

Abstract
Real-time molecular imaging to guide curative cancer surgeries is critical to ensure removal of all tumor cells; however, visualization of microscopic tumor foci remains challenging. Wide variation in both imager instrumentation and molecular labeling agents demands a common metric conveying the ability of a system to identify tumor cells. Microscopic disease, comprised of a small number of tumor cells, has a signal on par with the background, making the use of signal (or tumor) to background ratio inapplicable in this critical regime. Therefore, a metric that incorporates the ability to subtract out background, evaluating the signal itself relative to the sources of uncertainty, or noise is required. Here we introduce the signal to noise ratio (SNR) to characterize the ultimate sensitivity of an imaging system and optimize factors such as pixel size. Variation in the background (noise) is due to electronic sources, optical sources, and spatial sources (heterogeneity in tumor marker expression, fluorophore binding, and diffusion). Here, we investigate the impact of these noise sources and ways to limit its effect on SNR. We use empirical tumor and noise measurements to procedurally generate tumor images and run a Monte Carlo simulation of microscopic disease imaging to optimize parameters such as pixel size.
Keywords: molecular imaging, optical surgical navigation, intraoperative imaging, noise, fluorescence
Introduction
Knowledge of the presence of tumor cells is essential for cancer surgery. Small numbers of tumor cells, impossible to detect with the unaided eye or by touch, are often left behind, leading to positive margins that are strikingly common. Positive margins occur in 25% to 40% of breast cancer surgeries1,2 and 20% to 50% of high-risk prostate cancer surgeries.3 Positive margins, or microscopic residual disease (MRD), are consequential, significantly increasing the risk that cancer returns across cancer types, for example, doubling the recurrence in breast cancer leading to decreased survival.4 Similarly, MRD in prostate cancer increases the risk of recurrence 2 to 4 times5-8 Efforts to address MRD have long centered around physician judgment through preoperative imaging and intraoperative sight and touch. However, these techniques are limited to millimeter to centimeter scale resolution—equivalent to 104 to 109 cells, orders of magnitude above the needed threshold of detection to ensure a margin negative outcome. Gold-standard methods of margin detection rely on pathologic examination of the excised specimen, and if the specimen surface includes tumor cells (called a positive margin), additional therapy is performed at a later date—for example, re-excision for breast cancer and postoperative radiation for prostate cancer.
Current strategies for intraoperative tumor identification face challenges when assessing microscopic disease. Intraoperative specimen radiography is an established imaging technique in which the surgical specimen is removed from the patient and placed inside a self-contained imaging unit in the operation room for margin detection.9 This technique can thereby assist with verification of gross tumor removal in vitro 10 but not for portions of the tumor remaining in the patient, as the imager cannot access the surgical margin in vivo.11 Strategies for identifying tumor margin have focused on frozen section and touch prep analysis. Frozen section analysis, particularly challenging with the fatty tissue in breast cancer, is hindered by false negatives,12 requires a pathologist present at the time of surgery, is limited in the area that can be evaluated potentially missing disease, and significantly prolongs operative time.
Real-time molecular imaging on the other hand offers the opportunity to visualize MRD intraoperatively, directly in the tumor bed, enabling treatment of all disease at the time of the initial operation. Consequently, the need for imaging microscopic disease has driven the development of highly sensitive intraoperative imagers. Taking advantage of the growing armamentarium of fluorescently tagged molecular imaging agents, fluorescence imaging has moved to the forefront of intraoperative visualization techniques.13 While a wide range of intraoperative imagers exist, no standardized metric exists to evaluate their performance, particularly when coupled to a targeted molecular imaging agent. Therefore, a platform-independent method is needed to quantify the ability of imagers to detect microscopic disease intraoperatively.
The default method for identifying residual tumor using intraoperative imagers has been physician identification from an image. Efforts to define a common quantification metric for imaging tools have centered around the signal to background ratio (SBR).14,15 Implicit in this metric is a tumor signal significantly above background—true for larger tumor foci but not necessarily for microscopic disease that is often just above background contributed by nonspecific binding, autofluorescence, and other optical and electronic sources. However, to properly identify microscopic tumor foci in an image, the background must be accurately subtracted—often in software—using a combination of background subtraction and image recognition to achieve sensitivities far beyond human visual identification. This makes accurate determination of background critical, as any error in background estimation translates directly into an error in signal.
The background variation in biological systems can be analogized to measurement uncertainty in general, often called “noise” and for images is quantified as spatial noise. When combined with signal intensity, this leads to a quantifiable signal to noise ratio (SNR) for detecting microscopic disease in an imaging system.
Here we propose SNR as a figure of merit for optical detection of microscopic disease, which represents the fundamental limit of electronic and computer-aided detection. While recent literature increasingly considers SNR,16,17 methods to define signal and noise vary widely. A standardized quantification of signal and noise can be used to compare sensitivity across the imaging systems and define the ultimate limits of detection for a system.
To quantify SNR, we measure both the signal and the background as well as their variation. The signal is defined as the number of photons collected from a tumor foci, and this article addresses the identification and quantification of noise sources in the imaging system such as electronic noise and spatial noise. Key to accurate background subtraction (as shown in Figure 1), these factors are affected by the detector sensitivity, optical background rejection, properties of the imaging marker (antibody binding kinetics), antigen expression by the tumor and normal cells, and pixel size. The latter parameter is critical, as smaller pixels (higher resolution) are not always “better”—too small of a pixel may only sample noise with minimal signal, while too large of a pixel may washout tumor signal by averaging with background. Conversely, a pixel size larger than a single cell is still capable of single cell detection if the background is accurately subtracted. Thus, maximum SNR is intrinsically linked to pixel size.
Figure 1.

Sources of signal, background, and noise. A, A simulated image of microscopic disease including background and noise sources that obfuscate the tumor signal. Both the tumor area and background are procedurally generated. B, Without background subtraction and averaging, the tumor is hard to identify, while with background subtraction the tumor is more apparent.
Of the noise sources, this article focuses on defining and quantifying spatial noise so that an accurate SNR is defined. Electronic (eg, time varying) noise can be mitigated with sufficient image averaging (or equivalently, longer integration times), but spatial noise arising from variations in the underlying tissue and staining conditions cannot, thus driving a need to study the impact of spatial noise on the SNR for background subtraction. Analogous to time-varying noise sources, spatial noise is composed of both high- and low-frequency components. Similar in concept to averaging or longer integration time for time-varying noise, high-frequency spatial noise can be reduced by imaging a larger area for each pixel (eg, larger pixel size); however, this comes at the expense of spatial resolution and can lead to errors by integrating large fluctuations in slowly varying background intensity. Therefore, there is an ideal pixel size to optimize SNR for a given imaging system.
In this article, we outline a general method for evaluating the SNR of any optical imager in combination with a biologic labeling of tumor cells, and relate this to optimal pixel size. Since the analysis is based on the image itself, this method can be used as a platform independent metric to compare imagers (and biologics) in the evaluation of microscopic disease, essential for modern optical surgical navigation. Quantification of imaging systems using SNR allows single cell imaging, even with systems whose spatial resolution is below that of a single cell. We discuss the various contributions to the tumor signal and the sources of background and their inherent variability, which contributes noise as shown in Table 1.
Table 1.
Signal, Background and Noise Sources in a Fluorescence Image.
| Signal Sources | Background Sources | Noise Sources |
|---|---|---|
| Number tumor cells | Dark current | Electronic (shot noise) |
| Molecular labeling specificity | Nonspecific binding | Cell-to-cell variability |
| Molecular label concentration | Healthy cell antigen expression | Diffusion |
| Illumination intensity | Optical bleed through | Tissue heterogeneity |
| Fluorophore quantum efficiency | Autofluorescence | Tissue surface heterogeneity |
| Electronic responsivity | Illumination heterogeneity | |
| Pixel size |
To illustrate our methodology, we quantified spatial noise with immunofluorescence imaging of breast and prostate cancer cells, using both in vitro and in vivo molecular staining. For the purposes of molecular labeling, we used a model system of HER2-overexpressing (HER2+) breast cancer cell lines (SKBR3, HCC1569) compared against HER2-negative (HER2-) cell lines (S1, MDA-MB-231) with trastuzumab,18 an antibody targeting the HER2 receptor. Similarly, we use prostate-specific membrane antigen (PSMA)-overexpressing prostate cancer cell lines (LnCAP) and PSMA-negative (PC3) with J591, a humanized antibody against PSMA.19-22 As a demonstration of this technique, we quantified the signal from tumor, and the sources of noise in Table 1, with a fluorescence microscope. Figure 1 illustrates these sources of noise, drawing a distinction between high-frequency spatial noise and low-frequency spatial noise. High-frequency spatial noise, which varies rapidly over the image, consists of variations in antibody binding per cell and natural tissue (and tumor) heterogeneity.
Low-frequency spatial noise—which varies slowly over the entire image—can be a result of gradients in antibody concentration due to diffusion, tumor perfusion, vascularity, or nonuniform illumination. Noise due to non-uniform illumination can be reduced using spatial calibration,23 and any variations that cannot be subtracted out must be accounted as spatial noise. Using these measured metrics, we then randomly generate images of clusters of cells of specific size and quantify the SNR across varying arrangements of cell clusters. We use the characterization data obtained from real cell samples to make a simulated model of residual cancer tissue in order to determine the optimal pixel size for an intraoperative imager. The method and algorithm can be easily generalized for any cell line and antibody combination.
Methods
Cell Culture
Breast cancer cell lines
In vitro breast cancer cell cultures consisted of SKBR3 (HER2-overexpressing) and S1 (HER2-negative; from ATCC) cultured in Roswell Park Memorial Institute (RPMI) 1640 Medium with 10% fetal bovine serum (FBS).
Prostate cancer cell lines
Prostate cell cultures consisted of LnCAP cells (PSMA overexpressing) and PC3 cells (PSMA negative; from ATCC, Manassas, VA) cultured in RPMI with 10% FBS.
In Vivo Mouse Models
To determine the in vivo kinetic and spatial distribution of trastuzumab, we subcutaneously implanted HER2 overexpressing HCC1569 cells and MDA-MB-231 (HER2-negative) cells as a negative control in nude mice. Tumors were grown for 2 to 3 weeks until they were 1 to 1.5 cm in diameter. Mice were then injected with 1 mg of trastuzumab via intraperitoneal injection and sacrificed at 24, 48, and 96 hours, and tumor, kidney, muscle, and liver were removed and stained for trastuzumab binding.
Staining
Fixation
Mouse tissue sections were fixed with 2% paraformaldehyde in phosphate-buffered saline (PBS) solution for 20 minutes at room temperature. Slides are then washed with PBS and glycine solution.
Blocking
Tissue sections were blocked with 10% goat serum in immunofluorescence buffer.
Immunostaining
Sections were then further stained with anti-human FITC and the nuclear stain DAPI to simplify locating cells using the microscope.
Mounting
Coverslips were then mounted with Vectashield Storage Medium H1000 (Vector Labs, Burlingame, CA).
Imaging Procedure
Images were taken with Leica DMIRB, Wetzlar, Germany with 20× objective and standard FITC filter sets (Chroma) using a Hamamatsu ORCA-Flash4.0 V2, Bellows Falls, VT camera. Tissue images, used for in vivo binding quantification, were taken from the center of tissue samples. Background variation data were taken by imaging 66 µm × 66 µm areas, then shifting the slide by 59.4 µm and taking another image. This provided a 10% overlap between images, allowing for image stitching. The procedure was repeated across the entire tissue slice. The slide movement was precisely controlled using a Thorlabs XY Newton, New Jersey Mechanized Stage, and the stage and camera was controlled by Micro-Manager.24 Individual cells were identified using CellProfiler,25 and the total fluorescence intensity was quantified. The number of antibodies corresponding to the fluorescence intensity value was determined by imaging a set of reference dilutions of FITC-conjugated secondary antibody and comparing fluorescence intensity. A linear fit was established, defining the relationship between number of antibodies and and fluorescence intensity per pixel using the same objective and integration time. Using this calibration curve, the fluorescence intensity of each cell was converted to the number of antibodies bound as seen in Figures 2 and 3. Diffusion across tissue was estimated using MATLAB Natick, MA to determine average differences in intensity across an entire tissue slice as seen in Figure 4.
Figure 2.
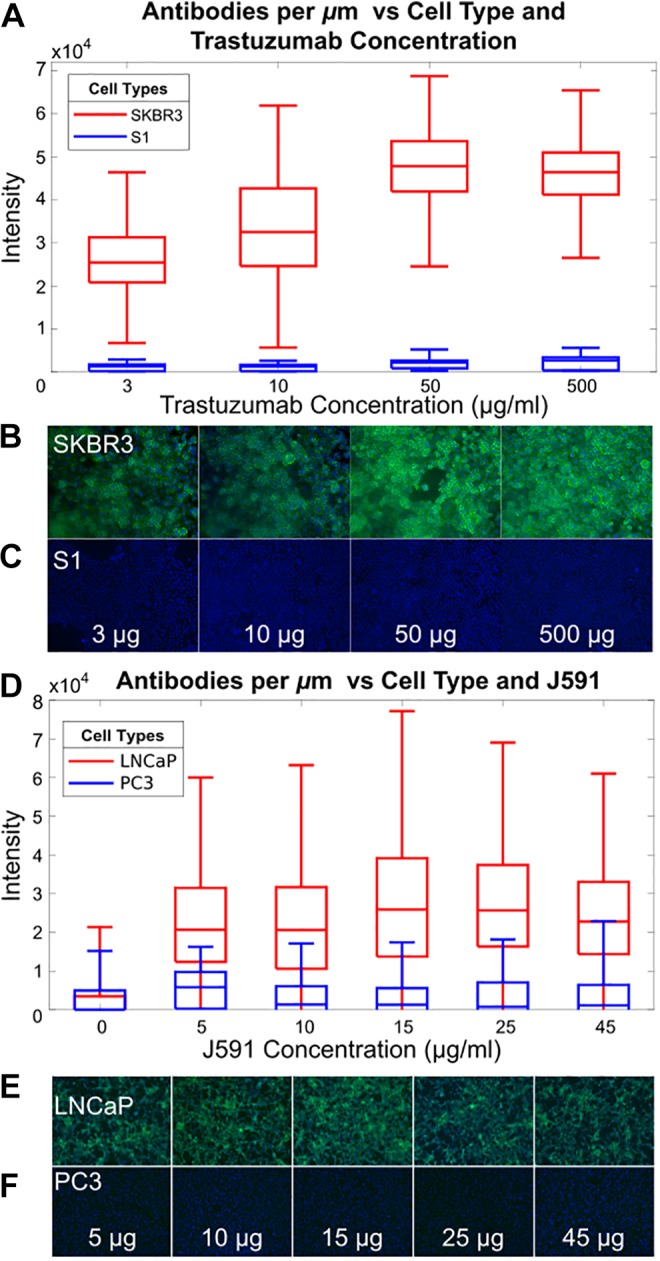
Quantification of signal and noise in vitro. A, Quantification of trastuzumab binding to SKBR3 (HER2+) cells shows average of 30 to 50 000 antibodies/μm2, while S1 (HER2-) cells are ×17 less. B, Cell staining of SKBR3 and (C) cell staining of S1 with trastuzumab (green = trastuzumab, blue = nucleus) with various concentrations of anti-HER2 antibody. D-F, Similar experiment with prostate cancer cell line LNCaP (PSMA+) and PC3 (PSMA-). D, Quantification of J591 binding to LNCaP cells shows 40 000 antibodies/μm2. (E and F) Cell staining with J591 (green = J591, blue = nucleus) with various concentrations of J591 with LNCaP and PC3, respectively.
Figure 3.

Quantification of signal and noise in vivo. A, binding of 1 mg of trastuzumab to HER2+ (HCC1569) and HER2− (triple negative, MDA-MB-231) tumors in nude mice versus time (24, 48, and 96 hours), stained with anti-human FITC (green) and DAPI (blue) nuclear counterstain. Binding to HER2+ cells increases with time. B, Tumor to background ratio is 4, 30, and 21 at 24, 48, and 96 hours post-injection. C, Representative images of tumor tissue are shown at 24, 48, and 96 hours in inset (i-iii).
Figure 4.
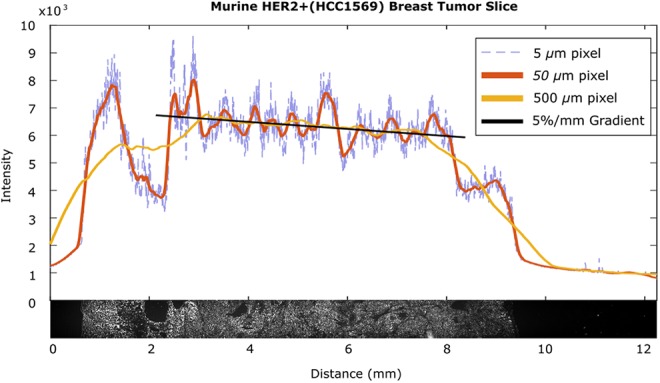
Low-frequency spatial variations in a tumor slice. A section of HER2+ (HCC1569) tumor alongside line scans at various spatial resolutions demonstrates the heterogeneities that make determining background difficult. The 5-µm pixel information demonstrates high-frequency variation between cells, the 50-µm pixel information demonstrates the background variation due to tissue physiology and the linear portion of the 500-µm pixel information between 3 mm and 7 mm demonstrates a 5%/mm gradient likely due to diffusion
Monte Carlo Simulation of SNR
We generated images of cell clusters to estimate the maximal SNR and optimal pixel size for our imaging sensor. Each image consists of a randomly generated cell cluster of ∼100 cells. The cell images are procedurally generated using Perlin noise26 to create a binary mask to demarcate tumor versus nontumor areas as seen in Figure 1A (tumor signal). To accurately replicate both the signal and the background intensity, we assign cell intensity and background on the values found in Figure 3 for HER2-overexpressing and HER2-negative cells, respectively.
Background is created as a random matrix with the same average intensity and variation (quantified as the standard deviation) as nonspecifically labeled cells imaged within the MDA-MB-231 (HER2-negative) tumor stained with trastuzumab. A 5%/mm intensity gradient is added to mimic the gradient measured in Figure 4. In addition, we simulate nonuniform illumination as background with a Gaussian radial gradient. Similarly, to accurately replicate the intensity and spatial noise of the tumor signal, a random matrix is created with the same average intensity and standard deviation as specifically labeled cells in the HCC1569 (HER2-overexpressing) tumor stained with trastuzumab. This matrix is then clipped (multiplied) by the binary mask defined earlier. The background matrix and tumor matrix are then summed resulting in a simulated image, where the background has the same variance and average intensity as HER2-negative tissue, and the tumor areas have the same variance and intensity as HER2-overexpressing tissue.
Receiver–Operator Characteristic
To determine an SNR threshold for detection, we analyzed the receiver–operator characteristic (ROC) of images at various spatial resolutions. First, random binary cell cluster images and noisy images are generated using the method described earlier. The SNR is calculated for each pixel in the noisy image and thresholded by SNRs ranging from 0 to 30. Pixels at or above the threshold are considered positives, while pixels below the threshold are negatives. The number of true-positive pixels is divided by the number of positive pixels in the initial binary tumor matrix to determine the sensitivity. The number of true negative pixels is divided by the number of negative pixels in the initial tumor matrix to determine specificity. 1-specificity is then plotted against the sensitivity for each threshold to create a ROC curve. We sampled 4 spatial resolutions, corresponding to pixel sizes of 5, 10, 20, and 50 microns to demonstrate how the ROC changes with spatial resolution.
Results
Signal to Noise Ratio as a Metric for Intraoperative Detection of Microscopic Disease
The SNR defines the detection limit of the complete imaging system and is defined as:
| 1 |
The MRD signal is defined as the total signal or number of photons, T, received by a pixel (gathered and converted to electrons by a pixel’s photosensitive element). This consists of both the photons emitted by the optically labeled tumor cells (called the signal, S) and the background, B, at that location (x, y) as seen in Figure 1. We can write this as:
| 2 |
To estimate background intensity for background subtraction, we measure the pixel intensity at a location away from the microscopic tumor (x + dx, y + dy), absent of tumor (S(x+dx, y+dy) = 0), such that:
| 3 |
The MRD signal alone can then be estimated from these 2 measurements as:
| 4 |
Recognizing that B(x, y) and B(x+dx, y+dy) may differ, this introduces the spatial noise in the system, and the minimum tumor signal detectable is then equal to this uncertainty, ΔB = B(x, y) − B(x+dx, y+dy). Here we have assumed that there is sufficient averaging to reduce electronic noise to below the level of the spatial noise and do not quantify its contribution. In this regime we find:
| 5 |
For small clusters of cells, the tumor signal is weak and is on the same order as the background intensity. We define the minimum number of detectable cells as that which gives an SNR >10, a value ensuring we identify tumor cells (true positives) without mistakenly identifying noise as tumor cells (false positives) across varying pixel sizes. This is supported by an ROC curve analysis (in the supplementary materials), wherein an SNR >10 does not result in false positives.
Quantification of Tumor Signal
The goal of intraoperative MRD imaging is to identify, and quantify, the number of tumor cells amidst a background of physiologically similar normal tissue cells. The signal from the tumor cells is proportional to
the number cells to be detected (Ncell);
the number of molecules labeled or bound to each cell (αbound);
the illumination (excitation) photon flux; and
the fluorophore efficiency of converting those illumination photons to Stokes shifted emitted photons.
The number of antibodies labeled per cell (αbound) is a function of the tumor biomarker binding affinity, biomarker expression level, and the labeling molecules exposure time to cells. The ratio of bound biomarker to tumor cells relative to healthy tissue cells is called the tumor to background ratio (TBR), used interchangeably here with SBR, and together the 2 quantities (TBR and αbound) can be used to quantitatively describe the biologics role in determining signal and therefore SNR. To demonstrate how to apply this technique, we have performed the following experiments determining TBR in vitro in both example breast (SKBR3, S1, HCC1569, MDA231) and prostate (LNCaP and PC3) cell lines and in vivo in the breast tumor model.
In vitro determination of TBR and αbound
Labeling tumor cells in vivo using a molecularly targeted imaging agent is the first step27 in translating the cell identifying procedures from the pathology laboratory into the real-time operating room environment. Identification of small foci (<200 cells) of fluorescently labeled MRD requires (1) accurate detection of the tumor focus and (2) differentiation of the tumor from the surrounding background, which can overwhelm and mask the small MRD signal. Here we quantify the binding of trastuzumab to HER2 overexpressing cell lines and J591 to PSMA overexpressing cell lines as model systems.
We quantify the number of fluorophore-labeled antibodies bound per tumor cell (αbound) and the relative background signal with the tumor to background ratio, TBR. Figure 2 illustrates in vitro quantification of HER2 labeling with increasing concentrations of trastuzumab. At 10 μg/mL of trastuzumab, SKBR3 cells bind ∼30,000 antibodies/μm2 (αbound=3.6 × 106/cell), while only 1700 antibodies/μm2 (8.5 × 103/cell) bind to S1 cells, for a TBR of 17. Higher concentrations of Trastuzumab saturate binding at 5 × 104 antibodies/μm2, although TBR is reduced due to increased background. Similar analysis on PSMA overexpressing prostate cancer cells demonstrates an αbound= 3.7 × 104/cell with a TBR of 28, consistent with the lower expression level of PSMA28 versus HER2.29
In vivo determination of TBR and α bound
To drive maximal signal for in vivo imaging, it is important to determine the optimal timing and concentration of a systemically injected imaging agent to maximize tumor binding (αbound). Studies30-34 demonstrate maximal TBR 24 to 72 hours after injection. To determine the in vivo kinetic and spatial distribution of trastuzumab, we subcutaneously implanted HER2 overexpressing HCC1569 cells and MDA-MB-231 (HER2-negative) cells as a negative control in nude mice and injected increasing amounts of trastuzumab via intraperitoneal injection.
Figure 3 shows selective binding of trastuzumab to HCC1569 cells in vivo (∼40 000 antibodies/μm2, αbound = 5 × 106/cell), with optimal TBR (30) at 48 hours post-injection. Tumor to background ratio in vivo exceeds in vitro due to receptor-mediated endocytosis of trastuzumab.35
These experiments show how to quantify TBR, capturing both the ratio of the biological labels (eg, antibodies) per tumor and normal tissue cell and the amount of labeling per cell. Tumor signal is the difference between the intensity of the tumor cells and background cells. The ratio between the tumor signal and the background (TBR), computed from the image, is the key driver of detection sensitivity, representing both the signal intensity and the background intensity. The TBR is a function of the biological system, relying on antibody-binding specificity and tumor receptor expression level. This quantitative description of biologic performance is agnostic to the imaging instrument itself (relying only on the final image). As such, we can analyze imager performance for a tumor-antibody system of a particular TBR and predict imager response for other TBRs. This allows us to predict (or simulate) sensitivity of the same imager across different biological (antibody cell) systems based on imaging just 1 biological system.
Additional determinants of the tumor signal include factors such as illumination intensity, fluorophore quantum efficiency, photon gathering, and elements of electronic detection, such as pixel responsivity, pixel capacity, and integration time. Here, we qualitatively describe their impact on the signal.
Illumination intensity
Illumination intensity proportionally increases the intensity of the fluorescence signal and the optical sources of background. However, given a fixed total imaging time to evaluate SNR, increasing illumination decreases electronic noise, as multiple images can be averaged in the same period due to the increased number of photons reaching the sensor per unit time (decreasing the integration time for each image). While illumination intensity cannot reduce spatial noise (since it is not varying with time), the illumination intensity must be increased to the level at which the net tumor signal is above the electronic noise of the detector. However, illumination intensity cannot be arbitrarily increased, as limits exist either due to safety requirements or photobleaching of organic fluorophores, representing an upper limit to illumination intensity and duration with estimates that each fluorophore can repeat the excitation–emission cycle 10,000 to 40,000 times before permanently photobleaching.36
Fluorophore quantum efficiency
Photons incident on a labeled cell interact with the bound fluorophore to produce a lower energy, stokes-shifted, fluorescently emitted photon. The efficiency of this process is directly proportional to the optical signal intensity. However, the relative low efficiency of this process requires illumination intensities 3 to 6 orders of magnitude larger than the fluorescence emission. This is driven by the small fluorophore absorption cross section, of the order of 10-16 cm2,37 which defines the area in which a photon can interact with a fluorophore, and the fluorescence quantum yield, which quantifies the probability that a photon interacting with a fluorophore will produce an emitted photon. The quantum yield is typically between 3% and 10% 38,39 for organic fluorophores used in in vivo applications.
Electronic detection (responsivity)
Each pixel converts incident photons to electrons, which in turn are converted to a voltage for electronic readout. Each pixel’s total capacity for integrated electrons is the sum of the signal and total background with electronic noise. Pixel responsivity quantifies the efficiency of converting received photons into electrons, which can then be converted to a voltage for electronic readout.
Pixel size
The maximal signal in a pixel can be obtained by matching the area imaged to the area subtended by the tumor cells being imaged. This optimizes signal detection as pixel capacity is not consumed by background photons from neighboring normal tissue. As an example, with a goal of imaging 100 cells, this area is approximately 10,000 μm2 or roughly a 120 µm × 120 µm for a 10 µm × 10 µm × 10 µm cell. However, maximizing signal in this manner comes with the sharp trade-off of resolution, which can compromise performance of automated image recognition and machine learning algorithms.
Quantification of Background
The background is comprised of electrons integrated by the pixel from sources other than the labeled tumor cells. Given all electrons are identical at the pixel level, electrons generated from signal and background are indistinguishable. Therefore, background must be accurately subtracted from the total pixel signal to yield the tumor signal. Here we describe the electronic, optical, and biological sources of background: dark current, optical bleed through, autofluorescence, on-target off tumor binding as well as nonspecific binding.
Dark current
Electronic sources of background are primarily due to the dark current.40-42 The magnitude of the dark current is dependent on the process used to manufacture the sensor and the sensor operating temperature. While assumed to be constant across pixels, fabrication mismatch between pixels and subtle integrated circuit fabrication process variations result in pixel-to-pixel variation, and this value is best measured per pixel (in darkness) and subtracted from the final readout. The relative contribution of this source of noise can be decreased through longer integration times or averaging multiple images.
Optical bleed through
Poor fluorophore efficiency necessitates illumination intensities orders of magnitude greater than the emitted light. Identifying fluorescently labeled cells thereby requires high performance optical filters that can reject light differing by ∼50 nm by 4 to 6 orders of magnitude. These filters inevitably allow some light through contributing to background, consuming pixel capacity, and increasing shot noise contributions.
Autofluorescence
Autofluorescence results from a broad-spectrum optical emission of higher wavelength light by molecules in tissue when excited by light. A portion of this emission falls within the emission band of the fluorophore (and therefore the selected optical filter) and, as such, is imaged along with the tumor signal. While autofluorescence is reduced using near infrared (NIR) illumination light, it represents a significant source of background, as each cell (both tumor and normal tissue) contributes to this background signal.
On-target, off tumor labeling
Healthy (eg, non-cancerous) tissue cells often express a baseline amount of biomarker which binds to the optical label and contributes directly to the background signal. This background is particularly problematic because it appears identical to the tumor signal, as both emit at the same wavelength and cannot be blocked by the filter.
Nonspecific binding
Imaging agent adhering to cells that do not express the surface marker or are not eliminated from the patient directly contribute to background. This nonspecific binding limits pixel capacity for signal and is a major hindrance for optical imaging of microscopic disease. This is addressed through increased pixel capacity to accommodate the additional background light. The penetration of light into tissue (as with NIR illumination) further adds to this as nonspecific binding to cells below the surface also contributes to background. This background can be reduced with a lower wavelength fluorophore, although this sacrifices penetration of superficial, overlying layers of blood that may be found intraoperatively. Some techniques, such as spatial frequency domain imaging, can reduce background by controlling illumination to calculate reflectance and scatter using both amplitude and phase information.43
Noise
Various sources of biochemical, physical, optical, and electronic features contribute to variation in the background, obfuscating the appropriate signal for background subtraction. Broadly speaking, electronic noise varies over time and therefore can be reduced by averaging multiple images. However, spatial noise is a function of the tissue itself and does not change with time (at least within the short interval of intraoperative imaging) and therefore cannot be reduced with averaging. Here, we describe biochemical and physical sources of noise as forms of spatial noise.
Time-varying electronic noise
Shot noise represents the fundamental physical limit of detection of counting electrons (generated from incident photons), including all sources of electrons such as optical background and dark current. Consequently, in the presence of a significant background signal, even if accurate background subtraction can be ensured, the noise from a large background signal (but not necessarily the background signal itself) can mask a small signal due to this noise source alone.
Spatial noise
The relative ease of subtracting a constant background would obviate the need for SNR considerations. However, background cannot be measured to an arbitrary precision, and variation can inhibit our ability to identify the signal of interest. This background variation can occur over a wide frequency scale. Notably, variations in antibody distribution and binding cannot be predicted a priori, prohibiting the use of a global threshold for MRD and current efforts at background subtraction are limited to centimeter-scale tumor foci.15 For example, the standard deviation for antibody binding per square micron in tissue labeled with an antibody in vivo (Figure 2) ranged from 1259 antibodies/μm2 in an HER2-negative tumor (eg, background variation) to 21 807 antibodies/μm2 in HER2-overexpressing cells (eg, tumor variation and heterogeneity). This drives the need for more accurate, patient (and tumor)-specific background measurements.
Spatial noise is divided into high-frequency (eg, rapidly varying) spatial noise and low-frequency spatial (eg, slowly varying) noise, wherein the high-frequency component is composed of cell-to-cell binding variations or tissue heterogeneity, and the low-frequency component is composed of gradients in the signal due to diffusion of the antibody. Both high- and low-frequency variations inhibit the ability to identify an accurate background to subtract and drive the need to quantify the optical pixel size for background subtraction.
High-frequency spatial noise can be addressed through averaging a larger area, driving the need for a larger pixel size, while low-frequency spatial noise precludes too large a pixel size to avoid integrating slowly varying intensity changes over the tissue surface. Thus, there is an optimal pixel size to minimize noise and maximize signal.
Noise quantification
To quantify both the high- and the low-frequency noise, we measured the variability of antibody staining across a tissue slice as a function of position on the slide. This tissue slice of an 8-mm HER2+ tumor, shown at the bottom of Figure 4, was resampled at various simulated pixel sizes to demonstrate the impact of noise at different resolutions. At a small pixel size (5 µm; Figure 4, dotted blue trace), high-frequency spatial noise fluctuations render local background identification impossible. At large pixel sizes (500 µm; Figure 4, yellow trace), low-frequency drift also impairs background identification in a focal area.
Electronic noise
In this example, we assume that both low-noise imager design (such as a cooled charge coupled device (CCD)) adequate averaging (or increased integration time) reduces electronic noise (relative to the signal) to below the noise level of the spatial noise and therefore disregard it. For example, the tissue free portions of Figure 4 have low variation with a small pixel size because the electronic noise contribution is very small relative to the spatial noise of the tissue.
Low-frequency spatial noise
When sampling background at large distances from the tumor, the background changes as a function of the distance from the tumor cluster being imaged and can be thought of as “low-frequency” spatial noise. For example, in Figure 4 (yellow trace), this can be as large as 5%/mm when sampling with a 500-µm pixel. This drives the need for high-spatial resolution so that a background measurement can be taken from a pixel close to the pixel imaging microscopic tumor, reducing this noise component.
High-frequency spatial noise
However, high-frequency variation puts an upper limit on the spatial resolution: As seen in Figure 4 (blue dashed trace), sampling using 5-µm pixels shows marked pixel-to-pixel variation due to cell-to-cell variations that can be quantified as the standard deviation in background. Hence, the optimal spatial resolution must sufficiently average the cell-to-cell variation without merely detecting the drift in intensity due to low-frequency noise effects such as antibody diffusion, justifying measurement of these variables for imaging of microscopic disease.
Calculation of SNR
To calculate the spatial noise as a function of pixel size, we find the variance across the pixels in the image by finding the mean of the square of differences between neighboring pixels as follows. If Px, y is the intensity of pixel at position x, y and Npixel is the number of pixels on the sensor, then noise ΔB is as follows
| 6 |
Variance is the more relevant measure of noise to determine SNR. To demonstrate this, we compute the variance using our metric at various spatial resolutions for the image in Figure 1 and plot the results in Figure 5. Below this plot, we show the image as it appears at given spatial resolutions (A-E). Variance across the image changes as a function of pixel size with the expected behavior of decreasing variance, as high-frequency noise components are reduced with increasing spatial averaging, until low-frequency noise dominates, and the variance begins to increase again. Thus, an intermediate pixel size (“C”) provides the optimal SNR for this system.
Figure 5.
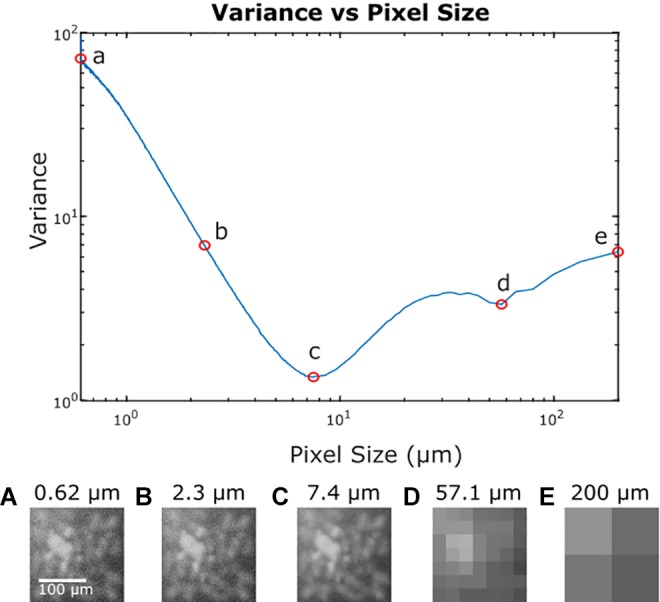
Variance in the image from Figure 1 at varying spatial resolutions. A, Represents the original image, (B) illustrates a reduction in high-frequency noise without significant loss in resolution. The minimum variance across the image occurs at point (C) at a pixel size that averages the high-frequency noise component without being dominated by low-frequency noise. Point (D) has increased variance due to averaging of low frequency noise components and in point (E), this low-frequency noise is the only feature visible.
Monte Carlo Simulation of Maximal SNR and Optimal Pixel Size
To ensure that our results are generalizable and not simply driven by our chosen sample, we ran a Monte Carlo simulation of 50 computer-generated cell images. Each image consists of randomly generated cell clusters with an average of 100 cells, with signal, background, and spatial noise derived from measured image data from the HER2 model as described. The simulation can represent any tumor model system by simply substituting the signal, background, and spatial noise obtained from an in vivo experiment of that system. Here we have chosen to use HER2+ breast cancer as the model system, given its relatively higher TBR.
In Figure 6, we plot the SNR over spatial resolutions corresponding to pixel sizes ranging from 0.61 to 200 µm for 50 random cell clusters. One of these random clusters is shown at select resolutions to illustrate how optimized SNR can enhance micro-tumor identification. In this instance, the optimal pixel size is in the range of 10 to 35 µm with a maximum SNR of 25.
Figure 6.

Monte Carlo simulation of procedurally generated tumor images. A, Example of randomly generated tumor followed by images at various spatial resolutions with simulated noise. The average background and noise are modeled after empirically determined distributions. The top and bottom row illustrate a linear and radial low-frequency noise gradient, respectively. B, Plot of signal to noise ratio (SNR) at various pixel sizes over 20 randomly generated tumors. Ten were simulated with linear low-frequency noise, where the blue squares indicate the data for the images in the top row of part (A). Ten were simulated with radial low-frequency noise, where the red circles indicate data for the bottom row in part (A). C, Variance across an image at various pixel sizes. Variance decreases as high-frequency noise is averaged away and then increases as low-frequency noise dominates.
Single-Cell Imaging with Relatively Large Pixels
Assuming a sparse distribution of tumor cells, a single cell with sufficient SNR can be detected by pixels much larger than the size of the cell. We simulated a 10-µm cell with TBRs from 1 to 30 to cover the real-world range we found in in vivo staining data shown in Figure 3. In Figure 7, we present an instance of this simulation with images of a single cancer cell at the center of a background of healthy tissue. This cell has a TBR of 10.4, while the healthy cells have random intensity with the same mean intensity and distribution as empirically determined. Even pixels an order of magnitude larger than the cell can uniquely locate a single cancerous cell; however, there is an upper bound to pixel sizes used to locate single cells. With an SNR below 10, there is an increasing chance that other pixels yield a greater intensity than the pixel over the tumor cell, rendering unique identification impossible. Establishing that the maximum size of the pixel that can reliably detect a single cell occurs when the SNR is greater than 10, we plot the SNR at various spatial resolutions for the example TBR of 10.4. In addition, we plot the largest pixel size that achieves an SNR of 10 for the full range of TBRs. Figure 7 illustrates that single-cell imaging is achievable even in an optical imaging system with resolution lower than that of a single cell. In Figure 7, panel B, the calculated SNR using the known cell location degenerates once the SNR is less than 10, demonstrating how detection becomes contingent on the initial distribution of noise in the image. However, the single cell is reliably detectable at higher SNRs. Figure 7, panel C, illustrates the effect of TBR on the ability to identify single cells. A greater TBR corresponds to a higher tumor signal, and as the signal intensity increasingly outweighs the integrated low-frequency noise (as larger pixels average out and lower the contribution of high frequency noise), relatively large pixels (multiple times the size of single cells) can adequately identify even single cells.
Figure 7.

Detecting a single cell at various spatial resolutions. In (A) we see the results of imaging a 10-micron diameter cell in healthy tissue with some noise and a tumor to background ratio of 10.4. We can still determine the location of the cell at the center of the imager as long as the signal to noise ratio (SNR) is greater than 10. Detecting a single cell does not require subcellular resolution contingent on the notion that the single cells are sparsely distributed and have a large tumor to background ratio. In (B) we see the SNR over a range of pixel sizes for 10 randomly generated samples given a tumor to background ratio (TBR) of 10.4. In (C) we plot the largest pixel that can detect a single cell at a given TBR with SNR greater than 10. The plot is not smooth because it is based on randomly generated samples with arbitrary noise.
Discussion
In this study, we have outlined and demonstrated a method to characterize the ability of optical imagers and targeted molecular imaging agents (TMIAs) to identify microscopic tumor foci, including single-cell residual disease. These small areas of tumor often exhibit intensity on the order of background, necessitating a metric beyond signal to background ratio. Furthermore, the advent of machine learning and automated image recognition algorithms lend themselves to a more quantitative evaluation of imaging system performance.
Through characterization of the signal intensity per cell, background intensity per cell, and the variation in background, the ultimate level of sensitivity for a given foci of tumor cells can be calculated. Furthermore, we demonstrate a methodology for simulating imaging of microscopic disease using computer-generated images, allowing evaluation of the sensitivity of an imaging system and companion TMIAs.
We can use this analysis to optimize the design parameters such as pixel size for future intraoperative imagers. The current paradigm is to pursue small pixels for high-resolution images. However, while higher resolution images can be binned to create a larger pixel size in postprocessing software, higher pixel density has intrinsic costs: Smaller pixels have relatively more temporal nose (as they integrate less signal), requiring longer averages; the fill factor is reduced (eg, CMOS-based imagers often include in-pixel electronics); and longer readout times are necessary to obtain the data from more pixels. Therefore, it is advantageous to have the optimal sized pixel within the system itself.
Pixels cannot be arbitrarily large either, both to retain resolution for accurate location data, and to prevent capturing low frequency spatial noise across the image. To image a microtumor with ∼100 µm diameter and background noise consistent with trastuzumab or J591 labeling, pixel sizes between 10 µm and 35 µm yielded optimal results (Figure 6). As pixel size increases up to 10 μm, the variance across the image decreases, while pixels over the tumor capture a greater portion of the signal, causing an increase in SNR. As pixel size continues to increase past the optimal range, the image comes to be dominated by any underlying antibody diffusion or tissue patterns that mask the tumor location.
In Figure 6, we see tumor location is not readily visible due to high-frequency noise with a small pixel size of 0.61 μm. Spatial averaging by increasing pixel size to 2.34 µm results in improved SNR, and tumor areas are better defined. However, there is still large variance across the image that may limit automatic detection. Peak SNR with sampling at 13.3 µm pixel size yields clear identification of microtumor areas. Tumor areas are still identifiable even at low resolution with a pixel size of 57.1 μm. High-frequency spatial noise is reduced until low-frequency spatial noise dominates at larger pixels such as 200 μm, where only the gradient is visible. Of particular note, a low pixel size may result in easily identifiable tumor areas, such is the case at a pixel size of 2.34 μm. However, thresholding for automatic tumor detection would not work well, as there are many background pixels with high intensities outside tumor areas, likely to generate false positives. Increased spatial averaging, either in postprocessing or at a hardware level with larger pixels, reduces this high-frequency noise at the expense of integrating (and therefore increasing) more low-frequency noise. We extend our technique to simulate varying patterns of low-frequency noise, such as a radial pattern—characteristic of non-uniform illumination sources. We also observe differences in SNR at various spatial resolutions between the linear low-frequency noise simulations and radial low-frequency noise simulations. In Figure 6, panel B, the SNR from the linear gradient simulations (marked with a blue square) is greater than the radial gradient simulations at optimal pixel sizes. This is an artifact of our cell cluster generation within the frame of the image. High background in the center of the image during radial gradient simulations reduces the signal. However, peak SNR remains consistent between these 2 low-frequency imaging conditions.
In the extreme condition of detecting a single tumor cell among a background of noncancerous tissue, detectors with pixels ranging from 10 µm to 250 µm can be used corresponding to TBRs ranging from 1 to 30 (Figure 7c). If a detector has pixels that are too large, then background areas distant from the tumor cell may have average intensities that appear to be tumor, as seen in Figure 7A, with a 210 µm pixel size. This results in a degeneration of SNR, where the area with a tumor cell cannot be uniquely identified.
While the metrics incorporated here address the image quality and ability to identify microscopic disease with optical imagers, they do not address other key metrics of intraoperative imagers, including imager size, mobility, and ability to fit within hard-to-access areas allowing visualization of all sides of a tumor cavity and within lymph node basins. For example, fiber optics has a fundamental trade-off between fiber diameter (which directly relates to the area visualized with each image) and flexibility, with a 1-cm bending radius achievable only with optical fibers of roughly 100-µm diameter. Similarly, imaging speed is important, as the entire surface area must be imaged rapidly to enable seamless integration into surgery and prevent the image from being degraded with hand motion.
Imaging small numbers of tumor cells with very low fluorescence levels has a large impact for guiding cancer surgery and requires the assistance of image processing algorithms.44 These techniques can be used synergistically with intraoperative imagers designed to image broad areas45 and guide gross resection. Following initial removal, tools adept at quantification and characterization of MRD can assist in the decision to further resect with the goal of achieving negative margins.
Conclusion
Detecting and removing microscopic disease in margins during tumor resection has significant impact on patient care and outcomes. A growing array of sensors, imagers, and optical labels address this problem of intraoperative imaging and are largely characterized by the TBR they can detect as a proxy for human detection. Here, we show that the spatial SNR is a fundamental limit of electronic image detection and describe techniques to quantify signal and noise in image systems as well as optimizations to improve SNR for the most accurate detection. We demonstrate our results using a Monte Carlo simulation of SNR in procedurally generated tumor images based on parameters of signal, background, and noise that we quantified from imaging HER2-overexpressing and HER2-negative cell lines with fluorescently labeled trastuzumab and PSMA-positive and PSMA-negative cell lines with fluorescently labeled J591 antibody. We extend this SNR analysis to optical imaging systems for single cell detection.
Supplemental Material
Supplementary_Material for Signal to Noise Ratio as a Cross-Platform Metric for Intraoperative Fluorescence Imaging by Asmaysinh Gharia, Efthymios P. Papageorgiou, Simeon Giverts, Catherine Park and Mekhail Anwar in Molecular Imaging
Acknowledgments
The authors would like to thank Hui Zhang for performing the in vitro and in vivo antibody staining and Dr. Neil Bander, Weill Cornell Medicine, Dept. of Urology, for the J591 antibody.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received the following financial support for the research, authorship, and/or publication of this article: MA was supported by NIH R21EB027238 and DOD PC141609. This project is supported by DOD PC141609 and NIH R21EB027238.
ORCID iD: Mekhail Anwar  https://orcid.org/0000-0002-6650-2594
https://orcid.org/0000-0002-6650-2594
Supplemental Material: Supplemental material for this article is available online.
References
- 1. McCahill LE, Single RM, Aiello Bowles EJ, et al. Variability in re excision following breast conservation surgery. JAMA. 2012;307(5):467–475. [DOI] [PubMed] [Google Scholar]
- 2. Chagpar AB, Killelea BK, Tsangaris TN, et al. A randomized, controlled trial of cavity shave margins in breast cancer. N Engl J Med. 2015;373(6):503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zietman AL, Shipley WU, Willett CG. Residual disease after radical surgery or radiation therapy for prostate cancer. Clinical significance and therapeutic implications. Cancer. 1993;71(S3):959–969. [DOI] [PubMed] [Google Scholar]
- 4. Early Breast Cancer Trialists’ Collaborative Group (EBCTCG), Darby S, McGale P, et al. Effect of radiotherapy after breast-conserving surgery on 10-year recurrence and 15-year breast cancer death: meta-analysis of individual patient data for 10,801 women in 17 randomised trials. Lancet. 2011;378(9804):1707–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hull GW, Rabbani F, Abbas F, Wheeler TM, Kattan MW, Scardino PT. Cancer control with radical prostatectomy alone in 1000 consecutive patients. J Urol. 2002;167(2 pt 1):528–534. [DOI] [PubMed] [Google Scholar]
- 6. Swindle P, Eastham JA, Ohori M, et al. Do margins matter? the prognostic significance of positive surgical margins in radical prostatectomy specimens. J Urol. 2008;179(5 suppl):S47–S51. [DOI] [PubMed] [Google Scholar]
- 7. Karakiewicz PI, Eastham JA, Graefen M, et al. Prognostic impact of positive surgical margins in surgically treated prostate cancer: multi-institutional assessment of 5831 patients. Urology. 2005;66(6):1245–1250. [DOI] [PubMed] [Google Scholar]
- 8. Yossepowitch O, Bjartell A, Eastham JA, et al. Positive surgical margins in radical prostatectomy: outlining the problem and its long-term consequences. Eur Urol. 2009;55(1):87–99. [DOI] [PubMed] [Google Scholar]
- 9. Kaufman CS, Jacobson L, Bachman BA, et al. Intraoperative digital specimen mammography: rapid, accurate results expedite surgery. Ann Surg Oncol. 2007;14(4):1478–1485. [DOI] [PubMed] [Google Scholar]
- 10. Kim SHH, Cornacchi SD, Heller B, Farrokhyar F, Babra M, Lovrics PJ. An evaluation of intraoperative digital specimen mammography versus conventional specimen radiography for the excision of nonpalpable breast lesions. Am J Surg. 2013;205(6):703–710. [DOI] [PubMed] [Google Scholar]
- 11. Maloney BW, McClatchy DM, Pogue BW, Paulsen KD, Wells WA, Barth RJ. Review of methods for intraoperative margin detection for breast conserving surgery. J Biomed Opt. 2018;23(10):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Riedl O, Fitzal F, Mader N, et al. Intraoperative frozen section analysis for breast-conserving therapy in 1016 patients with breast cancer. Eur J Surg Oncol. 2009;35(3):264–270. [DOI] [PubMed] [Google Scholar]
- 13. Frangioni JV. New technologies for human cancer imaging. J Clin Oncol. 2008;26(24):4012–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Warram JM, de Boer E, Moore LS, et al. A ratiometric threshold for determining presence of cancer during fluorescence-guided surgery. J Surg Oncol. 2015;112(1):2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Koch M, de Jong JS, Glatz J, et al. Threshold analysis and biodistribution of fluorescently labeled bevacizumab in human breast cancer. Cancer Res. 2017;77(3): 623–631. [DOI] [PubMed] [Google Scholar]
- 16. Bydlon TM, Kennedy SA, Richards LM, et al. Performance metrics of an optical spectral imaging system for intra-operative assessment of breast tumor margins. Opt Express. 2010;18(8):8058–8076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Papageorgiou EP, Zhang H, Giverts S, Park C, Boser BE, Anwar M. Real-time cancer detection with an integrated lensless fluorescence contact imager. Biomed Opt Express. 2018;9(8):3607–3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hudis CA. Trastuzumab—mechanism of action and use in clinical practice. N Engl J Med. 2007;357(1):39–51. [DOI] [PubMed] [Google Scholar]
- 19. Li Y, Tian Z, Rizvi SMA, Bander NH, Allen BJ. In vitro and preclinical targeted alpha therapy of human prostate cancer with Bi-213 labeled J591 antibody against the prostate specific membrane antigen. Prostate Cancer Prostatic Dis. 2002;5(1):36–46. [DOI] [PubMed] [Google Scholar]
- 20. Tagawa ST, Vallabhajosula S, Jhanwar Y, et al. Phase I dose-escalation study of225 Ac-J591 for progressive metastatic castration resistant prostate cancer (mCRPC). J Clin Oncol. 2018;36(6 suppl): TPS399–TPS399. [Google Scholar]
- 21. Vallabhajosula S, Kuji I, Hamacher KA, et al. Pharmacokinetics and biodistribution of 111In- and 177Lu-labeled J591 antibody specific for prostate-specific membrane antigen: prediction of 90Y-J591 radiation dosimetry based on 111In or 177Lu? J Nucl Med. 2005;46(4):634–641. [PubMed] [Google Scholar]
- 22. Nakajima T, Mitsunaga M, Bander NH, Heston WD, Choyke PL, Kobayashi H. Targeted, activatable, in vivo fluorescence imaging of prostate-specific membrane antigen (PSMA) positive tumors using the quenched humanized J591 antibody-indocyanine green (ICG) conjugate. Bioconjug Chem. 2011;22(8):1700–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee GS, Miele LF, Turhan A, et al. Spatial calibration of structured illumination fluorescence microscopy using capillary tissue phantoms. Microsc Res Tech. 2009;72(2):85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, Stuurman N. Advanced methods of microscope control using μManager software. J Biol Methods. 2014;1(2):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lamprecht MR, Sabatini DM, Carpenter AE. Cell profiler: free, versatile software for automated biological image analysis. BioTechniques. 2007;42(1):71–75. [DOI] [PubMed] [Google Scholar]
- 26. Perlin K. Improving noise. In Proceedings of the 29th annual conference on Computer graphics and interactive techniques, 1 July 2002, pp. 681–682. [Google Scholar]
- 27. Sampath L, Kwon S, Ke S, et al. Dual-labeled trastuzumab-based imaging agent for the detection of human epidermal growth factor receptor 2 overexpression in breast cancer. J Nucl Med. 2007;48(9):1501–1510. [DOI] [PubMed] [Google Scholar]
- 28. Smith-Jones PM, Vallabahajosula S, Goldsmith SJ, et al. In vitro characterization of radiolabeled monoclonal antibodies specific for the extracellular domain of prostate-specific membrane antigen. Cancer Res. 2000;60(18):5237–5243. [PubMed] [Google Scholar]
- 29. Gutierrez C, Schiff R. HER2: biology, detection, and clinical implications. Arch Pathol Lab Med. 2011;135(1):55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ballou B, Fisher GW, Waggoner AS, et al. Tumor labeling in vivo using cyanine-conjugated monoclonal antibodies. Cancer Immunol Immunother. 1995;41(4):257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gao X, Cui Y, Levenson RM, Chung LWK, Nie S. In vivo cancer targeting and imaging with semiconductor quantum dots. Nat Biotechnol. 2004;22(8):969–976. [DOI] [PubMed] [Google Scholar]
- 32. Tada H, Higuchi H, Wanatabe TM, Ohuchi N. In vivo real-time tracking of single quantum dots conjugated with monoclonal anti-HER2 antibody in tumors of mice. Cancer Res. 2007;67(3):1138–1144. [DOI] [PubMed] [Google Scholar]
- 33. Black RD, Bolick NG, Richardson RA, Dewhirst MW. In vivo monitoring of a fluorescently labeled antibody in mice with breast cancer xenografts. Sensors J, IEEE. 2008;8(1):81–88. [Google Scholar]
- 34. Kaushal S, McElroy MK, Luiken GA, et al. Fluorophore-conjugated anti-CEA antibody for the intraoperative imaging of pancreatic and colorectal cancer. J Gastrointest Surg. 2008;12(11):1938–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Austin CD, De Mazière AM, Pisacane PI, et al. Endocytosis and sorting of ErbB2 and the site of action of cancer therapeutics trastuzumab and geldanamycin. Mol Biol Cell. 2004;15(12):5268–5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lichtman JW, Conchello JA. Fluorescence microscopy. Nat Methods. 2005;2(12):910–919. [DOI] [PubMed] [Google Scholar]
- 37. Penzkofer A, Sperber P. Measurement of absorption cross sections in the long-wavelength region of the S0 S1 absorption band of dyes. Chem Phys. 1984;88(2):309–313. [Google Scholar]
- 38. Wang L, Gaigalas AK, Reipa V. Optical properties of Alexa 488 and Cy5 immobilized on a glass surface. BioTechniques. 2005;38(1):127–132. [DOI] [PubMed] [Google Scholar]
- 39. Hong G, Tabakman SM, Welsher K, et al. Near-infrared-fluorescence-enhanced molecular imaging of live cells on gold substrates. Angew Chem Int Ed Engl. 2011;50(20):4644–4648. [DOI] [PubMed] [Google Scholar]
- 40. Widenhorn R, Hartwig I, Dunlap JC, Bodegom E. Measurements of dark current in a CCD imager during light exposures In: Blouke MM, Bodegom E, eds. Sensors, Cameras, and Systems for Industrial/Scientific Applications IX. Bellingham, Washington: SPIE; 2008:68160B. [Google Scholar]
- 41. Porter WC, Kopp B, Dunlap JC, Widenhorn R, Bodegom E. Dark current measurements in a CMOS imager In: Blouke MM, Bodegom E, eds. Sensors, Cameras, and Systems for Industrial/Scientific Applications IX. Bellingham, Washington: SPIE; 2008:68160C. [Google Scholar]
- 42. Fowler B, Liu C, Mims S, et al. Wide dynamic range low light level CMOS image sensor. Proc Int Image Sensor Workshop. 2009;1. [Google Scholar]
- 43. Laughney AM, Krishnaswamy V, Rizzo EJ, et al. Spectral discrimination of breast pathologies in situ using spatial frequency domain imaging. Breast Cancer Res. 2013;15(4):R61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Carpenter AE, Jones TR, Lamprecht MR, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Troyan SL, Kianzad V, Gibbs-Strauss SL, et al. The FLARE intraoperative near-infrared fluorescence imaging system: a first-in-human clinical trial in breast cancer sentinel lymph node mapping. Ann Surg Oncol. 2009;16(10):2943–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary_Material for Signal to Noise Ratio as a Cross-Platform Metric for Intraoperative Fluorescence Imaging by Asmaysinh Gharia, Efthymios P. Papageorgiou, Simeon Giverts, Catherine Park and Mekhail Anwar in Molecular Imaging