

SUMMARY
Tissue development results from lineage-specific transcription factors (TFs) programming a dynamic chromatin landscape through progressive cell fate transitions. Here, we define epigenomic landscape during epidermal differentiation of human pluripotent stem cells (PSCs) and create inference networks that integrate gene expression, chromatin accessibility, and TF binding to define regulatory mechanisms during keratinocyte specification. We found two critical chromatin networks during surface ectoderm initiation and keratinocyte maturation, which are driven by TFAP2C and p63, respectively. Consistently, TFAP2C, but not p63, is sufficient to initiate surface ectoderm differentiation, and TFAP2C-initiated progenitor cells are capable of maturing into functional keratinocytes. Mechanistically, TFAP2C primes the surface ectoderm chromatin landscape and induces p63 expression and binding sites, thus allowing maturation factor p63 to positively autoregulate its own expression and close a subset of the TFAP2C-initiated surface ectoderm program. Our work provides a general framework to infer TF networks controlling chromatin transitions that will facilitate future regenerative medicine advances.
In Brief
Oro and colleagues describe epigenomic landscapes of epidermal lineage commitment from human PSCs. By delineating transcriptional and chromatin-regulatory networks, they identify TFAP2C and p63 as key factors for surface ectoderm initiation and keratinocyte maturation, respectively, and reveal TFAP2C-p63 feedback regulation of epigenetic transitions during lineage commitment.
Graphical Abstract
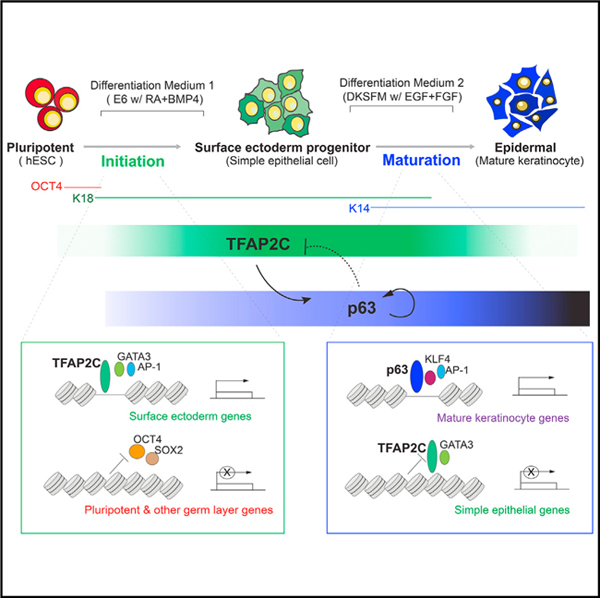
INTRODUCTION
Somatic tissue development, where pluripotent stem cells (PSCs) progressively commit into more specialized cell types, involves dynamic changes in gene expression and chromatin organization. Cells from different lineages possess specific chromatin accessibility patterns and cis-regulatory elements (REs) that instruct lineage-specific transcription factors (TFs) to precisely control their target genes (TGs). Although studies of individual TFs have elucidated discrete functions, detailed information is lacking about TF functions within a larger interconnected network. In addition, although lineage commitment requires an epigenetic transition from progenitor to terminally differentiated cells, a paucity of information exists how stage-specific TF networks interconnect to drive chromatin landscape maturation to the final committed state.
Stratified epidermal development is an ideal model system to investigate chromatin dynamic mechanisms. The epidermis represents a late ectoderm derivative, forming from lateral surface ectoderm initially specified by gradient morphogen induction by bone morphogenetic protein (BMP) and retinoic acid (RA) (Li et al., 2013; Metallo et al., 2008). Surface ectoderm is a single-layered epithelium expressing keratin 8 (K8) and keratin 18 (K18). In the presence of insulin, fibroblast growth factor (FGF), and epidermal growth factor (EGF), surface ectoderm commits to form stratified epidermal progenitors called basal keratinocytes expressing keratin 14 (K14) and keratin 5 (K5) that are capable of producing multi-layered skin (Koster and Roop, 2007). Previous efforts have begun to identify key TFs regulating skin differentiation. The p53 family member p63 regulates keratinocyte proliferation and epidermal stratification, and loss of p63 causes skin and limb hypoplasia (Mills et al., 1999; Yang et al., 1999). However, although the role of p63 during epidermal commitment is clear, how p63 connects with upstream transcription networks that drive surface ectoderm initiation and how it ensures forward differentiation and commitment remains unclear.
An important advance in understanding epidermal TF networks comes from the ability to drive PSCs, including embryonic stem cells (ESCs) or induced pluripotent stem cells (iPSCs) into keratinocytes (Metallo et al., 2008), thus enabling the collection of genome-wide regulatory information from cells at corresponding stages. Recently, we and others have used stem cell technologies to successfully generate patient-specific, genetically corrected iPSC-derived graftable keratinocyte sheets for treatment of epidermolysis bullosa, a genetic blistering disease caused by mutations in the COL7A1 gene (Sebastiano et al., 2014; Umegaki-Arao et al., 2014; Wenzel et al., 2014). Although these findings provide hope for tissue replacement therapies, a major roadblock remains the understanding and improvement of the differentiation process that will increase its efficiency and specificity to a level compatible with clinical manufacturing. Toward this end, dissecting the genome-wide regulatory landscape during differentiation remains critical for understanding lineage commitment in epidermal development.
Here, we use a defined feeder-free, xeno-free ESC differentiation system and propose a network inference modeling algorithm to identify the interconnecting TF networks during two major epigenetic transition periods. Subsequent functional studies uncover the surprising finding that a single factor, TFAP2C, drives skin differentiation by initiating the surface ectoderm chromatin landscape and inducing the maturation factor p63; p63, in turn, matures the chromatin landscape into stratified epithelium and inhibits select aspects of the TFAP2C surface ectoderm network. Our work defines the regulatory landscape during human epidermal lineage commitment and elucidates key regulatory principles that enable somatic tissue development and future stem-cell-based regenerative therapy.
RESULTS
Epigenomic Profiling Identifies Key Transitions during Epidermal Commitment
Using our defined 60-day differentiation protocol (Sebastiano et al., 2014), we detailed the transcription and chromatin dynamics at each stage of differentiation. RA and BMP4 induced ESCs into simple epithelium (K8+/K18+) after 7 days, followed by defined keratinocyte serum-free medium (DKSFM) (containing insulin, EGF, and FGF) that drove epidermal lineage maturation, consisting of cell death, migration, and epithelial colony formation of pure human ESC (hESC)-derived basal keratinocytes (H9KC; Figures 1A and S1A). H9KCs possessed similar morphology and marker gene expression as somatic foreskin normal human keratinocyte (NHK) (K14+p63+K18; Figures 1B and S1A) and formed stratified epidermal layers in organotypic cultures (K10+ and loricrin+; Figure 1C), demonstrating their functional capacity. We identified open chromatin REs and associated gene expression changes at six time points using assay for transposase accessible chromatin with high-throughput sequencing (ATAC-seq) (Buenrostro et al., 2013) and RNA sequencing (RNA-seq) methods (Figure S1B), with genome-wide profiling confirming a high similarity between H9KC and NHK (Figure S1H). Out of the total open chromatin REs across all samples, we selected 188,850 that were differentially enriched at specific stages (Figures 1D and S1C). Similarly, we characterized the stage-specific transcriptome and focused on 9,362 transcripts that defined each time point. Using the StepMiner algorithm (Sahoo et al., 2007) with D0 and H9KC data, we observed progressive closing of pluripotent-specific sites and opening of keratinocyte-specific sites over time (Figures S1F and S1G), with the associated-gene expression pattern following a similar trend (Figure S1E), indicating a gradual rather than stepwise maturation to keratinocytes.
Figure 1. Accessible Chromatin and Transcriptome Landscapes Identify Three Major Chromatin Stages during Epidermal Lineage Commitment.

(A) Schematic overview of epidermal differentiation from human embryonic stem cells (hESCs).
(B) Light image and immunofluorescence (IF) staining of keratinocytes derived from H9 hESCs (H9KC). Scale bar, 50 mm. K14, keratin 14; K18, keratin 18.
(C)Reconstruction of stratified epidermis with H9KC in organotypic culture. IF staining shows the maker localization in the organotypic epidermis. The nuclei were stained with DAPI (blue). Scale bar, 25 mm. LOR, loricrin; ColVII, collagen VII.
(D)Heatmap of differential open chromatin regulatory elements (REs) characterized from ATAC-seq. Hierarchical clustering yields three clusters of elements and three major groups of samples. The color bar shows the relative ATAC-seq signal (Z score of normalized read counts) as indicated.
(E)The trend of signal changes of the three clusters identified from ATAC-seq in (D).
(F)Heatmap of expression changes of the genes containing differential ATAC-seq signals at their promoters. The color bar shows the relative expression value (Z score of FPKM [fragments per kilobase of transcript per million mapped reads]) from the RNA-seq.
(G) Normalized ATAC-seq profiles at OCT4, K8-K18, and K5 loci, representing the dynamic changes of the three clusters identified in (D) and (E), respectively.
(H) Gene expression changes of OCT4, K18, and K5.
See also Figures S1 and S2.
The changing epigenetic landscape could be defined by three groups through hierarchical clustering: stage 1 (early stage: D0); stage 2 (middle stage: D7, D14, and D21); and stage 3 (late stage: D43 and H9KC; Figure 1D). The terms “initiation” and “maturation” describe the sequential transitions between pluripotent cell, surface ectoderm progenitor, and mature keratinocyte (Figure 1A). These three stages are tightly associated with three RE clusters. Cluster I, typified by OCT4 (POU5F1), contains elements with higher chromatin accessibility signals in pluripotent cells that close upon differentiation (Figures 1D–1H, S1D, and S1E). Cluster II, typified by K8 and K18, reflects the emergence of chromatin accessibility of genes associated with surface ectoderm initiation from D7 to D21 (Figures 1D–1H, S1D, and S1E). Finally, cluster III, typified by K5, contains elements with increased accessibility changes and higher associated gene expression during late stage of differentiation as definitive keratinocytes emerge (D43 and H9KC; Figures 1D–1H, S1D, and S1E). Similar analyses are seen in the differentiation of other independent PSCs (i.e., HUES6 hESCs and iPSCs from human fibroblast reprogramming; Macarthur et al., 2012) using the same or different differentiation protocols (Figures S2A–S2F), demonstrating the robustness of this model system and allowing us to conclude that addition of RA/BMP drives ESC/iPSC through 3 major stages of differentiation defined by transitions of initiation and maturation.
TF-Chromatin Transcriptional Regulatory Networks during Epidermal Commitment
To create a comprehensive TF network of keratinocyte differentiation, we developed an algorithm that links changes in accessible chromatin using ATAC-seq, the predicted REs for specific TFs, and the TF activity associated with a transcription start site as revealed through RNA-seq (Figure S3A). First, we identified 17 key TF motifs most correlated with the dynamic chromatin accessibility landscape (Figure 2A; Table S1). Next, we employed our recently described network inference framework that combines gene expression and chromatin accessibility data with accessible TF binding motifs (Duren et al., 2017). A major hurdle for network modeling comes from the discovery that chromatin changes during differentiation occur in the distal regulatory regions (enhancers) rather than the promoters, an observation we confirm (Figure S1C), making assignment of chromatin changes and RE to TG challenging. To overcome this challenge, we incorporated publicly available chromosome conformation data and co-occurrence information of distal and promoter DNase I hypersensitive sites (DHSs) across 79 diverse cell types from ENCODE (Rao et al., 2014; Thurman et al., 2012), significantly enhancing the accuracy of gene assignment (Figures 2B and S3B; Table S2).
Figure 2. Identification of Master TFs Driving Surface Ectoderm Initiation and Keratinocyte Maturation by the TF-Chromatin Transcriptional Regulatory Network.

(A)TF motifs identified from differential ATAC-seq peaks at each time point. The circle size represents different levels of motif enrichment, and the color represents the expression level of each TF in RNA-seq data.
(B)Schematic overview of the method for constructing TF-chromatin transcriptional regulatory network. (Top panel) Connections between TF and target gene (TG) are established through motifs present in open chromatin and publicly available chromatin conformation data. (Bottom panel) The triple elements, i.e., TF-accessible RE (i.e., ATAC-seq peaks)-TG expression, were ranked by the coherence among genomic features and extracted through a statistical model to build the regulatory network. We investigated and ranked their feature changes (Figures S3A–S3C) during two major transition events, initiation and maturation.
(C)TF networks identified from the chromatin regions gaining or losing accessibility in the initiation and maturation process. The red and blue nodes represent TF and TG, respectively; the gray edges represent the accessible RE, which was bound by TF to regulate TG expression. Larger size of TF nodes represents more TG connections. Top-ranked TFs are listed at the bottom of each network. Note: TF family name was used to represent several members.
See also Figure S3.
We focused our analysis on the two transition periods: the initiation phase (i.e., the transition from stage 1 to 2) and maturation phase (the transition from stage 2 to 3). Assuming that the rate of transcription of a TG depends on TFs bound to REs that are open (Figure 2B), we created TF-RE-TG triplet simplifications at each stage that allowed us to integrate all investigated genomic features over time. We then quantitatively ranked all the TF-RE-TG triplets and selected the high confident connection triplets to generate a network for representation (Figures 2B, S3A, and S3B; Table S3). During initiation, where ESCs lose pluripotency and differentiate into committed surface ectodermal cells, AP2 factors (TFAP2A and TFAP2C), GATA factors (e.g., GATA3), and GRHL factors (e.g., GRHL2) gain chromatin accessibility and transcriptional activity at the expense of OCT4, NANOG, and SOX2 (Figures 2C and S3C). By contrast, during maturation, p63, KLF4, and AP1 gain accessibility and transcriptional activity at their targets at the expense of the initiation network TFs TFAP2C and GATA3 (Figures 2C and S3C). We validated network predictions by TF chromatin immunoprecipitation sequencing (ChIP-seq) and RNA-seq (Figure S3D) and observed a consistent pattern of initiation and maturation TF network factors in other differentiated cell lines (Figures S2G–S2I). We conclude that inference network modeling provides a ranked TF network that describes key transitions during keratinocyte differentiation.
TFAP2C Alters Chromatin Landscape to Drive Surface Ectoderm Differentiation
To dissect the hierarchical relationships of TFs during surface ectoderm initiation and keratinocyte maturation, we established doxycycline (Dox)-inducible piggyBac ESC lines to investigate ectopic expression and CRISPR-edited lines to assay loss-of-function phenotypes with the goal of determining which TF could replace the addition of RA/BMP. Surprisingly, overexpression of TFAP2C (“TetO-TFAP2C”; Figure 3A) induced the flattened K18+ epithelial-like morphology seen with RA/BMP addition and led to much higher early network expression changes compared to other initiation TFs in the absence of RA/BMP (Figures 3B–3D, S4A, S4M, and S4N). Although sufficient to drive surface ectoderm differentiation, TFAP2C was also necessary. Attempts to remove TFAP2C in ESCs by CRISPR/Cas9 failed to generate homozygous knockouts due to a TFAP2C-dependent growth defect in ESCs (Figures S4B and S4C); however, heterozygous knockout cell lines (TFAP2C+/; 20% residual protein) show reduced K18 protein levels (Figure 3B) and reduced expression of many surface ectodermal genes (Figure 3C), supporting an essential role for TFAP2C in early commitment. Taken together, we conclude that TFAP2C is necessary and sufficient to induce the surface ectodermal phenotype.
Figure 3. TFAP2C Initiates the Chromatin Landscape to Induce Surface Ectoderm Differentiation.

(A) Schematic representation of the piggyBac TetO-TFAP2C inducible expression system in H9 hESC. TRE, tetracycline-responsive element; rtTA, reverse tetracycline transactivator.
(B) IF staining of K18 and TFAP2C in TetO-TFAP2C cells ± Dox and TFAP2C wild-type (WT) and heterozygous (+/−) cells with RA/BMP4 treatment. Scale bar, 20 μm.
(C)Gene expression changes in early differentiation upon TFAP2C loss of function. WT and TFAP2C +/− hESCs were induced into surface ectoderm progenitor cells by RA/BMP4 for 7 days. qRT-PCR values were normalized to the values in WT group.
(D) Gene expression changes in TetO-TFAP2C cells upon Dox induction. qRT-PCR values were normalized to the values of internal control GAPDH. In (C) and (D),mean ± SD is shown (n = 3; *p < 0.05; **p < 0.01; t test).
(E) Heatmap of germ-layer-specific gene expression from the RNA-seq in TetO-TFAP2C cells ± Dox and cells induced by RA/BMP4 for 7 days.(F and G) Scatterplot of gene expression from RNA-seq
(F) and of read counts from ATAC-seq
(G) in early differentiated cells by TFAP2C activation (TetO-TFAP2C Dox+) versus by RA/BMP4 induction at D7 with Pearson correlation (R) value displayed.
(H) Principal-component analysis (PCA) of ATAC-seq data from TetO-TFAP2C-D7 (Dox+) and samples at all time points during normal differentiation.
(I) Scatterplot of differential accessibility in TetO-TFAP2C-D7 Dox+ versus Dox cells. The values of log2 fold change > 1 or <−1 are labeled as “peaks gained” or “peaks lost” with respective percentage and color. CPM, count per million reads.
(J) ATAC-seq tracks show increased chromatin accessibility at K8-K18 and GATA3 loci upon TFAP2C-induced early differentiation (Dox+ versus Dox−).
(K) TF motif enrichment in the regions with decreased (left) or increased (right) chromatin accessibility in TetO-TFAP2C-D7 Dox+ versus Dox− cells. TF motifs from one family are labeled in the same color and are indicated in the top corners.
(L) Average enrichment of ATAC-seq chromatin accessibility within −1/+1 kb from TFAP2C binding sites in cells ± Dox induction.
(M) Average enrichment of histone marks within −1/+1 kb from TFAP2C binding sites in TFAP2C overexpressed cells.
See also Figure S4
As the TFAP2C-induced cell morphology mimicked early RA/ BMP-induced differentiation, we compared their expression and chromatin accessibility changes. First, TFAP2C induced expression of most surface ectodermal TFs (Figures 3D, 3E, S2J, and S2K) and enriched for associated genes (normalized enrichment score [NES] = 1.42; p < 0.001; Qu et al., 2016; Figure S3D), resulting in high gene expression correlation coefficients (R = 0.8967; p < 0.0001) compared to D7 cells (Figure 3F). Second, chromatin accessibility changes measured by principal-component analysis (PCA) between ATAC-seq signals from TFAP2C overexpressed cells (hereafter “TetO-TFAP2C-D7 Dox+”) at day 7 and the different stages of RA/BMP differentiation (Figure 3H) were highly correlated with open chromatin signatures between TetO-TFAP2C-D7 Dox+ and D7 cells (R = 0.7714; p < 0.0001; Figure 3G) or gene ontology (GO) enrichment terms (Figures 3I and S4F). Further analysis shows increased ATAC-seq signals around the transcription start site (TSS) in surface ectodermal genes genome-wide (Figure S4E) and at individual key loci, such as K8-K18 and the initiation network TF GATA3 (Figures 3E and 3J).
Importantly, TFAP2C not only induces the expression of TFs in the initiation network but also activates their binding sites. TF motif enrichment within the differential accessible regions (Figures 3K, S4G, and S4H) and foot printing analysis using protein interaction quantification (PIQ) software (Sherwood et al., 2014; Figure S4I) revealed a statistically significant increase in the binding of such initiation network TFs, including the AP-2 family (AP-2gamma and AP-2alpha), followed by TEAD family (TEAD4 and TEAD1), GATA family (GATA3 and GATA2), GRHL family (GRHL2), and bZIP family (JUN). The most closed sites are for TFs responsible for ESC maintenance or other lineage specification, such as SOX family (Sox3 and Sox2), Lhx family (Lhx2 and Lhx3), and Nanog (Figure 3K). TFAP2C action could be direct or indirect through inducing expression of initiation network TFs that could in turn open chromatin binding sites. To distinguish between these possibilities, we performed TFAP2C ChIP-seq analysis in D7 TFAP2C overexpressed cells and identified ~10,000 TFAP2C binding sites that are predominantly located in intergenic and intronic regions at a distance from the gene TSS (Figures S4J–S4L). Consistent with a direct role, we found increased chromatin accessibility around TFAP2C binding sites upon TFAP2C overexpression (Figure 3L). In addition, TFAP2C-bound regions have a higher enrichment of active histone marks, such as H3K4m1, H3K27ac, and H3K4m3 (Figure 3M). Collectively, these results support the conclusion that TFAP2C binds to active regulatory regions to increase genome-wide chromatin accessibility associated with surface ectoderm lineage commitment.
TFAP2C-Induced Surface Ectoderm Cells Produce Functional Keratinocytes
Because TFAP2C induces surface ectodermal differentiation, we determined whether the committed progenitor cells had the potential to develop into mature keratinocytes. TFAP2C-induced progenitor cells (hereafter “TetO-TFAP2C-D7”) grown in keratinocyte maturation medium (Figure 4A) possessed a 5-fold-higher yield of keratinocyte colonies (hereafter “TetOTFAP2C-KC”) than RA/BMP-induced keratinocytes (Figures 4B and 4C). In addition, fluorescence-activated cell sorting (FACS) (Figure S5A) shows that the majority of TetO-TFAP2C-KC cells are K14+K18 (95%), a pattern similar to that in NHK (K14+K18; 95%). Like foreskin and RA/BMP-induced keratinocytes, TetO-TFAP2C-KC cells respond to high calcium stimulation, which results in cellular structural changes and increased expression of stratification markers (Figures S5B and S5C), and were able to form all stratified layers in organotypic cultures (Figure 4D), indicative of the ability to create a stratified epidermal tissue.
Figure 4. TFAP2C-Induced Surface Ectoderm Progenitors Are Competent to Produce Functional Keratinocytes in Maturation Medium.

(A) Schematic diagram shows the experimental procedure to study whether TFAP2C-induced surface ectoderm progenitor cells (TetO-TFAP2C-D7) can further differentiate into functional keratinocytes (TetO-TFAP2C-KC) in maturation medium.
(B) Gene expression changes in late differentiation by comparing TetO-TFAP2C-KC versus TetO-TFAP2C-D7. qRT-PCR values were normalized to the values inTetO-TFAP2C-D7. Mean ± SD is shown (n = 3; **p < 0.01; t test).
(C) IF staining of selected markers in TFAP2C-induced keratinocytes. Scale bar, 25 μm.
(D) Reconstruction of stratified epidermis with TFAP2C-induced keratinocytes. IF staining of selected markers is shown. Scale bar, 25 μm.
(E) Hierarchical clustering of TetO-TFAP2C-KC and samples from normal differentiation using chromatin accessibility similarities from ATAC-seq analysis. Color denotes the three major clusters.
(F and G) Scatterplot of read counts from ATAC-seq (F) and of gene expression from RNA-seq (G) in TFAP2C-induced keratinocytes versus keratinocytes from normal differentiation with Pearson correlation (R) value displayed, respectively.
(H) Scatterplot of differential accessibility in TetO-TFAP2C-KC versus TetO-TFAP2C-D7 cells.
(I) TF motif enrichment in the regions with decreased (left) or increased (right) chromatin accessibility in TetO-TFAP2C-KC versus TetO-TFAP2C-D7. TF motifsfrom one family are labeled with the same color and are indicated in the top corner.
(J) ATAC-seq tracks show decreased chromatin accessibility at K8-K18 locus and increased accessibility at K14 locus in TFAP2C-induced mature keratinocytes (TetO-TFAP2C-KC) versus early progenitor cells (TetO-TFAP2C-D7).
See also Figure S5
Hierarchical clustering of ATAC-seq signals from RA/BMP-induced differentiation and TetO-TFAP2C-KC revealed that the chromatin landscape of TetO-TFAP2C-KC is close to that of late differentiated cells (D43 and H9KC) and distinct from cells at earlier stages (Figure 4E). ATAC-seq accessibility (R = 0.8736; p < 0.0001; Figure 4F) and RNA-seq signatures (R = 0.8252; p < 0.0001; Figure 4G) are highly correlated (Figures 4H, 4I, S5D, and S5E), and PIQ analysis reveals a statistically significant increase in the binding of most maturation network TFs in TetO-TFAP2C-KC (Figure S5F). Significantly, we found a decrease of ATAC-seq signal in the regulatory regions of K8-K18 locus and a strong increase in K14 locus in TetO-TFAP2CKC versus TetO-TFAP2C-D7 (Figure 4J). We conclude that TFAP2C-induced surface ectodermal progenitors are competent to further differentiate into functional keratinocytes, which is accomplished by the transition of the chromatin landscape from the initiation to the maturation network.
TFAP2C Requires p63 to Drive Keratinocyte Maturation
As the TetO-TFAP2C-KCs showed a decrease in initiation network TFs and an increase in the maturation network TFs (Figures 2C, 4I, S3C, and S5F), we functionally interrogated the maturation-associated TF required to mature cells into keratinocytes. Although we assayed other maturation network TFs, like KLF4 (Figures S6B–S6F), p63 solely demonstrated the ability to both induce keratinocyte maturation and repress portions of the initiation landscape. We knocked out the p63 gene in TetO-TFAP2C cells by CRISPR/Cas9 and evaluated its effect on TFAP2C-induced differentiation (Figure 5A). TetO-TFAP2C+ p63KO cells with Dox displayed typical epithelial morphology (D7; Figure 5B) and high gene expression of surface ectoderm markers by immunofluorescence (IF) and qRT-PCR (Figures 5C and 5D), indicating that p63 is not necessary for TFAP2C-induced early initiation. By contrast, when exposed to epidermal maturation media, TetO-TFAP2C+p63KO cells underwent apoptosis rather than keratinocyte maturation (D50; Figure 5B), demonstrating p63-dependent survival. Moreover, mutant cells exhibited persistent surface ectoderm markers and lack of mature keratinocyte markers (Figures 5E and 5F), further confirming the p63 dependence of the maturation process.
Figure 5. p63 Is Necessary for Keratinocyte Maturation during TFAP2C-Induced Epidermal Differentiation.

(A) Schematic illustration of the approach to functionally study p63 via CRISPR/Cas9-mediated gene knockout (KO) during TFAP2C-induced epidermal differentiation.
(B) Morphological changes in TFAP2C-induced epidermal differentiation upon p63 deletion. Scale bar, 200 μm.
(C and D) p63 loss of function does not affect surface ectoderm initiation at early differentiation.
(C) IF staining of the cells at D7. Scale bar, 50 mm.
(D) qRT-PCR shows increased expression of surface ectoderm markers in TetO-TFAP2C+p63KO cells upon Dox induction. qRT-PCR values were normalized tothe values of internal control GAPDH.
(E and F) p63 loss of function results in failure of keratinocyte maturation at late stage of differentiation.
(E) IF staining of the cells with genotypes labeled as TetO-TFAP2C (left) and TetO-TFAP2C+p63KO (right) at D21. Nuclei were stained by Hoechst. Scale bar, 40 μm.
(F) qRT-PCR shows higher expression level of surface ectoderm markers and lower level of mature keratinocyte markers upon p63 KO at D21. qRT-PCR values were normalized to the values from control cells (TetO-TFAP2C). In (D) and (F), mean ± SD is shown (n = 3; *p < 0.05; **p < 0.01; t test).
(G) Schematic illustration of the analysis of chromatin changes upon p63 KO at D21. The TetO-TFAP2C-D7(K8 and K18+)- and TetO-TFAP2C-KC(K5 and K14+)-specific accessible regions were utilized as the representative features of “progenitor” and “mature KC,” respectively, to evaluate the differentiation status of the cells at D21 with and without p63 (i.e., TetO-TFAP2C versus TetO-TFAP2C+p63KO).
(H) Hierarchical clustering of the ATAC-seq signals shows a close relationship between TetO-TFAP2C-D21 and TetO-TFAP2C-KC, and loss of p63 arrests cells at a more immature stage between TetO-TFAP2C-D7 and TetO-TFAP2C-KC.
(I) Loss of p63 results in a higher level of chromatin accessibility at initiation-stage-specific peaks and a lower level of accessibility at maturation-stage-specific peaks. Histogram shows ATAC-seq read counts distribution from the above two peak regions in TetO-TFAP2C versus TetO-TFAP2C+p63KO at D21.
(J) Genome browser tracks comparing ATAC-seq signal in TetO-TFAP2C versus TetO-TFAP2C+p63KO at K8-K18 and K5 loci.
See also Figure S6
To analyze p63-dependent gene expression and chromatin accessibility changes during the transition to the maturation network, we collected D21 cells, the time point at which we showed cells were capable of becoming keratinocytes (Figure 4), and checked gene expression and chromatin accessibility changes in TetO-TFAP2C+p63KO versus TetO-TFAP2C (Figure 5G). Hierarchical clustering shows that D21 TetO-TFAP2C (i.e., TetO-TFAP2C-D21), but not p63 mutant TetO-TFAP2C cells (i.e., TetO-TFAP2C+p63KO-D21), cluster with mature KC (TetOTFAP2C-KC), and p63 knockout (KO) cells cluster with cells from time points between TetO-TFAP2C-D7 and TetO-TFAP2C-KC (Figure 5H). This supports an arrest of the TFAP2C-driven differentiation process in the absence of p63. Next, we took stage-specific accessible REs from TetO-TFAP2C-D7 and TetO-TFAP2C-KC as the features for epidermal progenitor and mature KCs and evaluated their openness level in differentiated cells at D21. Consistent with arrested differentiation, greater read counts appeared in progenitor-associated, rather than keratinocyte-associated, elements in p63 KO cells (TetO-TFAP2C+ p63KO) compared to control cells (TetO-TFAP2C; Figure 5I). In addition, we found significantly more overlap with progenitor elements than mature KC elements in the peak regions highly accessible in TetO-TFAP2C+p63KO than TetO-TFAP2C (Figure S6A). To confirm such findings, we examined the local chromatin status at the loci of K8-K18 and K5, representative genes for epidermal progenitor and mature KCs, respectively, and found higher ATAC-seq signals around K8-K18 and fewer signals around K5 in TetO-TFAP2C+p63KO versus TetO-TFAP2C at D21 (Figure 5J). Therefore, we conclude that loss of p63 arrests the TFAP2C-driven transition from progenitor program to mature KC program.
Feedback Regulation between p63 and TFAP2C Drives the Epigenetic Transition
In the transition from an TFAP2-initiated landscape to a p63-matured landscape, TFAP2C levels initially increase with concomitant increases in p63 but then drop precipitously during maturation as p63 expression levels become TFAP2 independent (Figure 6A). To understand the mechanism of this transition, we first studied the transcriptional regulation of TFAP2C on the well-studied p63 locus (encoded by TP63). Previous murine and human ectodermal dysplasia patient studies demonstrate a critical RE that mediates p63-positive autoregulation, known as the C40 enhancer (Figure 6B; Antonini et al., 2015). Intriguingly, we found the p63 locus inaccessible in ESCs and D7 TetO-TFAP2C inducible cells in the absence of Dox. By contrast, addition of Dox at D7 opened several sites within the p63 locus, including the C40 enhancer (Figure 6B). In addition, TFAP2C ChIP-seq in TetO-TFAP2C D7 cells revealed several binding sites on the p63 locus (Figure 6B) and a marked increase in the ATAC-seq read counts above p63 binding sites (Figure 6C). To interrogate TFAP2C action on p63 binding sites genome-wide, we examined the chromatin status of p63 binding sites in keratinocytes (Zarnegar et al., 2012) and found that an overwhelming majority of the mature p63 binding sites that are closed in TetO-TFAP2C Dox- become more accessible when TFAP2C is overexpressed at D7 (Figure 6C). We conclude that TFAP2C turns on expression of endogenous p63 and increases chromatin accessibility surrounding p63 binding sites, allowing p63-dependent epidermal lineage maturation. By contrast, TFAP2C locus and its binding site accessibility declined during maturation (Figures S7A and S7B), suggesting a reciprocal regulation between the two factors.
Figure 6. Feedback Regulation between p63 and TFAP2C Drives Chromatin Transition from Progenitor to Mature Keratinocytes.

(A) Gene expression changes of TFAP2C and p63 during TFAP2C-induced epidermal differentiation.
(B) Genome browser tracks show ATAC-seq signal in TetO-TFAP2C cells ± Dox induction, relative to TFAP2C ChIP-seq (black bar highlights the peak region) fromD7 Dox-induced differentiation at TP63 locus. Note: two isoform types (full-length and deltaN-p63) are schematically shown. There are multiple regions gaining accessibility signal at TP63 locus upon Dox induction, including the p63 self-activation enhancer: C40 enhancer (black bar annotated).
(C) p63 binding sites become more accessible in TFAP2C-induced surface ectoderm progenitor cells. Histograms of read counts distribution (left) and average signal enrichment (right) of ATAC-seq in p63-bound regions in cells with and without TFAP2C induction are shown.
(D) Schematic illustration of identification of TF motifs associated with chromatin accessibility changes upon p63 loss of function.
(E) (Left) Scatterplot of differential accessibility in TetO-TFAP2C+p63KO versus TetO-TFAP2C at D21. (Right) Top enriched TF motifs identified from differential accessible regions are shown. TF motif name, TF family, and p values are presented.
(F) PIQ footprinting analysis indicates a lower likelihood of TF occupancy of p63 and a higher likelihood of TFAP2C in p63 KO cells (TetO-TFAP2C+p63KO). Significant difference with ****p < 0.0001 relative to control (TetO-TFAP2C) was determined by t test.
(G) A model diagram shows TFAP2C/p63 negative feedback regulation and p63 self-activation during epidermal lineage commitment.
(H) Heatmap shows chromatin accessibility changes within p63-bound regions during TFAP2C-induced epidermal differentiation. K-means clustering identifies three groups of changes (labeled as “a, b, and c” with respective numbers).
(I) Top enriched GO terms identified from the three clusters shown in (H).
(J and K) Comparison of ATAC-seq signal between TetO-TFAP2C+p63KO versus TetO-TFAP2C in the three groups of p63 binding sites. The heatmap (J) and average enrichment (K) of signal are shown.
(L) Heatmap shows chromatin accessibility changes within TFAP2C-bound regions during TFAP2C-induced epidermal differentiation. K-means clustering identifies two groups of chromatin changes (labeled as a and b with respective numbers).
(M) Top enriched GO terms identified from the two clusters in (L).
(N) Comparison of ATAC-seq signal changes between TetO-TFAP2+p63KO versus TetO-TFAP2C in the two groups of TFAP2C binding sites. The heatmap (N) and average enrichment (O) of signal are shown.
See also Figure S7
If p63 controls the maturation TF network, then loss of p63 at D21 should result in the reduction in the accessibility of the other maturation-associated TFs and a persistence of the initiation-associated TF binding sites. Indeed, analysis on differential accessibility peaks in TetO-TFAP2C+p63KO versus TetO-TFAP2C reveals that motifs for initiation TFs (AP-2gamma, AP-2alpha, GATA3, and TEAD) were enriched in higher accessible regions (Figures 6D–6F), and motifs for CTCF, BORIS, p63, p53, RFX, and KLF4 were enriched in lower accessible regions, along with a decreased occupancy probability (Figures 6D–6F and S6G) upon p63 deletion; this confirms the reciprocal feedback regulation between TFAP2C and p63 during late-stage differentiation (Figure 6G).
To identify which sets of genes are subjected to TFAP2C/p63 feedback regulation, we analyzed the p63 and TFAP2C ChIP-seq data, chromatin accessibility, and gene expression at TetO-TFAP2C-D7 Dox−, Dox+, and TetO-TFAP2C-KC during TFAP2C-induced keratinocyte differentiation. The inference model predicts the existence of at least three types of p63 binding sites: a, sites unchanged by TFAP2C or p63; b, sites opened by TFAP2C that stay open independent of p63; and c, sites opened by TFAP2C and opened further by elevated p63 protein levels. Indeed, analysis of the chromatin accessibility changes at p63 binding sites revealed the predicted three groups of regions (a, b, and c; Figures 6H, 6I, and S7C). Group a p63 binding sites are located in promoter-proximal regulatory regions of metabolic genes and are constitutively open in all three samples, and group b sites are located in both proximal and distal regions and depend only on early TFAP2C for accessibility. By contrast, group c sites are keratinocyte maturation genes containing GO terms “programmed cell death,” “response to wounding,” “skin development,” and “epidermis development.” Although they demonstrate a small increase in accessibility with TFAP2C expression, there is a strong dependence on high-level p63 expression to achieve the increased accessibility seen in mature keratinocytes. These findings indicate that key keratinocyte maturation genes are “primed” early by TFAP2C and then become fully accessible through increased p63 expression during maturation. A test of this prediction is that, in TetO-TFAP2C+p63KO cells at D21, group c sites should fail to open as TFAP2C levels fall and p63 is null. Indeed, although group a and b sites show no change in accessibility in the absence of p63, group c sites demonstrate decreased accessibility in the absence of p63 (Figures 6J and 6K).
Reciprocal regulation by p63 of TFAP2C D7 binding site accessibility illuminates p63 feedback regulation on a subset of the TFAP2C network. We identified two groups of TFAP2C binding sites (Figures 6L, 6M, and S7D): group a TFAP2C sites consist of metabolic genes that are located in both proximal and distal regulatory regions, and group b sites consist of genes with GO terms “response to hypoxia,” “Notch signaling pathway,” and “epithelial-to-mesenchymal transition” and are located in distal regions. In both groups, binding site accessibility increases with TFAP2C expression and decreases with decreasing TFAP2C levels in late differentiation (Figure 6L). Consistent with p63-negative feedback regulation of TFAP2C, the accessible signal of both groups increases upon p63 deletion, with group b demonstrating much greater sensitivity to p63 levels (Figures 6N and 6O). We conclude that p63 increases accessibility of key epidermal genes as its levels increase while shutting down the accessibility of a subset of TFAP2C binding sites associated with simple epithelium.
DISCUSSION
In this study, we use paired temporal chromatin accessibility and transcriptome data from hESCs undergoing epidermal differentiation to identify two distinct TF-chromatin regulatory networks responsible for epidermal lineage initiation and maturation. Through functional validation, we make the surprising finding that TFAP2C acts as the “initiation factor,” which induces the surface ectoderm chromatin landscape on which p63 subsequently acts as the “maturation factor,” modifying the prepatterned landscape into that of functional keratinocytes. Replicated in three distinct PSC lines, the two-step, feedforward autoregulation and negative feedback regulatory mechanisms between initiation and maturation factors provide an important and general logic to understand the chromatin dynamics of tissue lineage commitment (Figure 7).
Figure 7. A Model of Epigenomic Regulation during Epidermal Lineage Commitment.

A proposed model depicts the identified chromatin states and feedback regulation between TFAP2C- and p63-centered TF regulatory networks driving the chromatin transition during epidermal lineage commitment.
Our network model provides several advantages to understand lineage commitment at the genome-wide level. It integrates matched chromatin accessibility, TF binding, gene expression, and histone modifications into a “TF- accessible RE-TG” triplet, greatly facilitating accurate inference of gene regulatory relations. In addition, our approach incorporates publicly available chromatin connection data that allow accurate assignment of dynamically changing distal enhancer accessible sites to their TGs. However, we anticipate the incorporation of newer conformation technologies, such as HiChIP (Mumbach et al., 2016), will improve the coverage of RE and TG association and deepen the inferred TF network.
Gain and loss of functional studies reveal the surprising finding that the early and late networks can be collapsed into the actions of TFAP2C and p63. Previous studies have revealed important functions of TFAP2C in various biological processes, such as trophectodermal development (Auman et al., 2002) and breast cancer progression (Woodfield et al., 2010). In skin development, loss of Tfap2c results in disrupted epidermal gene expression and delayed skin development (Qiao et al., 2012), demonstrating an essential role in epidermal lineage commitment. Our ATAC-seq and ChIP-seq data support the hypothesis that TFAP2C acts directly on chromatin to alter chromatin accessibility, consistent with previous reports of TFAP2 family members possessing pioneer factor characteristics (Pihlajamaa et al., 2014). By contrast, TFAP2C in other systems requires chromatin regulators that act as co-activators or co-repressors in TFAP2C-mediated gene regulation (Braganca et al., 2003; Wong et al., 2012), leading us to speculate that TFAP2C might recruit specific chromatin-associated factors, such as p300/CBP or MLL/Set1. Although our work shows the necessity and sufficiency for TFAP2C to induce the initiation TF network, potential cross regulatory interactions with TFAP2C targets like GATA3 or GRHL2 prevent us from excluding a role for the network TFs in modulating or synergizing with TFAP2C to affect the ultimate composition of the dynamic chromatin landscape (Figures S4M and S4N). This includes a role for other TFAP2 family members, which share similar motif and cross-regulation of each other’s loci and can homodimerize with each other, complicating a straightforward genetic epistasis analysis.
A major insight of this work is the demonstration that TFAP2C prepares the underlying chromatin landscape for p63-dependent keratinocyte maturation. Our work tempers the long-held view that p63 is the master regulator of keratinocyte differentiation and places p63 as a critical but dependent partner to the TFAP2C-modified landscape. p63 plays a central role inthe development of a wide variety of ectodermal and endodermal-derived epithelia, including epidermal development, mammary, lung, and prostate (Crum and McKeon, 2010), but remarkably, loss of p63 in TFAP2C-induced surface ectoderm differentiation (Figures 5B–5D) or forced expression in hPSCs (Medawar et al., 2008) does not affect differentiation. Our results explain how a single lineage selector like p63 can direct such a panoply of transcriptional programs during development, as it is expressed in the context of distinct preestablished chromatin landscapes. The genomic context on which p63 acts ultimately determines the final differentiation product and suggests that small changes in diffusible morphogens RA/BMP could dramatically alter TFAP2C levels, the set of enhancer sites that are p63 sensitive (group c; Figure 6H), and the gene expression profile of the epidermis generated.
A surprising revelation from the network modeling was that p63 drives maturation both by positive autoregulation of its own locus and by inhibition of the immature surface epithelial landscape. In response to maturation medium, p63 levels are dramatically boosted by self-activation from the p63 C40 enhancer (Antonini et al., 2015), facilitating the activation of keratinocyte-specific targets. By contrast, p63 reduces the level and binding site accessibility of TFAP2C in at least two ways. First, p63 represses transcription of TFAP2C itself. We have identified a p63 binding site distal to the TSS of TFAP2C, and p63 inhibits expression through looping with this TSS (Figure S7A; unpublished data). Second, p63 antagonizes a subset of TFAP2C TG activated during initiation (Figures 6L–6O). Interestingly, TFAP2C remains required at particular loci for mature keratinocyte function (McDade et al., 2012), arguing that p63 action must repress only a subset of potential targets and that the balance between TFAP2C and p63 networks determines the nature of the mature end product.
Taken together, our work provides a general analysis strategy to depict epigenetic transition during lineage commitment through TF-chromatin network modeling and reveals an essential regulatory principle between lineage initiation and maturation factors to ensure faithful tissue generation. Such findings will bring more insight in understanding the logic of somatic tissue development and improve the assessment of stem cell competency and differentiation efficiency in tissue engineering and regenerative medicine.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to the Lead Contact, Anthony Oro (oro@stanford.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human ESC and iPSC culture
Human ESCs H9 (XX) and HUES6 (XX) and modified cell lines were seeded in a feeder-free system using Matrigel hESC-Qualified Matrix (BD Corning) and were maintained in Essential 8 media (Life Technologies). Cells were passaged every 3 days as clumps with 0.5 mM EDTA. The iPSC line (sex information is not available) was derived by sendai virus vector-mediated reprogramming in human fibroblast from Invitrogen (Macarthur et al., 2012). The iPSCs were cultured on mitomycin-treated CF1 Mouse Embryonic Fibroblasts (MEFs, Millipore, PMEF-CFL) in W8 medium (for making 250ml medium: DMEM/F12 Knockout medium 200ml, Knockout Serum Replacement 50ml, 100X L-Glutamine 1.25ml, 100X Non-Essential Amino Acids 2.5ml, 100X Beta-Mercaptoethanol 2.5ml, 10 mg/ml Basic FGF 200 μl).
METHOD DETAILS
Epidermal differentiation assays
In vitro differentiation of hESC and iPSC into keratinocyte
H9 hESCs were passed on to Matrigel coated plates and maintained in Essential 8 media. To induce differentiation into keratinocytes, the cells were first fed with “differentiation medium 1” containing 5 ng/ml BMP4 and 1 μM RA in Essential 6 media (Life Technologies) for seven days, thus forming surface ectodermal progenitor cells. After seven days, the medium was changed to “differentiation medium 2,” i.e., Defined Keratinocyte Serum Free Medium (DKSFM) with growth supplements containing EGF and FGF (Life Technologies), and the cells underwent selection and expansion for 2 months for maturation into keratinocytes. The resulting keratinocyte colonies were passed onto Corning PureCoat ECM Mimetic 6-well Collagen I Peptide Plate (Corning) and expanded in DKSFM medium.
The study of keratinocyte differentiation has been reproduced in two other pluripotent cell lines: HUES6 hESCs and iPSCs. For HUES6 hESCs: the cells were maintained and differentiated into keratinocytes using the same protocol for H9 hESC differentiation. The iPSC line was derived by sendai virus vector-mediated reprogramming in human fibroblast from Invitrogen (Macarthur et al., 2012). The cells were differentiated into keratinocytes using a different protocol revised from Sebastiano et al., 2014. Before differentiation, iPSCs were passaged onto CELLstart (Life Technologies, a xeno-free and defined coating matrix)-coated dishes in medium conditioned with MEFs for two passages. Then the iPSCs were formed into embryoid bodies, using AggreWell 400 plates and AggreWell medium (STEMCELL Technologies) supplemented with 1mg/ml RA and 10 mM ROCK inhibitor (STEMCELL Technologies) for 24 hours. Embryoid bodies were collected and cultured in suspension for 2 days and then plated onto gelatin-coated dishes in FAD medium (3:1 mixture of Dulbecco’s modified Eagle’s medium [DMEM] and Ham’s F12 media, 10% fetal calf serum, 5 mg/ml insulin, 0.5 mg/ml hydrocortisone, 10−10 mol/l cholera toxin, 1.37 ng/mL triiodothyronine, 24 μg/ml adenine, and 10 ng/ml recombinant human epidermal growth factor) for 4 days and then in N2 medium (DMEM/F12 supplemented with 1X N2 supplement [Invitrogen]) for 3 days. During this 7-day differentiation, the media were supplemented with RA (1mg/ml) and human recombinant BMP4 (25 ng/ml). The medium was then changed to DKSFM (Life Technologies), and the cells underwent selection and expansion for 2 months. The resulting keratinocyte colonies were passed onto Corning PureCoat ECM Mimetic 6-well Collagen I Peptide Plate (Corning) and expanded in DKSFM medium. The differentiation experiments were repeated multiple times (N > 5) with the cell lines at different passages by independent investigators. In each batch of differentiation, the cells were plated in over three individual wells/plates and were verified by morphological and gene expression criteria (IF and qRT-PCR) before being subject to study further.
Doxycycline (Dox) inducible TFAP2C (TetO-TFAP2C) human ESCs
To generate Doxycycline inducible expression cell lines, we used the PiggyBac Doxycycline inducible plasmid (PB/TW/CRB, courtesy of Yamanaka lab), which contains a CAG promoter driven rtTA-IRES-BSD cassette and pTRE (tetracycline response element) driven transgene expression cassette. Human TFAP2C cDNA (Sino Biological Inc. Cat. No. HG13115-G) was amplified and subcloned into the Piggybac plasmid (named as “PB-CRB-TFAP2C”) using In-Fusion HD Cloning Kit (Clontech). PB-CRB-TFAP2C expression plasmid and Transposase expression plasmid (System Biosciences) were mixed and co-transfected into H9 hESCs with Lipofectamine LTX Reagent with PLUS Reagent (Thermo Fisher Scientific,15338100) by standard protocol. Forty-eight hours after transfection, the cells were selected by blasticidin (4 mg/mL) and resistant ES clones were picked and expanded. TFAP2C induction was confirmed by IF and qRT-PCR after Dox treatment over 2 days.
In vitro differentiation of TetO-TFAP2C H9 hESCs
To study the function of TFAP2C in surface ectoderm differentiation, TetO-TFAP2C H9 hESCs were induced with 2 μg/ml Dox in E6 medium without RA/BMP4 for 7 days. The resulting cells were imaged and collected for subsequent analysis. The differentiation experiments were repeated over three times, and there are more than three individual plates of cells from each group (i.e., Dox+ and Dox−) in each batch.
To study long term epidermal differentiation ability, TetO-TFAP2C H9 hESCs were first induced into surface ectodermal cells by activation of TFAP2C expression with Dox in E6 medium for 7 days, and then were transitioned to DKSFM with Dox for 14 days and maintained in DKSFM-only until keratinocyte colonies were formed. The differentiated keratinocytes were passed and maintained onto Corning PureCoat ECM Mimetic 6-well Collagen I Peptide Plate (Corning) in DKSFM, and collected for additional analysis. This long-term differentiation experiment was repeated in multiple batches (n > 5) with at least three replicate wells, and only the wells containing well-formed keratinocyte colonies were chosen for the further study.
Gene Knockout and Knockdown in ESCs
CRISPR/Cas9-mediated targeted gene deletion/replacement via homologous recombination in hESCs
We used CRISPR/Cas9-mediated genome editing to generate our gene knockout hESC lines. We designed two gRNAs to target independent loci surrounding a 5 kb genomic region that we wished to delete. Donor sequences with 700 bp arms flanking left and right of the region to be deleted were also used to promote homologous recombination at the locus. Knockouts were designed to mimic the null mouse models, whereby the conserved DNA binding domains were targeted for deletion: for TFAP2C – exons 4–7; for p63 – exons 6–8 (more detail is shown in Pattison et al., 2018). Both gRNAs and donor sequences were synthesized as 5’-phosphorylated gene blocks (IDT), and the gRNAs were incorporated into a DNA fragment with all components necessary for gRNA expression. Constructs were transfected into the hESC cells alongside an hCas9 expression plasmid, using Lipofectamine LTX Reagent with PLUS Reagent (Thermo Fisher Scientific,15338100) and the recommended manufacturer’s protocol. After 48 hours, we added puromycin to the cells to select out positive cells for gRNA expression. hESC colonies were picked and expanded individually and knockouts were verified by genomic PCR, western blotting, and immunofluorescence. The sequences of gRNA are as follows:
TFAP2C-KO gRNA1: ATGCTTAAATGCCTCGTTAC
TFAP2C-KO gRNA2: ACAGAACCTCCACGGGGACT
p63-KO gRNA1: TAGTCATTTGATTCGAGTAG
p63-KO gRNA2: GTAAAGCTGTAGTACATGCC
Small interfering RNA (siRNA)-mediated gene knockdown in ESCs
To knockdown the expression level of TFAP2C, H9 hESCs were plated onto 6 well plates and were transfected with 30 pmol of siRNAs (Sigma-Aldrich) using Lipofectamine RNAiMAX Reagent (Invitrogen) with standard protocol. The siRNA oligonucleotides were designed, synthesized and fluorescent labeled by Sigma-Aldrich. To achieve higher knockdown efficiency and avoid off-target effect, we initially screened five individual siRNAs from different sites and chose the top two for the subsequent study. The transfection experiment was performed over three times, and the knockdown efficiency was measured by qRT-PCR. Only the samples containing successful target gene knockdown were used for additional gene expression analysis. In each batch of the experiment, there are two target gene siRNA and one control siRNA oligos transfected in three replicate wells. The sequences of siRNAs are as follows:
TFAP2C siRNA #1:sense (5’−3’): GUAAACCAGUGGCAGAAUA[dT][dT];
antisense (5’−3’): UAUUCUGCCACUGGUUUAC[dT][dT][Cyanine5];
TFAP2C siRNA #2: sense (5’−3’): CACAGAACUUCUCAGCCAA[dT][dT];
antisense (5’−3’): UUGGCUGAGAAGUUCUGUG[dT][dT][Cyanine5]
Control siRNA:
siRNA Fluorescent Universal Negative Control #1, Cyanine 5 (cat. No. SIC005–10NMOL, Sigma-Aldrich)
Short hairpin RNA (shRNA)-mediated gene knockdown in cells from hESC differentiation
To knockdown the expression level of KLF4 during epidermal differentiation, the TetO-TFAP2C H9 hESCs were induced into surface ectodermal cells by 7-days Dox treatment, and then were infected with lentivirus from a pGFP-C-shLenti vector (Origene, cat. No.TL316853) containing two cassettes for shRNA of KLF4 and GFP expression under different promoters. At differentiation day 21, infected cells were sorted based on GFP intensity and were subjected to expression analysis. Lentiviral particles were produced, and the cells were infected according to the manual from Origene (HUSH shRNA plasmids and lenti-particles application guide). We tested the knockdown efficiency in undifferentiated H9 hESCs by qRT-PCR and chose the top two from five candidate shRNAs for further differentiation study. The infection and differentiation experiments were repeated three times with one control, and two target gene shRNAs in two replicate wells. The sequences of shRNA are as follows:
KLF4 shRNA#1 (5’−3’): TGAGGCAGCCACCTGGCGAGTCTGACATG
KLF4 shRNA#2 (5’−3’): TCAGATGAACTGACCAGGCACTACCGTAA
Control shRNA: Scrambled negative control non-effective shRNA cassette in pGFP-C-shLenti plasmid (Cat. No. TR30021, Origene).
Assays for RNA and protein expression
RNA expression analysis
Total RNA from cells was extracted with the RNeasy mini kit (QIAGEN). Gene expression was measured using the One-Step RT-PCR SYBR green kit (Stratagene) according to manufacturer’s instructions and normalized to the internal control gene GAPDH. The qRT-PCR assay was performed with samples from multiple repeating experiments (three or more than three times). In each batch, three replicates were used for expression analysis. The qRT-PCR primer list is shown in Table S4.
Immunofluorescence staining
Cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100 in PBS for 30 min, and blocked with 10% horse serum (Vector Laboratories) in 0.2% Triton ×−100 in PBS for 30 min at room temperature. Cells were incubated overnight at 4C with primary antibodies. Primary antibodies are: K18 (1:800, R&D AF7619), K14 (1:800, BioLegend SIG-3476–100); AP-2γ (1:100, Cell Signaling 2320), p63 (1:100 Gene Tex GTX102425), ITGA6 (1:200, Millipore, MAB1378). Then the cells were incubated with Alexa 488, 555 and 647-conjugated secondary antibodies diluted with 0.2% Triton X-100 in PBS (1:500, Life Technologies) for 1 hour at room temperature. Following three washes with PBS, slides were mounted with the Prolong Gold mounting medium (Life Technologies). Prior to mounting, slides were incubated with 1:10000 Hoechst for 10 min. The fluorescence images were taken using the TCS SP2 confocal laser scanning microscope (Leica). In this assay, the cell culture was performed over three times and subsequent cell staining was done by different investigators. For each slides with same antibodies staining, the images were randomly taken from 5–10 individual regions using same parameters and the representative one was shown. Slides stained only with secondary antibodies were used as a control to determine the false positive signal.
Intracellular staining and FACS analysis
Cells were dissociated and washed twice using FACS buffer (2% BSA /PBS), then stained with Fc blocker (anti-mouse CD16/CD32) and live/dead Aqua 30min on ice. After washing, cells were then fixed and permeabilized with BD reagent (BD cytofix/cytoperm kit, Cat #554714). Subsequently, fluorophore conjugated antibodies: anti-K14 (FITC, CBL197F, Millipore), anti-K18 (PerCP, NB120–7797, Novus) antibodies or isotype control were incubated with cells at 4C for 30min (1:100 dilution). After washing, the samples were applied for FACS analysis. In this study, the TetO-TFAP2C differentiation assay were repeated three times, and samples at different stages were collected and subject to FACS analysis by different investigators without knowing detailed sample information. All three groups of samples (i.e., TetO-TFAP2C-D7, TetO-TFAP2C-KC and NHK) were stained and FACS analyzed with same parameters.
Functional analysis of ES-derived keratinocytes
Calcium induced differentiation
To mimic the stratification process in 2-D culture system, keratinocytes derived from TetO-TFAP2C H9 hESCs were treated with 1.2mM CaCl2 in the medium for 3 or 6 days at a full confluence. The cells at each time points were imaged by light microscope and lysed for qRT-PCR analysis on skin stratification associated genes. This treatment assay was repeated three times, and each batch of samples contain three replicates for qRT-PCR analysis. Light microscope images were taken randomly from different areas of the plate (n > 5), and the representative one is shown.
In vitro skin reconstitution assay
Generation of organotypic epidermis was performed by following the protocol described previously (Sebastiano et al., 2014) with minor modification. In brief, 1×106 keratinocytes derived from normal differentiation with H9 hESC or from TFAP2C-induced differentiation were collected and seeded on top of devitalized dermis and cultured submerged in DKSF medium for 5 days. The medium was then gradually changed to Keratinocyte Growth Medium (KGM, Lonza) for 7 days, after which stratification was induced by raising the dermis to air-liquid interface. After 2 weeks, the reconstituted epidermis was collected, fixed in 4% paraformaldehyde, and embedded in optimum cutting temperature compound (OCT) and paraffin for IF staining. The primary antibodies are: K14(1:2000, COVANCE, PRB-155P), K10 (1:500, COVANCE, PRB-159P), Loricrin (1:500, Covance, PRB-145P), Collagen VII (1:250, Millipore MAB1345). The differentiation experiments with H9 hESCs or TetO-TFAP2C cells were repeated over five times. The plates containing keratinocyte colonies of high quality were chosen for skin reconstitution assay. The reconstitution assay was repeated over 3 times, and each repeat assay included 2–3 replicates depending on the total number of collected keratinocyte. The slides were pre-examined by morphology and H&E staining to make sure the skin structure was normal and then were subjected to IF staining. Sections from adjacent regions were stained and compared. For each slide with the same antibodies staining, the images were randomly taken from 5–10 individual areas using the same parameters, and the representative one was shown. Slides stained only with secondary antibodies were used as a control to determine the false positive signal.
RNA-seq, ChIP-seq and ATAC-seq
RNA-seq library preparation and data processing
RNA extraction was performed using RNeasy Plus (QIAGEN) from the samples reported in this research. Ribosomal RNA was removed from each RNA extraction using Ribo-Zero Gold rRNA Removal kit (Illumina). The RNA-seq libraries were constructed by TruSeq Stranded mRNA Library Prep kit (Illumina) and sequenced on Illumina Hiseq2000 or NextSeq sequencers. Sequencing reads were mapped to hg19 using TopHat. FPKM values were called using Cufflinks. For the time series dataset, the genes having a FPKM value of 0 across all time points were filtered out. Differential gene expression was performed by looking at log2 fold change > 1. For the TFAP2C samples, differential expression was performed using Cuffdiff. Differentially expressed genes have a p value < 0.01 as well as a log2 fold change > 1. The RNA-seq data are from two biological samples. We confirmed the sample quality by verifying the representative gene expression by qRT-PCR and IF staining from the same batch of differentiation experiments (n > 3).
ChIP-seq library preparation and data processing
ChIP assay was performed following previously described method (Calo et al., 2015) with minor modification. Briefly, TetO-TFAP2C H9 hESCs were induced with Dox for 7 days, after which cells were crosslinked in 1% formaldehyde for 10 min, followed by quenching with 0.125M glycine for 5min at room temperature (RT). Chromatin was sheared to an average 100–300bp in the Covaris sonicator (Covaris). Sonicated chromatin solution was aliquoted and incubated overnight at 4°C with the specific antibodies. The antibodies used are as follows: AP-2γ (H-77) (sc-8977X, Santa Cruz); H3K4m3 (39159, Active Motif); H3K4m1 (ab8895, Abcam); H3K27ac (39133, Active Motif); H3K27m3 (39155, Active Motif); H3K9m3 (ab8898, Abcam). For TFAP2C ChIP-seq, we used chromatin extracted from 30 million cells for each experiment. For histone ChIP-seq, we used 10 million cells for each experiment. Immunocomplexes were captured by protein-G Dynal magnetic beads (Life Technologies) followed by stringent washes and elution. The eluted samples were reverse cross-linked at 65C overnight, and were digested by RNase A at 37°C for 2 hours and proteinase K at 55C for 30 min to deplete the RNA and proteins. The DNA was purified by QIAquick PCR purification Kit (QIAGEN). ChIP-seq libraries were prepared following the NEBNext protocol and sequenced on Illumina NextSeq sequencers. Sequencing reads were mapped to hg19 using Bowtie1.1.2 (Langmead et al., 2009) with parameters –best,–strata and –m 1 to allow for only one alignment. Duplicates are then removed using Samtools rmdup (http://samtools.sourceforge.net/). Peaks were identified using MACS1.4 (Zhang et al., 2008) with an FDR threshold 0.01. The ChIP-seq deep sequencing data are derived from two biological samples. Each library was sequenced initially on a MiSeq sequencer to determine the quality. Only libraries containing higher signal-to-noise ratio were chosen for deeper sequencing.
ATAC-Seq Library preparation, sequencing, and data preprocessing
The ATAC-seq was performed as described (Buenrostro et al., 2013). Briefly, 50,000 cells were collected and washed with cold PBS and lysed using 0.1% NP40 in resuspension buffer. Tn5 transposition of nuclei pellets was carried out at 37°C for 30 min, using the DNA Sample Preparation Kit from Nextera (Illumina). The reaction was purified using QIAGEN MinElute columns and then amplified for 8–15 cycles to produce libraries for sequencing. ATAC-seq libraries were sequenced on Illumina Hiseq2000 or NextSeq sequencers. ATAC-Seq pair-end reads were trimmed for Illumina adaptor sequences and transposase sequences using a customized script and mapped to hg19 using Bowtie v0.12.9 (Langmead et al., 2009) with parameters –S –X2000 –m1. Duplicate reads were discarded with Samtools v0.1.18. Peaks were identified using HOTSPOT with default parameters (https://github.com/rthurman/hotspot). HOTSPOT analysis generates two types of peaks: narrow peak and hotspot regions (broad peak). In this study, we used the narrow peaks for all the analysis. Overlapping peaks from all samples were merged into a unique peak list, and raw read counts mapped to each peak [using bedtools multicov (https://bedtools.readthedocs.io/en/latest/)] for each individual sample were quantified. The samples for ATAC-seq were initially examined by expression analysis (qRT-PCR and IF staining) from the same batch of differentiation (n > 3). In one batch, we made multiple libraries (n > = 4) for two biological samples (aliquoting every 50,000 cells for each library). The libraries were initially sequenced on a Miseq sequencer and analyzed using a custom script to determine the enrichment score by calculating the ratio of signal over background at TSS over a 2-kb window. Only libraries that had the highest score above the threshold (> 5) were chosen for deeper sequencing. Two independent, biological replicates were sequenced per sample.
Deep sequencing data analysis
Irreproducible Discovery Rate (IDR) analysis
IDR (https://bioconductor.org/packages/release/bioc/html/edgeR.html) analysis was performed on ATAC-seq and ChIP-seq peaks. In both cases, IDR was called on the p value of the original peaks from two biological samples.
Differential ATAC-seq peak analysis
Differential peak analysis in paired samples: Differentially accessible peaks from the merged union peak list were selected with the edgeR package from Bioconductor using raw read counts of each samples in the union peak list using a log2 fold change > 1, and p value < 0.01.
Differential peak analysis in time-series ATAC-seq from normal differentiation: Peaks with significantly changing signal were selected using the edgeR package from Bioconductor. All dispersions were estimated. An ANOVA-like test was conducted by specifying all pairwise contrasts with an arbitrarily selected time point, relative to a 1.5-fold change, and a significance threshold of FDR 0.05 was used. The read counts of the differential peaks in each sample were further normalized by Z-score transformation. Hierarchical clustering was used to cluster the peaks and samples. The result was presented as a heatmap by Java TreeView.
Genomic annotation of peaks from ATAC-seq or ChIP-seq
Peak genomic annotation was performed by a customized script assigning peaks to specific regions using the following criteria: “promoter”: regions from 2kb to 1kb of transcription start sites (TSS); “TSS-proximal enhancer”: regions from 10kb to 2kb of TSS; “gene tail”: regions from 2kb to transcription end sites; “exon” and “intron” regions in the gene body and defined in Refseq annotation; “intergenic”: any other genomic regions.
Gene ontology analysis
Gene ontology enrichment analysis was performed using GREAT (McLean et al., 2010) to calculate statistics by associating genomic regions with nearby genes (here we chose the parameter: “two nearest genes within 100kb”) and applying the gene annotations to the regions.
Motif analysis of peaks from ATAC-seq and ChIP-seq
Motif analysis on peak regions was performed using HOMER function (http://homer.ucsd.edu/homer/motif/) “findMotifsGenome.pl” with default parameters to calculate the occurrence of a TF motif in peak regions compared to that in background regions. We used “−log10 (p-value)” to rank the enrichment level of TF motifs.
PIQ footprinting analysis
We used PIQ, a machine-learning algorithm (Sherwood et al., 2014), to identify the TF binding footprints at corresponding motifs from ATAC-seq data. PIQ outputs the purity scores to evaluate the probability of true TF binding. In this study, we investigated the binding activity of a list of specific TFs in several group comparisons. For each TF, the motif position weight matrix (PWM) were from JASPAR database (http://jaspar.genereg.net) and HOMER (http://homer.ucsd.edu/homer/motif/). To maximize the detection sensitivity, we merged the ATAC-seq reads from two replicates. To eliminate the detection bias, we sampled from the higher-depth merged library to achieve equal depth for next-step analysis if the sequencing depth was quite different among the samples from one group of comparison. After running PIQ algorithm, the purity score of one given TF from different samples were compared by paired t test.
ATAC-seq signal intensity around TSS
A window from –2k upstream to +2k downstream of TSS was taken and divided into eighty 50bp equally sized bins. The number of uniquely mapped and properly paired ATAC-seq tags overlapping each bin was counted by a customized script. The average fragment count in each bin of all the TSS regions was plotted.
ATAC-seq signal intensity around transcription factor bound regions
TF ChIP-seq data were either generated in this study or download from public resource. In each TF analysis, a window from 1k upstream to +1k downstream of ChIP-seq peak summits was taken and divided into forty 50bp equally sized bins. The number of uniquely mapped and properly paired ATAC-seq tags overlapping each bin was counted, normalized by library size and log-transformed using an in-house script. Therefore, we generated a matrix with each row representing a peak region, and each column containing the normalized tag counts from a 50 bp bin in a consecutive manner within the 2kb window. To visualize the signal intensity across all the TF bound regions, the data matrix was presented as a heatmap by Java TreeView. To get an average intensity, we took the mean of fragment count in each bin of all the binding regions and plotted the result as a line graph or heatmap. To compare the intensity among different samples, we merged the individual data matrix from each group into one single file, and performed k-means clustering on peak regions and presented the result in a heatmap. Each cluster of peak regions was taken and compared among all the samples. The associated GO terms and distance to TSS were analyzed based on each cluster of peak regions.
Gene set enrichment analysis (GSEA)
GSEA was performed to determine whether a priori defined set of genes shows statistically significant difference between biological samples. In this study, we generated a gene set of surface ectoderm from previous research (Qu et al., 2016). Gene expression values from the RNA-seq analysis of the samples “TetO-TFAP2C Dox+” and “TetO-TFAP2C Dox-” were used for comparison. GSEA was performed following the developer’s protocol (https://www.broadinstitute.org/gsea/). The normalized enrichment score, nominal p value, and FDR q value are presented to determine the significance of enrichment level.
TF-Chromatin Transcriptional Regulatory Networks
We initially selected key TFs by integrating expression and motif enrichment, then predicted target genes for ATAC-seq union peaks using publicly available chromosome conformation data and co-occurrence information of distal and promoter DNase I hypersensitive sites across diverse cell types from ENCODE, and finally performed TF-RE-TG triplet inference modeling with multiple investigated features. For details on algorithm and parameters used for generating TF regulatory networks, see Methods S1: The computational method for TF-chromatin transcriptional regulatory networks, Related to STAR Methods.
QUANTIFICATION AND STATISTICAL ANALYSIS
All Data represent similar results from three or above independent, biological samples and cell cultures, unless otherwise described. Deep sequencing data are from two biological samples, except NHK RNA-seq from one biological sample. Bar chart results were presented as the mean with SD. Student’s t test was used to determine the significance of differences with the annotations: *p < 0.05, **p < 0.01 and **** p < 0.0001. Details of statistical methods for specific analysis are descried in the corresponding methods sections.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Antibodies | |||||||||||||
| K14 antibody for IF (section) | Covance | Cat #PRB-155P RRID:AB_292096 | |||||||||||
| K10 antibody for IF (section) | Covance | Cat #PRB-159P RRID:AB_291580 | |||||||||||
| Loricrin antibody for IF (section) | Covance | Cat #PRB-145P RRID:AB_292095 | |||||||||||
| Collagen VII antibody for IF (section) | Millipore | Cat #MAB1345 RRID:AB_11210494 | |||||||||||
| Anti-K14-FITC antibody for FACS | Millipore | Cat #CBL197F RRID:AB_11212136 | |||||||||||
| Anti-K18-FITC antibody for FACS | Novus | Cat #NB120–7797 RRID:AB_2133302 | |||||||||||
| Purified anti-human CD16 antibody for FACS | BioLegend | Cat #302002 RRID:AB_314202 | |||||||||||
| Purified anti-human CD32 antibody for FACS | BioLegend | Cat #303202 RRID:AB_314334 | |||||||||||
| K14 antibody for IF | BioLegend | Cat #SIG-3476–100 RRID:AB_10718041 | |||||||||||
| K18 antibody for IF | R&D | Cat #AF7619 RRID: N/A | |||||||||||
| AP-2γ antibody for IF | Cell Signaling | Cat #2320S RRID:AB_10695101 | |||||||||||
| p63 antibody for IF | GeneTex | Cat #GTX102425 RRID:AB_1952344 | |||||||||||
| ITGA6 antibody for IF | Millipore | Cat #MAB1378 RRID:AB_11210794 | |||||||||||
| TFAP2C antibody for ChIP | Santa Cruz | Cat #sc-8977X RRID:AB_2286995 | |||||||||||
| H3K4m3 antibody for ChIP | Active Motif | Cat #39159 RRID:AB_2615077 | |||||||||||
| H3K4m1 antibody for ChIP | Abcam | Cat #ab8895 RRID:AB_306847 | |||||||||||
| H3K27ac antibody for ChIP | Active Motif | Cat #39133 RRID:AB_2561016 | |||||||||||
| H3K27m3 antibody for ChIP | Active Motif | Cat #39155 RRID:AB_2561020 | |||||||||||
| H3K9m3 antibody for ChIP | Abcam | Cat #ab8898 RRID:AB_306848 | |||||||||||
| Chemicals, Peptides, and Recombinant Proteins | |||||||||||||
| BMP4 | R&D Systems | Cat #314-BP-050 | |||||||||||
| RA | Sigma | Cat #R2625 | |||||||||||
| Doxycycline | Sigma | Cat #D9891 | |||||||||||
| Blasticidin | ThermoFisher | Cat #A1113903 | |||||||||||
| 16% Formaldehyde solution | ThermoFisher | Cat #28906 | |||||||||||
| Essential 8 (E8) Medium | Life Technologies | Cat #A1517001 | |||||||||||
| Essential 6 (E6) Medium | Life Technologies | Cat #A1516401 | |||||||||||
| Defined Keratinocyte-SFM | Life Technologies | Cat #10744–019 | |||||||||||
| AggreWell EB Formation Medium | StemCell Technologies | Cat #05893 | |||||||||||
| Knockout DMEM/F12 | GIBCO/invitrogen | Cat #12660–012 | |||||||||||
| Knockout Serum Replacement | GIBCO/invitrogen | Cat #10828–028 | |||||||||||
| MEM Non-Essential Amino Acids Solution 10 mM (100X) | GIBCO/invitrogen | Cat #11140050 | |||||||||||
| EmbryoMax® ES Cell Qualified 2-Mercaptoethanol (100X) | Millipore | Cat #ES-007-E | |||||||||||
| L-Glutamine-200mM 200X | GIBCO/invitrogen | Cat #25030–081 | |||||||||||
| KGM Keratinocyte Growth Medium | Lonza | Cat #CC-3111 | |||||||||||
| Corning PureCoat ECM Mimetic 6-well Collagen I Peptide Plate |
Corning | Cat #356270 | |||||||||||
| BD Matrigel hESC-qualified Matrix | Fisher | Cat #354277 | |||||||||||
| CELLstart Substrate | Life Technologies | Cat #A1014201 | |||||||||||
| recombinant human fibroblast growth factor-2 | PeproTech | Cat #100–18B | |||||||||||
| Y-27632 ROCK Inhibitor | StemCell Technologies | Cat #07172 | |||||||||||
| Normal Horse Serum Blocking Solution | Vector Laboratories | Cat #S-2000 | |||||||||||
| ProLong Gold Antifade Mountant | Life Technologies | Cat #P36930 | |||||||||||
| Fisher Healthcare Tissue-Plus O.C.T. Compound | FisherScientific | Cat #23–730-571 | |||||||||||
| Mitomycin C | Sigma | Cat #M4287–2MG | |||||||||||
| HyClone FetalClone II Serum | ThermoScientific | Cat #SH30066.03 | |||||||||||
| Insulin | Sigma | Cat #I5500–100MG | |||||||||||
| Hydrocortisone 21-hemisuccinate sodium salt | Sigma | Cat #H2270 | |||||||||||
| Cholera Toxin | Calbiochem | Cat #227035 | |||||||||||
| 3,3’,5-Triiodo-L-Thyronine Sodium Salt | Sigma | Cat #T2752–100MG | |||||||||||
| Adenine Hydrochloride | Sigma | Cat #A9795–1g | |||||||||||
| Human Recombinant Epidermal Growth Factor | Sigma | Cat #E9644 | |||||||||||
| Critical Commercial Assays | |||||||||||||
| TruSeq Stranded mRNA Library Prep kit | Illumina | Cat #20020594 | |||||||||||
| Ribo-Zero Gold rRNA Removal kit | Illumina | Cat #MRZG12324 | |||||||||||
| NEBNext ChIP-Seq Library Prep kit | NEB | Cat #E6240S/L | |||||||||||
| Nextera DNA Library Prep Kit | Illumina | Cat #FC-121–1030 | |||||||||||
| Lipofectamine LTX Reagent with PLUS Reagent | ThermoFisher Scientific | Cat #15338100 | |||||||||||
| Lipofectamine RNAiMAX Reagent | ThermoFisher Scientific | Cat #13778075 | |||||||||||
| In-Fusion HD Cloning Kit | Clontech | Cat #639650 | |||||||||||
| One-Step RT-PCR SYBR green kit | Stratagene | Cat #600826 | |||||||||||
| RNeasy mini kit | QIAGEN | Cat #74106 | |||||||||||
| QIAquick PCR purification Kit | QIAGEN | Cat #28106 | |||||||||||
| protein-G Dynal magnetic beads | Life Technologies | Cat #10004D | |||||||||||
| Fixation/Permeabilization Solution Kit | BD | Cat #554714 | |||||||||||
| Live/dead Fixable Aqua Dead cell stain kit | ThermoFisher Scientific | Cat #L34966 | |||||||||||
| Deposited Data | |||||||||||||
| Deep sequencing data | This study | GEO: GSE108248 | |||||||||||
| Experimental Models: Cell Lines | |||||||||||||
| H9 (WA09) hESC line | Stanford Stem Cell Bank | NIHhESC-10–0062 | |||||||||||
| HUES6 hESC line | HSCI iPS Core | NIHhESC-09–0019 | |||||||||||
| iPSC | invitrogen | Macarthur et al., 2012 | |||||||||||
| CF1 Mouse Embryonic Fibroblast | Millipore | PMEF-CFL | |||||||||||
| Oligonucleotides | |||||||||||||
| Real time qRT-PCR primers are listed in Table S4. | This study | N/A | |||||||||||
| Oligonucleotides for sgRNA for CRISPR-Cas9 | This study | N/A | |||||||||||
| TFAP2C-KO gRNA1 | This study | ATGCTTAAATGCCTCGTTAC | |||||||||||
| TFAP2C-KO gRNA2 | This study | ACAGAACCTCCACGGGGACT | |||||||||||
| p63-KO gRNA1 | This study | TAGTCATTTGATTCGAGTAG | |||||||||||
| p63-KO gRNA2 | This study | GTAAAGCTGTAGTACATGCC | |||||||||||
| Oligonucleotides for siRNA | This study | N/A | |||||||||||
| TFAP2C siRNA #1: sense (50−30) | This Study | GUAAACCAGUGGCAGAAUA[dT][dT] | |||||||||||
| TFAP2C siRNA #1: antisense (50−30) | This Study | UAUUCUGCCACUGGUUUAC[dT][dT][Cyanine5] | |||||||||||
| TFAP2C siRNA #2: sense (50−30): | This Study | CACAGAACUUCUCAGCCAA[dT][dT]; | |||||||||||
| TFAP2C siRNA #2: antisense (50−30): | This Study | UUGGCUGAGAAGUUCUGUG[dT][dT][Cyanine5] | |||||||||||
| siRNA Fluorescent Universal Negative Control #1, Cyanine 5 | Sigma | Cat #SIC005–10NMOL | |||||||||||
| Oligonucleotides for shRNA | This study | N/A | |||||||||||
| KLF4 shRNA#1 (50−30) | This Study | TGAGGCAGCCACCTGGCGAGTCTGACATG | |||||||||||
| KLF4 shRNA#2 (50−30) | This Study | tcagatgaactgaccaggcactaccgtaa | |||||||||||
| Scrambled negative control non-effective shRNA | Origene | Cat #TR30021 | |||||||||||
| Recombinant DNA | |||||||||||||
| PiggyBac Doxycycline inducible plasmid (PB/TW/CRB) | courtesy of Yamanaka lab | N/A | |||||||||||
| Transposase expression plasmid | System Biosciences | Cat #PB210PA-1 | |||||||||||
| Human TFAP2C cDNA | Sino Biological Inc. | Cat #HG13115-G | |||||||||||
| PB-CRB-TFAP2C | This Study | N/A | |||||||||||
| PB-CRB-GATA3 | This Study | N/A | |||||||||||
| PB-CRB-GRHL2 | This Study | N/A | |||||||||||
| PB-CRB-KLF4 | This Study | N/A | |||||||||||
| Transposase expression plasmid | System Biosciences | Cat #PB210PA-1 | |||||||||||
| hCas9 expression plasmid | Addgene | Cat #41815 | |||||||||||
| KLF4 Human shRNA Plasmid Kit | Origene | Cat #TL316853 | |||||||||||
| Non-effective 29-mer Scrambled shRNA Cassette in pGFP-C-shLenti Vector | Origene | Cat #TR30021 | |||||||||||
| Software and Algorithms | |||||||||||||
| Tophat 2.1.0 | Kim et al., 2013 | https://ccb.jhu.edu/software/tophat/index.shtml | |||||||||||
| Cufflinks 2.2.1 | Trapnell et al., 2010 | https://github.com/cole-trapnell-lab/cufflinks | |||||||||||
| PIQ | Sherwood et al., 2014 | http://piq.csail.mit.edu/ | |||||||||||
| GREAT | McLean et al., 2010 | http://great.stanford.edu/public/html/ | |||||||||||
| EdgeR | Robinson et al., 2010 | https://bioconductor.org/packages/release/ bioc/html/edgeR.html | |||||||||||
| MACS | Zhang et al., 2008 | https://github.com/taoliu/MACS | |||||||||||
| Bowtie | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml | |||||||||||
| StepMiner | Sahoo et al., 2007 | http://genedesk.ucsd.edu/home/public/StepMiner/ | |||||||||||
| Other | |||||||||||||
| OCT4 ChIP-seq | ENCODE | GEO: GSM803438 | |||||||||||
| p63 ChIP-seq | Zarnegar et al., 2012 | GEO: GSE33571 | |||||||||||
| NHEK Hi-C | Rao et al., 2014 | GEO: GSE63525 | |||||||||||
| DNase co-accessibility correlation across tissues | Thurman et al., 2012 | Table S7 in Thurman et al., 2012 | |||||||||||
Highlights.
Epigenome landscape maps reveal key transitions during epidermal lineage commitment
Network modeling identifies master regulators of lineage initiation and maturation
TFAP2C drives chromatin dynamics during initiation and primes p63-dependent maturation
Crosstalk between TFAP2C and p63 drives epigenetic transitions during differentiation
ACKNOWLEDGMENTS
We thank S. Yamanaka for sharing PiggyBac inducible expression plasmid and E. Bashkirova, M. Ameen, S. Tao, and K. Qu for technical support. We thank P. Khavari, A. Christiano, E. Fuchs, and Q. Luo for pre-submission review and other Oro and Chang laboratory members for discussion. This work was supported by NIH grants P50-HG007735 (to H.Y.C. and W.H.W.), R01GM109836 (to W.H.W.), and F32AR070565 (to J.M.P.); by California Institute for Regenerative Medicine tools grant RT3–07796 (to A.E.O.); by EB Research Partnership (to A.E.O.); by National Natural Science Foundation of China grants 61671444, 61621003, and 91730301 (to Y.W.); and by Strategic Priority Research Program of the Chinese Academy of Sciences XDB13000000 (to Y.W.). H.Y.C. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures, four tables, and one methods file and can be found with this article online at https://doi.org/10.1016/j.stem.2018.12.012.
DECLARATION OF INTERESTS
H.Y.C. is co-founder of Accent Therapeutics and an advisor to 10X Genomics and Spring Discovery.
DATA AND SOFTWARE AVAILABILITY
The accession number for deep sequencing data generated in this paper is GEO: GSE108248. Other publicly available deep sequencing datasets used in this paper are listed in the Key Resources Table.
REFERENCES
- Antonini D, Sirico A, Aberdam E, Ambrosio R, Campanile C, Fagoonee S, Altruda F, Aberdam D, Brissette JL, and Missero C. (2015). A composite enhancer regulates p63 gene expression in epidermal morphogenesis and in keratinocyte differentiation by multiple mechanisms. Nucleic Acids Res. 43, 862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auman HJ, Nottoli T, Lakiza O, Winger Q, Donaldson S, and Williams T. (2002). Transcription factor AP-2gamma is essential in the extra-embryonic lineages for early postimplantation development. Development 129, 2733–2747. [DOI] [PubMed] [Google Scholar]
- Bragança J, Eloranta JJ, Bamforth SD, Ibbitt JC, Hurst HC, and Bhattacharya S. (2003). Physical and functional interactions among AP-2 transcription factors, p300/CREB-binding protein, and CITED2. J. Biol. Chem. 278, 16021–16029. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, Flynn RA, Martin L, Spitale RC, Chang HY, and Wysocka J. (2015). RNA helicase DDX21 coordinates transcription and ribosomal RNA processing. Nature 518, 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crum CP, and McKeon FD (2010). p63 in epithelial survival, germ cell surveillance, and neoplasia. Annu. Rev. Pathol. 5, 349–371. [DOI] [PubMed] [Google Scholar]
- Duren Z, Chen X, Jiang R, Wang Y, and Wong WH (2017). Modeling gene regulation from paired expression and chromatin accessibility data. Proc. Natl. Acad. Sci. USA 114, E4914–E4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster MI, and Roop DR (2007). Mechanisms regulating epithelial stratification. Annu. Rev. Cell Dev. Biol. 23, 93–113. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Liu C, Biechele S, Zhu Q, Song L, Lanner F, Jing N, and Rossant J. (2013). Location of transient ectodermal progenitor potential in mouse development. Development 140, 4533–4543. [DOI] [PubMed] [Google Scholar]
- Macarthur CC, Fontes A, Ravinder N, Kuninger D, Kaur J, Bailey M, Taliana A, Vemuri MC, and Lieu PT (2012). Generation of human-induced pluripotent stem cells by a nonintegrating RNA Sendai virus vector in feeder-free or xeno-free conditions. Stem Cells Int. 2012, 564612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDade SS, Henry AE, Pivato GP, Kozarewa I, Mitsopoulos C, Fenwick K, Assiotis I, Hakas J, Zvelebil M, Orr N, et al. (2012). Genome-wide analysis of p63 binding sites identifies AP-2 factors as co-regulators of epidermal differentiation. Nucleic Acids Res. 40, 7190–7206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G. (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medawar A, Virolle T, Rostagno P, de la Forest-Divonne S, Gambaro K, Rouleau M, and Aberdam D. (2008). DeltaNp63 is essential for epidermal commitment of embryonic stem cells. PLoS ONE 3, e3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metallo CM, Ji L, de Pablo JJ, and Palecek SP (2008). Retinoic acid and bone morphogenetic protein signaling synergize to efficiently direct epithelial differentiation of human embryonic stem cells. Stem Cells 26, 372–380. [DOI] [PubMed] [Google Scholar]
- Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, and Bradley A. (1999). p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 398, 708–713. [DOI] [PubMed] [Google Scholar]
- Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, and Chang HY (2016). HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 13, 919–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattison JM, Melo SP, Piekos SN, Torkelson JL, Bashkirova E, Mumbach MR, Rajasingh C, Zhen HH, Li L, Liaw E, et al. (2018). Retinoic acid and BMP4 cooperate with p63 to alter chromatin dynamics during surface epithelial commitment. Nat. Genet. 50, 1658–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihlajamaa P, Sahu B, Lyly L, Aittomäki V, Hautaniemi S, and Jänne OA. (2014). Tissue-specific pioneer factors associate with androgen receptor cistromes and transcription programs. EMBO J. 33, 312–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Y, Zhu Y, Sheng N, Chen J, Tao R, Zhu Q, Zhang T, Qian C, and Jing N. (2012). AP2g regulates neural and epidermal development downstream of the BMP pathway at early stages of ectodermal patterning. Cell Res. 22, 1546–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, Zhou B, Yang W, Han B, Yu-Rice Y, Gao B, Johnson J, Svendsen CN, Freeman MR, Giuliano AE, et al. (2016). Transcriptome and proteome characterization of surface ectoderm cells differentiated from human iPSCs. Sci. Rep. 6, 32007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, and Aiden EL (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo D, Dill DL, Tibshirani R, and Plevritis SK (2007). Extracting binary signals from microarray time-course data. Nucleic Acids Res. 35, 3705–3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastiano V, Zhen HH, Haddad B, Bashkirova E, Melo SP, Wang P, Leung TL, Siprashvili Z, Tichy A, Li J, et al. (2014). Human COL7A1-corrected induced pluripotent stem cells for the treatment of recessive dystrophic epidermolysis bullosa. Sci. Transl. Med. 6, 264ra163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood RI, Hashimoto T, O’Donnell CW, Lewis S, Barkal AA, van Hoff JP, Karun V, Jaakkola T, and Gifford DK (2014). Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nat. Biotechnol. 32, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. (2012). The accessible chromatin landscape of the human genome. Nature 489, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L. (2010). Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umegaki-Arao N, Pasmooij AM, Itoh M, Cerise JE, Guo Z, Levy B, Gostynski A, Rothman LR, Jonkman MF, and Christiano AM (2014). Induced pluripotent stem cells from human revertant keratinocytes for the treatment of epidermolysis bullosa. Sci. Transl. Med. 6, 264ra164. [DOI] [PubMed] [Google Scholar]
- Wenzel D, Bayerl J, Nyströ m A, Bruckner-Tuderman L, Meixner A, and Penninger JM (2014). Genetically corrected iPSCs as cell therapy for recessive dystrophic epidermolysis bullosa. Sci. Transl. Med. 6, 264ra165. [DOI] [PubMed] [Google Scholar]
- Wong PP, Miranda F, Chan KV, Berlato C, Hurst HC, and Scibetta AG (2012). Histone demethylase KDM5B collaborates with TFAP2C and Myc to repress the cell cycle inhibitor p21(cip) (CDKN1A). Mol. Cell. Biol. 32, 1633–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodfield GW, Chen Y, Bair TB, Domann FE, and Weigel RJ (2010). Identification of primary gene targets of TFAP2C in hormone responsive breast carcinoma cells. Genes Chromosomes Cancer 49, 948–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, Tabin C, Sharpe A, Caput D, Crum C, and McKeon F. (1999). p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 398, 714–718. [DOI] [PubMed] [Google Scholar]
- Zarnegar BJ, Webster DE, Lopez-Pajares V, Vander Stoep Hunt B, Qu K, Yan KJ, Berk DR, Sen GL, and Khavari PA (2012). Genomic profiling of a human organotypic model of AEC syndrome reveals ZNF750 as an essential downstream target of mutant TP63. Am. J. Hum. Genet. 91, 435–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Modelbased analysis of ChIP-seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.