

Abstract
Objective
To estimate the prevalence of Joubert syndrome (JS) in Italy applying standards of descriptive epidemiology and to provide a molecular characterization of the described patient cohort.
Methods
We enrolled all patients with a neuroradiologically confirmed diagnosis of JS who resided in Italy in 2018 and calculated age and sex prevalence, assuming a Poisson distribution. We also investigated the correlation between proband chronological age and age at diagnosis and performed next-generation sequencing (NGS) analysis on probands' DNA when available.
Results
We identified 284 patients with JS: the overall, female- and male-specific population-based prevalence rates were 0.47 (95% confidence interval [CI] 0.41–0.53), 0.41 (95% CI 0.32–0.49), and 0.53 (95% CI 0.45–0.61) per 100,000 population, respectively. When we considered only patients in the age range from 0 to 19 years, the corresponding population-based prevalence rates rose to 1.7 (95% CI 1.49–1.97), 1.62 (95% CI 1.31–1.99), and 1.80 (95% CI 1.49–2.18) per 100,000 population. NGS analysis allowed identifying the genetic cause in 131 of 219 screened probands. Age at diagnosis was available for 223 probands, with a mean of 6.67 ± 8.10 years, and showed a statistically significant linear relationship with chronological age (r2 = 0.79; p < 0.001).
Conclusions
We estimated for the first time the age and sex prevalence of JS in Italy and investigated the patients’ genetic profile. The obtained population-based prevalence rate was ≈10 times higher than that available in literature for children population.
Joubert syndrome (JS) is a rare congenital ataxia characterized by a pathognomonic midhindbrain malformation (the molar tooth sign [MTS]) and multiorgan involvement.1 More than 35 associated genes are known with autosomal or X-linked recessive inheritance,2 overall accounting for ≈60% of cases.3 To date, descriptive epidemiology data such as population-based prevalence rates are almost completely lacking. The commonly reported range of 1:80,000 to 100,000 livebirths4 is probably underestimated due to low awareness of MTS in historical texts.1 Furthermore, it does not reflect the presence of the disease at later ages because medical complications in JS can lead to early death.5
A recent systematic review attempted to estimate the prevalence of childhood ataxia across World Health Organization regions.6 Among the 115 articles included, only 1 refers to JS: the estimated prevalence in Swedish children at December 31, 1992, was 0.17 per 100,000 (based on a single JS case).7
Our aim was to estimate the age and sex prevalence of JS in Italy, applying standards of descriptive epidemiology.8
Methods
We have established a network of 46 Italian centers active in the diagnosis, care, and research of JS and connected to the Italian Association for Joubert Syndrome and Congenital Ataxias and have created a comprehensive clinical-genetic database.
Patients had a neuroradiologically confirmed diagnosis of JS and variable features ranging from pure neurologic phenotype (developmental delay, hypotonia, and ataxia) to more severe cases with multiorgan involvement (including ocular, renal, hepatic, skeletal, and orofacial defects).5 Age at diagnosis among probands, inferred from both medical records and brain MRIs, was defined as age at first detection of the MTS.
Only patients with JS resident in Italy (60,483,973 inhabitants as of 2018) on October 8, 2018, entered a point-prevalence rate epidemiologic study. The crude age- and sex-specific prevalence was calculated, assuming a Poisson distribution. The 95% confidence intervals (CIs) were computed with the formula 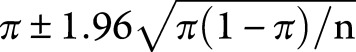 , where π means the prevalence and n the absolute number of inhabitants.9 The database of the Italian National Institute of Statistics provided population data for 2018 (demo.istat.it/pop2018/index.html).
, where π means the prevalence and n the absolute number of inhabitants.9 The database of the Italian National Institute of Statistics provided population data for 2018 (demo.istat.it/pop2018/index.html).
Statistical analyses were performed with the t test for age comparison. Linear regression analysis was used to test the relationship between age at point prevalence and age at diagnosis. Data were analyzed with SPSS (version 25.0, SPSS Inc, Chicago, IL). A value of p ≤ 0.05 was considered statistically significant, with Bonferroni correction for multiple comparisons.
Next-generation sequencing (NGS) panel analysis of 34 JS genes was conducted on 219 probands (data available from Dryad, table e-1, doi.org/10.5061/dryad.2220d7m). Identified variants were classified as pathogenic, likely pathogenic, of uncertain significance, likely benign, or benign, according to current guidelines.10 Validation and segregation of pathogenic and likely pathogenic variants were performed by Sanger.
Standard protocol approvals, registrations, and patient consents
The study was approved by the ethics committee of National Institute of Health. Participant families provided informed consent.
Data availability
All data are available from the corresponding authors. Tables e-1 through e-3 are also available from Dryad (doi.org/10.5061/dryad.2220d7m).
Results
We ascertained 284 patients with JS (220 white) from 251 families. The male/female ratio was 1.22 (156/128). The overall mean age at point prevalence was 16.7 ± 10.4 years (median 14.7 years, range 1–60 years, interquartile range [IQR] 8.3–23.2 years). The mean male- and female-specific ages at point prevalence were 17.5 ± 11.2 years (median 15 years, range 2–60 years, IQR 8.8–23.5 years) and 15.7 ± 9.2 years (median 14.1 years; range 1–39 years; IQR 8.8–23.5 years), respectively, without significant differences between sexes.
Among patients, 124 (44%) presented exclusive neurologic manifestations and 134 (47%) had additional multiorgan involvement, while data were missing in 26 cases (9.1%).
The crude prevalences of JS in Italy on October 8, 2018, for total, females, and males were 0.47 (95% CI 0.41–0.53), 0.41 (95% CI 0.32–0.49), and 0.53 (95% CI 0.45–0.61) per 100,000 population, respectively (table 1). When we focused on patients 0 to 19 years of age, the crude prevalences for total, females, and males increased to 1.7 (95% CI 1.49–1.97), 1.62 (95% CI 1.31–1.99), and 1.80 (95% CI 1.49–2.18) per 100,000 population (table 1).
Table 1.
Population-based prevalence rate of JS in Italy (per 100,000 population, year 2018)

Two hundred of the 251 (80%) probands have both parents of Italian origin; 44 (17.5%) have at least 1 non-Italian parent; and 23 (9.1%) descend from consanguineous unions. Table e-2 available from Dryad (doi.org/10.5061/dryad.2220d7m) summarizes the proband genetic status. Among the 219 tested probands, 131 had a confirmed molecular diagnosis, resulting in a mutation rate of 60%, in accordance with the literature.3 The commonest mutated genes are AHI1 (9.1%), CPLANE1 (8.7%), TMEM67 (8.2%), and CEP290 (6.9%), similarly distributed between sexes (figure 1). When we also considered affected siblings, molecularly confirmed cases increased to 152. In this latter group, the crude prevalences for total, females, and males were 0.25 (95% CI 0.21–0.29), 0.24 (95% CI 0.20–0.31), and 0.26 (95% CI 0.21–0.32) per 100,000 population (data available from Dryad, table e-3, doi.org/10.5061/dryad.2220d7m).
Figure 1. Mutational frequency in probands.

Absolute number and percentage of probands mutated in different Joubert syndrome genes. For each gene, the percentage was calculated on the total number of tested probands (n = 219).
Age at diagnosis was available for 223 probands, with a mean of 6.67 ± 8.10 years (median 3.18 years, range 0–46.49 years, IQR 1.06–10.37 years). The mean sex-specific age at diagnosis was 7.24 ± 9.08 years for males and 6.01 ± 6.76 years for females, without a statistically significant difference between groups. Finally, the linear relationship between age at point prevalence and age at diagnosis was statistically significant (r2 = 0.79; p < 0.001) (figure 2).
Figure 2. Age at point prevalence vs age at diagnosis in probands.
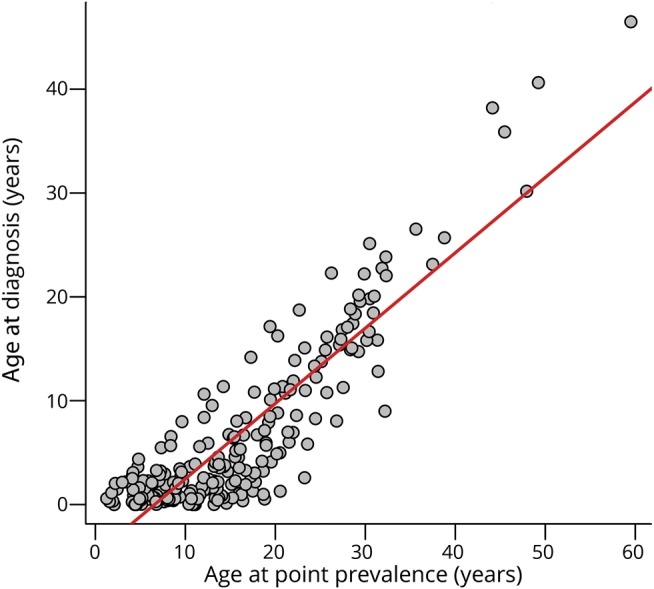
Linear regression analysis between age at point prevalence and age at diagnosis. Data refer to the 223 probands for whom both ages were available, independently of their genetic status.
Discussion
Here, we show that the creation of a national network represents a valuable strategy for estimating the prevalence of rare diseases such as JS, defining their impact on the population and allowing personalized rehabilitative interventions to be planned at the national level.
Because the network has not been created for epidemiologic purposes, the main limitation of our approach consists of a possible underrepresentation of patients with JS. First, we are not able to provide information about the referring process or the systematic use of MRI in ataxic patients diagnosed outside the Italian network. Second, we may have missed some other cases due to the wide clinical variability of the disease. Indeed, patients with a very mild phenotype might not undergo brain MRI, remaining undiagnosed. For all these reasons, we consider our data minimum prevalence estimates. However, it must be noted that the distribution of our cohort among the 3 macro-areas (North, Center, South Islands) is in accordance with that reported in the general population.
This study considerably increases knowledge of JS epidemiology, demonstrating an overall population-based crude prevalence rate of 0.47 per 100,000 population. This rate is approximately halved for molecularly confirmed cases (ratio 0.5 = 0.25/0.47 per 100,000 population) (data available from Dryad, tables e-1 and e-3, doi.org/10.5061/dryad.2220d7m). When we considered pediatric age, JS prevalence was 1.7 per 100,000 population, 10 times higher than the single report published to date (0.17 per 100,000 population).7 However, this comparison is not completely appropriate because the latter study was performed before the MTS identification. In addition, a comparison with the figure reported in that study4 may be complicated, because it describes an incidence for live birth.
Because the probability of diagnosis might increase with a previously identified case in the family, age at diagnosis was considered only for probands, showing a linear relationship with age at point prevalence. A possible explanation is that elderly patients exhibited first signs of disease at a time of lower awareness of JS, leading to delayed diagnosis. However, such a delay could also relate to the existence of mild phenotypes, not recognizable until later ages. A detailed clinical characterization of patients is required to clarify this issue.
A genetic diagnosis of JS was achieved in 60% patients, confirming literature data.3 Single heterozygous pathogenic/likely pathogenic variants have been detected in 35 probands (16% of tested cases). For these patients, the potential role of variants of uncertain significance and the existence of a second variant missed by NGS (e.g., variants outside coding regions or large rearrangements) should be considered, requiring complementary diagnostic strategies.
We propose a minimum prevalence estimate of JS in Italy and provide a molecular characterization of this cohort. Our findings, including age-standardized prevalence and sex and age distribution, define for the first time the epidemiology of this rare disease.
Acknowledgment
The authors thank all patients and clinicians for enthusiastically encouraging this study and R. Cutigni for her contribution to the data collection.
Glossary
- CI
confidence interval
- IQR
interquartile range
- JS
Joubert syndrome
- MTS
molar tooth sign
- NGS
next-generation sequencing
Appendix. Authors
Footnotes
Editorial, page 337
Study funding
Supported by the Ministry of Health (Ricerca Finalizzata NET-2013-02356160, and Ricerca Corrente 2019 to IRCCS Eugenio Medea), European Research Council (ERC Starting Grant 260888), Italian Ministry of University and Research (Progetto Dipartimenti di Eccellenza), and Pierfranco and Luisa Mariani Foundation (PADAPORT project).
Disclosure
S. Nuovo is the recipient of a PhD bursary financed by Associazione Italiana Syndrome di Joubert e Atassie Congenite. I. Bacigalupo, M. Ginevrino, and R. Battini report no disclosures relevant to the manuscript. E. Bertini is funded by the Ministry of Health (grant Ricerca Finalizzata NET-2013-02356160). R. Borgatti is funded by the Ministry of Health (grant Ricerca Finalizzata NET-2013-02356160). A. Casella, A. Micalizzi, and M. Nardella report no disclosures relevant to the manuscript. R. Romaniello is funded by the Ministry of Health (Ricerca Corrente 2019). V. Serpieri and G. Zanni report no disclosures relevant to the manuscript. E. Valente is funded by the Ministry of Health (grant Ricerca Finalizzata NET-2013-02356160), the European Research Council (ERC Starting Grant 260888), the Italian Ministry of University and Research (Progetto Dipartimenti di Eccellenza), and the Pierfranco and Luisa Mariani Foundation (PADAPORT project). N. Vanacore is funded by Ministry of Health (grant Ricerca Finalizzata NET-2013-02356160). Go to Neurology.org/N for full disclosures.
References
- 1.Romani M, Micalizzi A, Valente EM. Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol 2013;12:894–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nuovo S, Fuiano L, Micalizzi A, et al. Impaired urinary concentration ability is a sensitive predictor of renal disease progression in Joubert syndrome. Nephrol Dial Transpl Epub 2018 Nov 6. [DOI] [PMC free article] [PubMed]
- 3.Bachmann-Gagescu R, Dempsey JC, Phelps IG, et al. Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. J Med Genet 2015;52:514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parisi MA, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders) (OMIM 213300). Eur J Hum Genet 2007;15:511–521. [DOI] [PubMed] [Google Scholar]
- 5.Dempsey JC, Phelps IG, Bachmann-Gagescu R, Glass IA, Tully HM, Doherty D. Mortality in Joubert syndrome. Am J Med Genet A 2017;173:1237–1242. [DOI] [PubMed] [Google Scholar]
- 6.Musselman KE, Stoyanov CT, Marasigan R, et al. Prevalence of ataxia in children: a systematic review. Neurology 2014;82:80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Esscher E, Flodmark O, Hagberg G, Hagberg B. Non-progressive ataxia: origins, brain pathology and impairments in 78 Swedish children. Dev Med Child Neurol 1996;38:285–296. [DOI] [PubMed] [Google Scholar]
- 8.Khoury MJ, Gwinn M, Clyne M, Yu W. Genetic epidemiology with a capital E, ten years after. Genet Epidemiol 2011;35:845–852. [DOI] [PubMed] [Google Scholar]
- 9.Schoenberg BS. Calculating confidence intervals for rates and ratios. Simplified method utilizing tabular values based on the Poisson distribution. Neuroepidemiology 1983;2:257–265. [Google Scholar]
- 10.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are available from the corresponding authors. Tables e-1 through e-3 are also available from Dryad (doi.org/10.5061/dryad.2220d7m).