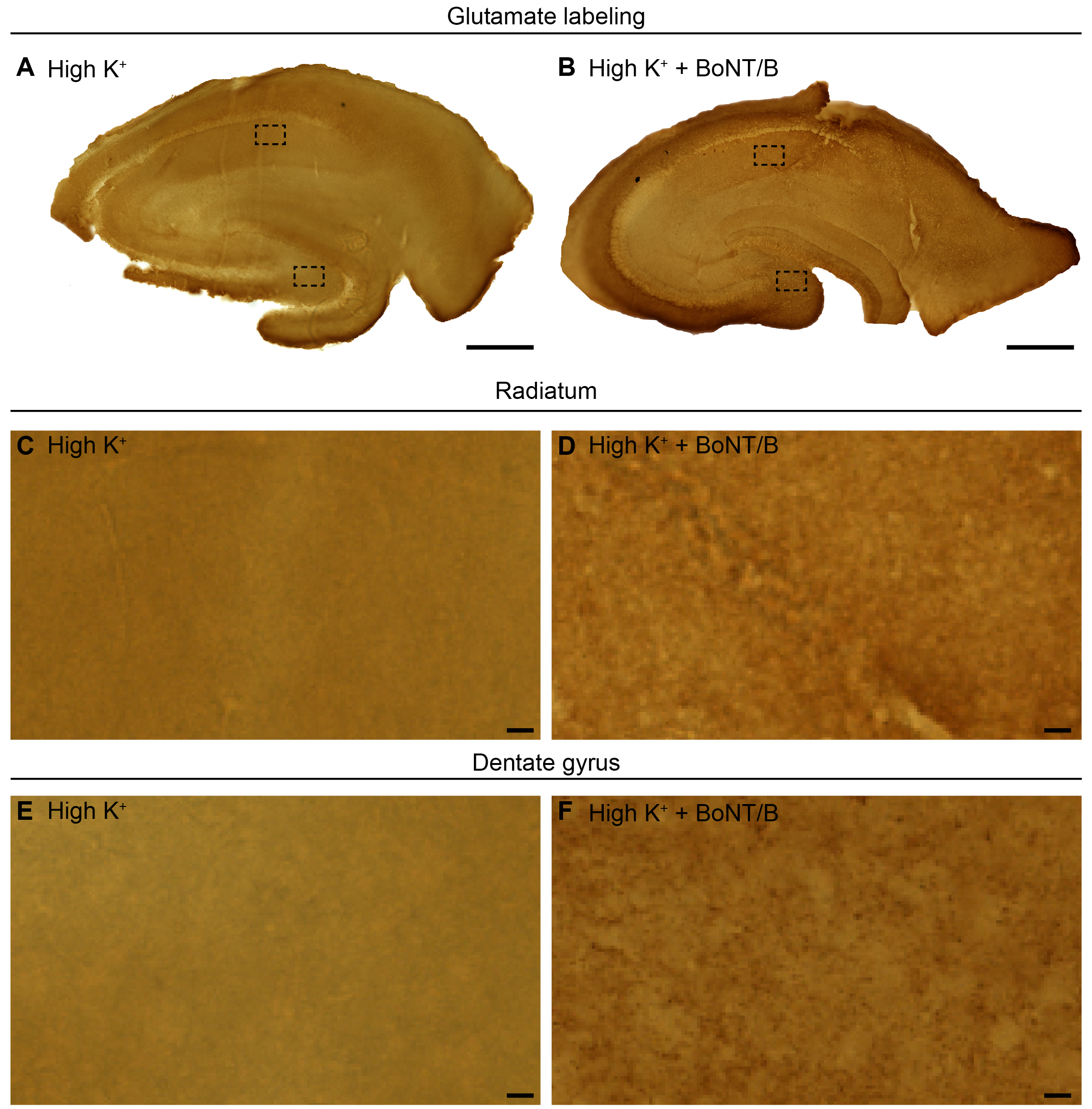
The accumulation of NAAG labeling over postsynaptic vesicles prompted the question whether NAAG underwent vesicular release from the postsynaptic spines/thorns. To elucidate the mechanism whereby NAAG escapes from dendritic spines/thorns, we stimulated slices with GluR agonists and inhibited exocytosis either by removing Ca2+ from the extracellular medium or by treatment with BoNT/B. BoNT/B blocks vesicular fusion by cleaving VAMP2 (Pellizzari et al. 1996), a SNARE protein reported to regulate postsynaptic exocytosis (Jurado et al. 2013). To determine the efficacy of BoNT/B in our set-up, we first evaluated the effect of BoNT/B on glutamate depletion during membrane stimulation. In hippocampal slices depolarized with K+ (55 mM) BoNT/B clearly inhibited the depletion of glutamate from nerve terminals (see Supplementary Fig. S3). Thus, having tested that the BoNT/B concentration used in our experiments was sufficient to block exocytosis, we proceeded to analyzing the effect of BoNT/B on the depletion of NAAG. In hippocampal slices stimulated with GluR agonists, BoNT/B or zero Ca2+ blocked depletion of NAAG from postsynaptic CA1 dendritic spines (Fig. 4B–E) and mossy fiber thorn synapses (Fig. 4F–I). Inhibiting exocytosis by BoNT/B did not affect the content of NAAG immunogold particles in the nerve terminals (Fig. 4D,H) or in dendritic shafts (data not shown) during membrane stimulation. Interestingly, in postsynaptic spines/thorns subjected to membrane depolarization combined with inhibited exocytosis (in particular BoNT/B treatment), the NAAG signal was increased above resting values (Fig. 4B vs. Fig. 4D; Fig. 4J,F vs. Fig. 4H,K), P < 0.01, one-way ANOVA, Tukey’s multiple comparisons test, GraphPad.
Figure 4.
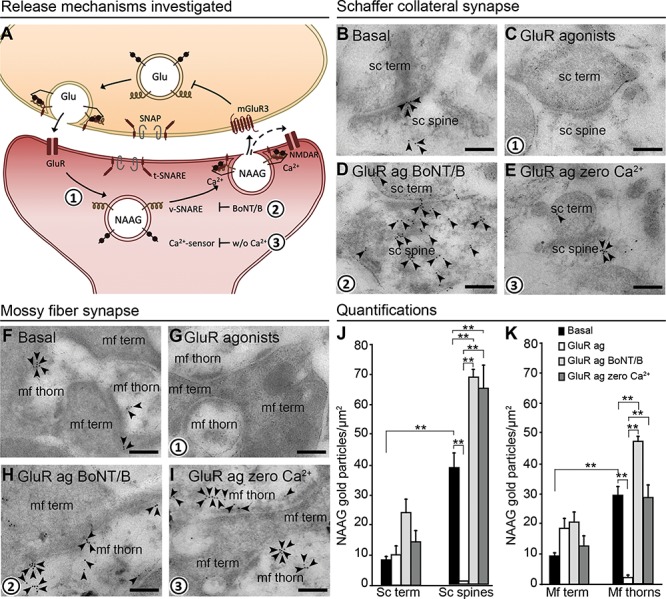
NAAG is depleted by exocytosis from postsynaptic dendrites. The schematic drawing (A) shows the proposed mechanism of action and regulation of NAAG release (numbers in bold indicate mechanisms studied by us). Presynaptic membrane fusion of transmitter-containing vesicles is mediated by members of the SNARE family: the v-SNARE, target membrane SNARE (t-SNARE), and synaptosomal-associated protein (SNAP), in addition to Ca2+ sensors. We investigated whether the same mechanism is responsible for the postsynaptic NAAG release. Glutamate released from the nerve terminal stimulates dendritic ionotropic glutamate receptors (GluR) (1), which triggers Ca2+-dependent (2) and v-SNARE-dependent (3) release of NAAG. NAAG then acts on presynaptic mGluR3/postsynaptic NMDAR (mixed agonist/antagonist), the former inhibiting Glu release from the nerve terminal. The electron micrographs (B–I) show how inhibiting exocytosis during membrane depolarization affects the depletion of NAAG immunogold particles from postsynaptic dendritic spines/thorns. During basal conditions (2.5 mM K+), gold particles representing NAAG were present postsynaptically in dendritic spines and thorns belonging to Schaffer collaterals (B, sc spine) and mossy fibers (F, mf thorn) and with very low levels in the presynaptic terminals (sc term and mf term). When postsynaptic depolarization was elicited by GluR agonists, NAAG was depleted from the postsynaptic spines (C) and thorns (G). When v-SNARE was degraded by BoNT/B, NAAG was trapped postsynaptically despite GluR agonist activation (D and H). Finally, in the absence of Ca2+ (zero Ca2+), NAAG was also trapped postsynaptically despite the presence of GluR agonists (E and I). Scale bars, 100 nm. Quantitative results of NAAG immunogold particles in different tissue profiles are shown in (J) for the Schaffer collateral synapses of the stratum radiatum and (K) for the mossy fiber synapses of the dentate gyrus. Resting conditons; 2.5 mM K+ (basal, black bars), GluR agonists (GluR ag., white bars), GluR agonists in the presence of botulinum toxin B (GluR ag BoNT/B, light gray bars), and GluR agonists in the absence of Ca2+ (GluR ag zero Ca2+, dark gray bars). The bars represent the average number of NAAG immunogold particles/μm2 + SD in Schaffer collateral spines (sc spines) and nerve terminals (sc term) belonging to Schaffer collateral synapses in the stratum radiatum (J), and thorns (mf thorns) and mossy fiber terminals (mf term) belonging to hilar mossy fiber synapses (K). **: P < 0.01, one-way ANOVA, Tukey’s multiple comparisons test, GraphPad. The numbers of tissue profiles per slice that was included in the quantifications were as follows: sc spines and sc terminals: 20–26 (basal), 23–29 (GluR agonists), 21–25 (GluR agonists zero Ca2+), 19–25 (GluR agonists BoNT/B); mf thorns: 37–63 (basal), 41–71 (GluR agonists), 53–57 (GluR agonists zero Ca2+), 47–73 (GluR agonists BoNT/B); mf terminals: 19–25 (basal), 20–29 (GluR agonists), 20–21 (GluR agonists zero Ca2+), 19–22 (GluR agonists BoNT/B), (slice experiment from two rats (pooled), n = 4–5 slices in each condition).
Postsynaptic Accumulation of NAAG and Depletion Upon Depolarization Did Not Occur in GABA Synapses
In GABAergic nerve terminals, K+-induced membrane depolarization led to depletion of GABA (P < 0.05, unpaired t-test, two tails, GraphPad) as predicted (Gundersen et al. 2004), but there was no depletion of NAAG (P > 0.05, unpaired t-test, two tails, GraphPad) (Fig. 5). Hence, postsynaptic release of NAAG appears to be restrained to the glutamatergic system.
Figure 5.
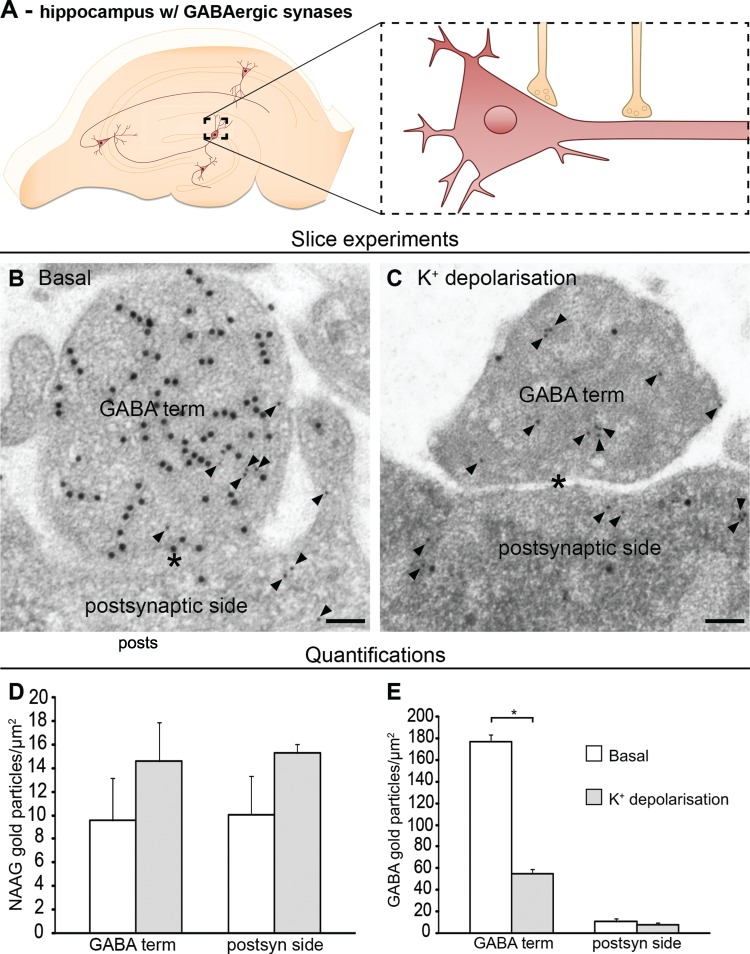
NAAG is neither released from GABA-positive nerve terminals nor their postsynaptic sides. In GABAergic synapses in the molecular layer in dentate gyrus of the hippocampal formation (A), NAAG was neither depleted from GABA-positive nerve terminals nor their postsynaptic sides (B and C) in response to depolarization. GABA synapses are colabeled for NAAG (small gold particles, arrowheads) and GABA (large gold particles). The figure shows representative electron micrographs of a slice incubated at 2.5 mM K+ (B) or at 55 mM K+(C). Labeled nerve terminals (GABA term) contact granule cell bodies (postsynaptic side) (B and C). Scale bars, 100 nm. Bar charts show the density of NAAG (D) and GABA (E) labeling (average number of gold particles/μm2 ± SD, n = 4 basal slices and six depolarized slices, slice experiment from one adult Wistar rat) in nerve terminals containing GABA gold particles (GABA terminal) and the opposing postsynaptic elements (granule cell bodies or proximal dendrites [postsynaptic]). Asterisk indicates that the GABA values in terminals at 55 mM K+ were significantly different from those in terminals at 2.5 mM K+ (P < 0.05, unpaired t-test, two tails, GraphPad). In contrast, NAAG labeling in GABA terminals and postsynaptic element was unaffected by depolarization (P > 0.05, unpaired t-test, two tails, GraphPad). The total numbers of tissue profiles included in the quantifications were as follows: Basal GABA terminal, 95; depolarized GABA terminal, 138; basal postsynaptic elements, 64; depolarized postsynaptic element, 99.
High Ratio of NAAG Between Post- and Presynaptic Compartments was Consistent Between Experimental Conditions
Although the staining intensity may vary between different staining experiments, the staining pattern was stable. The high ratio between sc. spines and sc. terminals was high both from perfusion-fixed rat hippocampus 4.98 (95% confidence interval [CI] 3.27–6.69, n = 3 rats) to acute hippocampal slices from in vitro experiments 2.82 (95% CI 2.18–3.45, n = 8 slices from 4 rats). The same is true for the high NAAG-density in mossy fiber thorns and low density in mossy fiber terminals, where the mean ratio between mf. thorns and mf. terminals was 3.45 (95% CI 2.13–4.77, n = 3 rats) in perfusion-fixed brain and 3.88 (95% CI 1.91–5.85, n = 9 slices from 4 rats) in acute hippocampal slices.
The high postsynaptic:presynaptic NAAG-ratio was eradicated after depolarization with high potassium or GluR agonists with mean ratio at 0.89 (95% CI −0.32–2.09, n = 17 slices from 4 rats) in the Shaffer collateral synapse and 0.66 (95% CI 0.40–0.91, n = 17 slices from 4 rats) in the mossy fiber synapse.
Discussion
The present study shows that NAAG is predominantly localized to postsynaptic compartments, with background levels in other neuronal compartments, including in presynaptic terminals. NAAG immunogold particles were spatially related to vesicular structures in these profiles; such vesicles have previously been described in postsynaptic spines in the hippocampus (Spacek and Harris 1997). In acute hippocampal slices, we demonstrate that NAAG is depleted from the spines in response to neuronal stimulation. The finding of high postsynaptic NAAG-content is very robust, with comparable post-to-presynaptic ratios in rat hippocampus fixed in vivo and acute hippocampal slices fixed after 2 h in vitro. The same goes for the depletion of postsynaptic NAAG content in response to depolarization. The depletion was dependent on Ca2+ and sensitive to BoNT/B, strongly suggesting that NAAG undergoes vesicular release from the postsynaptic spine and acts as a retrograde transmitter in the brain. The observation that inhibited exocytosis by BoNT/B during depolarization gave postsynaptic NAAG levels above what was found in the resting condition suggesting a basal release of NAAG. The NAAG content in stem dendrites is not altered in the experimental conditions tested in this study.
The presence of BoNT/B, which cleaves the vesicular SNARE (v-SNARE) VAMP2, completely blocked depletion of NAAG from postsynaptic spines/thorns. This is in line with a recent report showing that VAMP2 is important for regulated postsynaptic exocytosis (Jurado et al. 2013) and strongly suggests that NAAG undergoes VAMP-mediated exocytosis from postsynaptic elements. How BoNT/B is internalized in the postsynaptic element is not totally clear, but gangliosides and other proteins in the postsynaptic membrane have recently been shown to mediate internalization of BoNTs (Rummel 2013). For instance, the ganglioside GD1a seems to be especially important for binding BoNT/B (Ochanda et al. 1986). Furthermore, translocation of the BoNT/B light chain requires a pH of 4.5–6 (Pirazzini et al. 2011). The lumen of recycled synaptic vesicles, with an estimated pH of ~ 5.8, (Miesenbock et al. 1998), therefore provides an appropriate environment where light chain translocation can be induced. Therefore, a postsynaptic effect of BoNT/B after extracellular application of the drug could be expected. Concentration curves for the extracellular concentration of BoNT/B that effectively inhibits Ca2+-dependent exocytosis from postsynaptic dendritic spines are not available. The concentration used in the present study (133 nM) is one-third higher than the concentration previously been shown to effectively inhibit Ca2+-dependent glutamate exocytosis from synaptosomes (100 nM) (McMahon et al. 1992).
Indeed, a postsynaptic localization of VAMP2 has been reported (Hussain and Davanger 2015) and that SNARE proteins are located in dendritic “synaptic-like vesicles” (Hussain and Davanger 2015; Hussain et al. 2017; 2019). The dendritic vesicles observed by us are morphologically similar to synaptic vesicles (Fig. 1D,E) and similar to those presented by Hussain and Davanger (2015) and Hussain et al. (2017), (2019). Supporting vesicular accumulation of NAAG, a vesicular transport mechanism for NAAG through the sialic acid transporter sialin has been suggested (Lodder-Gadaczek 2013). However, this finding is yet to be confirmed.
Calcium-dependent postsynaptic release of NAAG suggests that the influx of Ca2+ from the extracellular space takes part in postsynaptic exocytosis. In the hippocampus, release of signal molecules from postsynaptic dendrites has been shown to rely on L-type Ca2+ channels (Simmons et al. 1995; Ovsepian and Dolly 2011), through which Ca2+ influx could be sufficient to directly induce exocytosis. However, also store-operating Ca2+ channels may contribute to Ca2+ signals in dendrites. In addition, Ca2+ influx through NMDA receptors can also trigger dendritic transmitter release (Isaacson and Strowbridge 1998), although this may not be a direct effect, but takes place through elevation of cAMP and activation of protein kinase A (Chetkovich et al. 1991). Our results further suggest that there are postsynaptic Ca2+ sensors. Little is known about the identity and function of these sensors in the brain, but recently synaptotagmin-1 was localized in postsynaptic dendritic spines of hippocampal Schaffer collateral synapses (Hussain et al. 2017).
Postsynaptic vesicular release of transmitters has been suggested before for glutamate (Zilberter 2000; Harkany et al. 2004; Jenstad et al. 2009), serotonin (Chazal and Ralston 1987), and ATP (Zhang et al. 2007; Larsson et al. 2012). All these transmitters, however, are also released from nerve terminals. Therefore, to the best of our knowledge, NAAG is the only transmitter that may be released exclusively from dendrites in the brain.
NAAG activates mGluR3 (Wroblewska et al. 1997; Neale 2011) and has both antagonist and agonist actions at NMDA receptors (Westbrook et al. 1986; Valivullah et al. 1994; Fricker et al. 2009; Kolodziejczyk et al. 2009), depending on subunit composition and tissue pH (Khacho et al. 2015). This supports the idea that NAAG can act as a retrograde transmitter through binding to one or both of these receptors. One possibility is that NAAG provides the postsynaptic neuron with the ability to control its own inputs both at high and low activation levels. When the spine/thorn receives very low glutamate signals, low levels of NAAG are released, consistent with the increased NAAG levels found in the spines/thorns after exposure to BoNT/B during GluR depolarization. When the glutamate signal is low, NAAG has been shown to activate the synaptic, GluN2A-containing NMDARs, while inhibiting the extrasynaptic GluN2B-containing NMDARs (Khacho et al. 2015), thereby enhancing the synaptic glutamate signal. In contrast, in situations of extensive glutamatergic signaling and a subsequent lowering of tissue pH, NAAG released from the postsynapse could reduce excitotoxicity by inhibiting both intra- and extrasynaptic NMDARs. The activation of mGluR3 would further strengthen this neuroprotective effect by reducing glutamate release from the nerve terminal. In support of this hypothesis, knock-out or pharmacological inhibition of glutamate carboxypeptidase II, which breaks down synaptically released NAAG (Robinson and Coyle 1987; Stauch et al. 1989; Berger et al. 1999; Bergeron et al. 2007), is protective in animal models of ischemia (Slusher et al. 1999; Tortella et al. 2000; Jackson et al. 2001; Williams et al. 2001; Bacich et al. 2005; Long et al. 2005; Zhang et al. 2016) and epilepsy (Witkin et al. 2002). With this said, we should emphasize that our means of inducing membrane depolarization is not physiologic. Thus, the exact effects of NAAG release from postsynaptic dendrites during physiological conditions must await further studies.
A neurotransmitter role of NAAG has long been suggested, despite lack of direct evidence for presynaptic localization and release. Such evidence could only be obtained by immune-electron microscopic detection of NAAG in stimulated and unstimulated brain tissue. The commonly used aldehyde fixatives preserve good morphology for ultrastructural studies. NAAG, however, lacking free amine groups, is not fixed by aldehydes (Moffett et al. 1993). For fixation of NAAG, the less common, water soluble carbodiimide fixation agent EDC must be used. It acts by forming amine bonds between carboxyl groups or primary amines and proteins (Bauminger and Wilchek 1980). Despite this, a previous study (Renno et al. 1997) investigated the ultrastructural localization of NAAG in the cerebellum, using only aldehyde fixation. This strongly suggests that their NAAG labeling was not selective.
Using EDC fixation, selective detection of NAAG is possible, but EDC fixation produces inferior morphology compared to tissue fixed with aldehydes. Further adding to a suboptimal morphologic result is that we used tissue embedding in an acrylic resin (Lowicryl HM20) over an epoxy resin. Low temperature embedding with Lowicryl HM20 preserves the antigens to a large extent and results in a superior immunogold sensitivity, but the ultrastructure is inferior compared with epoxy-embedded tissue (van Lookeren Campagne et al. 1991; Newman and Hobot 1999). Our group has successfully used Lowicryl HM20 in immunogold cytochemistry for many years (Bergersen et al. 2008 Nat Prot). In the present study, the morphology of the perfusion-fixed tissue (Fig. 1D,E) was better compared to the morphology of the immersion-fixed tissue from acute brain slices (Figs 2, A–CE–GI–K,3B,C and 4B–I). Perfusion fixation gives a rapid and uniform penetration of the fixative into the tissue, reducing the risk of ischemic damage. This method is therefore the preferred method for morphology preservation (Gonzalezaguilar and Derobertis 1963). For in vitro experiments, like acute brain slices, immersion fixation must be performed, which gives a slower tissue fixation. Thus, some morphological changes are known to happen during slice incubation (Schurr et al. 1984; Fiala et al. 2003), although all precautions were made to preserve the morphology (Schurr et al. 1984; Fiala et al. 2003). However, in the present material, pre- and postsynaptic elements were clearly visible, making the identification of nerve terminals and postsynaptic dendritic spines/thorns reliable. To ensure that we could distinguish between pre- and postsynaptic elements, we double-labeled some tissue for NAAG and glutamate and consistently found that NAAG was localized in the postsynaptic compartment low in glutamate (unpublished results; KN, CM, VG).
Conclusion
The present study shows evidence for exocytotic postsynaptic release of NAAG, implying that fine-tuning of the glutamate signal occurs through retrograde rather than anterograde NAAG signaling. To our knowledge, this is the first demonstration of vesicular release of a transmitter exclusively from dendrites.
Funding
This work was supported by the Norwegian South-Eastern Health Authority; Research Council of Norway.
Notes
The authors acknowledge Joseph H Neale (Department of Biology, Georgetown University, Washington DC, USA) and John Moffett (Department of Anatomy, Physiology and Genetics; Neuroscience Program, Uniformed Services University of the Health Sciences, Bethesda, USA) for kindly providing the NAAG antibodies. Conflict of Interest: None declared.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
References
- Anderson KJ, Monaghan DT, Cangro CB, Namboodiri MA, Neale JH, Cotman CW. 1986. Localization of N-acetylaspartylglutamate-like immunoreactivity in selected areas of the rat brain. Neurosci Lett. 72:14–20. [DOI] [PubMed] [Google Scholar]
- Bacich DJ, Wozniak KM, Lu XC, O’Keefe DS, Callizot N, Heston WD, Slusher BS. 2005. Mice lacking glutamate carboxypeptidase II are protected from peripheral neuropathy and ischemic brain injury. J Neurochem. 95:314–323. [DOI] [PubMed] [Google Scholar]
- Bauminger S, Wilchek M. 1980. The use of carbodiimides in the preparation of immunizing conjugates. Methods Enzymol. 70:151–159. [DOI] [PubMed] [Google Scholar]
- Berger UV, Luthi-Carter R, Passani LA, Elkabes S, Black I, Konradi C, Coyle JT. 1999. Glutamate carboxypeptidase II is expressed by astrocytes in the adult rat nervous system. J Comp Neurol. 415:52–64. [DOI] [PubMed] [Google Scholar]
- Bergeron R, Coyle JT. 2012. NAAG, NMDA receptor and psychosis. Curr Med Chem. 19:1360–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron R, Imamura Y, Frangioni JV, Greene RW, Coyle JT. 2007. Endogenous N-acetylaspartylglutamate reduced NMDA receptor-dependent current neurotransmission in the CA1 area of the hippocampus. J Neurochem. 100:346–357. [DOI] [PubMed] [Google Scholar]
- Bergersen LH, Storm-Mathisen J, Gundersen V. 2008. Immunogold quantification of amino acids and proteins in complex subcellular compartments. Nat Protoc. 3:144–152. [DOI] [PubMed] [Google Scholar]
- Cangro CB, Namboodiri MA, Sklar LA, Corigliano-Murphy A, Neale JH. 1987. Immunohistochemistry and biosynthesis of N-acetylaspartylglutamate in spinal sensory ganglia. J Neurochem. 49:1579–1588. [DOI] [PubMed] [Google Scholar]
- Chazal G, Ralston HJ 3rd. 1987. Serotonin-containing structures in the nucleus raphe dorsalis of the cat: an ultrastructural analysis of dendrites, presynaptic dendrites, and axon terminals. J Comp Neurol. 259:317–329. [DOI] [PubMed] [Google Scholar]
- Cheramy A, Leviel V, Glowinski J. 1981. Dendritic release of dopamine in the substantia nigra. Nature. 289:537–542. [DOI] [PubMed] [Google Scholar]
- Chetkovich DM, Gray R, Johnston D, Sweatt JD. 1991. N-methyl-D-aspartate receptor activation increases cAMP levels and voltage-gated Ca2+ channel activity in area CA1 of hippocampus. Proc Natl Acad Sci USA. 88:6467–6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Iorio P, Battaglia G, Ciccarelli R, Ballerini P, Giuliani P, Poli A, Nicoletti F, Caciagli F. 1996. Interaction between A1 adenosine and class II metabotropic glutamate receptors in the regulation of purine and glutamate release from rat hippocampal slices. J Neurochem. 67:302–309. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Kirov SA, Feinberg MD, Petrak LJ, George P, Goddard CA, Harris KM. 2003. Timing of neuronal and glial ultrastructure disruption during brain slice preparation and recovery in vitro. J Comp Neurol. 465:90–103. [DOI] [PubMed] [Google Scholar]
- Fricker AC, Mok MH, de la Flor R, Shah AJ, Woolley M, Dawson LA, Kew JN. 2009. Effects of N-acetylaspartylglutamate (NAAG) at group II mGluRs and NMDAR. Neuropharmacology. 56:1060–1067. [DOI] [PubMed] [Google Scholar]
- Frondoza CG, Logan S, Forloni G, Coyle JT. 1990. Production and characterization of monoclonal antibodies to N-acetyl-aspartyl-glutamate. J Histochem Cytochem. 38:493–502. [DOI] [PubMed] [Google Scholar]
- Gonzalezaguilar F, Derobertis E. 1963. A formalin-perfusion fixation method for histophysiological study of the central nervous system with the electron microscope. Neurology. 13:758–771. [DOI] [PubMed] [Google Scholar]
- Guarda AS, Robinson MB, Ory-Lavollee L, Forloni GL, Blakely RD, Coyle JT. 1988. Quantitation of N-acetyl-aspartyl-glutamate in microdissected rat brain nuclei and peripheral tissues: findings with a novel liquid phase radioimmunoassay. Brain Res. 427:223–231. [DOI] [PubMed] [Google Scholar]
- Gundersen V, Holten AT, Storm-Mathisen J. 2004. GABAergic synapses in hippocampus exocytose aspartate on to NMDA receptors: quantitative immunogold evidence for co-transmission. Mol Cell Neurosci. 26:156–165. [DOI] [PubMed] [Google Scholar]
- Gundersen V, Ottersen OP, Storm-Mathisen J. 1991. Aspartate- and glutamate-like immunoreactivities in rat hippocampal slices: depolarization-induced redistribution and effects of precursors. Eur J Neurosci. 3:1281–1299. [DOI] [PubMed] [Google Scholar]
- Harkany T, Holmgren C, Hartig W, Qureshi T, Chaudhry FA, Storm-Mathisen J, Dobszay MB, Berghuis P, Schulte G, Sousa KM et al. . 2004. Endocannabinoid-independent retrograde signaling at inhibitory synapses in layer 2/3 of neocortex: involvement of vesicular glutamate transporter 3. J Neurosci. 24:4978–4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holten AT, Danbolt NC, Shimamoto K, Gundersen V. 2008. Low-affinity excitatory amino acid uptake in hippocampal astrocytes: a possible role of Na+/dicarboxylate cotransporters. Glia. 56:990–997. [DOI] [PubMed] [Google Scholar]
- Hsu SM, Raine L, Fanger H. 1981. The use of antiavidin antibody and avidin-biotin-peroxidase complex in immunoperoxidase technics. Am J Clin Pathol. 75:816–821. [DOI] [PubMed] [Google Scholar]
- Hussain S, Davanger S. 2015. Postsynaptic VAMP/aynaptobrevin facilitates differential vesicle trafficking of GluA1 and GluA2 AMPA receptor subunits. PLoS One. 10:e0140868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain S, Egbenya DL, Lai YC, Dosa ZJ, Sorensen JB, Anderson AE, Davanger S. 2017. The calcium sensor synaptotagmin 1 is expressed and regulated in hippocampal postsynaptic spines. Hippocampus. 27:1168–1177. [DOI] [PubMed] [Google Scholar]
- Hussain S, Ringsevjen H, Schupps M, Hvalby O, Sorensen JB, Jensen V, Davanger S. 2019. A possible postsynaptic role for SNAP-25 in hippocampal synapses. Brain Struct Funct. 224:521–532. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Strowbridge BW. 1998. Olfactory reciprocal synapses: dendritic signaling in the CNS. Neuron. 20:749–761. [DOI] [PubMed] [Google Scholar]
- Jackson PF, Tays KL, Maclin KM, Ko YS, Li W, Vitharana D, Tsukamoto T, Stoermer D, Lu XC, Wozniak K et al. . 2001. Design and pharmacological activity of phosphinic acid based NAALADase inhibitors. J Med Chem. 44:4170–4175. [DOI] [PubMed] [Google Scholar]
- Jenstad M, Quazi AZ, Zilberter M, Haglerod C, Berghuis P, Saddique N, Goiny M, Buntup D, Davanger S, Fm SH et al. . 2009. System A transporter SAT2 mediates replenishment of dendritic glutamate pools controlling retrograde signaling by glutamate. Cereb Cortex. 19:1092–1106. [DOI] [PubMed] [Google Scholar]
- Jurado S, Goswami D, Zhang Y, Molina AJ, Sudhof TC, Malenka RC. 2013. LTP requires a unique postsynaptic SNARE fusion machinery. Neuron. 77:542–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ, Davison IG, Robinson CG, Ehlers MD. 2010. Syntaxin-4 defines a domain for activity-dependent exocytosis in dendritic spines. Cell. 141:524–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khacho P, Wang B, Ahlskog N, Hristova E, Bergeron R. 2015. Differential effects of N-acetyl-aspartyl-glutamate on synaptic and extrasynaptic NMDA receptors are subunit- and pH-dependent in the CA1 region of the mouse hippocampus. Neurobiol Dis. 82:580–592. [DOI] [PubMed] [Google Scholar]
- Koller KJ, Zaczek R, Coyle JT. 1984. N-acetyl-aspartyl-glutamate: regional levels in rat brain and the effects of brain lesions as determined by a new HPLC method. J Neurochem. 43:1136–1142. [DOI] [PubMed] [Google Scholar]
- Kolodziejczyk K, Hamilton NB, Wade A, Karadottir R, Attwell D. 2009. The effect of N-acetyl-aspartyl-glutamate and N-acetyl-aspartate on white matter oligodendrocytes. Brain. 132:1496–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson M, Sawada K, Morland C, Hiasa M, Ormel L, Moriyama Y, Gundersen V. 2012. Functional and anatomical identification of a vesicular transporter mediating neuronal ATP release. Cereb Cortex. 22:1203–1214. [DOI] [PubMed] [Google Scholar]
- Larsson M, Broman J. 2005. Different basal levels of CaMKII phosphorylated at Thr286/287 at nociceptive and low-threshold primary afferent synapses. Eur J Neurosci. 21:2445–2458. [DOI] [PubMed] [Google Scholar]
- Lau CG, Takayasu Y, Rodenas-Ruano A, Paternain AV, Lerma J, Bennett MV, Zukin RS. 2010. SNAP-25 is a target of protein kinase C phosphorylation critical to NMDA receptor trafficking. J Neurosci. 30:242–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodder-Gadaczek J, Gieselmann V, Eckhardt M. 2013. Vesicular uptake of N-acetylaspartylglutamate is catalysed by sialin (SLC17A5). Biochem J. 454:31–38. [DOI] [PubMed] [Google Scholar]
- Long JB, Yourick DL, Slusher BS, Robinson MB, Meyerhoff JL. 2005. Inhibition of glutamate carboxypeptidase II (NAALADase) protects against dynorphin A-induced ischemic spinal cord injury in rats. Eur J Pharmacol. 508:115–122. [DOI] [PubMed] [Google Scholar]
- McMahon HT, Foran P, Dolly JO, Verhage M, Wiegant VM, Nicholls DG. 1992. Tetanus toxin and botulinum toxins type A and B inhibit glutamate, gamma-aminobutyric acid, aspartate, and met-enkephalin release from synaptosomes. Clues to the locus of action. J Biol Chem. 267:21338–21343. [PubMed] [Google Scholar]
- Miesenbock G, De Angelis DA, Rothman JE. 1998. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 394:192–195. [DOI] [PubMed] [Google Scholar]
- Miyamoto E, Kakimoto Y, Sano I. 1966. Identification of N-acetyl-alpha-aspartylglutamic acid in the bovine brain. J Neurochem. 13:999–1003. [DOI] [PubMed] [Google Scholar]
- Moffett JR, Namboodiri MA, Neale JH. 1993. Enhanced carbodiimide fixation for immunohistochemistry: application to the comparative distributions of N-acetylaspartylglutamate and N-acetylaspartate immunoreactivities in rat brain. J Histochem Cytochem. 41:559–570. [DOI] [PubMed] [Google Scholar]
- Morland C, Nordengen K, Larsson M, Prolo LM, Farzampour Z, Reimer RJ, Gundersen V. 2013. Vesicular uptake and exocytosis of L-aspartate is independent of sialin. FASEB J. 27:1264–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale JH. 2011. N-acetylaspartylglutamate is an agonist at mGluR(3) in vivo and in vitro. J Neurochem. 119:891–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman GR, Hobot JA. 1999. Resins for combined light and electron microscopy: a half century of development. Histochem J. 31:495–505. [DOI] [PubMed] [Google Scholar]
- Ochanda JO, Syuto B, Ohishi I, Naiki M, Kubo S. 1986. Binding of clostridium botulinum neurotoxin to gangliosides. J Biochem. 100:27–33. [DOI] [PubMed] [Google Scholar]
- Ottersen OP, Storm-Mathisen J, Bramham C, Torp R, Laake J, Gundersen V. 1990. A quantitative electron microscopic immunocytochemical study of the distribution and synaptic handling of glutamate in rat hippocampus. Prog Brain Res. 83:99–114. [DOI] [PubMed] [Google Scholar]
- Ottersen OP, Zhang N, Walberg F. 1992. Metabolic compartmentation of glutamate and glutamine: morphological evidence obtained by quantitative immunocytochemistry in rat cerebellum. Neuroscience. 46:519–534. [DOI] [PubMed] [Google Scholar]
- Ovsepian SV, Dolly JO. 2011. Dendritic SNAREs add a new twist to the old neuron theory. Proc Natl Acad Sci U S A. 108:19113–19120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellizzari R, Rossetto O, Lozzi L, Giovedi S, Johnson E, Shone CC, Montecucco C. 1996. Structural determinants of the specificity for synaptic vesicle-associated membrane protein/synaptobrevin of tetanus and botulinum type B and G neurotoxins. J Biol Chem. 271:20353–20358. [DOI] [PubMed] [Google Scholar]
- Pirazzini M, Rossetto O, Bolognese P, Shone CC, Montecucco C. 2011. Double anchorage to the membrane and intact inter-chain disulfide bond are required for the low pH induced entry of tetanus and botulinum neurotoxins into neurons. Cell Microbiol. 13:1731–1743. [DOI] [PubMed] [Google Scholar]
- Pittaluga A, Barbeito L, Serval V, Godeheu G, Artaud F, Glowinski J, Cheramy A. 1988. Depolarization-evoked release of N-acetyl-L-aspartyl-L-glutamate from rat brain synaptosomes. Eur J Pharmacol. 158:263–266. [DOI] [PubMed] [Google Scholar]
- Regehr WG, Carey MR, Best AR. 2009. Activity-dependent regulation of synapses by retrograde messengers. Neuron. 63:154–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renno WM, Lee JH, Beitz AJ. 1997. Light and electron microscopic immunohistochemical localization of N-acetylaspartylglutamate (NAAG) in the olivocerebellar pathway of the rat. Synapse. 26:140–154. [DOI] [PubMed] [Google Scholar]
- Robinson MB, Coyle JT. 1987. Glutamate and related acidic excitatory neurotransmitters: from basic science to clinical application. FASEB J. 1:446–455. [DOI] [PubMed] [Google Scholar]
- Rummel A. 2013. Double receptor anchorage of botulinum neurotoxins accounts for their exquisite neurospecificity. Curr Top Microbiol Immunol. 364:61–90. [DOI] [PubMed] [Google Scholar]
- Sanabria ER, Wozniak KM, Slusher BS, Keller A. 2004. GCP II (NAALADase) inhibition suppresses mossy fiber-CA3 synaptic neurotransmission by a presynaptic mechanism. J Neurophysiol. 91:182–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffhauser H, Richards JG, Cartmell J, Chaboz S, Kemp JA, Klingelschmidt A, Messer J, Stadler H, Woltering T, Mutel V. 1998. In vitro binding characteristics of a new selective group II metabotropic glutamate receptor radioligand, [3H]LY354740, in rat brain. Mol Pharmacol. 53:228–233. [PubMed] [Google Scholar]
- Schurr A, Reid KH, Tseng MT, Edmonds HL Jr. 1984. The stability of the hippocampal slice preparation: an electrophysiological and ultrastructural analysis. Brain Res. 297:357–362. [DOI] [PubMed] [Google Scholar]
- Shave E, Pliss L, Lawrance ML, FitzGibbon T, Stastny F, Balcar VJ. 2001. Regional distribution and pharmacological characteristics of [3H]N-acetyl-aspartyl-glutamate (NAAG) binding sites in rat brain. Neurochem Int. 38:53–62. [DOI] [PubMed] [Google Scholar]
- Simmons ML, Terman GW, Gibbs SM, Chavkin C. 1995. L-type calcium channels mediate dynorphin neuropeptide release from dendrites but not axons of hippocampal granule cells. Neuron. 14:1265–1272. [DOI] [PubMed] [Google Scholar]
- Slusher BS, Vornov JJ, Thomas AG, Hurn PD, Harukuni I, Bhardwaj A, Traystman RJ, Robinson MB, Britton P, Lu XC et al. . 1999. Selective inhibition of NAALADase, which converts NAAG to glutamate, reduces ischemic brain injury. Nat Med. 5:1396–1402. [DOI] [PubMed] [Google Scholar]
- Spacek J, Harris KM. 1997. Three-dimensional organization of smooth endoplasmic reticulum in hippocampal CA1 dendrites and dendritic spines of the immature and mature rat. J Neurosci. 17:190–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauch BL, Robinson MB, Forloni G, Tsai G, Coyle JT. 1989. The effects of N-acetylated alpha-linked acidic dipeptidase (NAALADase) inhibitors on [3H] NAAG catabolism in vivo. Neurosci Lett. 100:295–300. [DOI] [PubMed] [Google Scholar]
- Storm-Mathisen J, Leknes AK, Bore AT, Vaaland JL, Edminson P, Haug FM, Ottersen OP. 1983. First visualization of glutamate and GABA in neurones by immunocytochemistry. Nature. 301:517–520. [DOI] [PubMed] [Google Scholar]
- Suh YH, Terashima A, Petralia RS, Wenthold RJ, Isaac JT, Roche KW, Roche PA. 2010. A neuronal role for SNAP-23 in postsynaptic glutamate receptor trafficking. Nat Neurosci. 13:338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suresh A, Dunaevsky A. 2015. Preparation of synaptosomes from the motor cortex of motor skill trained mice. Bio-protocol. 5:e1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortella FC, Lin Y, Ved H, Slusher BS, Dave JR. 2000. Neuroprotection produced by the NAALADase inhibitor 2-PMPA in rat cerebellar neurons. Eur J Pharmacol. 402:31–37. [DOI] [PubMed] [Google Scholar]
- Tsai G, Slusher BS, Sim L, Coyle JT. 1993. Immunocytochemical distribution of N-acetylaspartylglutamate in the rat forebrain and glutamatergic pathways. J Chem Neuroanat. 6:277–292. [DOI] [PubMed] [Google Scholar]
- Valivullah HM, Lancaster J, Sweetnam PM, Neale JH. 1994. Interactions between N-acetylaspartylglutamate and AMPA, kainate, and NMDA binding sites. J Neurochem. 63:1714–1719. [DOI] [PubMed] [Google Scholar]
- van Lookeren Campagne M, Oestreicher AB, van der Krift TP, Gispen WH, Verkleij AJ. 1991. Freeze-substitution and Lowicryl HM20 embedding of fixed rat brain: suitability for immunogold ultrastructural localization of neural antigens. J Histochem Cytochem. 39:1267–1279. [DOI] [PubMed] [Google Scholar]
- Volgyi K, Gulyassy P, Todorov MI, Puska G, Badics K, Hlatky D, Kekesi KA, Nyitrai G, Czurko A, Drahos L et al. . 2018. Chronic cerebral hypoperfusion induced synaptic proteome changes in the rat cerebral cortex. Mol Neurobiol. 55:4253–4266. [DOI] [PubMed] [Google Scholar]
- Wang BL, Larsson LI. 1985. Simultaneous demonstration of multiple antigens by indirect immunofluorescence or immunogold staining. Novel light and electron microscopical double and triple staining method employing primary antibodies from the same species. Histochemistry. 83:47–56. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Sugiura Y, Sugiyama E, Narita M, Navratilova E, Kondo T, Uchiyama N, Yamanaka A, Kuzumaki N, Porreca F et al. . 2018. Extracellular N-acetylaspartylglutamate released in the nucleus accumbens modulates the pain sensation: analysis using a microdialysis/mass spectrometry integrated system. Mol Pain. 14:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westbrook GL, Mayer ML, Namboodiri MA, Neale JH. 1986. High concentrations of N-acetylaspartylglutamate (NAAG) selectively activate NMDA receptors on mouse spinal cord neurons in cell culture. J Neurosci. 6:3385–3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AJ, Lu XM, Slusher B, Tortella FC. 2001. Electroencephalogram analysis and neuroprotective profile of the N-acetylated-alpha-linked acidic dipeptidase inhibitor, GPI5232, in normal and brain-injured rats. J Pharmacol Exp Ther. 299:48–57. [PubMed] [Google Scholar]
- Williamson LC, Neale JH. 1988. Ultrastructural localization of N-acetylaspartylglutamate in synaptic vesicles of retinal neurons. Brain Res. 456:375–381. [DOI] [PubMed] [Google Scholar]
- Witkin JM, Gasior M, Schad C, Zapata A, Shippenberg T, Hartman T, Slusher BS. 2002. NAALADase (GCP II) inhibition prevents cocaine-kindled seizures. Neuropharmacology. 43:348–356. [DOI] [PubMed] [Google Scholar]
- Wroblewska B, Wroblewski JT, Pshenichkin S, Surin A, Sullivan SE, Neale JH. 1997. N-acetylaspartylglutamate selectively activates mGluR3 receptors in transfected cells. J Neurochem. 69:174–181. [DOI] [PubMed] [Google Scholar]
- Zhang W, Zhang Z, Wu L, Qiu Y, Lin Y. 2016. Suppression of glutamate carboxypeptidase II ameliorates neuronal apoptosis from ischemic brain injury. J Stroke Cerebrovasc Dis. 25:1599–1605. [DOI] [PubMed] [Google Scholar]
- Zhang X, Chen Y, Wang C, Huang LY. 2007. Neuronal somatic ATP release triggers neuron-satellite glial cell communication in dorsal root ganglia. Proc Natl Acad Sci USA. 104:9864–9869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong C, Zhao X, Van KC, Bzdega T, Smyth A, Zhou J, Kozikowski AP, Jiang J, O’Connor WT, Berman RF et al. . 2006. NAAG peptidase inhibitor increases dialysate NAAG and reduces glutamate, aspartate and GABA levels in the dorsal hippocampus following fluid percussion injury in the rat. J Neurochem. 97:1015–1025. [DOI] [PubMed] [Google Scholar]
- Zilberter Y. 2000. Dendritic release of glutamate suppresses synaptic inhibition of pyramidal neurons in rat neocortex. J Physiol. 528:489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zollinger M, Brauchli-Theotokis J, Gutteck-Amsler U, Do KQ, Streit P, Cuenod M. 1994. Release of N-acetylaspartylglutamate from slices of rat cerebellum, striatum, and spinal cord, and the effect of climbing fiber deprivation. J Neurochem. 63:1133–1142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.