

Abstract
Genetic studies have revealed that autoimmune susceptibility variants are over-represented in memory CD4+ T cell regulatory elements1–3. Understanding how genetic variation affects gene expression in different T cell physiological states is essential for deciphering genetic mechanisms of autoimmunity4,5. Here we characterized the dynamics of genetic regulatory effects at eight time points during memory CD4+ T cell activation with high depth RNA-seq in healthy individuals. We discovered widespread dynamic allele-specific expression across the genome, where the balance of alleles changes over time. These genes were four-fold enriched within autoimmune loci. We found pervasive dynamic regulatory effects within six HLA genes. HLA-DQB1 alleles had one of three distinct transcriptional regulatory programs. Using CRISPR/Cas9 genomic editing we demonstrated that a promoter variant is causal for T cell-specific control of HLA-DQB1 expression. Our study shows that genetic variation in cis regulatory elements affects gene expression in a lymphocyte activation status-dependent manner contributing to the inter-individual complexity of immune responses.
Memory CD4+ T cells are essential orchestrators of immune response. Hence, it is crucial to study how genetic variation affects their gene expression patterns to unravel the complex dynamics of regulation. Previous T cell studies analyzed a limited number of cell states and genes6–12, and an understanding of how the transcriptome is influenced by genetic regulatory variation in multiple physiological states is lacking. Allele-specific expression (ASE) of genes can detect context-specific cis regulatory effects13,14, here we applied this in a high resolution time series.
We stimulated memory CD4+ T cells from 24 genotyped individuals of European ancestry (Supplementary Fig. 1) with anti-CD3/CD28 beads. We performed RNA-seq at 0, 2, 4, 8, 12, 24, 48 and 72 hours after stimulation (Fig. 1a). Gene expression principal component analysis showed that the 200 samples separated by time (Fig. 1b, Supplementary Fig. 2), and gene clustering identified activation and repression clusters (Supplementary Fig. 3). Using a logistic regression framework, we identified dynamic ASE (dynASE) events at heterozygous SNPs. At these dynASE sites, the imbalance of the two expressed alleles is time dependent. First, for each heterozygous site in an individual, we merged counts from all time points and identified significant ASE. We queried a total of 207,519 sites, representing 38,890 unique SNPs in 8,322 genes and some in transcribed intergenic regions (3%) (Supplementary Fig. 4). We observed a total of 15,268 ASE (intercept P<2.4×10−7=0.05/207,519 tests) events within 2,147 genes. Next, we checked ASE sites for dynASE events by fitting a second order polynomial model, thus modeling linear and non-linear ASE effects with respect to time. To account for over-dispersion of allelic counts15,16, we incorporated sample-to-sample variability with a random intercepts effect. We observed 561 significant dynASE events across the genome (P<3.2×10−3, <5% FDR), representing 356 SNPs in 186 genes and seven intergenic sites (Fig. 1c, Supplementary Table 1). These dynASE events show high reproducibility and expanded upon known cis regulatory genetic effects in T cells6,8,9 (Supplementary Fig. 5–6, Extended Data Fig. 1–4, Supplementary Note). For example, we observed dynASE in F11R and CXCR5 (Fig. 1d), where the reference and alternative alleles were dynamically regulated in time.
Fig. 1. Dynamic allele-specific expression during T cell activation.

(a) Study design. (b) Principal Component Analysis on top 1,070 most variable genes. Shown are PC1 and PC2 scores for the 200 samples colored by time point. (c) Plot showing position across the genome of dynamic allele-specific expression (ASE) events, with y-axis indicating FDR (N = 24 individuals). In red, highlighted examples. (d) Examples of dynamic ASE events in two genes, each in a different individual. For each time point, we show allele counts for the SNP (top) and fraction of reads with the reference allele (bottom).
It is possible that differences in regulatory complex activity over time influences both expression of a gene and results in differential binding of alleles in regulatory elements. If so, we would expect to see an association between changes in level of expression of the gene and changes in allelic imbalance (i.e. the absolute difference between the observed allelic fraction and 0.5) over time. As expected, when we calculated Spearman rank correlation at each dynASE site between its allelic imbalance and expression over time, we observed a bimodal distribution of positive and negative associations (Fig. 2a). For example, SNP rs6021:A>G in gene F5 has a negative correlation between allelic imbalance and gene expression, with allelic imbalance starting high at 0 hours (red circle in Fig. 2b), going down as expression increases, and going back up as expression decreases. In contrast, SNP rs41306241:A>G in the gene CTSL1 has a positive correlation between allelic imbalance and gene expression (Fig. 2b). Hence, dynamic ASE suggests complex differential usage of cis regulatory elements during T cell activation affected by genetic variation.
Fig. 2. Dynamic allele-specific expression patterns and enrichment in autoimmune disease loci.

(a) Spearman correlation coefficient between gene expression levels (log2(tpm +1)) and SNP allelic imbalance (distance to 0.5 reference fraction) across time for all dynASE events at 5% FDR. (b) Specific examples of allelic imbalance for a heterozygous SNP in an individual, associated with the expression levels log2(tpm+1) of its gene. Red dot indicates 0 hour time point. Consecutive time points are connected by solid lines, arrows indicate direction of time. (c-d) DynASE examples for two autoimmune disease genes. N = 24 for gene expression, 1 for ASE. (e) Fold enrichment of dynASE genes (blue) in risk loci for autoimmune diseases and 3 non-immune mediated diseases, using 1000 null sets of loci matched by number of loci per disease and number of genes per locus. Same for genes with significant ASE at 0 hours (green), and naive T cell eQTL genes (pink). Filled circles mark permutation P < 0.01, empty circles mark permutation P > 0.01.
We found that 31 of our dynASE genes were within autoimmune disease loci outside of the MHC, including UBASH3A and IL10 (Fig. 2c,d). DynASE genes are significantly enriched for risk loci for ankylosing spondylitis (OR = 5.7, P = 0.008), celiac disease (OR = 5.4, P = 0.004), vitiligo (OR = 5, P = 0.004), type 1 diabetes (T1D, OR = 4.5, P = 0.002), inflammatory bowel disease (OR = 3.7, P = 0.001), rheumatoid arthritis (OR = 3.6, P = 0.005), and multiple sclerosis (OR = 3.1, P = 0.003), but not for non-immune mediated diseases (Fig. 2e, Methods, Supplementary Note, Supplementary Fig. 7). We compared these enrichments to genes with ASE at 0 hours only and with published eQTLs in resting CD4+ naïve T cells9. We observed that our dynASE genes, spanning up to 8 different cellular states, had the highest enrichment for autoimmune loci (Fig. 2e).
We observed 182 dynASE events within the MHC locus (Fig. 1c), with 15 events in HLA-DQB1 (examples in Supplementary Fig. 8, Supplementary Table 1), which harbors most of the genetic risk for T1D and celiac disease17,18. HLA-DQB1 is part of HLA class II, which codes for antigen presenting proteins typically expressed by professional antigen presenting cells. In human T cells, these genes are recognized as activation markers, and are expressed by T cell subsets expanded in patients with autoimmune disorders19–23. Literature suggests that T cells may present antigens to alter immune responses24–26. However, the function of HLA class II in T cells remains controversial.
To define the relationship between HLA classical alleles (a combination of multiple coding variants) and regulatory variation, we performed high resolution typing of HLA-DQB1, HLA-DRB1, HLA-DQA1, HLA-A, HLA-B, and HLA-C, for all 24 individuals. To robustly identify dynASE in this highly polymorphic region, we built an HLA-personalized genome for each individual (Extended Data Fig. 5) and quantified the number of uniquely mapped reads to each HLA allele per individual. Among the 48 HLA-DQB1 ~780bp sequences, there were 14 HLA-DQB1 4-digit classical alleles. Our HLA-personalized genome strategy quantified expression of HLA-DQB1 alleles by taking advantage of >20 SNPs in four exons (Fig. 3a-b, replication in Extended Data Fig. 6). With this strategy, we determined that most (15/24) individuals have significant dynASE for HLA-DQB1 (P<0.002=0.05/24 tests; examples in Fig. 3c).
Fig. 3. HLA-DQB1 dynamic allele-specific expression at mRNA and protein levels.

(a) Scheme of HLA-DQB1 alleles: their properties (highly divergent within each individual) and their expression quantification. (b) HLA-DQB1 allele counts for an individual over time. (c) Normalized allelic expression for HLA-DQB1 (top, in log2 scale), and allelic fraction (bottom) for three individuals. (d) Heatplot shows normalized allelic expression levels (in log2 scale) for each of the 48 HLA-DQB1 alleles in our cohort. Allelic profiles were clustered into three cis Regulatory Programs, for which the average expression profile is shown on the right with a black line, and total expression area occupied by all alleles in that cluster is shown with the colored ribbon. (e) Left panel shows normalized allelic mRNA expression levels (in log2 scale, values from (d), for alleles present in protein validation cohort). Middle panel shows protein levels (median fluorescence intensity of HLA-DQ+ CD4+ memory T cells) for 5 homozygous individuals for alleles within the Late-Spike regulatory program (blue) and 5 homozygous individuals for alleles in Constant-Low or Fluctuating programs (yellow). Right panel shows log2 fold change in HLA-DQ MFI between day 0 and day 7. P-value from Wilcoxon one-tailed test.
Allelic expression profiles of HLA-DQB1 4-digit classical alleles clustered together more than expected by chance (permutation P<0.001, Supplementary Fig. 9), suggesting that cis regulatory effects segregate with HLA-DQB1 classical coding alleles. We observed three transcriptional cis regulatory programs: Late-Spike, Constant-Low, and Fluctuating (Fig. 3d, Extended Data Fig. 7). To our knowledge, the identification of three distinct transcriptional profiles in HLA-DQB1 expression over time is the first description of such complex and variable regulation in any gene.
We wanted to confirm that the drastic mRNA up-regulation in the Late-Spike cis regulatory program affected cell-surface protein expression levels. We recruited five independent homozygous individuals with HLA-DQB1 classical alleles with the Late-Spike cis regulatory program, and five homozygous individuals for HLA-DQB1 classical alleles with Constant-Low or Fluctuating cis regulatory programs (Supplementary Fig. 10). After seven days of activating memory CD4+ T cells, we observed that the fold change increase in HLA-DQ cell surface expression was significantly higher in Late-Spike homozygotes (P=0.03, Wilcoxon test, Fig. 3e, Supplementary Fig. 11).
We then sought to identify the genetic variant driving the Late-Spike cis regulatory program with genetic and epigenetic fine-mapping tools. We called SNP genotypes and 4-digit classical HLA alleles in 2,198 fully sequenced whole genomes from Estonia27. We identified SNPs in tight linkage disequilibrium (LD) with the Late-Spike HLA-DQB1 classical alleles, that together represent what we henceforth call the Late-Spike haplotype. While most of the SNPs in highest LD with this haplotype (r2 ≥ 0.96) were within the HLA-DQB1 gene (89%), only six were intergenic (Fig. 4a, Extended Data Fig. 8a). The same six variants emerged in an eQTL analysis in our 24 individual cohort and in an independent analysis in another European descent cohort from United States28 (N = 1,603) (Supplementary Note, Extended Data Fig. 8b-d). We applied ATAC-seq in memory CD4+ T cells after 72 hours of stimulation to identify open chromatin regions. Among these six SNPs, we observed that the rs71542466:C>G SNP overlapped the highest ATAC-seq peak. We also observed in public data that this SNP overlapped other regulatory element marks and peaks for HLA class II regulators29–31 (Fig. 4a, Supplementary Fig. 12).
Fig. 4. Validation of causal variant for Late-Spike cis regulatory program.

(a) Location of 6 fine-mapped non-coding SNPs around HLA-DQB1. Tracks showing open chromatin regions (ATAC-seq) or regions marked by histone modifications (ChIP-seq). (b) CRISPR/Cas9 cuts at or near six fine-mapped SNPs in HH T cell lines. Left, experiment scheme. Middle, representative example of HLA-DQ expression levels. Right, HLA-DQ median fluorescence intensity relative to control, for each of the 6 SNPs, in triplicate. (c) Validation of causal SNP rs71542466 with CRISPR/Ca9 and ssDNA oligo HDR template. Left, experiment scheme. Middle, mRNA HLA-DQB1 quantification with qPCR Taqman assay for 7 wild type (WT, homozygous for C allele), and 7 SNP edited (ALT, homozygous for G allele) expanded clones for rs71542466, as well as a cell line clone with an indel at the same target position. Right, HLA-DQ protein levels measured with flow cytometry. N = 5 WT and 4 ALT clones. (d) Electrophoretic Mobility Shift Assay using nuclear extract of three cell lines, with biotin labeled probes with reference (REF) or alternative (ALT) alleles for rs71542466. Blots were cropped from original shown in Supplementary Figures. (e) Luciferase assay in three cell lines. Cntrl, control. REF, reference allele. ALT, alternative allele. All P-values from Mann-Whitney one-tailed test. Error bars are S.E.M.
We hypothesized that one of the 6 intergenic SNPs in phase with the Late-Spike haplotype were causal and influenced gene regulation. If so, (1) the causal SNP should be in regions that have regulatory activity and (2) that editing the allele of the causal SNP itself should influence gene expression. To this end, we used CRISPR/Cas9 editing in HH, an HLA class II expressing T cell line, to test the regions around all 6 candidate SNPs for regulatory activity (Supplementary Fig. 13). After cutting near the 6 SNPs, only editing near rs71542466, located 39bp upstream of the transcription start site, caused significant decrease in HLA-DQ expression (Fig. 4b, Extended Data Fig. 9). Next, we applied targeted homology directed repair with CRISRPR-Cas9 to rs71542466 in order to convert the reference C allele to the alternative G allele. We predicted that HH T cell line clones homozygous for the rs71542466 reference allele should have lower HLA-DQB1 expression than base-edited clones homozygous for the alternative allele. We identified 7 clones homozygous for the alternative G allele, 7 clones homozygous for the reference C allele, and one clone with a 104bp deletion (Extended Data Fig. 10). As predicted, HLA-DQB1 was higher in alternative allele clones by real-time PCR (P = 0.003, Fig. 4c). After extended culture, surviving alternative allele clones had also higher expression of HLA-DQ protein by flow cytometry (P = 0.03, Fig. 4c). These results confirmed that the rs71542466 promoter SNP changes HLA-DQB1 expression and that it accounts for the Late-Spike cis regulatory program. We cannot rule out, however, that additional variants inside HLA-DQB1 (such as those within the UTR or intragenic enhancers) may also contribute.
After identifying rs71542466 as an HLA-DQB1 condition-specific regulatory variant, we considered whether its effect is T cell-specific. Notably, rs71542466 SNP was not in LD with reported eQTL SNPs for HLA-DQB1 in B cell derived lymphoblastoid cell lines (LCLs), monocytes, and resting and infected macrophages (Supplementary Table 2, r2≤0.27))8,9,32–34. Luciferase assays, Electrophoretic Mobility Shift Assays (EMSA) in cell lines and flow cytometry data in primary B cells further confirmed the activation-dependent regulatory effect is T cell specific (Fig. 4d,e; Supplementary Figs. 15,16; Supplementary Note, Source Data Fig. 4, Source Data Supplementary Fig. 15).
We observed dynASE in 5 HLA genes besides HLA-DQB1. Within HLA class II genes, HLA-DRB1 had significant dynASE in 9 individuals (P< 2.1 ×10−3=0.05/24 tests), and HLA-DQA1 in 6 (P< 2.2×10−3=0.05/23 tests). Within HLA class I genes, HLA-B and HLA-C had significant dynASE in 12 individuals (P<2.3 ×10−3=0.05/22 tests and P<2.4 ×10−3=0.05/21 tests) and HLA-A in 6 (P < 2.3 ×10−3=0.05/22). However, for these 5 genes, the magnitude of change in allelic fraction across time was more modest than for HLA-DQB1 (Supplementary Fig. 17). We noticed that individuals with a Late-Spike HLA-DQB1 allele often have a similar pattern of expression over time for the HLA-DRB1 allele on the same chromosome (based on phasing), with upregulation of expression at 48 and 72 hours (examples in Supplementary Fig. 18a). This suggests that cis co-regulation between these two genes could be mediated through promoter-promoter interactions between HLA-DQB1 and HLA-DRB1, consistent with promoter capture HiC data (Supplementary Fig. 18b)35,36.
Here, we observed that allelic imbalance in expression is highly dependent on time after stimulation of memory CD4+ T cells. In most cases, one of the two alleles over time gradually increased expression over the other allele (e.g. UBASH3A, CXCR5). We suspect these cases are a consequence of a regulatory complex interacting with a single genetic variant altering gene expression; where the status of the regulatory complex may vary depending on the environmental context over time. However, in many instances, we observed that the dominant expressed allele switched as stimulation progressed (e.g. HLA-DQB1, F11R), raising the possibility of multiple regulatory variants or complexes. For example, we identified distinct driving variants for HLA-DQB1 at 0 and 72 hours after stimulation (LD r2=0.01, Supplementary Table 2). Overall, this widespread dynamic allelic imbalance across the genome illustrates the continuously changing regulatory landscape of genes during T cell activation. We predict that future studies with larger samples sizes ascertaining additional cellular states will reveal that dynamic ASE is even more widespread than what we conservatively identified in this study.
We found that dynASE genes were highly enriched in autoimmune disease loci, suggesting that autoimmune risk alleles may affect the expression of its target gene under very specific conditions. Indeed, an autoimmune risk variant in an IL2RA enhancer has already been shown to act in a time-dependent manner, and influencing polarization of T cells into an inflammatory subtype (Th17) instead of the regulatory population (Treg)4. These results may explain why investigators have found limited shared genetic effects between autoimmune susceptibility variants and eQTL variants at resting state3,12,37 , despite the presence of autoimmune susceptibility variants in regulatory elements1–3,38 .
Intriguingly, we identified the most dramatic T cell and condition dependent cis regulatory variation within a major autoimmune disease gene: HLA-DQB1. This raises the question of whether, and to what extent, genetic regulatory variation controlling HLA gene expression could affect disease susceptibility or disease penetrance, as has been highlighted for other loci and traits39. For most autoimmune diseases, the MHC region is the major contributing locus to disease risk. In this study, three of the four Late-Spike HLA-DQB1 classical alleles are protective for T1D (OR 0.045–0.732), while the other two regulatory programs represent a mixture of risk and protective alleles17. While amino acid changes causing differential antigen display may be the primary autoimmune mechanism at the HLA locus17,40, our data underscores the possibility that expression levels of HLA class II may also play a crucial and unappreciated role41,42. Over the past several decades, there has been literature suggesting variation in expression among different HLA alleles34,43–45 – but to date the idea that this regulation changes with cell-state has not been established.
Broadly, class II expression has been well characterized as a marker for both late activation in CD4+ T cells and suppressive capacity in T regulatory cells46–48. However, the exact mechanisms and functional implications remain to be defined. Our work shows that not only do CD4+ T cells express high levels of HLA class II, but that its expression is regulated in a cell-type specific manner and varies between individuals. This suggests that during immune responses, expression of HLA II on CD4+ T cells is dynamically controlled and may be important to modulating function.
Methods
Study design
The goal of this study was to characterize cell state-dependent regulatory effects in memory CD4+ T cells to obtain new insights into autoimmune disease mechanisms. In initial experiments, we observed the most dramatic dynamic allele-specific expression effects in HLA-DQB1. To investigate this phenomenon in an optimal way, we recruited individuals heterozygous for common HLA-DQB1 classical alleles (characterized by a combination of coding single nucleotide alleles), with highly divergent alleles (at least a 20 mismatch differences between the two alleles). We used the de-identified genome-wide genotypes available from the individuals at the Genotype and Phenotype (GaP) Registry at The Feinstein Institute for Medical Research, to impute HLA classical alleles with SNP2HLA49 and select individuals. The GaP Registry provided de-identified cryopreserved PBMCs from 24 donors with no autoimmune disease, 20–50 years old, and of European ancestry. Donors provided fresh, de-identified human peripheral blood mononuclear cells (PBMCs); blood was collected from subjects under an IRB-approved protocol (IRB# 09–081) and processed to isolate PBMCs. The GaP is a sub-protocol of the Tissue Donation Program (TDP) at Northwell Health and a national resource for genotype-phenotype studies50. HLA classical alleles were subsequently experimentally confirmed with HLA typing (see below).
Similarly, for the protein level validation experiments, we recruited through the GaP Registry individuals homozygous for HLA-DQB1 classical alleles pertaining to the Late-Spike regulatory program (N = 5) or other programs (N = 5). These individuals were also between 20 to 50 years old, with no reported autoimmune disease, and of European ancestry.
Additional details can be found in the Life Sciences Reporting Summary form associated with this article.
Memory CD4+ T cell stimulation time course
PBMCs were thawed and resuspended in pre-warmed complete RPMI (cRPMI) (RPMI 1640, supplemented with 10% heat inactivated FBS, and 1% non-essential amino acids, sodium pyruvate, HEPES, L-Glutamine, Penicillin & Streptomycin, and 0.1% β-mercaptoethanol). Memory CD4+ T cells were isolated by magnetic selection (Miltenyi, Memory CD4+ T cell Isolation Kit human). One million cells per well were plated in sterile 48 well plates (Corning) and rested at 37°C overnight. Twelve hours after the beginning of the rest marked the first time point (0 hours). At this time, cells were spun down and resuspended in 350 μL of RLT lysis buffer (Qiagen) containing β-mercaptoethanol and stored at −80°C. To the remaining wells, 500 μL cRPMI with human T-Activator CD3/CD28 Dynabeads (Gibco) were added to each well at a ratio of 2 cells : 1 bead. Cells were collected at 2, 4, 8, 12, 24, 48, and 72 hours after stimulation. Once all cell pellets were collected, resuspended in RLT and frozen, the RNA was isolated using an RNeasy mini kit (Qiagen) and stored at −80°C until submitted for sequencing at the Broad Institute in Cambridge, MA.
Library construction and RNA sequencing
RNA-seq library preparations were performed with Illumina TruSeq stranded mRNA sample preparation kit, and 101bp paired-end reads were sequenced at a mean depth of 41 million fragments (read pairs). Additional details found in Supplementary Note.
Gene expression analyses
We mapped reads to the hg19 reference genome with subread v1.5.151 (with parameters: -u -Q -D 100000 -t 0 -T 4) and quantified expression levels using featureCounts (with parameters: -T 4 -Q 20 -C -s 2 -p -P -D 100000) and GENCODE52 v19 annotation. For PCA we took 1,070 genes with standard deviation > 1 and mean expression > 3 log2(tpm+1), we scaled gene to mean zero and variance one and performed PCA with the R53 function prcomp. For gene clustering, we used k-means. Enrichment of MsigDB hallmarks v6.254,55 was performed with the enricher function of the R package clusterProfiler56.
In all boxplots in the manuscript, boxes show the first to third quartile with median, whiskers encompass 1.5× the interquartile range, and data beyond that threshold are indicated as outliers.
Variant genotyping, imputation and filtering
Individuals were genotyped genome-wide using the Global Screening Array (GSA) assaying 647K SNPs. For pre-imputation QC, we used plink v1.90b3w57 to filter out variants with missing call frequencies greater than 0.05, Hardy Weinberg Equilibrium (HWE) threshold P < 1e-05, MAF 0.03, keeping a total of 339,333 variants. We imputed variants into the 1000 Genomes reference panel58 using SHAPEIT v2.r83759 and IMPUTE2 v2.3.260. We filtered out variants with info score < 0.9, multi-allelic, HWE threshold P < 1e-05, non-polymorphic within our 24 individuals, and with MAF <1% in Europeans of the 1000 Genomes reference panel, and indels. This yielded a total of 5,144,453 SNPs. When selecting heterozygous SNPs per individual, we further required a genotype probability > 0.9; a total of ~1.5M heterozygous SNPs per individual remained.
Dynamic allele-specific expression analysis
We used subread v1.5.1 to align reads to the hg19 reference genome and filtered out reads with mapping quality < 10. We used WASP15 to filter out reads that had mapping bias at heterozygous sites and to remove duplicates. For quantifying allele counts at heterozygous sites, we used GATK61 v3.8 ASEReadCounter requiring a minimum read mapping quality of 10 and a minimum base quality of 10 for the site in question (with parameters --minMappingQuality 10, --minBaseQuality 10, -U ALLOW_N_CIGAR_READS), and followed recommended best practices62. For initial QC (Supplementary Fig. 4), for each sample we took all heterozygous sites with at least 10 reads (6,496–44,864 sites per sample). The mean coverage across sites per sample ranged from 50–127. All samples had >95% of both alleles observed at included heterozygous sites. The mean reference fraction was close to 0.5 for all samples (mean 0.5098, range from 0.5045 to 0.5159). For a given heterozygous site, the reference fraction refers to the number of reads with the reference allele divided by the total number of reads overlapping the site. Allelic imbalance is the distance to 0.5 reference fraction (i.e. absolute value of: the reference fraction minus 0.5).
To identify sites with dynamic ASE, we used a nested approach using a logistic regression framework on a per individual, per heterozygous site basis, with the lme463 R package. Each read is encoded according to the following: 1 if it contains a reference allele or 0 if it contains the alternative allele. For each time course per individual, we included sites with at least 20 reads in at least 4 time points and required that both reference and alternative alleles are seen in all included samples. First, we identified sites with evidence of ASE by merging data from all time points and testing if the intercept is significantly different from zero (assuming a standard normal distribution and using a z-test, two-sided) and used a Bonferroni threshold to determine significance (0.05 divided by the number of tests). Then, we tested which of these sites had ASE that changes with time by fitting a second order polynomial model, which allowed to model non-linear ASE effects with respect to time. Time was coded as time points 1 through 8 (or maximum number of time points) and scaling to mean zero variance one. We controlled for overdispersion of allelic counts due to technical or biological sample to sample variability64 by incorporating a random intercept effect, coding sample ID as a factor. We tested for the effects of time by performing a likelihood ratio test between the two nested models using R anova function (two-sided). The null model H0 and alternative model H1 for a given SNP i in a given individual j are detailed below:
Where is the probability of observing the reference allele, α is the intercept, μ is the random intercept effect across samples, t is time, and β is the effect of time on the log-odds of observing the reference allele. We calculated the FDR per test using the qvalue R package65, and called significant all events with q<0.05 (<5% FDR), unless otherwise stated. Using a quadratic function identified twice as many events as a linear function, while capturing most of the linear events (87%).
HLA typing
Purified DNA from the 24 individuals was sent to the NHS Blood and Transplant, UK, where HLA typing was performed. Next generation sequencing was done for HLA-DQB1, HLA-A, HLA-B, HLA-C, and for HLA-DRB1. PCR-SSOP was done for HLA-DQA1 in all individuals, and for HLA-DRB1 in 6 individuals for which limited DNA was available. These typing methods yielded classical allele calls for the six genes at 4 to 8-digit resolution.
Allele-specific expression in HLA genes
To prepare an HLA personalized genome for each individual we first took the HLA 6-digit classical allele calls for each of the 6 HLA genes (12 alleles total per individual) and downloaded the corresponding cDNA sequence from the IPD-IMGT/HLA database66. Next, we added the 12 HLA allele cDNA sequences to the hg19 reference genome, each encoded as a separate chromosome. We masked the exonic regions of the 6 HLA genes on chromosome 6 in the original hg19 reference (by replacing the A, T, C and G bases with Ns), to remove redundancy with the added cDNA sequences (so that reads will map to only the cDNA sequences and not the original reference genome sequences). For each individual, we aligned per sample the reads to the HLA-personalized genome with subread requiring uniquely mapped reads. We removed PCR duplicates with Picard Tools v1.119. We counted the number of uniquely mapped reads to each HLA allele with featureCounts, requiring a minimum mapping quality of 40, using a personalized gtf annotation file per individual. This way, we got quantifications of each of the 6 HLA alleles, as though each was a separate gene.
To identify dynASE for each HLA gene, we used the same statistical approach mentioned above. Instead of using allele counts for a single SNP, we used counts for the whole cDNA per HLA allele (usually encompassing 3–4 exons, 552–1119 bp). To compare HLA allelic expression levels between samples, we normalized the HLA allele counts by library size and cDNA size (FPKM). For the two individuals for which we had full time course replicates, we used the mean of the FPKM values.
For testing whether allelic profiles of HLA-DQB1 4-digit classical allele groups are more similar to each other than expected by chance, we calculated the sum of squares within 4-digit allele groups, and total sum of squares (observed values). Then, we permuted the 4-digit allele groups 10,000 times, and repeated the sum of squares calculations (Supplementary Fig. 9). For this analysis, we excluded four allelic profiles with 4-digit alleles that were present only once in our 48 allelic profiles (DQB1*04:02, DQB1*06:01, DQB1*06:09 and DQB1*05:03).
For HLA-DQB1 allelic profile grouping with k-means clustering and PCA, we removed the 12 hour time point due to its high number of missing values (caused by an insufficient number of cells obtained for some individuals), and further excluded another 2 individuals with missing values, resulting in a total of 44 allelic profiles (2×22 individuals). For both of these analyses we used log2(FPKM+1) values. Three clusters captured 64% of the variation in our data, defined by the ratio of between group sum of squares and total sum of squares. Two clusters captured a significantly lower amount of between group variability (37%), and four clusters (70%) had a modest increase from three. PCA was performed on allelic expression profiles with prcomp with center = TRUE, and independently showed that the three clusters identified with k-means separate well (Extended Data Fig. 7).
HLA-DQ protein level validations on memory CD4+ T cells
Ten additional individuals were recruited through the GaP Registry (see Study Design). Five individuals were homozygous for Late-Spike alleles: one for DQB1*05:03 and four for DQB1*05:01. Five individuals were homozygous for alleles in the other two cis regulatory programs (Constant-Low, and Fluctuating): one for DQB1*03:02, one for DQB1*02:01, one for DQB1*02:02 and two for DQB1*03:01. Memory CD4+ T were isolated and stimulated as explained above. T cells were assayed for expression of HLA-DQ by flow cytometry on day 0 (unstimulated) and days 1, 3, and 7. Additional details for protein quantifications in T cells and B cells in Supplementary Note.
Regulatory variant fine-mapping
To look for genetic variants in LD with the Late-Spike haplotype, we called Single Nucleotide Variants (SNVs), small INsertions and DELetions (INDELs) and classical HLA variants using whole genome sequences of 2,244 healthy volunteers recruited from the Estonian Genome Project27 sequenced at 25X coverage. We performed high-resolution (G-group) HLA calling of three class-I HLA genes (HLA-A, -B and -C) and three class-II HLA genes (HLA-DRB1, -DQA1 and -DQB1) using the HLA*PRG algorithm67. We called SNVs and INDELs using GATK version 3.6 according to the best practices for variant discovery68. In total we called 246,505 variants in the extended MHC region (29–34 Mb on chromosome 6, NCBI Build 37).
To check if any SNVs are in high LD with the Late-Spike haplotype, we first used the Estonian reference panel and restricted our analyses to individuals who carried the alleles present in our 24 individuals (N = 2,198). Namely, there were 58 individuals with two HLA-DQB1 alleles (at 4-digit resolution) pertaining to the Late-Spike haplotype (i.e. DQB1*05:01, 05:02, 05:03 or 06:01), 616 with one Late-Spike haplotype allele, and 1,524 individuals with zero Late-Spike haplotype alleles. To filter out possible false-positive variants, we next restricted the analyses to SNPs with minor allele frequency (MAF) ≥ 0.05 and within 1 Mb region of the HLA-DQB1 gene (N = 27,210). Next, we computed the r2 between Late-Spike haplotype dosage (0, 1 or 2) and individual SNP genotypes. Refseq gene annotations were used to determine start and end of HLA-DQB1, HLA-DRB1 and HLA-DQA1.
ATAC-seq experiments and data processing
Memory CD4+ T cells from one new PBMC donor from the GaP Registry were purified and cultured as described above for 72 hours with anti-CD3/CD28 stimulation beads. Open chromatin was assessed with ATAC-seq69,70 as detailed in Supplementary Note.
Cell lines
HH cutaneous T cell lines (ATCC: CRL-2105), Jurkat E6–1 (ATCC: TIB-152), and Daudi (ATCC: CCL-213, provided by Dr. Michael Brenner), were cultured in complete RPMI as previously described. The HH cell line was chosen for CRISPR/Cas9 experiments because it is a T cell line that expressed HLA class II and has a transcriptome similar to the 72-hour time point in our dataset, at which the largest effect of the Late-Spike haplotype occurs (Supplementary Fig. 13).
Bulk CRISPR/Cas9 editing
To investigate regulatory regions around HLA-DQ, the nearest sgRNA to the SNP of interest was selected using Deskgen online tools (www.deskgen.com). Distances of designed sgRNA to the nearest SNP are shown in Extended Data Fig. 9. To confirm the sequence of the region, genomic DNA around HLA-DQB1 was PCR amplified and Sanger sequenced. CRISPR/Cas9 RNP complexes were assembled as previously described71. Briefly, 40 μM Cas9 protein (QB3 Mircolabs) was mixed with equal volumes of 40 μM modified sgRNA (Synthego) and incubated at 37°C for 15 minutes to form ribonuclear protein (RNP) complexes. HH cells were nucleofected with 2μL of RNPs in an Amaxa 4D nucleofector (SE protocol: CL-120). Cells were immediately transferred to 24 well plates with pre-warmed media and cultured. After 7–10 days, HLA-DQ expression was assessed by flow cytometry. Editing was confirmed by PCR amplifying genomic DNA around HLA-DQB1 and sequences analyzed by Tracking of Indels by Decompostiion (TIDE) analysis (tide.deskgen.com). All primer and sgRNA sequences are in Supplementary Table.
CRISPR/Cas9 base-editing of rs71542466 in HH cells
For generation of base-edited cell lines, HH cells were nucleofected with RNPs using sgRNA targeting near rs71542466 and asymmetrical ssDNA donors as previously described72. Modified cells were grown for 7–10 days then single cell sorted using a BD Aria II into 96 well U bottom plates. After 2–3 months of outgrowth, DNA from surviving clones (194/1200) was isolated using DNA quick extract solution (Lucigen) following a modified protocol. Briefly, 100 μL of cell culture was spun down, washed once with PBS, and then re-suspended in 20 μL of DNA extraction solution. Solution was heated in a thermocycler to 65°C for 15 minutes, 68°C for 15 minutes, 98°C for 10 minutes, and stored at 4 degrees. After DNA extraction, solution was diluted 1:20 and 5 μL used in a standard 50 μL PCR reaction using Q5 enzyme (NEB). PCR products were Sanger sequenced and analyzed using SnapGene to identify SNP corrected clones (7/192), wild type HH clones (7/192), and a single (1/194) insertion / deletions clone. Sequences are shown in Extended Data Fig. 10. All sgRNA and HDR donor sequences are found in Supplementary Table.
Real-time PCR and flow cytometry analysis of HH clones
For analysis of HLA-DQB1 expression on HH WT (C/C) and ALT (G/G) clones, RNA was extracted from clones using a Monarch Total RNA extraction kit (NEB). cDNA was synthesized using MaximaH RT (NEB) enzyme following manufacturer’s protocol and oligoDT primers. cDNA was diluted 1 in 4 with HLA-DQB1 (Assay ID:Hs00409790) and actinB (Assay ID:Hs01060665) probes and Taqman MasterMix (Thermofisher). Samples were run on an ARIAmx qPCR machine (Agilent) and data analyzed with Aria 1.5 (Agilent) software. Expression is represented as 2-delta(HLA-DQB1 Ct - ActinB Ct). For analysis of protein expression, clones that survived after 3–4 months of culture were washed with PBS and stained with FITC anti-HLA-DQ (Biolegend, Clone: HLADQ1) for 30 minutes on ice and cell surface expression assessed by flow cytometry. Data was analyzed using Flowjo and Graphpad PRISM.
Electrophoretic Mobility Shift Assays
EMSAs were performed using the LightShift Chemiluminescent EMSA Kit (Thermo Scientific) and single-stranded biotinylated oligonucleotides and complementary sequences corresponding to 31 nucleotides (15 nucleotides flanking the SNP of interest). Nuclear extract from Jurkat, HH, and Daudi cells was isolated using the NE-PER nuclear and cytoplasmic extraction kit (Thermofisher Scientific) with slight modification. Binding reactions were incubated at room temperature for 30 min and loaded onto a 6% polyacrylamide 0.5× Criterion precast TBE gel (Biorad). After sample electrophoresis and transfer to a nylon membrane, DNA was crosslinked for 10 min, and biotinylated probes detected by chemiluminescence followed by film exposure. Details in Supplementary Note, Supplementary Figures, Supplementary Table and Source Data files.
Luciferase assay
A double-stranded oligonucleotide containing the SNP of interest (31nt + restriction enzyme sites) was ordered and annealed as described above. See Supplementary Note for vector preparation details. 3 × 105 Jurkat, HH, and Daudi cells were nucleofected with 0.9 μg of pGL3-Promoter vector along with 0.1 μg of pRL-TK Renilla luciferase vector (Promega) in 16 well strips in a 4D nucleofector with the following protocols and buffers in 20 μL of total volume: Jurkat, SE buffer, CL-120 protocol; HH, SE buffer, CL-120 protocol; Daudi SF buffer, CA-137 protocol. After nucleofection, 180 μL of complete RPMI was added and cells cultured in 96 well flat bottom plates (Falcon). After 48 hours, cells were spun down, resuspended in 75 of fresh complete media, and luciferase/renilla activity measured using the Dual-Glo Luciferase Assay System (Promega). Firefly luciferase activity was expressed as relative luciferase units (RLU) after correction for Renilla luciferase activity. Data were normalized to those cells transfected with empty pGL3-Promoter vector. Each dot represents an independent nucleofection reaction.
Autoimmune disease enrichment analyses
We downloaded SNPs from the GWAS catalogue on July 17, 2018. We selected SNPs with P < 5e-08 for 11 autoimmune diseases and 3 non-immune mediated diseases that served as a negative control (schizophrenia, type 2 diabetes, coronary heart disease). We used SNPsea to capture genes within disease loci based on LD and recombination interval information73. We then assessed how many of the dynASE genes overlap genes in disease loci for each disease (observed overlap). To assess whether this overlap represented a significant enrichment, for each disease we created 1000 null sets of N random regions in the genome (N = number of disease loci), which were matched by the number of genes per locus (within 15% of each disease locus). We then calculated the ratio of observed overlap with the mean overlap of our 1000 null sets (fold enrichment). We calculated the P-value as: (number of null overlaps larger than observed overlap + 1)/100174. We took the same approach for genes with significant ASE at 0 hours (N = 501), and eQTL genes of naïve T cells reported by the Blueprint Consortium (N = 5,688)9.
Extended Data
Extended Data Fig. 1. Replication of dynamic ASE in two pilot individuals.

For two individuals, we performed full time-course replicates (from the same CD4+ memory T cell isolation batch, but independent stimulation experiment and RNA-seq library preparation). From the dynamic ASE events called significant in replicate A at 5% FDR (as explained in main text and Methods), we asked how do the P-values and betas look in replicate B. Left plots show distribution of P-values in replicate B, middle plots show correlation of betas for time, right plots show correlation of betas for time squared. (a) Individual TB03072560. (b) Individual TB03073798.
Extended Data Fig. 2. Replication examples of dynamic ASE in two pilot individuals.

Examples of a dynamic ASE event significant in individual TB03072560 (a) and TB03073798 (b). Shown are allelic counts for heterozygous SNP (left) and reference fraction over time (right) for replicate A (top panels) and replicate B (bottom panels).
Extended Data Fig. 3. Reproducibility of dynASE across heterozygous individuals for the same SNP.

Here we wanted to assess whether dynamic ASE replicates well in different heterozygous individuals for the same SNP. First, from the 561 dynASE events at 5% FDR we took the top 356 unique SNPs (ensuring one heterozygous individual per SNP), and then asked how do the P-values look in other heterozygous individuals for those 356 SNPs. (a) Qqplot depicting the observed P-values in the other heterozygous individuals (y-axis), compared to the expected uniform distribution of P-values (x-axis). (b) Next, within all 561 significant events at 5% FDR, we evaluated the correlation of betas for time (left) and time squared (right) for all pairwise combinations of heterozygous individuals for the same SNP, i.e. het1 and het2 in x and y axis labels.
Extended Data Fig. 4. DynASE examples for SNPs with two or more heterozygous individuals.

(a-c) Shown are gene expression levels across 24 individuals (left), and allele counts (SNP and individual indicated) and reference fraction (P-value and FDR for dynASE indicated) for heterozygous SNPs in corresponding gene.
Extended Data Fig. 5. Scheme depicting HLA allelic expression quantification with HLA-personalized genome.

In order to quantify robustly allele-specific expression in the highly polymorphic HLA genes, we first create an HLA-personalized genome per individual. We do this by inserting into the reference genome the cDNA sequences of each HLA allele as separate sequences (12 in total given that we sequenced or typed 6 HLA genes), and masking the exonic sequences corresponding to those cDNAs in chromosome 6 of the reference genome. Next, we map the RNA-seq reads to this HLA-personalized genome, we remove PCR duplicates and we count the number of uniquely mapped reads to each HLA cDNA allele.
Extended Data Fig. 6. Allelic fraction replication in HLA gene quantifications.

Allelic fraction over time for the 3 HLA class II genes (a) and 3 HLA class I genes (b), for the two pilot individuals with full time course replicates. Replicate A in black, replicate B in blue.
Extended Data Fig. 7. Principal component analysis of HLA-DQB1 allelic profiles over time.

PCA performed for 48 HLA-DQB1 allelic expression profiles of 24 individuals (log2(FPKM+1) values over time. Allelic profiles are colored by 4-digit classical HLA-DQB1 allele (a), and by the k-means cluster to which they belong (b). Average allelic expression was computed for samples with replicates. Twelve hour time point was removed because of high number of missing values. These plots depict how 4-digit alleles group near each other (a), and how PCA also captures the three distinct cis regulatory programs (Fluctuating, Constant-Low and Late-Spike) (b).
Extended Data Fig. 8. Mapping variants associated with Late-Spike haplotype.
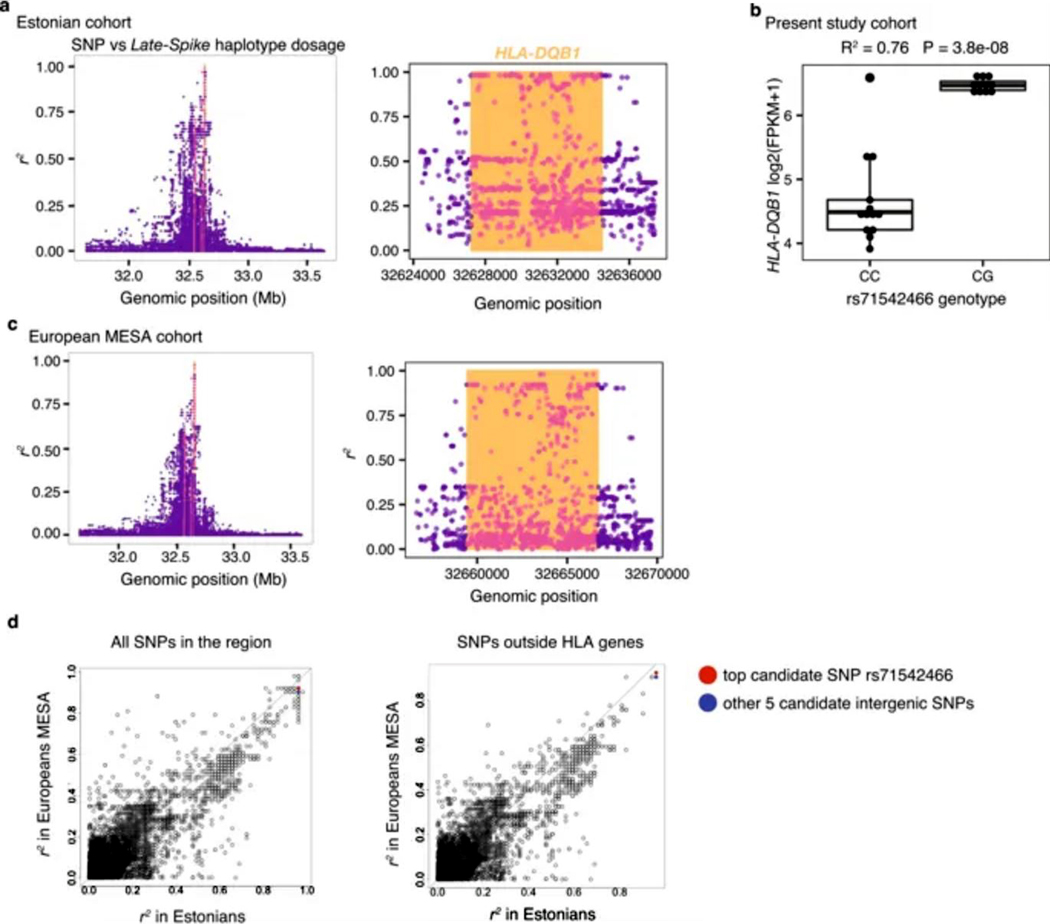
(a) r2 between Late-Spike haplotype dosage and SNPs within 1Mb of HLA-DQB1 in Estonian cohort. Orange vertical lines indicate location of HLA-DQB1. Dots that are colored pink are intragenic SNPs in HLA-DQB1, HLA-DRB1, and HLA-DQA1. Right plot is zoomed in on HLA-DQB1 region to show top SNPs (reference genome hg19). (b) HLA-DQB1 gene expression levels (log2(FPKM+1)) at 72 hours after stimulation for individuals separated by their rs71542466 genotype. (c) Same as in (a) but in European MESA cohort (reference genome GRCh38). (d) r2 comparison between Estonian and European MESA cohort, for all SNPs in the region (left) or the subset of SNPs in the regions that do not overlap HLA-DQB1, HLA-DRB1 or HLA-DQA1 start-end genomic coordinates (right). The 6 intergenic SNPs with top r2 in Estonians are highlighted, with 3 of them having top r2 in the European MESA cohort too. Identity line marked. These results show that our top candidate SNP rs71542466 (and the other candidate SNPs) tracks well with the Late-Spike haplotype in both the Estonian and the MESA cohort of individuals of European ancestry recruited in the United States.
Extended Data Fig. 9. Genomic location of nearest gRNAs to tested causal SNPs and representative flow cytometry plot of CRISPR-Cas9 edited HH cells.

(a) Location of SNPs (red) is shown in reference to the nearest exon (blue) both upstream and downstream of HLA-DQB1. The nearest gRNA sequences used for targeting the regions are highlighted with their corresponding colors (rs71542466 - dark green, rs71542467 - light purple, rs71542468 - purple, rs72844401 - beige/orange, rs4279477 - blue, rs28451423 - light green). Alignments were plotted using SnapGene(v3.2.1). (b) Representative staining of HLA-DQ on CRISPR-Cas9 modified HH cells. Cells were modified with proximal gRNA as shown in (a) and labelled accordingly. Cells stained 7–10 days after modification with HLA-DQ antibodies as a bulk population.
Extended Data Fig. 10. Sanger sequencing alignment of HH reference and base-edited clones reveal seamless editing.

Genomic DNA from expanded clones was sequenced and aligned to the reference (hg38) and visualized using SnapGene(v3.2.1). Red colored nucleotide indicates the location of the rs71542466 SNP in the reference. Highlighted red nucleotides indicate mismatches from the reference and yellow colored nucleotides indicate unresolved/heterozygous sequences.
Supplementary Material
Acknowledgments
We are indebted to Gila Klein RN for her outstanding management of the Genotype and Phenotype (GaP) registry at the Feinstein Institute, to the Raychaudhuri laboratory members for critical discussions and feedback, and to Henry Long and Paloma Cejas for support on primary T cell ATAC-seq experiments. This work was supported by the National Institutes of Health (U19AI111224, U01GM092691, U01HG009379 and R01AR063759 to S.R., NHGRI T32 HG002295 to T.A.), the Swiss National Science Foundation (Early Postdoc Mobility Fellowship to M.G.-A.), the Broad Institute through the SPARC mechanism (S.R.), the Estonian Research Council (PUT1660 to T.E.), the European Union Horizon 2020 (MP1GI18418R to T.E.). Whole genome sequencing (WGS) for the Trans-Omics in Precision Medicine (TOPMed) program was supported by the National Heart, Lung and Blood Institute (NHLBI). WGS for “NHLBI TOPMed: Multi-Ethnic Study of Atherosclerosis (MESA)” (phs001416.v1.p1) was performed at the Broad Institute of MIT and Harvard (3U54HG003067-13S1). Centralized read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract HHSN268201800002I). Phenotype harmonization, data management, sample-identity QC, and general study coordination were provided by the TOPMed Data Coordinating Center (3R01HL-120393-02S1; contract HHSN268201800001I). We gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed. MESA and the MESA SHARe project are conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with MESA investigators. Support for MESA is provided by contracts HHSN268201500003I, N01-HC-95159, N01-HC-95160, N01-HC-95161, N01-HC-95162, N01-HC-95163, N01-HC-95164, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-TR-000040, UL1-TR-001079, UL1-TR-001420. The provision of genotyping data was supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR001881, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center. The full authorship list for the NHLBI TOPMed consortium can be found in https://www.nhlbiwgs.org/topmed-banner-authorship.
Footnotes
Competing Interests
Authors declare no competing interests.
Data availability
The RNA-seq data supporting this publication is available at GEO, with accession number GSE140244. The flow cytometry data supporting this publication is available at ImmPort (https://www.immport.org) under study accession SDY1555.
Code availability
Code for key analyses in this study are publicly available in GitHub (https://github.com/immunogenomics/dynamicASE) or upon request to authors.
Full list of members in Supplementary Note
References
- 1.Trynka G. et al. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat. Genet 45, 124–130 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Onengut-Gumuscu S. et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat. Genet 47, 381–386 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farh KK-H et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518, 337–343 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simeonov DR et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature 549, 111–115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gutierrez-Arcelus M, Rich SS & Raychaudhuri S. Autoimmune diseases [mdash] connecting risk alleles with molecular traits of the immune system. Nat. Rev. Genet 17, 160–174 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raj T. et al. Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science 344, 519–523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dimas AS et al. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science 325, 1246–1250 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gutierrez-Arcelus M. et al. Passive and active DNA methylation and the interplay with genetic variation in gene regulation. Elife 2, e00523 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen L. et al. Genetic Drivers of Epigenetic and Transcriptional Variation in Human Immune Cells. Cell 167, 1398–1414.e24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishigaki K. et al. Polygenic burdens on cell-specific pathways underlie the risk of rheumatoid arthritis. Nat. Genet 49, 1120–1125 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Ye CJ et al. Intersection of population variation and autoimmunity genetics in human T cell activation. Science 345, 1254665 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu X. et al. Regulation of gene expression in autoimmune disease loci and the genetic basis of proliferation in CD4+ effector memory T cells. PLoS Genet. 10, e1004404 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buil A. et al. Gene-gene and gene-environment interactions detected by transcriptome sequence analysis in twins. Nat. Genet 47, 88–91 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hauff N, Zhou X, Wen X, Pique-Regi R. & Luca F. High-throughput allele-specific expression across 250 environmental conditions. Genome (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van de Geijn B, McVicker G, Gilad Y. & Pritchard JK WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat. Methods 12, 1061–1063 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moyerbrailean GA et al. High-throughput allele-specific expression across 250 environmental conditions. Genome Res. 26, 1627–1638 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu X. et al. Additive and interaction effects at three amino acid positions in HLA-DQ and HLA-DR molecules drive type 1 diabetes risk. Nat. Genet 47, 898–905 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sollid LM et al. Evidence for a primary association of celiac disease to a particular HLA-DQ alpha/beta heterodimer. J. Exp. Med 169, 345–350 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burmester GR, Yu DT, Irani AM, Kunkel HG & Winchester RJ Ia+ T cells in synovial fluid and tissues of patients with rheumatoid arthritis. Arthritis Rheum. 24, 1370–1376 (1981). [DOI] [PubMed] [Google Scholar]
- 20.Yu DT et al. Peripheral blood Ia-positive T cells. Increases in certain diseases and after immunization. J. Exp. Med 151, 91–100 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ko HS Ia determinants on stimulated human T lymphocytes. Occurrence on mitogen- and antigen-activated T cells. Journal of Experimental Medicine 150, 246–255 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rao DA et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 542, 110–114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fonseka CY et al. Mixed-effects association of single cells identifies an expanded effector CD4+ T cell subset in rheumatoid arthritis. Sci. Transl. Med 10, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lanzavecchia A, Roosnek E, Gregory T, Berman P. & Abrignani S. T cells can present antigens such as HIV gp120 targeted to their own surface molecules. Nature 334, 530–532 (1988). [DOI] [PubMed] [Google Scholar]
- 25.LaSalle JM, Tolentino PJ, Freeman GJ, Nadler LM & Hafler DA Early signaling defects in human T cells anergized by T cell presentation of autoantigen. J. Exp. Med 176, 177–186 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brandes M, Willimann K. & Moser B. Professional antigen-presentation function by human gammadelta T Cells. Science 309, 264–268 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Guo MH et al. Comprehensive population-based genome sequencing provides insight into hematopoietic regulatory mechanisms. Proc. Natl. Acad. Sci. U. S. A 114, E327–E336 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bild DE et al. Multi-Ethnic Study of Atherosclerosis: objectives and design. Am. J. Epidemiol 156, 871–881 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Roadmap Epigenomics Consortium et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wong D. et al. Genomic mapping of the MHC transactivator CIITA using an integrated ChIP-seq and genetical genomics approach. Genome Biol. 15, 494 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.GTEx Consortium. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nédélec Y. et al. Genetic Ancestry and Natural Selection Drive Population Differences in Immune Responses to Pathogens. Cell 167, 657–669.e21 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Aguiar VRC, César J, Delaneau O, Dermitzakis ET. & Meyer D. Expression estimation and eQTL mapping for HLA genes with a personalized pipeline. PLoS Genet. 15, e1008091 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Javierre BM et al. Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters. Cell 167, 1369–1384.e19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schofield EC et al. CHiCP: a web-based tool for the integrative and interactive visualization of promoter capture Hi-C datasets. Bioinformatics 32, 2511–2513 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chun S. et al. Limited statistical evidence for shared genetic effects of eQTLs and autoimmune-disease-associated loci in three major immune-cell types. Nat. Genet 49, 600 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Finucane HK et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet 47, 1228–1235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Castel SE et al. Modified penetrance of coding variants by cis-regulatory variation contributes to disease risk. Nat. Genet 50, 1327–1334 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raychaudhuri S. et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat. Genet 44, 291–296 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raj P. et al. Regulatory polymorphisms modulate the expression of HLA class II molecules and promote autoimmunity. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cavalli G. et al. MHC class II super-enhancer increases surface expression of HLA-DR and HLA-DQ and affects cytokine production in autoimmune vitiligo. Proc. Natl. Acad. Sci. U. S. A 113, 1363–1368 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vandiedonck C. et al. Pervasive haplotypic variation in the spliceo-transcriptome of the human major histocompatibility complex. Genome Res. 21, 1042–1054 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pelikan RC et al. Enhancer histone-QTLs are enriched on autoimmune risk haplotypes and influence gene expression within chromatin networks. Nat. Commun 9, 2905 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Senju S. et al. Allele-specific expression of the cytoplasmic exon of HLA-DQB1 gene. Immunogenetics 36, (1992). [DOI] [PubMed] [Google Scholar]
- 46.Baecher-Allan C, Wolf E. & Hafler DA MHC class II expression identifies functionally distinct human regulatory T cells. J. Immunol 176, 4622–4631 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Reinherz EL et al. Ia determinants on human T-cell subsets defined by monoclonal antibody. Activation stimuli required for expression. J. Exp. Med 150, 1472–1482 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Engleman EG, Benike CJ & Charron DJ Ia antigen on peripheral blood mononuclear leukocytes in man. II. Functional studies of HLA-DR-positive T cells activated in mixed lymphocyte reactions. J. Exp. Med 152, 114s–126s (1980). [PubMed] [Google Scholar]
Methods-only References
- 49.Jia X. et al. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS One 8, e64683 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.GAP Registry | The Feinstein Institute for Medical Research. The Feinstein Institute for Medical Research Available at: https://www.feinsteininstitute.org/robert-s-boas-center-for-genomics-and-human-genetics/gap-registry/ (Accessed: 27th February 2019)
- 51.Liao Y, Smyth GK & Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frankish A. et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.R Core Team, R. & Others. R: A language and environment for statistical computing. (2013). [Google Scholar]
- 54.Subramanian A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liberzon A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu G, Wang L-G, Han Y. & He Q-Y clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Purcell S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Delaneau O, Marchini J. & Zagury J-F A linear complexity phasing method for thousands of genomes. Nat. Methods 9, 179–181 (2011). [DOI] [PubMed] [Google Scholar]
- 60.Howie BN, Donnelly P. & Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 5, e1000529 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McKenna A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Castel SE, Levy-Moonshine A, Mohammadi P, Banks E. & Lappalainen T. Tools and best practices for data processing in allelic expression analysis. Genome Biol. 16, 195 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bates D, Mächler M, Bolker B. & Walker S. Fitting Linear Mixed-Effects Models Using lme4. Journal of Statistical Software, Articles 67, 1–48 (2015). [Google Scholar]
- 64.Knowles DA et al. Allele-specific expression reveals interactions between genetic variation and environment. Nat. Methods 14, 699–702 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Storey JD & Tibshirani R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U. S. A 100, 9440–9445 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Robinson J. et al. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res. 43, D423–31 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dilthey A, Cox C, Iqbal Z, Nelson MR & McVean G. Improved genome inference in the MHC using a population reference graph. Nat. Genet 47, 682–688 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Poplin R. et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 201178 (2017). doi:10.1101/201178 [Google Scholar]
- 69.Corces MR et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Buenrostro JD, Wu B, Chang HY. & Greenleaf WJ ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Current Protocols in Molecular Biology 2129.1–21.29.9 (2015). doi: 10.1002/0471142727.mb2129s109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schumann K. et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proceedings of the National Academy of Sciences 112, 10437–10442 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Richardson CD, Ray GJ, DeWitt MA, Curie GL & Corn JE Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol 34, 339–344 (2016). [DOI] [PubMed] [Google Scholar]
- 73.Slowikowski K, Hu X. & Raychaudhuri S. SNPsea: an algorithm to identify cell types, tissues and pathways affected by risk loci. Bioinformatics 30, 2496–2497 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Phipson B. & Smyth GK Permutation P-values should never be zero: calculating exact P-values when permutations are randomly drawn. Stat. Appl. Genet. Mol. Biol 9, Article39 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.