

Abstract
Boronic acids are centrally important functional motifs and synthetic precursors. Visible light-induced borylation may provide access to structurally diverse boronates, but a broadly efficient photocatalytic borylation method that can effect borylation of a wide range of substrates, including strong C-O bonds, remains elusive. Herein, we report a general, metal-free visible light-induced photocatalytic borylation platform that enables borylation of electron rich derivatives of phenols and anilines, chloroarenes, as well as other haloarenes. The reaction exhibits excellent functional group tolerance, as demonstrated by the borylation of a range of structurally complex substrates. Remarkably, the reaction is catalyzed by phenothiazine, - a simple organic photocatalyst with MW<200 that mediates the previously unachievable visible light-induced single electron reduction of phenol derivatives with reduction potentials as negative as ~-3 V vs SCE by a proton-coupled electron transfer mechanism. Mechanistic studies point to the crucial role of the photocatalyst-base interaction.
Introduction
The importance of boronic acids continues to grow, as new applications emerge in the areas of organic synthesis,1 catalysis,2 materials science,3 drug discovery,4 and analytical chemistry.5 Although significant advances have recently been made in synthetic approaches to boronic acids based on transition metal catalysis,6 main group chemistry7 and photochemistry,8–13 various challenges persist and efforts to expand the scope, structural diversity, functional group tolerance, and catalyst availability of emerging organoboron methodologies remain at the forefront of several areas of chemistry. Given the importance of aromatic boronic acids, it is desirable to have a single catalytic platform that enables borylation of all common carbon-heteroatom bonds, including C-O, C-N, and C-X (Cl, Br, I) bonds.
Since the initial reports on the UV light-induced Ar-X and Ar-N borylation,8 a number of methods were developed that allow for borylation of some of the aryl-heteroatom bonds, in particular, by the groups of Li,9 Jiao,10 Schelter,11 and Studer.12 Despite the impressive progress, further work is needed to develop a broad photocatalytic platform that encompasses a wide array of diverse C-O, C-N, and C-X bonds and is powered by visible light as a more sustainable and functional group-tolerant source of energy. A desirable photocatalytic platform that can address these limitations would be based on a single, readily available organic photocatalyst that enables borylations of phenols, anilines, as well as chloro-, bromo- and iodoarenes under visible light. Development of such a broad, visible light-mediated, photocatalytic platform is a major challenge, because of the wide spectrum of reactivities of the aromatic precursors.
For example, phenols with their strong C-O bond (BDE(C-O) = 110.8 kcal/mol),14 present a particularly challenging problem for visible light-induced C-O-functionalization. Conversion of phenols to sulfonates (e.g., triflates) is commonly used in transition metal catalysis to facilitate the C-O bond cleavage, but this activation method may be of limited value in photoinduced reactions, because sulfonates tend to undergo the competing S-O bond scission upon photoexcitation or photoinduced single electron transfer, regenerating phenols.15 In addition, they have low reduction potentials, placing them outside the range of typical photoredox catalysts and requiring exceptionally strongly reducing photocatalysts with suitable photophysical properties that remain to be developed. As a result, a visible light-mediated C-O-borylation has remained elusive, despite its high potential synthetic value. Given these challenges, a conceptually distinct approach to activation and cleavage of the Ar-O bond is required to achieve the C-O-borylation under visible light, even before any progress is made towards a general photocatalytic borylation platform. On the other hand, in the haloarene series, iodoarenes have relatively weak C-I bonds and less negative reduction potentials (e.g., Ered= −2.39 V vs SCE for p-iodotoluene),16 while chloroarenes, like derivatives of phenols, are much more difficult to reduce (e.g., Ered = −2.84 V for p-chlorotoluene). Hence, the development of a single photocatalytic system that can avoid excessively fast production of aryl radicals from aryl iodides, while also being capable of efficient borylation of chlorides is a significant hurdle.
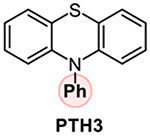
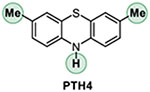
Addressing the C-O borylation problem first, we turned our attention to aryl phosphate esters as a platform for visible light-mediated activation of the C-O bond in phenols, hypothesizing that they are less likely to undergo a SET-induced P-O bond scission, due to the stronger P-O bond. However, aryl phosphate esters have very negative reduction potentials (Ered= −2.97 V vs SCE for p-tolyl diethyl phosphate), necessitating a design of a new photocatalytic system with an enhanced reduction scope. We, therefore, hypothesized that a reductive ability of an appropriately selected organic photocatalyst can be enhanced by a proton-coupled electron transfer (PCET), enabling a photoinduced single electron transfer to substrates with very negative reduction potentials, including electron-rich aryl phosphate esters and chloroarenes (Figure 1). Recent work by Knowles and other groups on proton-coupled electron transfer in photocatalysis has led to the development of new synthetic methods for functionalization of strong bonds.17 In search of a suitable photocatalyst, we turned our attention to phenothiazine PTH1 - a common and inexpensive commodity chemical. Although phenothiazine has not been used as a photocatalyst, W-alkyl and aryl substituted phenothiazines (e.g., PTH2 and PTH3, Table 1) have recently attracted attention as efficient metal-free photocatalysts in polymer chemistry and organic synthesis.18 W-Substituted phenothiazines PTH2 and PTH3 have relatively strongly reducing singlet excited states (Table 1) that allow for production of alkyl and aryl radicals from more easily reducible C-X bonds (e.g, electron deficient haloarenes and α-haloesters). Although phenothiazine PTH1 and the N-substituted analogues PTH2 and PTH3 have very close excited state reduction potentials (Table 1), we hypothesized that phenothiazine PTH1 can engage in a photoinduced PCET process with aryl phosphates in the presence of an appropriate base, effectively enhancing its reduction strength via this distinctly different photoactivation mechanism, and enabling the downstream borylation via a homolytic substitution with a diboron reagent. Given the potentially broad utility of a visible light-driven metal-free photocatalytic system with an adjustable reduction potential, PTH1 photocatalysis can inspire the development of other catalytic systems based on this concept.
Figure 1.
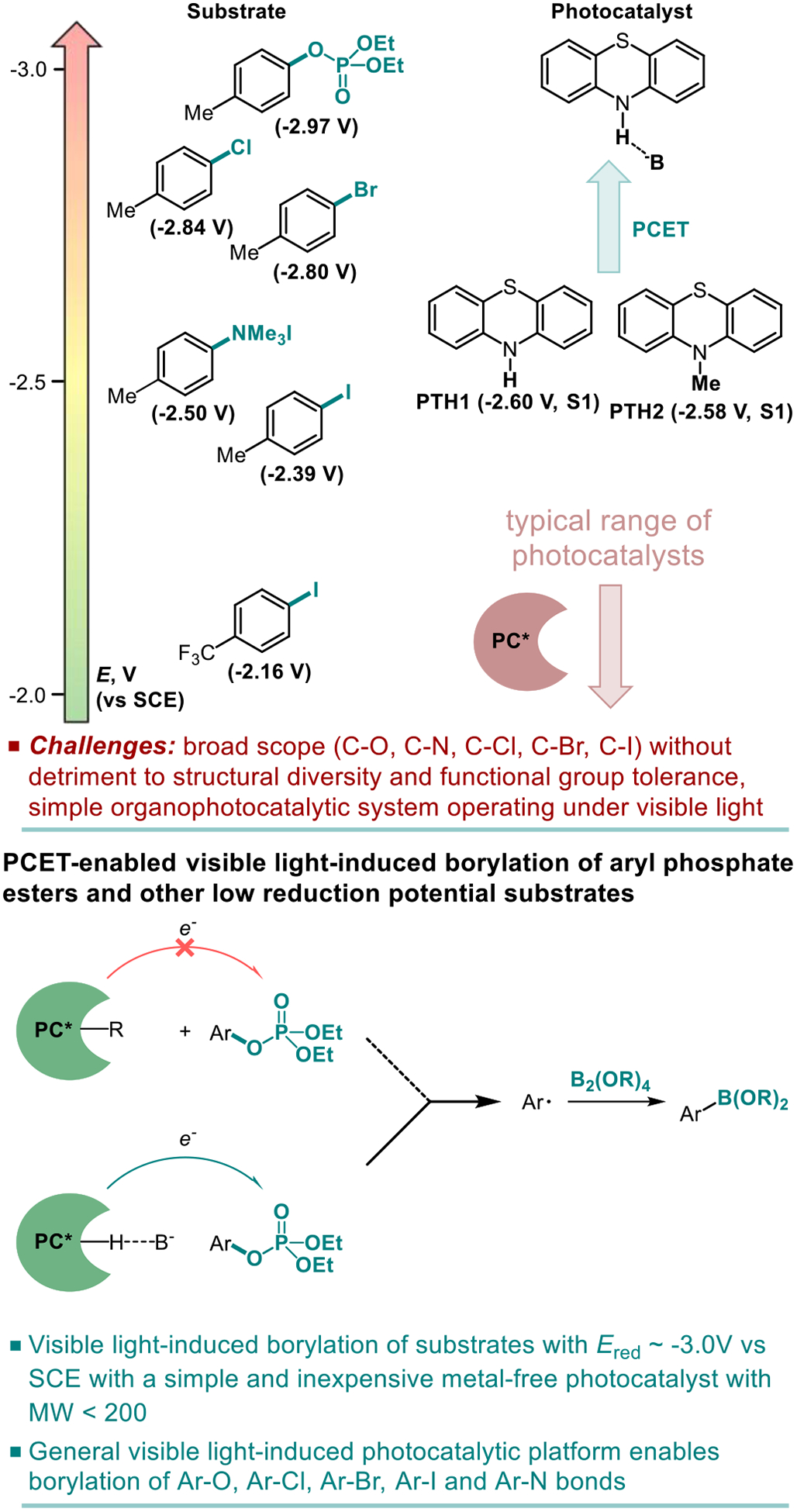
Visible light-induced phenothiazine-catalyzed borylation of substrates with low reduction potentials.
Table 1.
Influence of the Substituents in PTH1–4 on the Catalyst Performance.a
 | ||
|---|---|---|
| Catalyst | Eox(PTH+/1PTH*), V (SCE) | Yield, % |
 |
−2.60 | 64 |
 |
−2.58 | 12 |
 |
−2.51 | 10 |
 |
−2.71 | 68 |
Reaction conditions: phosphate 1 (0.2 mmol), PTH1–4 (10 mol%), B2Pin2 (0.6 mmol), Cs2CO3 (0.6 mmol), MeCN (2 mL), LED light (400 nm), 20 h. Yields were determined by 1H NMR spectroscopy with 1,4-dimethoxybezene as an internal standard.
Results and Discussion
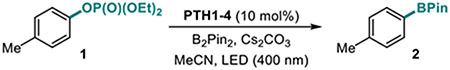
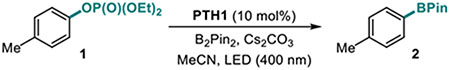
Our studies revealed that the unsubstituted catalyst PTH1 was uniquely active as a photocatalyst for the borylation of aryl phosphate 1, with Cs2CO3 as a base (Table 1), while a mere substitution of the N-hydrogen atom with a methyl or phenyl group (PTH2 and PTH3) led to a substantial decrease in the catalytic activity. This result is striking, given the comparable excited state redox properties of the N-substituted phenothiazines (Table 1). The structurally similar WH-phenothiazine PTH4 showed a slightly improved performance, likely due to the stronger reducing character of the more electron rich core. Notably, none of the photocatalysts PTH1–4 has a sufficiently strongly reducing singlet excited state to effect the reduction of the electron-rich phosphate 1 (Ered= −2.97 V). The borylation product 2 was not formed in the absence of light, PTH1, or cesium carbonate (Table 2). Hypothesizing that addition of reagents that improved solubility and phase transfer of the base would accelerate the borylation, we tested water and 18-crown-6 and observed a faster reaction (Table 2, entries 5 and 6, also see SI) and a higher yield (Table 3, product 2), pointing to the important role of the base in the catalytic process. Other bases were also able to mediate the borylation (entry 7), with cesium carbonate showing an optimal performance. The borylation can be carried out a lower loading of B2Pin2, albeit with a reduced conversion (entry 8). As anticipated, replacing phosphate 1 with the corresponding triflate led to a substantially reduced yield and a predominant S-O bond scission (entry 9).
Table 2.
Reaction Conditions for the Visible Light-Driven C-O Borylation.a
 | ||
|---|---|---|
| Entry | Variations from standard conditions | Yield, % |
| 1 | none | 64 (78b, 83c) |
| 2 | no light | 0 |
| 3 | no PTH1 | 0 |
| 4 | no Cs2CO3 | 0 |
| 5 | with water (1 equiv.), 10 h | 63 |
| 6 | with 18-crown-6 (1 equiv.), 10 h | 60 |
| 7 | K3PO4 instead of Cs2CO3 | 30 |
| 8 | 2 equiv. B2pin2 | 52 |
| 9 | Tolyl triflate instead of 1 | 37d |
Reaction conditions: see footnote for Table 1.
Isolated yield for a reaction in the presence of water (1 equiv.).
Isolated yield after 36 h.
p-Cresol was also formed in 54% yield.
Table 3.
Scope of the Visible Light-Induced C-O Borylation Reaction.a
|
Reaction conditions: phosphate ester (0.2 mmol), B2Pin2 (0.6 mmol), Cs2CO3 (0.6 mmol), PTH1 or PTH4 (2–12 mol%), MeCN (2 mL), LED light (400 or 420 nm).
B2Pin2 (0.4 mmol).
In the presence of water (1 equiv.).
PTH4 was used.
B2Pin2 (0.3 mmol).
The keto group in the phosphate ester was protected as an acetal that was removed concomitantly with conversion to trifluoroborate 31. g 450 nm.
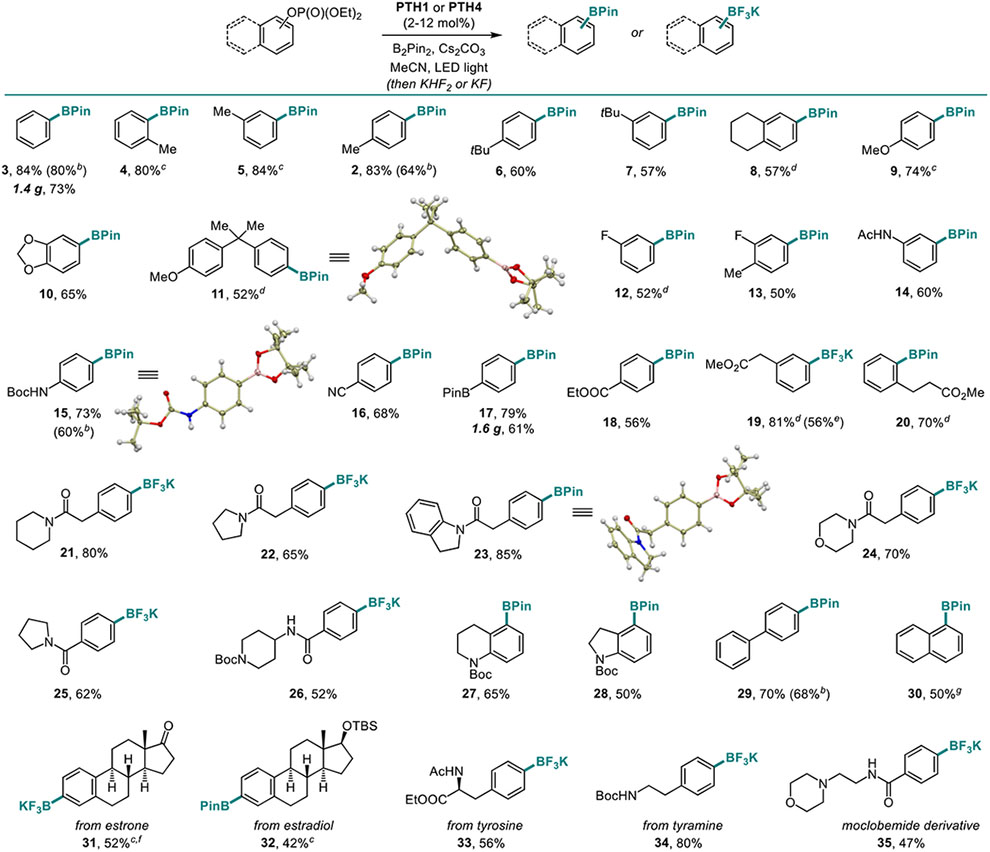
We further proceeded with the evaluation of the scope of the photocatalytic C-O borylation of phosphate esters (Table 3). Electron-rich phosphates, e.g., 2–11 were readily borylated, including those bearing strongly donating alkoxy groups. Medicinally relevant fluoro group (12 and 13), as well as amide (14) and carbamate (15) were also tolerated.
Phosphates with electron-withdrawing groups (e.g., cyano, boryl, ester groups, 16–18) were equally suitable substrates. All substitution patterns (o-, m-, p-) were tolerated (18–20), and gram-scale syntheses were readily performed (3 and 17). Similarly, phosphates bearing various N-heterocyclic substituents were converted to the corresponding boronic esters 21–28 in good yields. Further more, biphenyl and naphthalene-derived phosphates were readily borylated (29–30), suggesting that the reaction can be applicable to polyaromatic systems. Derivatives of estrone, estradiol, tyrosine, tyramine and moclobemide were subjected to the photoinduced C-O borylation and afforded boronates 31–35, indicating that the new method can be used for borylation of naturally-occurring and medicinally-relevant phenols. Catalysts PTH1 and PTH4 both performed well, and longer wavelength LED light (420 and 450 nm) was also used with success. The optimal catalyst loading was in the 10–12 mol% range, but a lower (2 mol%) loading is possible for more reactive substrates (e.g., 16). Furthermore, the borylation products can be isolated as organotrifluoroborate salts19 as a part of the work-up procedure, further enhancing the practicality of the method.
With the C-O borylation optimized, we were curious to test the performance of the new photocatalytic method on other strong carbon-heteroatom bonds. In particular, functionalization of the strong Ar-N bonds has remained a challenge. Our previous work showed that the C-N-borylation of quaternary arylammonium salts is feasible under UV light irradiation.8 However, a visible light-mediated C-N-borylation of quaternary arylammonium salts has remained elusive. Initial experiments with phenyltrimethylammonium halides (chlorides, bromides and iodides) demonstrated that the PTH1-photocatalyzed C-N-borylation proceeds smoothly (3, Table 4). Furthermore, in contrast to the UV light-induced reaction that required iodide and bromide as counteranions, the visible light-mediated reaction is insensitive to the counteranion, and proceeds equally well with all halide salts, as well as triflates and methylsulfates. Other N-alkyl groups were also tolerated in the ammonium residue. We further investigated the scope of the reaction with a variety of quaternary arylammonium salts. Substituents in the ortho, meta, and para positions were well-tolerated (4, 9, 37–41). Substrates bearing medicinally-relevant fluorine-containing groups (CF3, CF3O, F2HCO) were readily borylated (39–41), in addition to ester and sulfone groups (42, 43), as well as a naphthalene salt (30). Heterocycle-containing products 44–46 were obtained in good yields. Furthermore, the C-N-borylation performed well in the molecular setting of active pharmaceutical ingredients (API), allowing for preparation of borylated derivatives of aminoglutethimide (Cytadren, 47) and neostigmine (Prostigmin, 48). The prerequisite quaternary arylammonium salts can be prepared in situ by brief pretreatment of the corresponding aniline or W-alkylaniline with MeOTf. The reaction can be readily performed on gram scale (e.g., boronates 4 and 30). As with the C-O-borylation, the products can be isolated as organotrifluoroborate salts or boronic acids. Quaternary arylammonium salts were generally more reactive than phosphate esters, and 1–5 mol% PTH1 was sufficient to effect the borylation for most of the substrates. On the mechanistic level, the insensitivity of the visible light-mediated borylation of the quaternary ammonium salts to the counteranion indicated that the reaction does not proceed by the charge transfer from the anion, as it was observed in the UV light-induced reaction.8
Table 4.
Scope of the Visible Light-Induced C-N Borylation Reaction of Quaternary Ammonium Salts.a
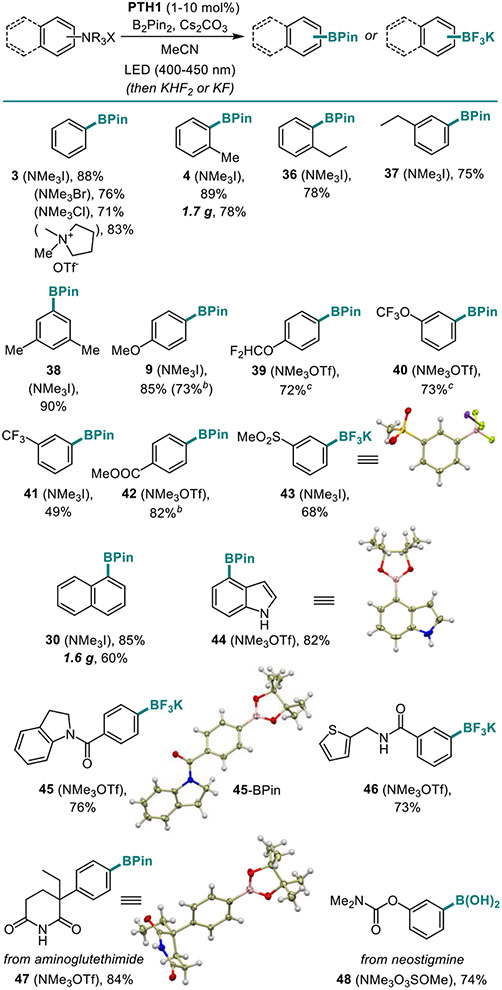
|
Reaction conditions: see footnote for Table 3.
B2Pin2 (0.3 mmol).
The salt was prepared in situ by stirring with MeOTf (3.2 equiv.) for 20 min at rt before adding B2Pin2 and PTH1.
Haloarenes are the most commonly used and readily accessible precursors to boronic acids, and it is important to have a broadly synthetically useful borylation method that can effect the borylation of the three most abundant classes of haloarenes, - chloro-, bromo-, and iodoarenes, in addition to phenols and anilines. We, therefore, tested the performance of the new visible light-mediated PTH1-photocatalyzed borylation reaction with chlorobenzene, as well as para-chloro- bromo- and iodotoluene (Table 5). Remarkably, an efficient borylation was observed with as little as 0.2–2 mol% PTH1 for all three classes of halides. Chlorobenzene and para-chlorotoluene were converted to the corresponding boronic esters 3 and 2 in good yields, despite their very negative reduction potentials (−2.83 and −2.84 V, respectively). Other alkyl-substituted aryl chlorides, bromides, and iodides were also borylated efficiently (4–6, 38, 49, 50), including the electron-rich ortho,para-substituted 4-chloro-m-xylene (Ered = −2.92 V, boronate 50). The reaction allows for production of fluorinated boronic esters (12, 13, 39, 40, 51–57), as well as for selective borylation of a more readily reduced C-X bond (58). Electron-rich 4-chloroanisole was readily reacted, as well as other alkoxy group-containing haloarenes (9, 10, 59). Boryl, thio, unprotected amino, amido, ester, and other electron-withdrawing groups (16, 17, 60–67) were well-tolerated. A variety of biaryl and polycyclic haloarenes (29, 30, 68–74) were converted to the corresponding boronic esters in good yields, independently of the halogen leaving group. Some reactive haloarenes required as little as 0.2 mol% of PTH1, and most of the reactions were performed with 1.2–2 equiv. B2Pin2. The reaction can be readily carried out on a gram scale (16, 61).
Table 5.
Scope of the Visible Light-Induced C-X Borylation Reaction.a
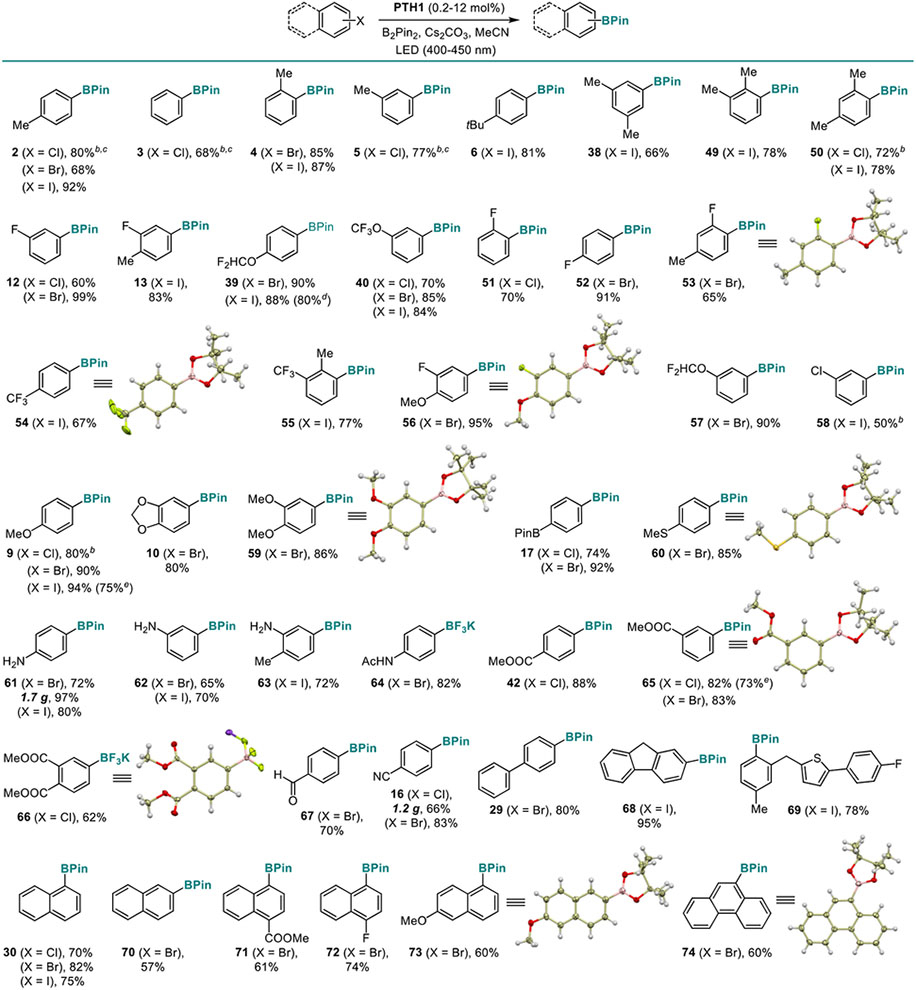
|
Reaction conditions: see footnote for Table 3 with 2 equiv. B2Pin2.
In the presence of water (1 equiv.).
3 equiv. B2Pin2.
1.2 equiv. B2Pin2.
1.5 equiv. B2Pin2.
Heterocyclic boronates are key precursors for many cross-coupling reactions that are difficult to produce by conventional borylation strategies, due to the interference of electrophilic or coordinating heteroatom-centered residues. We, therefore, evaluated the performance of the new catalytic system with a wide range of heterocyclic precursors (Table 6). A wide range of substituted pyridine-boronates (75–84), including those bearing unprotected amino group (75), fluoro (78 and 79), ester, cyano, trifluoromethyl and amido groups (80–84) were readily accessed. Borylated quinolines (85–87), indoles, and carbazoles (88–91) were also synthesized in good yields. Since heterocycles that contain multiple nitrogen atoms present a challenge for conventional borylation methods, we evaluated the reaction performance with borylated heterocycles 92–95 and observed smooth conversion. Oxygen- and sulfur-containing heterocycles were also tolerated (96–100).
Table 6.
Scope of the Visible Light-Induced C-X Borylation Reaction of Haloheteroarenes.a
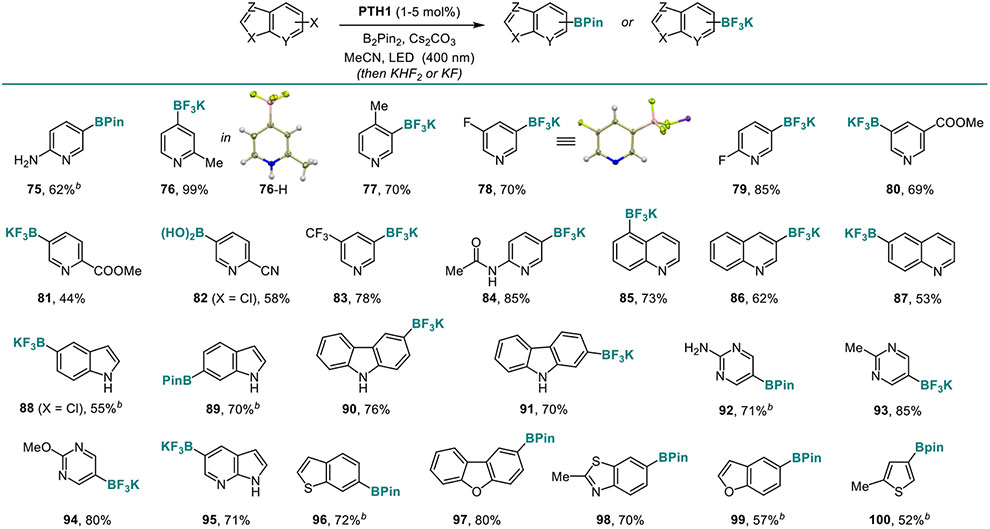
|
Reaction conditions: see footnote for Table 3 with 2 equiv. B2Pin2, X = Br unless otherwise specified.
3 equiv. B2Pin2.
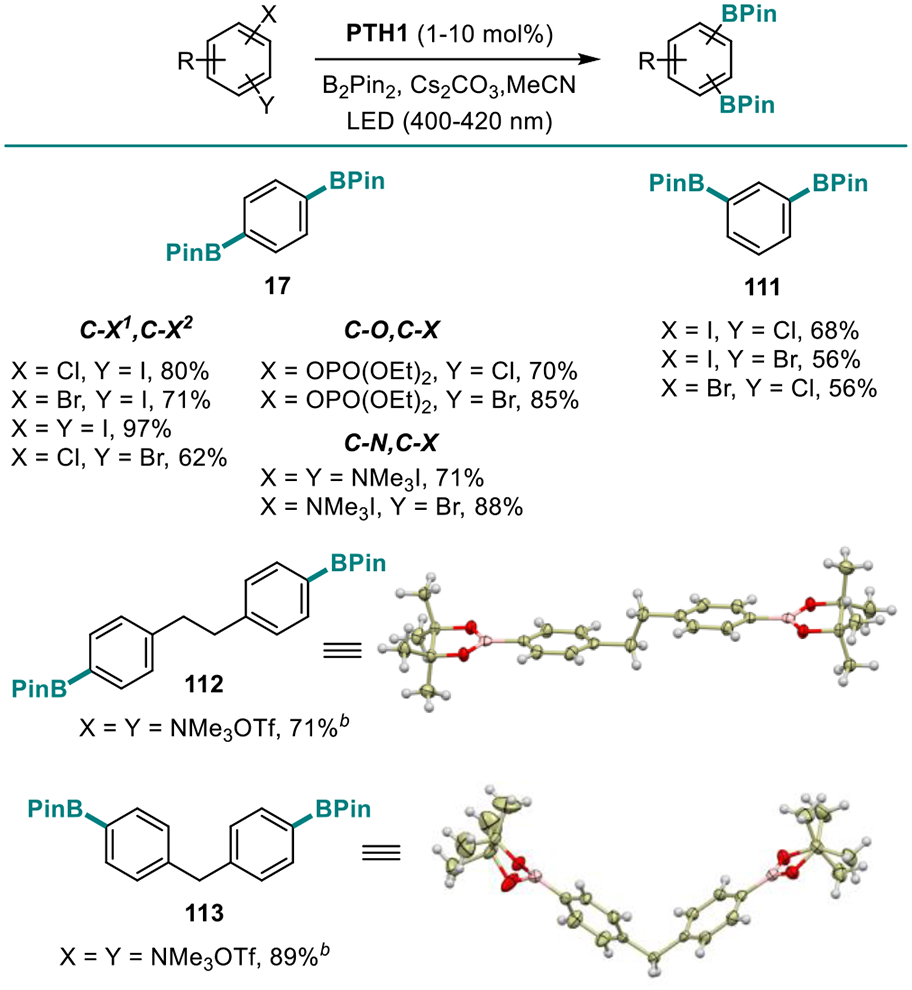
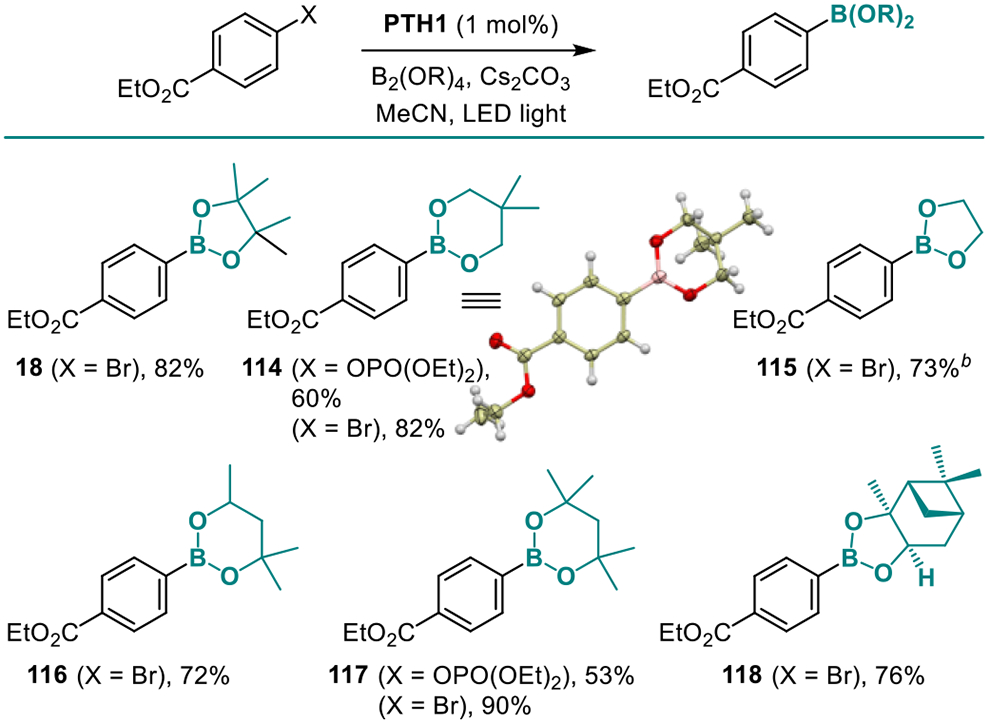
The new borylation preformed equally well in the more complex structural settings of natural products and APIs (Table 7). Boronates derived from salicine (101), strychnine (102), as well as a variety of APIs and estrone (35, 103–110) bearing basic heteroatoms, polar groups, and heterocyclic frameworks were readily synthesized, confirming versatility of the new method. The ability of the new catalytic system to effect the borylation of all five major classes of C-X bonds can be exploited for the simultaneous introduction of two boryl groups, en route to diborylarenes that have found applications as linchpins and phosphors in materials science.3 Thus, various combinations of C-O, C-Cl, C-Br, C-I, and C-N bonds were simultaneously borylated, providing a straightforward access to regioisomeric diborylarenes from a variety of halogenated phenols, anilines, as well as dihaloarenes, by-passing intermediates (Table 8, 17, 111–113). Additionally, we tested the performance of the catalytic system with a range of diboron esters (Table 9), since generation of various boryl esters using a single borylation protocol presents remains a challenge and is generally available only with UV-induced protocols. In all cases, a uniformly smooth performance was observed, and the corresponding products (18, 114–118) were obtained in good yields. In contrast, B2(OH)4 proved to be unsuitable, due to insolubility in acetonitrile. Boronic acids can still be prepared using the present method, as exemplified by product 48, by a simple modification in the workup procedure (See SI).
Table 7.
Scope of the Visible Light-Induced C-X Borylation Reaction of APIs and Natural Products.a
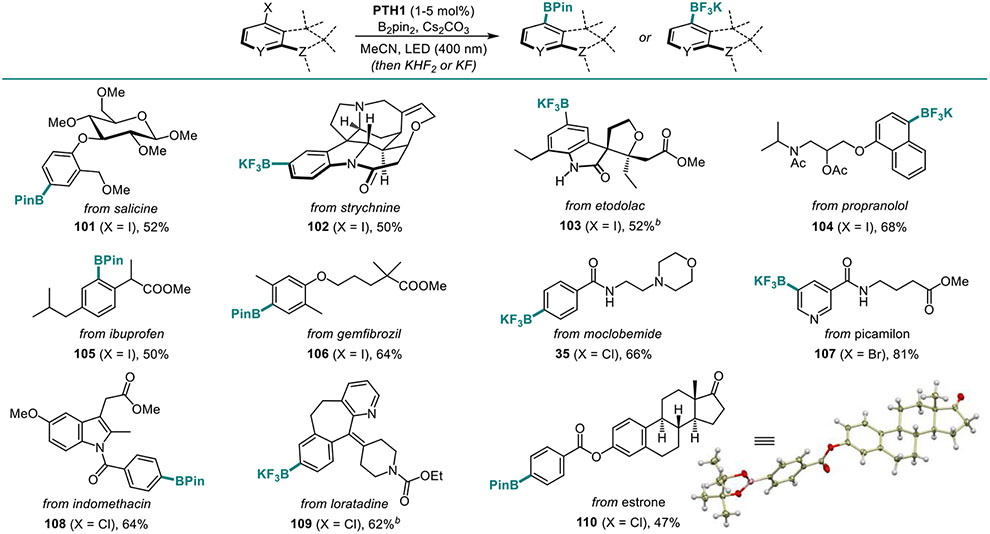
|
Reaction conditions: see footnote for Table 3 with 2 equiv. B2Pin2.
3 equiv. B2Pin2.
Table 8.
Scope of the Visible Light-Induced Dual Borylation.a
|
Reaction conditions: see footnote for Table 3.
The salt was prepared in situ by stirring the aniline precursor with MeOTf for 30 min at rt before adding B2PM2 and PTH1.
Table 9.
Scope of the Boronate Esters.a
|
Reaction conditions: see footnote for Table 3 with 2 equiv. B2Pin2.
Isolated as boronic acid.
Initial experimental observations of the pronounced W-substitution effects on the catalytic performance of phenothiazines PTH1–3 provided a strong indication of the crucial role of the N-H group for the photoredox catalysis by PTH1 (Table 1) that was also previously observed for other PCET-enabled catalytic systems.3f,g Furthermore, the acceleration observed with reagents that improve solubility and phase transfer of cesium carbonate also points to the equally important role of the base. We, therefore, conducted a series of experiments in order to clarify the mechanistic details of the photocatalysis. First, the aryl radical intermediacy was confirmed in radical clock experiments with phosphate 119 that produced boronates 120 and 121 (Figure 2.A). The bimolecular rate constant of the reaction of the aryl radical with B2pin2 (k = 1.1·108 M−1·s−1) was comparable with the value reported by Studer (7.4·107 M−1·s−1) for the reaction of the catechol-derived diboron reagent B2Cat2 with the aryl radical produced from the corresponding iodoarene.12 We next examined the roles of cesium carbonate and the diboron reagent in the photoredox catalysis by PTH1 by means of 1H NMR spectroscopy. First, formation of the phenothiazine anion from PTH1 in the presence of cesium carbonate was ruled out (Figure 2.B, PTH1-K, potassium salt of phenothiazine was used, cf. traces a-c, h), indicating that the reaction does not proceed by a stepwise mechanism with the phenothiazine anion acting as an electron donor in a photoinduced electron transfer. This conclusion was independently corroborated by UV/Vis studies of the PTH1/Cs2CO3/B2Pin2 system (see SI). Further, no significant changes in the 1H NMR spectrum of PTH1 were observed in the presence of B2Pin2, and only a minor shift of the N-H signal was observed in the PTH1-Cs2CO3 system, as expected from the poor solubility of Cs2CO3 in acetonitrile (cf. traces a,c, d). Significantly, addition of B2Pin2 to the PTH1-Cs2CO3 system led to a downfield shift of the N-H signal (cf. traces c and h). The same shift was observed for the PTH1-Cs2CO3 system in the presence of crown ethers (cf. traces f,g, and h). No interaction between PTH1 and B2PM2 or crown ethers was observed in the absence of Cs2CO3 (cf. traces d, e and g, h). These observations suggest that, in addition to the primary role as a borylation reagent, B2Pin2 also serves as a phase transfer mediator for cesium carbonate, mimicking the behavior of crown ethers. In support of this conclusion, the B2Pin−Cs+ species was detected in the solution of PTH1 in the presence of Cs2CO3 and B2Pin2 by HRMS (MW = 387.0920), while no signals corresponding to the sp3-adduct of B2Pin2 and Cs2CO3 were detected by 11B NMR spectroscopy, indicating that B2Pin2 primarily interacts with the base via the cesium ions. Taken together, these results suggest a hydrogen bonding interaction of PTH1 and Cs2CO3 that is assisted by B2Pin2. The role of the PTH1‒Cs2CO3 interaction in the borylation was further probed by fluorescence quenching experiments with representative substrates (Ar-LG, LG = O2P(OEt)2, Cl, Br, I, NMe3I) (Figure 3). For the purpose of the fluorescence quenching experiments, 18-crown-6 was used as a phase transfer mediator in place of B2Pin2 to ensure that the quenching is not affected by the concomitant borylation, and given the similar behavior of the crown ester and B2Pin2 (vide supra). As expected, no PTH1 fluorescence quenching was observed with phosphate ester 12220 in the absence of Cs2CO3 and the crown ether.21 However, the fluorescence was quenched in the PTH1-Cs2CO3-18-crown-6 system, pointing to the involvement of a PTH1-carbonate-enabled PCET process in the borylation. p-Chlorotoluene and p-bromotoluene exhibited similar quenching behavior, consistent with their low reduction potentials. Iodoarenes and arylammonium salts, on the other hand, have less negative reduction potentials, and direct photoinduced electron transfer from excited PTH1 was anticipated to contribute to the borylation process. Indeed, PTH1 fluorescence quenching was observed with p-iodotoluene. In line with the expected contribution of the PTH1-carbonate-driven PCET process, stronger quenching was observed in the PTH1-Cs2CO3-crown ether system. The same quenching behavior was observed for phenyltrimethylammonium iodide, as anticipated for the more readily reducible substrate. Taken together, the pronounced N-substitution effects combined with the results of the 1H NMR and fluorescence quenching studies suggest that the photoredox catalysis by PTH1 is enabled by a proton-coupled electron transfer process arising from the PTH1-carbonate interaction exemplified by complex 123 (Figure 4), resulting in the borylation of substrates that have previously not been suitable for visible light-induced photoredox catalysis.
Figure 2.
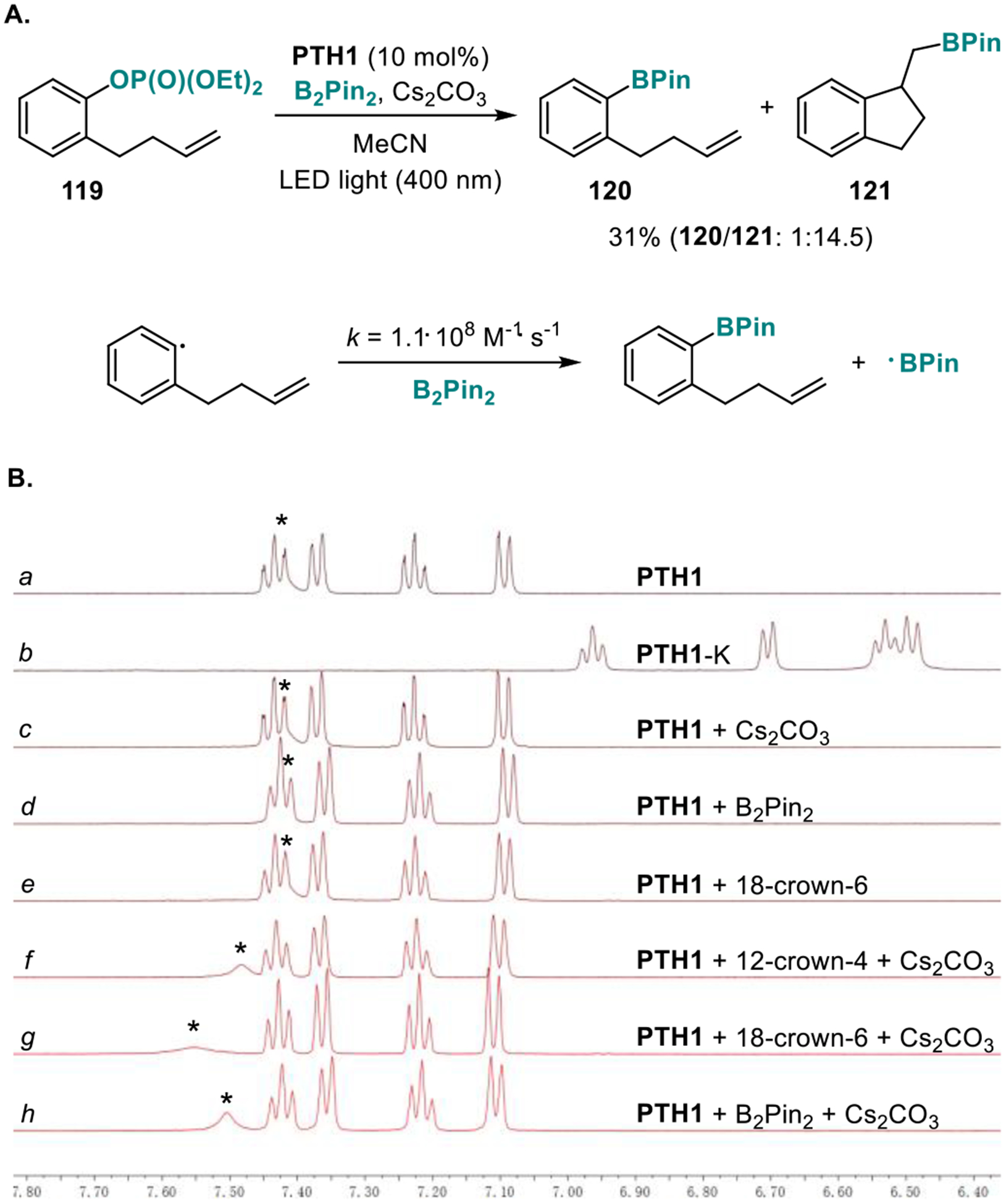
Mechanistic studies of the PTH1 photoredox catalyzed borylation reaction. A. Radical clock experiments with phosphate ester 119. B. 1H NMR spectra of PTH1 (a), potassium salt of PTH1 (PTH1-K, b), as well as PTH1 in the presence of Cs2CO3, B2Pin2, and crown ethers (c-h), in acetonitrile with a sealed coaxial capillary containing d12-cyclohexane.
Figure 3.
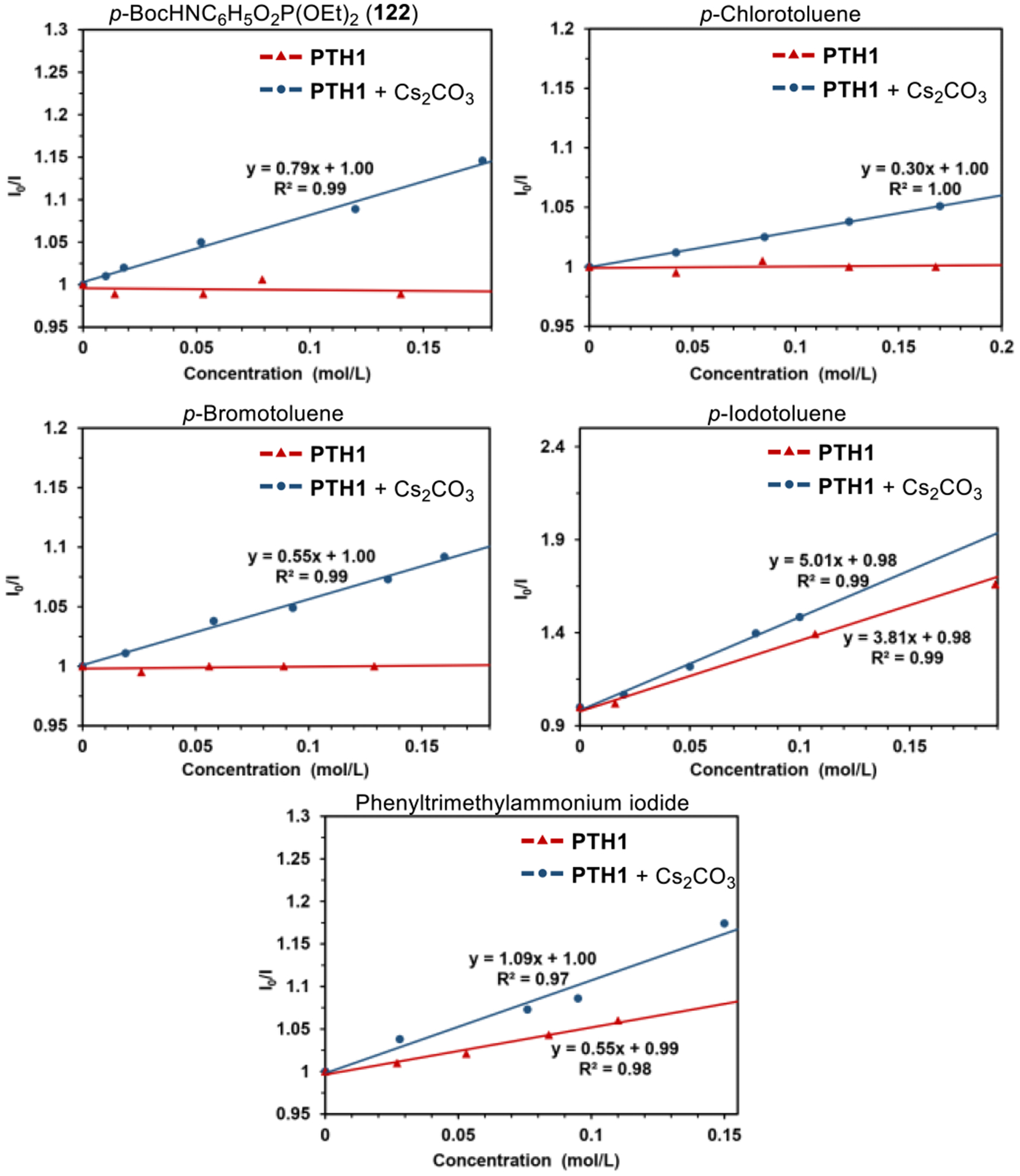
Fluorescence quenching experiments for PTH1 in the presence and in the absence of cesium carbonate and 18-crown-6 with representative borylation substrates.
Figure 4.
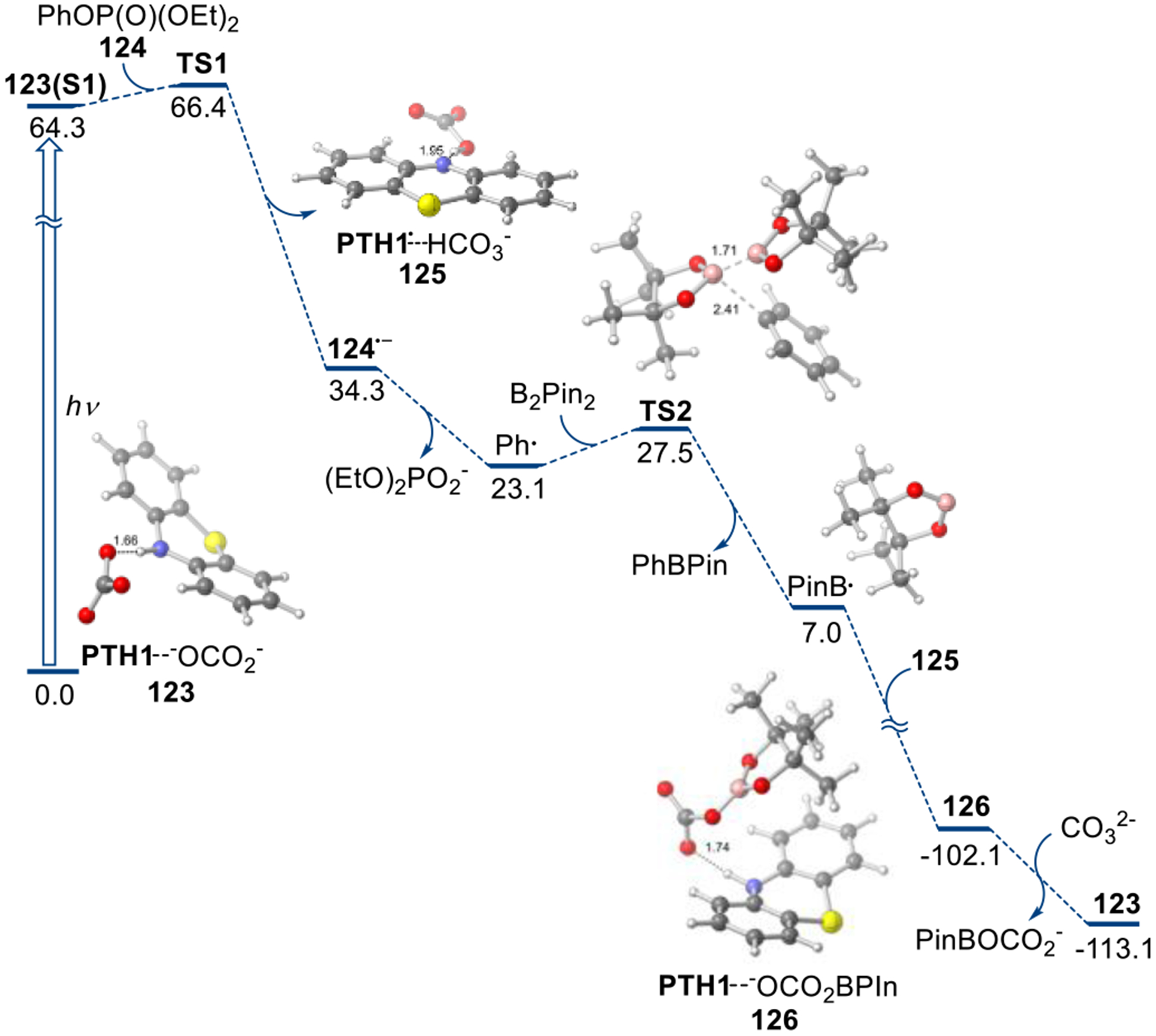
Computed Gibbs free energy profile of the PTH1-photoredox catalyzed borylation reaction of phosphate esters, ΔG, kcal/mol.
Thermodynamic and kinetic feasibility of the PTH1-carbonate photocatalytic borylation was further assessed computationally. (TD)DFT calculations show that the photoinduced proton-coupled electron transfer renders the one-electron reduction of aryl phosphate esters thermodynamically favorable from the singlet excited state of complex 123 (Figure 4, modelled with phosphate ester 124). Furthermore, Marcus theory-based estimations suggest that the reduction may occur by a stepwise dissociative process, proceeding with a low activation barrier (2.1 kcal/mol) via radical anion intermediate 124−. The subsequent homolytic substitution reaction of the generated aryl radical with B2Pin2 also proceeds with a low barrier of 4.4 kcal/mol. Importantly, the ensuing barrierless and exergonic reaction of the intermediate boryl radical with the oxidized photocatalyst complex 125 (i.e., phenothiazinyl radical - hydrogen carbonate complex) provides a pathway for the regeneration of catalytically active complex 123. The computational conclusions are consistent with the experimental observations and provide further support to the borylation mechanism.
Conclusion
In conclusion, we have developed a metal-free, visible light-induced photocatalytic borylation platform that enables borylation of a range of carbon-heteroatom bonds, including, for the first time, C-O borylation of electron rich phenol derivatives, chloroarenes, and C-N borylation of aniline-derived quaternary ammonium salts. The scope and functional group tolerance of the method were further demonstrated on a number of representative carbocyclic and heterocyclic substrates, as well as natural products and medicinally relevant compounds. The photocatalysis is enabled by the simple, abundant and low molecular weight heterocyclic commodity chemical phenothiazine PTH1 that effects single electron reduction of substrates with strong bonds and low reduction potentials by a proton-coupled electron transfer mechanism.
Supplementary Material
ACKNOWLEDGMENT
Financial support by the Welch Foundation (AX-1788), NSF (CHE-1455061) and NIGMS (GM134371) is gratefully acknowledged. UTSA NMR and X-ray crystallography facilities were supported by NSF (CHE-1625963 and CHE-1920057). The authors acknowledge the Texas Advanced Computing Center (TACC) at UT Austin for providing computational resources.
Footnotes
Supporting Information
Experimental and spectral details for all new compounds and all reactions reported. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Suzuki A; Brown HC Organic Syntheses Via Boranes; Aldrich Chemical Company: Milwakee, 2003; Vol. 3. [Google Scholar]; (b) Hall DG Boronic Acids, 2nd ed.; Wiley-VCH: Weinheim, 2011. [Google Scholar]
- (2).(a) Corey EJ Catalytic Enantioselective Diels-Alder Reactions: Methods, Mechanistic Fundamentals, Pathways, and Applications. Angew. Chem., Int. Ed 2002, 41, 1650. [DOI] [PubMed] [Google Scholar]; (b) Ishihara K Synthesis and Application of Organoboron Compounds. Top. Organomet. Chem 2015, 49, 243. [Google Scholar]
- (3).(a) Shoji Y; Ikabata Y; Wang Q; Nemoto D; Sakamoto A; Tanaka N; Seino J; Nakai H; Fukushima T Unveiling a New Aspect of Simple Arylboronic Esters: Long-Lived Room-Tempera-ture Phosphorescence from Heavy-Atom-Free Molecules. J. Am. Chem. Soc 2017, 139, 2728. [DOI] [PubMed] [Google Scholar]; (b) Huang N; Ding X; Kim J; Ihee H; Jiang D A Photoresponsive Smart Covalent Organic Framework. Angew. Chem., Int. Ed 2015, 54, 8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Ban HS; Nakamura H Boron-Based Drug Design. Chem. Rec 2015, 15, 616. [DOI] [PubMed] [Google Scholar]
- (5).Wu J; Kwon B; Liu W; Anslyn EV; Wang P; Kim JS Chromogenic/Fluorogenic Ensemble Chemosensing Systems. Chem. Rev 2015, 115, 7893. [DOI] [PubMed] [Google Scholar]
- (6).(a) Cho J-Y; Tse MK; Holmes D; Maleczka RE; Smith MR Remarkably Selective Iridium Catalysts for the Elaboration of Aromatic C-H Bonds. Science 2002, 295, 305. [DOI] [PubMed] [Google Scholar]; (b) Mazzacano TJ; Mankad NP Base Metal Catalysts for Photochemical C-H Borylation That Utilize Metal-Metal Co-operativity. J. Am. Chem. Soc 2013, 135, 17258. [DOI] [PubMed] [Google Scholar]; (c) Molander GA; Trice SLJ; Dreher SD Palladium-Catalyzed, Direct Boronic Acid Synthesis from Aryl Chlorides: A Simplified Route to Diverse Boronate Ester Derivatives. J. Am. Chem. Soc 2010, 132, 17701. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Huang K; Yu D-G; Zheng S-F; Wu Z-H; Shi Z-J Borylation of Aryl and Alkenyl Carbamates through Ni-Catalyzed C-O Activation. Chem. Eur. J 2011, 17, 786. [DOI] [PubMed] [Google Scholar]; (e) Dudnik AS; Fu GC Nickel-Catalyzed Coupling Reactions of Alkyl Electrophiles, Including Unactivated Tertiary Halides, To Generate Carbon-Boron Bonds. J. Am. Chem. Soc 2012, 134, 10693. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Nakamura K; Tobisu M; Chatani N Nickel-Catalyzed Formal Homocoupling of Methoxyarenes for the Synthesis of Symmetrical Biaryls via C-O Bond Cleavage. Org. Lett 2015, 17, 6142. [DOI] [PubMed] [Google Scholar]; (g) Zarate C; Manzano R; Martin R Ipso-Borylation of Aryl Ethers via Ni-Catalyzed C-OMe Cleavage. J. Am. Chem. Soc 2015, 137, 6754. [DOI] [PubMed] [Google Scholar]; (h) Atack TC; Cook SP Manganese-Catalyzed Borylation of Unactivated Alkyl Chlorides. J. Am. Chem. Soc 2016, 138, 6139. [DOI] [PubMed] [Google Scholar]; (i) Yoshida T; Ilies L; Nakamura E Iron-catalyzed Borylation of Aryl Chlorides in the Presence of Potassium t-Butoxide. ACS Catal. 2017, 7, 3199. [Google Scholar]; (j) Li C; Wang J; Barton LM; Yu S; Tian M; Peters DS; Kumar M; Yu AW; Johnson KA; Chatterjee AK; Yan M; Baran PS Decarboxylative Borylation. Science 2017, 356, eaam7355. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Arevalo R; Chirik PJ Enabling Two-Electron Pathways with Iron and Cobalt: From Ligand Design to Catalytic Applications. J. Am. Chem. Soc 2019, 141, 9106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Malapit CA; Bour JR; Laursen SR; Sanford MS Mechanism and Scope of Nickel-Catalyzed Decarbonylative Borylation of Carboxylic Acid Fluorides. J. Am. Chem. Soc 2019, 141, 17322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Nagashima Y; Takita R; Yoshida K; Hirano K; Uchiyama M Design, Generation, and Synthetic Application of Borylzincate: Borylation of Aryl Halides and Borylzincation of Ben-zynes/Terminal Alkyne. J. Am. Chem. Soc 2013, 135, 18730. [DOI] [PubMed] [Google Scholar]; (b) Bose SK; Fucke K; Liu L; Steel PG; Marder TB Zinc-Catalyzed Borylation of Primary, Secondary and Tertiary Alkyl Halides with Alkoxy Diboron Reagents at Room Temperature. Angew. Chem., Int. Ed 2014, 53, 1799. [DOI] [PubMed] [Google Scholar]; (c) Warner AJ; Lawson JR; Fasano V; Ingleson MJ Formation of C(sp2)-Boronate Esters by Borylative Cyclization of Alkynes Using BCl3. Angew. Chem., Int. Ed 2015, 54, 11245. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Legare MA; Courtemanche MA; Rochette E; Fontaine FG Metal-Free Catalytic C-H Bond Activation and Borylation of Heteroarenes. Science 2015, 349, 513. [DOI] [PubMed] [Google Scholar]; (e) Mo F; Qiu D; Zhang Y; Wang J Renaissance of Sandmeyer-Type Reactions: Conversion of Aromatic C-N Bonds into C-X Bonds (X = B, Sn, P, or CF3). Acc. Chem. Res 2018, 51, 496. [DOI] [PubMed] [Google Scholar]; (f) Zhang L; Jiao L Pyridine-Catalyzed Radical Borylation of Aryl Halides. J. Am. Chem. Soc 2017, 139, 607. [DOI] [PubMed] [Google Scholar]; (g) Zhang L; Jiao L Super Electron Donors Derived from Diboron. Chem. Sci 2018, 9, 2711. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Su Y; Cao D; Do H; Li Y; Kinjo R Metal-Free Selective Borylation of Arenes by a Diazadiborinine via C-H/C-F Bond Activation and Dearomatization. J. Am. Chem. Soc 2019, 141, 13729. [DOI] [PubMed] [Google Scholar]; (i) Lv J; Chen X; Xue X-S; Zhao B; Liang Y; Wang M; Jin L; Yuan Y; Han Y; Zhao Y; Lu Y; Zhao J; Sun W-Y; Houk KN; Shi Z Metal-Free Directed sp2-C-H Borylation. Wature 2019, 575, 336. [DOI] [PubMed] [Google Scholar]
- (8).(a) Mfuh AM; Doyle JD; Chhetri B; Arman HD; Larionov _OV Scalable, Metal-and Additive-Free, Photoinduced Borylation of Haloarenes and Quaternary Arylammonium Salts. J. Am. Chem. Soc 2016, 138, 2985. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen K; Zhang S; He P; Li P Efficient Metal-Free Photochemical Borylation of Aryl Halides under Batch and Continuous-Flow Conditions. Chem. Sci 2016, 7, 3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Liu W; Yang X; Gao Y; Li C-J Simple and Efficient Generation of Aryl Radicals from Aryl Triflates: Synthesis of Aryl Boronates and Aryl Iodides at Room Temperature. J. Am. Chem. Soc 2017, 139, 8621. [DOI] [PubMed] [Google Scholar]
- (10).Zhang L; Jiao L Visible-Light-Induced Organocatalytic Borylation of Aryl Chlorides. J. Am. Chem. Soc 2019, 141, 9124. [DOI] [PubMed] [Google Scholar]
- (11).Qiao Y; Yang Q; Schelter E Photoinduced Miyaura Borylation by a Rare Earth Photoreduct-ant: the Hexachlorocerate(III) Anion. Angew. Chem., Int. Ed 2018, 57, 10999. [DOI] [PubMed] [Google Scholar]
- (12).Cheng Y; Muck-Lichtenfeld C; Studer A Metal-Free Radical Borylation of Alkyl and Aryl Iodides. Angew. Chem. Int. Ed 2018, 57, 16832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Cheung MS; Lin Z; Li P Metal-Free Borylation of Electron-Rich Aryl (Pseudo)halides under Continuous-Flow Photolytic Conditions. Org. Chem. Front 2016, 3, 875. [Google Scholar]; (b) Candish L; Teders M; Glorius F Transition-Metal-Free, Visible-Light-Enabled Decarboxylative Borylation of Aryl W-Hydroxyphthalimide Esters. J. Am. Chem. Soc 2017, 139, 7440. [DOI] [PubMed] [Google Scholar]; (c) Fawcett A; Pradeilles J; Wang Y; Mutsuga T; Myers EL; Aggarwal VK Photoinduced Decarboxylative Borylation of Carboxylic Acids. Science 2017, 357, 283. [DOI] [PubMed] [Google Scholar]; (d) Jin S; Nguyen VT; Dang HT; Nguyen DP; Arman HD; Larionov OV Photoinduced Carboborative Ring Contraction Enables Regio- and Stereoselective Synthesis of Multiply Substituted Five-Membered Carbocycles and Heterocycles. J. Am. Chem. Soc 2017, 139, 11365. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Hu D; Wang L; Li P Decarboxylative Borylation of Aliphatic Esters under Visible-Light Photoredox Conditions. Org. Lett 2017, 19, 2770. [DOI] [PubMed] [Google Scholar]; (f) Wu J; He L; Noble A; Aggarwal VK Photoinduced Deaminative Borylation of Alkylamines. J. Am. Chem. Soc 2018, 140, 10700. [DOI] [PubMed] [Google Scholar]; (g) Cheng Y; Muck-Lichtenfeld C; Studer A Transition Metal-Free 1,2-Carboboration of Unactivated Alkenes. J. Am. Chem. Soc 2018, 140, 6221. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Friese FW; Studer A Deoxygenative Borylation of Secondary and Tertiary Alcohols. Angew. Chem. Int. Ed 2019, 58, 9561. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Wu J; Baer R; Guo L; Noble A; Aggarwal VK Photoinduced Deoxygenative Borylations of Aliphatic Alcohols. Angew. Chem. Int. Ed 2019, 58, 18830. [DOI] [PubMed] [Google Scholar]; (j) Mazzarella D; Magagnano G; Schweitzer-Chaput B; Melchiorre P Photochemical Organocatalytic Borylation of Alkyl Chlorides, Bromides, and Sulfonates. ACS Catal. 2019, 97, 5876. [Google Scholar]; For reviews, see:; (k) Nguyen VD; Nguyen VT; Jin S; Dang HT; Larionov OV Organoboron Chemistry Comes to Light: Recent Advances in Photoinduced Synthetic Approaches to Organoboron Compounds. Tetrahedron 2019, 75, 584. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Friese FW; Studer A New Avenues for C-B Bond Formation via Radical Intermediates. Chem. Sci 2019, 10, 8503. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Liu W; Li J; Huang C-Y; Li C-J Aromatic Chemistry in the Excited State: Facilitating Metal-Free Substitutions and Cross-Couplings. Angew. Chem. Int. Ed 2019, DOI: 10.1002/anie.201909138. [DOI] [PubMed] [Google Scholar]
- (14).Luo Y-R Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, FL, 2007. [Google Scholar]
- (15).Photochemistry of Phosphate and Sulfonate Esters. Ravelli D; Fagnoni M In CRC Handbook of Organic Photochemistry and Photobiology, 3rd Ed.; Griesbeck AG; Oelgemoller M; Ghetti F; Eds.; CRC Press: Boca Raton, 2012. [Google Scholar]
- (16).All reduction potentials are reported vs SCE in acetonitrile.
- (17).(a) Choi GJ; Zhu Q; Miller DC; Gu CJ; Knowles RR Catalytic Alkylation of Remote C-H Bonds Enabled by Proton-Coupled Electron Transfer. Wature 2016, 539, 268. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yayla HG; Wang H; Tarantino KT; Orbe HS; Knowles RR Catalytic Ring-Opening of Cyclic Alcohols Enabled by PCET Activation of Strong O-H Bonds. J. Am. Chem. Soc 2016, 138, 10794. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chen M; Zhao X; Yang C; Xia W Visible-light-triggered directly reductive arylation of carbonyl/iminyl derivatives through photocatalytic PCET. Org. Lett 2017, 19, 3807. [DOI] [PubMed] [Google Scholar]; (d) Nguyen VD; Nguyen VT; Haug GC; Dang HT; Jin S; Li Z; Flores-Hansen C; Benavides B; Arman HD; Larionov OV ACS Catal. 2019, 9, 9485. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Morton CM; Zhu Q; Ripberger H; Troian-Gautier L; Toa ZSD; Knowles RR; Alexanian EJ C-H Alkylation via Multisite Proton-Coupled Electron Transfer of an Aliphatic C-H Bond. J. Am. Chem. Soc 2019, 141, 13253. [DOI] [PMC free article] [PubMed] [Google Scholar]; For reviews, see:; (f) Gentry EC; Knowles RR Synthetic Applications of Proton-Coupled Electron Transfer. Acc. Chem. Res 2016, 49, 1546. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Hoffmann N Proton-Coupled Electron Transfer in Photoredox Catalytic Reactions. Eur. J. Org. Chem 2017, 15, 1982. [Google Scholar]
- (18).(a) Treat NJ; Sprafke H; Kramer JW; Clark PG; Barton BE; Read de Alaniz J; Fors BP; Hawker CJ Metal-Free Atom Transfer Radical Polymerization. J. Am. Chem. Soc 2014, 136, 16096. [DOI] [PubMed] [Google Scholar]; (b) Pan X; Lamson M; Yan J; Matyjaszewski K Photoinduced Metal-Free Atom Transfer Radical Polymerization of Acrylonitrile. ACS Macro Lett. 2015, 4, 192. [DOI] [PubMed] [Google Scholar]; (c) Discekici EH; Treat NJ; Poelma SO; Mattson KM; Hudson ZM; Luo Y; Hawker CJ; Read de Alaniz J A Highly Reducing Metal-Free Photoredox Catalyst: Design and Application in Radical Dehalogenation. Chem. Comm 2015, 51, 11705. [DOI] [PubMed] [Google Scholar]; (d) Martinez-Gualda AM; Cano R; Marzo L; Perez-Ruiz R; Luis-Barrera J; Mas-Balleste R; Fraile A; de la Pena O’Shea VA; Aleman J Chromoselective Access to Z- or E- Allylated Amines and Heterocycles by a Photocatalytic Allylation Reaction. Wat. Commun 2019, 10, 2634. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Boyington AJ; Seath CP; Zearfoss AM; Xu Z; Jui NT Catalytic Strategy for Regioselective Arylethylamine Synthesis. J. Am. Chem. Soc 2019, 141, 4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Molander GA; Ellis N Organotrifluoroborates: Protected Boronic Acids that Expand the Versatility of the Suzuki Coupling Reaction. Acc. Chem. Res 2007, 40, 275. [DOI] [PubMed] [Google Scholar]
- (20).Phosphate 122 was used because of the interference of phosphate 1-induced fluorescence at higher concentrations.
- (21).No quenching was observed when either Cs2CO3 or 18-crown-6 was absent.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.