

Abstract
Exogenous and endogenous chemicals can react with DNA to produce DNA lesions that may block DNA replication. Not much is known about the roles of polymerase (Pol) ν and Pol θ in translesion synthesis (TLS) in cells. Here we examined the functions of these two polymerases in bypassing major-groove O6-alkyl-2′-deoxyguanosine (O6-alkyl-dG) and minor-groove N2-alkyl-dG lesions in human cells, where the alkyl groups are ethyl, n-butyl (nBu), and, for O6-alkyl-dG, pyridyloxobutyl. We found that Pol ν and Pol θ promote TLS across major-groove O6-alkyl-dG lesions. O6-alkyl-dG lesions mainly induced G→A mutations that were modulated by the two TLS polymerases and the structures of the alkyl groups. Simultaneous ablation of Pol ν and Pol θ resulted in diminished mutation frequencies for all three O6-alkyl-dG lesions. Depletion of Pol ν alone reduced mutations only for O6-nBu-dG, and sole loss of Pol θ attenuated the mutation rates for O6-nBu-dG and O6-pyridyloxobutyl-dG. Replication across the two N2-alkyl-dG lesions was error-free, and Pol ν and Pol θ were dispensable for their replicative bypass. Together, our results provide critical knowledge about the involvement of Pol ν and Pol θ in bypassing alkylated guanine lesions in human cells.
Keywords: DNA damage, DNA polymerase, DNA replication, MS, mutagenesis, mutagenesis mechanism, DNA alkylation, polymerase ν, polymerase θ
Introduction
As the carrier of genetic information, DNA's chemical integrity has to be maintained for normal cellular function. Cells are continuously exposed to exogenous and endogenous genotoxic agents that give rise to numerous DNA lesions on a daily basis (1). Although cells have multiple repair mechanisms to remove DNA damage, some lesions escape from repair and can be encountered by DNA replication machinery (2). The nuclear replicative polymerases (Pol2 α, Pol δ, and Pol ϵ) have evolved to replicate undamaged DNA templates with high accuracy and processivity; DNA synthesis mediated by these polymerases is, however, often hindered by DNA lesions. To overcome replication blockage, cells are equipped with translesion synthesis (TLS) Pols to assist replication across DNA lesions (3).
Several DNA polymerases have been shown to be capable of promoting TLS (4). In this vein, the fidelities and efficiencies of B- and Y-family TLS Pols in bypassing various DNA lesions have been studied extensively over the past two decades. Although some TLS Pols can efficiently and accurately bypass specific DNA lesions (5, 6), this damage tolerance mechanism is often error-prone (7, 8), which may confer increased risks of mutagenesis and carcinogenesis (9). For instance, human Pol η has been shown to accurately and efficiently bypass the cyclobutane thymine dimer (10); nevertheless, its replication across other DNA lesions (e.g. O2-alkyl-dT, O4-alkyl-dT, and O6-alkyl-dG) is inaccurate (11–13). Another Y-family DNA polymerase, Pol κ, is involved in error-free bypass of minor-groove N2-alkyl-dG DNA adducts (14–17), but it catalyzes mutagenic bypass of many other DNA lesions (18).
Recent studies also documented the roles of Pol ν and Pol θ, two A-family polymerases, in DNA damage tolerance and repair (19–26). Thymine glycol (Tg) is one of the most prevalent oxidation products of thymine, and this lesion strongly blocks DNA synthesis (27). Biochemical studies revealed that Pol ν and Pol θ can elicit error-free and mutagenic TLS across Tg, respectively (22, 23). In addition, recombinant human Pol ν exhibits robust TLS activity across O6-methyl-2′-deoxyguanosine with reasonably high fidelity (24), and Pol θ–mediated TLS has been shown to safeguard against UV light–induced pyrimidine(6-4)pyrimidone photoproducts and DNA double-strand breaks (25, 26). Apart from TLS, Pol ν participates in interstrand cross-link repair (21), and Pol θ–induced microhomology-mediated end joining provides an alternative pathway to repair DNA double-strand breaks (28). However, the roles of these polymerases in bypassing alkylated DNA lesions in human cells remain poorly explored.
In this study, we set out to examine the involvement of Pol ν and Pol θ in TLS across major-groove O6-alkyl-dG and minor-groove N2-alkyl-dG lesions in human cells (Fig. 1). We observed that these two polymerases modulate the efficiency and fidelity of replication past the O6-alkyl-dG lesions; absence of these two polymerases, however, does not affect TLS of N2-alkyl-dG adducts.
Figure 1.
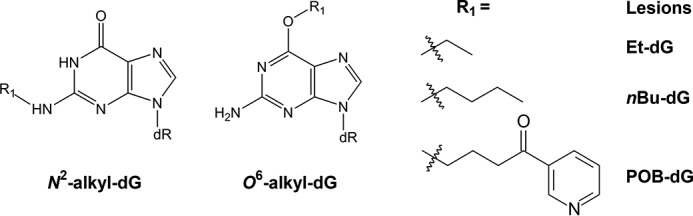
The O6- and N2-alkyl-dG lesions investigated in this study. dR, 2-deoxyribose.
Results
The O6 and N2 positions of dG are susceptible to alkylation by various environmental agents and endogenous metabolic byproducts (29, 30). In this study, we set out to explore the roles of two under-investigated A-family DNA polymerases, Pol ν and Pol θ, in promoting TLS across major-groove O6-alkyl-dG and minor-groove N2-alkyl-dG lesions in human cells. Because our previous results showed that the inhibitory effects of DNA lesions on replication are significantly influenced by adduct size (13, 31), we chose the ethyl (Et) and n-butyl (nBu) adducts for the investigation. In addition, a previous biochemical study suggested an inability of Pol ν to bypass the bulky O6-POB-dG lesion in vitro (24); hence, we also included O6-POB-dG in this study.
We employed the strand-specific PCR-competitive replication and adduct bypass (SSPCR-CRAB) assay to determine how the efficiencies and fidelities of replication past the alkylated guanine lesions are modulated by CRISPR-mediated genetic ablation of Pol ν and/or Pol θ in human cells (Fig. S1). In the SSPCR-CRAB assay, the forward P1 primer contains a G as the terminal 3′ nucleotide matching the C/C mismatch locus, which is used to specifically amplify the progeny genomes from the replication of the initial lesion-situated strand. Furthermore, a C/A mismatch is incorporated in the P1 primer at the third nucleotide from its 3′ end to afford better specificity for SSPCR, as described elsewhere (32, 33). The PCR amplicons were subsequently digested with two restriction enzymes, and the ensuing restriction fragments containing the initial damage site were interrogated by denaturing PAGE and LC-MS/MS to determine the identities and quantities of the replication products (Figs. 2 and 3 and Figs. S2–S11).
Figure 2.

Restriction digestion and post-labeling method for determining the bypass efficiency and mutation frequency of O6- and N2-alkyl-dG lesions in HEK293T cells and isogenic TLS polymerase-deficient cells. A, schematic showing restriction digestion of the PCR amplicon with NcoI and SfaNI and the post-labeling assay. B, representative gel image showing the NcoI/SfaNI-produced restriction fragments of interest in PCR products of progeny genomes of the indicated lesion- or control dG-containing plasmids isolated from HEK293T cells. The restriction fragment arising from the competitor vector, 5′-CATGGCGATATGCTGT-3′, is designated 16-mer. 13-mer A, 13-mer G, 13-mer T, and 13-mer C denote the standard synthetic ODNs 5′-CATGGCGMGCTGT-3′, where M is A, G, T, and C, respectively. p* indicates a 32P-labeled phosphate group.
Figure 3.

LC-MS/MS for identification of restriction digestion products (with NcoI and SfaNI) of PCR products of the progeny genomes arising from replication of the O6-nBu-dG–containing plasmid in Pol ν and Pol θ double-knockout cells. A, higher-resolution ultra-zoom scan ESI-MS results for the 13-mer restriction fragments. B–D, MS/MS for monitoring fragmentations of the [M-3H]3− ions of 5′-CATGGCGGGCTGT-3′ (WT product), 5′-CATGGCGAGCTGT-3′ (with G→A mutation), and 5′-CATGGCGTGCTGT-3′ (with G→T mutation).
The roles of Pol ν and Pol θ in bypassing O6-alkyl-dG lesions
Our PAGE analysis results revealed that both O6-nBu-dG and O6-POB-dG impede DNA replication in HEK293T cells, with the bypass efficiencies being 43.3% and 74.4%, respectively (Figs. 2B and 4A and Table S1). The smaller O6-Et-dG, however, does not appreciably inhibit DNA replication (Figs. 2B and 4A and Table S1). Genetic ablation of Pol ν attenuates the bypass efficiency (BE) of all the three O6-alkyl-dG lesions. Removal of Pol θ induces similar reductions in BE for O6-Et-dG and O6-nBu-dG as depletion of Pol ν (Fig. 4A and Fig. S2B and Table S1). No pronounced alteration in BE, however, was found for O6-POB-dG upon genetic ablation of Pol θ (Fig. 4A and Fig. S2C and Table S1), underscoring the different roles of Pol ν and Pol θ in bypassing this bulky lesion. Furthermore, we did not observe significant further attenuations in BE for O6-alkyl-dG lesions in Pol ν and Pol θ double-knockout cells relative to the two single knockout cells (Fig. 4A Fig. S2D and Table S1).
Figure 4.

A–C, the bypass efficiencies (A) and G→A (B) and G→T (C) mutation frequencies of alkyl-dG lesions in HEK293T cells and isogenic cells deficient in Pol ν and/or Pol θ. D, proposed roles of Pol ν and Pol θ in bypassing O6-alkyl-dG and N2-alkyl-dG lesions. The bypass efficiency and mutation frequency were quantified from PAGE analysis results, and the data represent the means and standard deviations of results obtained from three independent replication experiments. The p values were calculated using unpaired two-tailed Student's t test: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Depletion of Pol ν or Pol θ results in diminished G→A mutation for O6-alkyl-dG lesions
Our previous study showed that O6-alkyl-dG lesions (except O6-POB-dG) induce exclusively G→A mutation in HEK293T cells and isogenic cells deficient in B- and Y-family TLS polymerases, where loss of some TLS polymerases diminished the G→A mutation frequencies (MFs) (13). Here we observed reduced G→A mutation rates for some O6-alkyl-dG lesions in isogenic HEK293T cells deficient in Pol ν, Pol θ, or both (Fig. 4B and Figs. S5–S9 and Table S2), revealing the roles of the two polymerases in promoting error-prone TLS of these DNA adducts. Specifically, single ablation of Pol ν or Pol θ did not alter the MF of O6-Et-dG, whereas the rate of this mutation was pronouncedly decreased (from 73% to 40%) in double-knockout cells (Fig. 4B and Table S2). Genetic depletion of Pol ν and Pol θ, alone or in combination, reduced the frequency of G→A mutation for O6-nBu-dG (Fig. 4B and Table S2). For the bulkier O6-POB-dG, loss of Pol θ, by itself or in conjunction with Pol ν, led to a significant diminution in the frequency of G→A mutation, although the rate of this mutation was not altered upon depletion of Pol ν alone (Fig. 4B and Fig. S3 and Table S2).
Aside from the G→A transition, O6-POB-dG also induces G→T transversion, which is in line with what was observed previously (32, 34). However, individual or concurrent depletion of Pol ν and Pol θ did not change the frequency of this mutation (Fig. 4C and Fig. S3 and Table S2). Furthermore, appreciable frequencies of G→T mutation were detected for O6-nBu-dG in all three knockout backgrounds (Fig. 4C and Fig. S2 and Table S2), suggesting roles of Pol ν and Pol θ in minimizing the G→T mutation for this lesion.
Removal of Pol ν or Pol θ did not alter the efficiency or fidelity of replication across N2-alkyl-dG lesions
Minor-groove N2-alkyl-dG lesions are more blocking to DNA replication than the corresponding O6-alkyl-dG lesions, with the BEs for N2-Et-dG and N2-nBu-dG being 26.2% and 16.7%, respectively (Figs. 2 and 4 and Table S1). Additionally, the replication bypass efficiency was not altered upon depletion of Pol ν and/or Pol θ (Table S1). Moreover, replication across minor-groove N2-alkyl-dG lesions was error-free in HEK293T cells or isogenic cells with Pol ν and/or Pol θ being genetically ablated (Figs. S2, S10, and S11).
Discussion
Exposure to endogenous and exogenous genotoxic agents can lead to formation of various DNA lesions (35), many of which block replicative DNA polymerases and require TLS Pols for their replicative bypass. Extensive studies in the past two decades revealed the roles of B- and Y-family polymerases in modulating the cytotoxic and mutagenic properties of DNA lesions (36, 37). More recently, two A-family polymerases, Pol ν and Pol θ, have been shown to promote TLS in vitro (23, 24, 26, 38). Very few studies, however, have been conducted to explore their capabilities in TLS in cells (23, 25).
Many alkylating agents can conjugate with the O6 and N2 positions of guanine to yield stable alkylation adducts (39, 40). In this study, we examined the contributions of Pol ν and Pol θ in bypassing three major-groove O6-alkyl-dG lesions (alkyl groups = Et, nBu, and POB) and two minor-groove N2-alkyl-dG (alkyl groups = Et and nBu) adducts in human cells (Fig. 1). We unveiled that Pol ν and Pol θ promote G→A mutation during replication of the major-groove O6-alkyl-dG adducts, and we also revealed the lack of appreciable involvement of the two polymerases in bypassing minor-groove N2-alkyl-dG lesions.
Genetic depletion of Pol ν resulted in reduced bypass efficiencies for all O6-alkyl-dG lesions (Fig. 4A), underscoring the role of this polymerase in overcoming the replication blockage imposed by these lesions. Previous in vitro biochemical studies showed that Pol ν is able to bypass Tg along with major-groove O6-methyl-2′-deoxyguanosine and N6-dA interstrand cross-link lesions (22, 24, 38). Our cellular replication experiment revealed the involvement of Pol ν in bypassing the bulkier major-groove O6-alkyl-dG lesions. In this vein, O6-POB-dG has been shown previously to be a poor substrate for recombinant human Pol ν in vitro (24). Our results, however, support that Pol ν has an appreciable role in TLS across this lesion in cells (Fig. 4A). The exact reason for the different results obtained from in vitro and cellular experiments is unknown, although Pol ν's function in replicative bypass of O6-POB-dG may be modulated through its interaction with other protein(s) and/or its posttranslational modifications. Along this line, the efficiency and fidelity of DNA Pol η in replicating across DNA lesions are known to be stimulated by the presence of proliferating cell nuclear antigen (41, 42).
Biochemical and cellular studies have suggested the importance of Pol θ in protecting against replication blockage elicited by various DNA lesions (23, 25, 26, 43). We found that genetic ablation of Pol θ results in decreased BEs for O6-Et-dG and O6-nBu-dG to similar extents as what were observed in Pol ν–deficient cells. On the other hand, the BEs of O6-POB-dG were not affected by single ablation of Pol θ (Fig. 4A). This observation, along with the result that simultaneous knockout of Pol ν and Pol θ did not forge more pronounced decreases in BE than the corresponding single knockouts (Fig. 4A), indicates that these two polymerases may function in the same pathway for the TLS across O6-alkyl-dG lesions.
Previous studies have suggested that Pol ν functions only in the insertion step of TLS, whereas Pol θ can efficiently catalyze the insertion and extension steps of TLS (26, 31, 43). Therefore, we postulate that Pol θ's main role in bypassing O6-alkyl-dG adducts resides in the extension step. In this vein, Pol ν can perhaps incorporate a nucleotide opposite the lesion site but cannot extend the nascent primer beyond the damage site, which might be attributed to the distortion in DNA structure imposed by the mismatched terminus at the active site of the DNA polymerase (44, 45). Results from a previous biochemical study showed that Pol θ was able to tolerate distorted DNA structure and extend from a nonstandard terminus, facilitating lesion bypass (26). This cooperative TLS mediated by Pol ν and Pol θ is reminiscent of the sequential lesion bypass mediated by Y- and B-family polymerases, where Pol η, Pol κ, and Pol ι function in the insertion step (5, 47) and Pol ζ in the extension step (48).
Similar to what was reported previously (13), all three O6-alkyl-dG adducts are highly mutagenic in HEK293T cells. For O6-Et-dG, the G→A mutation rate was markedly decreased in double knockout cells, although no significant change in mutation frequency was observed in Pol ν or Pol θ single-knockout cells (Fig. 4B). Others showed that Pol ν has a strong bias in inserting dTTP opposite modified guanine bases (49), and Pol θ has been found to be responsible for elevated mutations at C:G base pairs (20, 50). Our previous study revealed that depletion of Pol η, Pol κ, or Pol ι did not affect the MF of O6-Et-dG, whereas removal of Pol ζ slightly increased the G→A mutation frequency (13). On the basis of these results, we propose that Y-family polymerases insert the correct dCMP and the incorrect dTMP opposite O6-Et-dG, followed by extension of the nascent strand by Pol ζ with a preference for the O6-Et-dG:dC terminus. However, O6-Et-dG can also be bypassed by Pol ν, with preferential insertion of dTTP, and the ensuing O6-Et-dG:dT base pair may distort DNA and require Pol θ for efficient extension beyond the lesion site.
The observation of reduced G→A MF for O6-nBu-dG in Pol ν and Pol θ knockout cells further revealed the poor fidelity of these two polymerases in bypassing O6-alkyl-dG lesions (Fig. 4B). Interestingly, appreciable levels of G→T transversion were also detected for this lesion in cells lacking Pol ν, Pol θ, or both (Fig. 4C). This type of mutation was not detected for O6-alkyl-dG lesions in HEK293T cells or isogenic cells deficient in Pol η, Pol ι, Pol κ, or Pol ζ (13). In light of the fact that Pol η could misincorporate dAMP opposite O6-alkyl-dG adducts (51), we reason that the ensuing O6-nBu-dG:dA mispair might be difficult to be extended in the presence of Pol θ. In this vein, Pol θ, perhaps in cooperation with Pol ν, serves predominantly as an extender and may preferentially extend the nascent strand, with a dT being incorporated opposite O6-nBu-dG. In the absence of Pol ν and Pol θ, other polymerases may extend the daughter strand more readily, with a dA being inserted opposite O6-nBu-dG.
Our previous study showed decreased BEs, along with the lack of alteration in MFs, of O6-POB-dG upon depletion of any of the Y-family polymerases (32). A similar effect was found in Pol ν–deficient cells (Fig. 4B), suggesting that Pol ν and Y-family polymerases exhibit similar selectivity in nucleotide insertion opposite the O6-POB-dG. Among our CRISPR-Cas9–based TLS Pol knockout systems, depletion of Pol θ seems to be the only case that alters the mutagenic properties of O6-POB-dG with a reduced frequency of G→A mutation (Fig. 4B), although the absence of Pol θ did not change the BE of the lesion (Fig. 4A). We reason that the O6-POB-dG:dT mismatch, similar to the O6-Et-dG:dT and O6-nBu-dG:dT pairs at the nascent primer/template junction, can be favorably extended by Pol θ. In the absence of Pol θ, the nascent strand is perhaps extended by Pol ζ in an unbiased manner, which results in diminished G→A mutation.
The accurate bypass of N2-alkyl-dG adducts in HEK293T cells and isogenic Pol ν– and/or Pol θ–deficient cells (Fig. 2B and Fig. S2) revealed different miscoding properties of O6- and N2-alkyl-dG lesions, which may be attributed to their chemical structures and base-pairing properties. Alkylation of the O6 position of guanine renders the nucleobase favoring thymine when base-pairing (52, 53). TLS Pols, including Pol η, Pol ι, and Pol κ, preferentially incorporate dTTP opposite O6-alkyl-dG lesions (51). On the other hand, alkylation of the N2 of guanine does not strongly impair the base-pairing property of the nucleobase, as supported by biochemical evidence showing that Pol κ and Pol ι can incorporate the correct dCTP opposite N2-alkyl-dG lesions (14, 16).
The fact that ablation of Pol ν and Pol θ did not affect the BE or MF of N2-alkyl-dG adducts indicates the incapability of these two A-family polymerases in bypassing minor-groove guanine lesions. Our finding is in line with biochemical results revealing the difficulty of Pol ν in bypassing various minor-groove O2-alkyl-dT and N2-dG cross-link lesions in vitro (24, 38). Along this line, Wu et al. (15) showed recently that, although individual depletion of Pol η or Pol ζ did not exert any apparent effect on the BE or MF of N2-alkyl-dG lesions in human cells, error-free bypass of N2-alkyl-dG lesions in human cells necessitates Pol κ, Pol ι, and REV1.
In summary, we assessed the roles of Pol ν and Pol θ in TLS across O6-alkyl-dG and N2-alkyl-dG lesions in human cells. Our findings revealed, for the first time, the involvement of these two A-family polymerases in error-prone TLS past major-groove O6-alkyl-dG lesions in human cells; these two polymerases, however, are dispensable for bypassing minor-groove N2-alkyl-dG adducts in human cells (Fig. 4D). In addition, Pol ν and Pol θ may function in the same genetic pathway, which parallels the sequential actions of Y- and B-family TLS polymerases in bypassing other DNA lesions. Hence, the results from this study improve our understanding of the involvement of mammalian TLS DNA polymerases in coping with alkylated DNA adducts. It would be interesting to explore the possible cooperative and/or redundant roles of A-, B-, and Y-family polymerases in TLS in the future.
Experimental procedures
All chemicals, unless otherwise specified, were from Sigma-Aldrich (St. Louis, MO), and all enzymes were from New England Biolabs (Ipswich, MA). 1,1,1,3,3,3-Hexafluoro-2-propanol and [γ-32P]ATP were obtained from Oakwood Products Inc. (West Columbia, SC) and PerkinElmer Life Sciences (Piscataway, NJ). All unmodified oligodeoxyribonucleotides (ODNs) were from Integrated DNA Technologies (Coralville, IA). All 12-mer O6-alkyl-dG– and N2-alkyl-dG–containing ODNs employed in this study were synthesized and characterized previously as described in our published studies (13, 15, 32).
HEK293T cells deficient in POLN, POLQ, or both were produced by using the CRISPR-Cas9 genome editing method (54). Successful knockout of these genes was confirmed by Sanger sequencing (Fig. S1). The guide sequences used in this study were as follows (protospacer adjacent motif sequences are underlined): human POLN gene (Pol ν), AATTGCGATAAAAATACCAGTGG; human POLQ gene (Pol θ), CTGACTCCAAAAGCGGTACAGGG.
Construction of lesion-containing and lesion-free control plasmids
The lesion-containing and lesion-free control/competitor genomes were prepared following procedures published previously (55). Briefly, the parental vector was cleaved with Nt.BstNBI to yield a gapped plasmid, followed by removal of the resultant 25-mer single-stranded ODN by annealing with a 25-mer complementary ODN that was 100-fold in excess. The gapped plasmid was then isolated from the mixture by using 100-kDa-cutoff ultracentrifugal filter units (Millipore). A 5′-phosphorylated 13-mer lesion-free ODN (5′-AATTGAGTCGATG-3′) and a 5′-phosphorylated 12-mer lesion-containing or lesion-free control ODN (5′-ATGGCGXGCTAT-3′, X = O6-alkyl-dG, N2-alkyl-dG, or G) were subsequently ligated into the purified gapped plasmid.
Cell culture, transfection, and plasmid isolation
Replication experiments were conducted using the previously published SSPCR-CRAB assay (33). The lesion-containing and the respective control plasmids were premixed individually with the competitor plasmid and transfected into HEK293T cells or isogenic polymerase-deficient cells. The molar ratios of the competitor to the control and lesion-bearing genome were 1:3 and 1:9, respectively. The cells (1 × 105) were seeded in 24-well plates and cultured overnight at 37 °C in a 5% CO2 atmosphere. They were then transfected with 300 ng of the mixed genomes by using TransIT-2020 (Mirus Bio), following the vendor's recommended procedures. The cells were harvested a24 h following transfection, and the progeny plasmids arising from cellular replication were isolated using the GeneJET Plasmid Miniprep Kit (Thermo Fisher Scientific). The residual unreplicated plasmids in the mixture were removed by DpnI digestion and exonuclease III cleavage, as described previously (46).
PCR amplification and PAGE analyses
A previously reported nested PCR method was employed to amplify the progeny plasmids (32). The final PCR amplicons were resolved using 1% agarose gel, purified using the GeneJET Gel Extraction Kit (Thermo Fisher Scientific, San Jose, CA) and stored at −20 °C until use. For PAGE analysis, 150 ng of the PCR fragments was incubated with 5 units of NcoI and 1 unit of shrimp alkaline phosphatase at 37 °C in 10 μl of New England Biolabs (NEB) buffer 3 for 1 h, followed by heating at 80 °C for 20 min to deactivate the enzymes. To the above mixture were then added 1.25 μCi (0.5 pmol) of [γ-32P]ATP and 5 units of T4 polynucleotide kinase (Fig. 2A and Fig. S2). The reaction was continued at 37 °C for 30 min, followed by heating at 75 °C for 20 min to deactivate the T4 polynucleotide kinase. To the above mixture was then added 2.0 units of SfaNI, and the solution was incubated at 37 °C for 1.5 h. The digestion was subsequently terminated by addition of 20 μl of formamide gel loading buffer. The above restriction cleavage and post-labeling procedures yielded a 16-mer 5′ 32P-labeled fragment for the progeny of the competitor genome and 13-mer 5′ 32P-labeled fragment(s) for the progeny of the control or lesion-carrying genome (Fig. 2B). The digestion products were separated using 30% native polyacrylamide gel (acrylamide:bisacrylamide 19:1) and quantified by phosphorimaging analysis. The bypass efficiency and mutation frequency were then calculated to gauge the effects of DNA lesions on replication efficiency and fidelity, respectively, following procedures published previously (33).
Identification of mutagenic products by LC-MS/MS
The replication products were also identified by LC-MS/MS. Briefly, 2 μg of the above-described PCR products was digested with 50 units of NcoI and 15 units of shrimp alkaline phosphatase in 150 μl of NEB buffer 3 at 37 °C for 2 h, followed by deactivating the phosphatase by heating at 80 °C for 20 min. To the mixture was added 30 units of SfaNI, and the solution was incubated at 37 °C for another 2 h. The resulting solution was extracted once with phenol:chloroform:isoamyl alcohol (25:24:1 (v/v)), and to the mixture were subsequently added 2.5 volumes of ethanol and 0.1 volume of 3.0 m sodium acetate to precipitate the DNA. The DNA pellet was then reconstituted in water and subjected to LC-MS and MS/MS analysis. An Agilent 1200 capillary HPLC system (Agilent Technologies, Santa Clara, CA) and an LTQ linear ion trap mass spectrometer (Thermo Fisher Scientific) were used for all LC-MS and MS/MS experiments. The mass spectrometer was set up to acquire the higher-resolution “ultra-zoom scan” MS and MS/MS for the [M-3H]3− ions of the 13-mer 5′-CATGGCGNGCTGT-3′, where N′ represents A, T, C, or G (Fig. 3 and Figs. S4–S11).
Author contributions
H. D. data curation; H. D. investigation; H. D. writing-original draft; P. W., J. W., and X. H. resources; Y. W. conceptualization; Y. W. supervision; Y. W. funding acquisition; Y. W. writing-review and editing.
Supplementary Material
This work was supported by National Institutes of Health Grant R01 ES029749 (to Y. W.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S11 and Tables S1 and S2.
- Pol
- polymerase
- TLS
- translesion synthesis
- Tg
- thymine glycol
- Et
- ethyl
- nBu
- n-butyl
- POB
- pyridyloxobutyl
- SSPCR-CRAB
- strand-specific PCR-competitive replication and adduct bypass
- BE
- bypass efficiency
- MF
- mutation frequency
- ODN
- oligodeoxyribonucleotide.
References
- 1. Hoeijmakers J. H. (2009) DNA damage, aging, and cancer. N. Engl. J. Med. 361, 1475–1485 10.1056/NEJMra0804615 [DOI] [PubMed] [Google Scholar]
- 2. Friedberg E. C. (2003) DNA damage and repair. Nature 421, 436–440 10.1038/nature01408 [DOI] [PubMed] [Google Scholar]
- 3. Waters L. S., Minesinger B. K., Wiltrout M. E., D'Souza S., Woodruff R. V., and Walker G. C. (2009) Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev. 73, 134–154 10.1128/MMBR.00034-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yang W., and Gao Y. (2018) Translesion and repair DNA polymerases: diverse structure and mechanism. Annu. Rev. Biochem. 87, 239–261 10.1146/annurev-biochem-062917-012405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shachar S., Ziv O., Avkin S., Adar S., Wittschieben J., Reissner T., Chaney S., Friedberg E. C., Wang Z., Carell T., Geacintov N., and Livneh Z. (2009) Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 28, 383–393 10.1038/emboj.2008.281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yoon J. H., Bhatia G., Prakash S., and Prakash L. (2010) Error-free replicative bypass of thymine glycol by the combined action of DNA polymerases κ and ζ in human cells. Proc. Natl. Acad. Sci. U.S.A. 107, 14116–14121 10.1073/pnas.1007795107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li Z., Zhang H., McManus T. P., McCormick J. J., Lawrence C. W., and Maher V. M. (2002) hREV3 is essential for error-prone translesion synthesis past UV or benzo[α]pyrene diol epoxide-induced DNA lesions in human fibroblasts. Mutat. Res. 510, 71–80 10.1016/S0027-5107(02)00253-1 [DOI] [PubMed] [Google Scholar]
- 8. Ohashi E., Ogi T., Kusumoto R., Iwai S., Masutani C., Hanaoka F., and Ohmori H. (2000) Error-prone bypass of certain DNA lesions by the human DNA polymerase κ. Genes Dev. 14, 1589–1594 [PMC free article] [PubMed] [Google Scholar]
- 9. Goodman M. F., and Woodgate R. (2013) Translesion DNA polymerases. Cold Spring Harb. Perspect. Biol. 5, a010363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hendel A., Ziv O., Gueranger Q., Geacintov N., and Livneh Z. (2008) Reduced efficiency and increased mutagenicity of translesion DNA synthesis across a TT cyclobutane pyrimidine dimer, but not a TT 6-4 photoproduct, in human cells lacking DNA polymerase η. DNA Repair 7, 1636–1646 10.1016/j.dnarep.2008.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu J., Wang P., Li L., You C., and Wang Y. (2018) Cytotoxic and mutagenic properties of minor-groove O2-alkylthymidine lesions in human cells. J. Biol. Chem. 293, 8638–8644 10.1074/jbc.RA118.003133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu J., Li L., Wang P., You C., Williams N. L., and Wang Y. (2016) Translesion synthesis of O4-alkylthymidine lesions in human cells. Nucleic Acids Res. 44, 9256–9265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Du H., Wang P., Li L., and Wang Y. (2019) Repair and translesion synthesis of O6-alkylguanine DNA lesions in human cells. J. Biol. Chem. 294, 11144–11153 10.1074/jbc.RA119.009054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yuan B., You C., Andersen N., Jiang Y., Moriya M., O'Connor T. R., and Wang Y. (2011) The roles of DNA polymerases κ and ι in the error-free bypass of N2-carboxyalkyl-2′-deoxyguanosine lesions in mammalian cells. J. Biol. Chem. 286, 17503–17511 10.1074/jbc.M111.232835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu J., Du H., Li L., Price N. E., Liu X., and Wang Y. (2019) The impact of minor-groove N2-alkyl-2′-deoxyguanosine lesions on DNA replication in human cells. ACS Chem. Biol. 14, 1708–1716 10.1021/acschembio.9b00129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Choi J. Y., Angel K. C., and Guengerich F. P. (2006) Translesion synthesis across bulky N2-alkyl guanine DNA adducts by human DNA polymerase κ. J. Biol. Chem. 281, 21062–21072 10.1074/jbc.M602246200 [DOI] [PubMed] [Google Scholar]
- 17. Minko I. G., Yamanaka K., Kozekov I. D., Kozekova A., Indiani C., O'Donnell M. E., Jiang Q., Goodman M. F., Rizzo C. J., and Lloyd R. S. (2008) Replication bypass of the acrolein-mediated deoxyguanine DNA-peptide cross-links by DNA polymerases of the DinB family. Chem. Res. Toxicol. 21, 1983–1990 10.1021/tx800174a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ziv O., Geacintov N., Nakajima S., Yasui A., and Livneh Z. (2009) DNA polymerase ζ cooperates with polymerases κ and ι in translesion DNA synthesis across pyrimidine photodimers in cells from XPV patients. Proc. Natl. Acad. Sci. U.S.A. 106, 11552–11557 10.1073/pnas.0812548106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chan S. H., Yu A. M., and McVey M. (2010) Dual roles for DNA polymerase θ in alternative end-joining repair of double-strand breaks in Drosophila. PLoS Genet. 6, e1001005 10.1371/journal.pgen.1001005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arana M. E., Seki M., Wood R. D., Rogozin I. B., and Kunkel T. A. (2008) Low-fidelity DNA synthesis by human DNA polymerase θ. Nucleic Acids Res. 36, 3847–3856 10.1093/nar/gkn310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moldovan G. L., Madhavan M. V., Mirchandani K. D., McCaffrey R. M., Vinciguerra P., and D'Andrea A. D. (2010) DNA polymerase POLN participates in cross-link repair and homologous recombination. Mol. Cell. Biol. 30, 1088–1096 10.1128/MCB.01124-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takata K., Shimizu T., Iwai S., and Wood R. D. (2006) Human DNA polymerase N (POLN) is a low-fidelity enzyme capable of error-free bypass of 5S-thymine glycol. J. Biol. Chem. 281, 23445–23455 10.1074/jbc.M604317200 [DOI] [PubMed] [Google Scholar]
- 23. Yoon J. H., Roy Choudhury J., Park J., Prakash S., and Prakash L. (2014) A role for DNA polymerase θ in promoting replication through oxidative DNA lesion, thymine glycol, in human cells. J. Biol. Chem. 289, 13177–13185 10.1074/jbc.M114.556977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gowda A. S., and Spratt T. E. (2016) DNA polymerase ν rapidly bypasses O6-methyl-dG but not O6-[4-(3-pyridyl)-4-oxobutyl-dG and O2-alkyl-dTs. Chem. Res. Toxicol. 29, 1894–1900 10.1021/acs.chemrestox.6b00318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yoon J. H., McArthur M. J., Park J., Basu D., Wakamiya M., Prakash L., and Prakash S. (2019) Error-prone replication through UV lesions by DNA polymerase θ protects against skin cancers. Cell 176, 1295–1309.e15 10.1016/j.cell.2019.01.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Seki M., and Wood R. D. (2008) DNA polymerase θ (POLQ) can extend from mismatches and from bases opposite a (6-4) photoproduct. DNA Repair 7, 119–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kryston T. B., Georgiev A. B., Pissis P., and Georgakilas A. G. (2011) Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 711, 193–201 10.1016/j.mrfmmm.2010.12.016 [DOI] [PubMed] [Google Scholar]
- 28. Kent T., Chandramouly G., McDevitt S. M., Ozdemir A. Y., and Pomerantz R. T. (2015) Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase θ. Nat. Struct. Mol. Biol. 22, 230–237 10.1038/nsmb.2961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tamae D., Lim P., Wuenschell G. E., and Termini J. (2011) Mutagenesis and repair induced by the DNA advanced glycation end product N2-1-(carboxyethyl)-2′-deoxyguanosine in human cells. Biochemistry 50, 2321–2329 10.1021/bi101933p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jansen J. G., Vrieling H., van Teijlingen C. M., Mohn G. R., Tates A. D., and van Zeeland A. A. (1995) Marked differences in the role of O6-alkylguanine in hprt mutagenesis in T-lymphocytes of rats exposed in vivo to ethylmethanesulfonate, N-(2-hydroxyethyl)-N-nitrosourea, or N-ethyl-N-nitrosourea. Cancer Res. 55, 1875–1882 [PubMed] [Google Scholar]
- 31. Masuda K., Ouchida R., Hikida M., Kurosaki T., Yokoi M., Masutani C., Seki M., Wood R. D., Hanaoka F., and O-Wang J. (2007) DNA polymerases η and θ function in the same genetic pathway to generate mutations at A/T during somatic hypermutation of Ig genes. J. Biol. Chem. 282, 17387–17394 10.1074/jbc.M611849200 [DOI] [PubMed] [Google Scholar]
- 32. Du H., Leng J., Wang P., Li L., and Wang Y. (2018) Impact of tobacco-specific nitrosamine-derived DNA adducts on the efficiency and fidelity of DNA replication in human cells. J. Biol. Chem. 293, 11100–11108 10.1074/jbc.RA118.003477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. You C., Swanson A. L., Dai X., Yuan B., Wang J., and Wang Y. (2013) Translesion synthesis of 8,5′-cyclopurine-2′-deoxynucleosides by DNA polymerases η, ι, and ζ. J. Biol. Chem. 288, 28548–28556 10.1074/jbc.M113.480459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pauly G. T., Peterson L. A., and Moschel R. C. (2002) Mutagenesis by O6-[4-oxo-4-(3-pyridyl)butyl]guanine in Escherichia coli and human cells. Chem. Res. Toxicol. 15, 165–169 10.1021/tx0101245 [DOI] [PubMed] [Google Scholar]
- 35. Bauer N. C., Corbett A. H., and Doetsch P. W. (2015) The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res. 43, 10083–10101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sale J. E., Lehmann A. R., and Woodgate R. (2012) Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 13, 141–152 10.1038/nrm3289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gan G. N., Wittschieben J. P., Wittschieben B. Ø., and Wood R. D. (2008) DNA polymerase ζ (pol ζ) in higher eukaryotes. Cell Res. 18, 174–183 10.1038/cr.2007.117 [DOI] [PubMed] [Google Scholar]
- 38. Yamanaka K., Minko I. G., Takata K., Kolbanovskiy A., Kozekov I. D., Wood R. D., Rizzo C. J., and Lloyd R. S. (2010) Novel enzymatic function of DNA polymerase ν in translesion DNA synthesis past major groove DNA-peptide and DNA-DNA cross-links. Chem. Res. Toxicol. 23, 689–695 10.1021/tx900449u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shrivastav N., Li D., and Essigmann J. M. (2010) Chemical biology of mutagenesis and DNA repair: cellular responses to DNA alkylation. Carcinogenesis 31, 59–70 10.1093/carcin/bgp262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu S., and Wang Y. (2015) Mass spectrometry for the assessment of the occurrence and biological consequences of DNA adducts. Chem. Soc. Rev. 44, 7829–7854 10.1039/C5CS00316D [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Acharya N., Yoon J. H., Gali H., Unk I., Haracska L., Johnson R. E., Hurwitz J., Prakash L., and Prakash S. (2008) Roles of PCNA-binding and ubiquitin-binding domains in human DNA polymerase η in translesion DNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 105, 17724–17729 10.1073/pnas.0809844105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Haracska L., Kondratick C. M., Unk I., Prakash S., and Prakash L. (2001) Interaction with PCNA is essential for yeast DNA polymerase η function. Mol. Cell 8, 407–415 10.1016/S1097-2765(01)00319-7 [DOI] [PubMed] [Google Scholar]
- 43. Seki M., Masutani C., Yang L. W., Schuffert A., Iwai S., Bahar I., and Wood R. D. (2004) High-efficiency bypass of DNA damage by human DNA polymerase Q. EMBO J. 23, 4484–4494 10.1038/sj.emboj.7600424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pagès V., and Fuchs R. P. (2002) How DNA lesions are turned into mutations within cells? Oncogene 21, 8957–8966 10.1038/sj.onc.1206006 [DOI] [PubMed] [Google Scholar]
- 45. Johnson S. J., and Beese L. S. (2004) Structures of mismatch replication errors observed in a DNA polymerase. Cell 116, 803–816 10.1016/S0092-8674(04)00252-1 [DOI] [PubMed] [Google Scholar]
- 46. Burns J. A., Dreij K., Cartularo L., and Scicchitano D. A. (2010) O6-methylguanine induces altered proteins at the level of transcription in human cells. Nucleic Acids Res. 38, 8178–8187 10.1093/nar/gkq706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Washington M. T., Johnson R. E., Prakash S., and Prakash L. (2000) Accuracy of thymine-thymine dimer bypass by Saccharomyces cerevisiae DNA polymerase η. Proc. Natl. Acad. Sci. U.S.A. 97, 3094–3099 10.1073/pnas.050491997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee Y. S., Gregory M. T., and Yang W. (2014) Human Pol ζ purified with accessory subunits is active in translesion DNA synthesis and complements Pol η in cisplatin bypass. Proc. Natl. Acad. Sci. U.S.A. 111, 2954–2959 10.1073/pnas.1324001111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Arana M. E., Takata K., Garcia-Diaz M., Wood R. D., and Kunkel T. A. (2007) A unique error signature for human DNA polymerase ν. DNA Repair 6, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Masuda K., Ouchida R., Takeuchi A., Saito T., Koseki H., Kawamura K., Tagawa M., Tokuhisa T., Azuma T., and O-Wang J. (2005) DNA polymerase θ contributes to the generation of C/G mutations during somatic hypermutation of Ig genes. Proc. Natl. Acad. Sci. U.S.A. 102, 13986–13991 10.1073/pnas.0505636102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Choi J. Y., Chowdhury G., Zang H., Angel K. C., Vu C. C., Peterson L. A., and Guengerich F. P. (2006) Translesion synthesis across O6-alkylguanine DNA adducts by recombinant human DNA polymerases. J. Biol. Chem. 281, 38244–38256 10.1074/jbc.M608369200 [DOI] [PubMed] [Google Scholar]
- 52. Swann P. F. (1990) Why do O6-alkylguanine and O4-alkylthymine miscode: the relationship between the structure of DNA containing O6-alkylguanine and O4-alkylthymine and the mutagenic properties of these bases. Mutat. Res. 233, 81–94 10.1016/0027-5107(90)90153-U [DOI] [PubMed] [Google Scholar]
- 53. Parthasarathy R., and Fridey S. M. (1986) Conformation of O6-alkylguanosines: molecular mechanism of mutagenesis. Carcinogenesis 7, 221–227 10.1093/carcin/7.2.221 [DOI] [PubMed] [Google Scholar]
- 54. Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., and Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yuan B., O'Connor T. R., and Wang Y. (2010) 6-Thioguanine and S6-methylthioguanine are mutagenic in human cells. ACS Chem. Biol. 5, 1021–1027 10.1021/cb100214b [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.