

Abstract
Aminopeptidase N (APN) is a zinc metallopeptidase involved in the inactivation of biologically active peptides. The knowledge of its precise distribution is crucial to investigate its physiological role. This requires the use of appropriate probes such as the recently developed highly potent and selective radiolabeled APN inhibitor 2(S)‐benzyl‐3‐[hydroxy(1′(R)‐aminoethyl)phosphinyl]propanoyl‐l‐3‐[125I]iodotyrosine ([125I]RB 129). Its binding properties were investigated using rat brain homogenates (K d=3.4 nM) or APN expressed in COS‐7 cells (K d=0.9 nM). The specific binding was 95% at [K d], and preliminary autoradiography in intestine is promising. The decreased affinity of [125I]RB 129 (=10−6 M) for the E350D APN mutant, supports the critical role of E350 in the amino‐exopeptidase action of APN.
Keywords: Aminopeptidase N, Inhibitor, Binding, Rat brain membrane, Recombinant APN
1. Introduction
Aminopeptidase N (APN; EC 3.4.11.2) is a membrane‐bound zinc metallopeptidase involved in the extracellular metabolism of biologically active peptides. APN is expressed in many tissues, with highest levels in intestinal and kidney brush border membranes [1], but also found in lower quantity in brain, lung, blood vessels, and primary culture of fibroblasts [2]. Furthermore, APN has been shown to be identical to human lymphocyte surface cluster differentiation antigen CD13 [3], and has been reported to act as a major receptor for transmissible gastroenteritis virus [4] and for human coronavirus 229E [5]. APN exhibits a broad specificity for peptides with an N‐terminal neutral or basic amino acid, and has been shown to be involved in enkephalins and nociceptin catabolism in the central nervous system [6, 7] and to participate in the enzymatic cascade of the renin‐angiotensin system in brain and peripheral organs by cleaving angiotensin III [8]. All these findings make this enzyme an interesting target for possible therapeutic applications [9]. The knowledge of the precise distribution of APN is therefore crucial to investigate its physiological role. Few studies on the localization of APN have been done exclusively by using antibodies. In brain APN has been reported to be essentially located on blood vessels [10, 11, 12], a distribution which is hardly reconcilable with its peptide metabolizing role. Radioactive inhibitors have the advantage over antibodies to label the active site quantitatively in all tissues and species, and to reveal even low levels of a biological target [13]. However, it is essential to verify that the labeled inhibitor has a good affinity and is specific for the enzyme, as zinc metallopeptidases have many active site features in common [14]. Another advantage of radioactive inhibitors is that their in vivo distribution can easily be followed after different routes of administration [15], an important factor, considering the potential clinical applications of such molecules.
We have recently described the first highly potent and selective APN inhibitor which belongs to the class of α‐aminophosphonic compounds [16, 17]. Due to its nanomolar inhibitory potency towards APN and its selectivity versus other metallopeptidases of the same family, RB 129 (2(S)‐benzyl‐3‐[hydroxy(1′(R)‐aminoethyl)phosphinyl]propanoyl‐l‐3‐iodotyrosine) has been radiolabeled [18] to carry out a complete characterization of the biochemical and pharmacological properties of APN. In this paper we report the binding properties of this radiolabeled inhibitor on the rat brain membranes, recombinant APN and one of its mutants expressed in COS‐7 cells [19].
2. Materials and methods
2.1. Rat brain membrane preparations
Rat brains minus cerebellum were homogenized in 10 vol of 50 mM Tris–HCl pH 7.4, and centrifuged at 1000×g for 10 min. The supernatant was then centrifuged at 100 000×g for 40 min. The pellet was resuspended in 50 mM Tris–HCl buffer, pH 7.4 containing 0.02% bacitracin. Binding assays were performed in the same buffer in a final volume of 1 ml containing 0.4–0.6 mg of protein and the iodinated ligand.
2.2. Expression of recombinant pig APN
Recombinant pig APN was expressed in COS‐7 cells. The plasmid used contained the full‐length cDNA encoding pig APN inserted into an expression vector pcDNA3 (Invitrogen). The APN mutant E350A was obtained by double‐strand mutagenesis using the Transformer™ Site Directed Mutagenesis kit (Stratagene) following the manufacturer's instructions. The presence of the expected mutation and the absence of non‐specific mutations were confirmed by sequencing the complete coding sequence [19].
COS‐7 cells were grown in Dulbecco's modified Eagle's medium complemented with 10% fetal calf serum and 10×106 cells were transfected with 50 μg of plasmid by electroporation (250 V and 1000 microfarads, Biorad electroporator). Each pool of transfected cells was incubated in a 10 cm Petri dish at 37°C for 48 h. The cells were then washed twice and harvested by scraping in phosphate‐buffered saline. After rapid centrifugation at 2000×g, the cell pellet was resuspended in ice‐cold 50 mM Tris–HCl, pH 7.4, and membranes were prepared by homogenizing the cells in a Teflon‐glass homogenizer followed by centrifugation at 100 000×g for 30 min at 4°C. The final membrane pellet was solubilized in 50 mM Tris–HCl, pH 7.4 containing 0.02% bacitracin. Binding assays were performed with 50–100 μg of protein/tube in a final volume of 1 ml.
2.3. Binding assays
Incubations were carried out at 35°C and were terminated by filtration through Whatman GF/B filters. The filters were rinsed twice with 5 ml of ice‐cold buffer and the radioactivity was measured. For kinetic studies [125I]RB 129 was used at 100 pM and dissociation was initiated by addition of 1 μM cold iodinated ligand RB 129 after a 120 min incubation. For saturation studies, incubations were for 80 min and the radioactive ligand was used from 0.075 nM to 8 nM. For competition experiments a fixed concentration of 10 pM [125I]RB 129 was used in presence of various concentrations of different APN inhibitors. Non‐specific binding was determined with 1 μM RB 129. Moreover, all experiments were performed in presence of thiorphan to avoid non‐specific binding to NEP, for which the ligand has a K i value of 23.6 nM when tested in an enzymatic assay [17]. Competition curves were analyzed, and kinetic and saturation binding parameters were determined with the computer program EBDA. K i values were determined with the Cheng‐Prussof equation: K i=IC50/[1+(radioligand concentration/K d of the radioligand)].
2.4. Western blotting
Proteins from membranes of COS‐7 cells were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and electroblotted onto nitrocellulose filters. The blots were rinsed with TBS‐Tween 20 buffer then incubated with superblock blocking buffer (Pierce), antiserum raised against pig APN and finally with an anti‐mouse Ig, horseradish peroxidase‐linked antibody from sheep (Amersham Corp.). Peroxidase activity was revealed with a chemoluminescent detection kit from Amersham Corp. Purified pig APN was used as a control to verify apparent molecular mass.
2.5. Autoradiography
After rat decapitation, the intestine was rapidly removed and frozen in isopentane at −45°C. Sections (16 μm thick) were cut on a Bright cryostat at −17°C, thaw‐mounted onto gelatin‐coated slides, and stored at −80°C. All sections were warmed to room temperature just prior to incubation with ligands. The slides were preincubated for 30 min at room temperature in 50 mM Tris–HCl (pH 7.4), 16 mM saccharose, 0.2% BSA. Then slides were incubated with 1 nM [125I]RB 129 in presence of thiorphan in 50 mM Tris–HCl (pH 7.4), 5 mM MgCl2 for 60 min at room temperature. For non‐specific binding, the slides were incubated as above but in presence of 1 μM of cold RB 129. At the end of the incubation, the sections were washed twice in ice‐cold buffer for 10 min, followed by a rapid rinse in ice‐cold water and then dried under a cold airstream. Then, the films were exposed for 3 days at 4°C and developed.
2.6. Materials
The synthesis of RB 129 and of its 125I‐radioiodinated analogue were performed as previously described [17, 18]. Thiorphan [20], Phe‐thiol [21, 22] and the hydroxamate RB 38A N‐[3(R)hydroxyaminocarbonyl)‐2‐benzyl‐1‐oxopropyl]‐l‐phenylalanine [23] used in this study were prepared in the laboratory as previously described.
3. Results
3.1. General binding characteristics of [125I]RB 129
Total, specific and non‐specific binding of [125I]RB 129 increased linearly with the concentration of membrane protein up to 2 mg/ml. At the final protein concentrations used routinely in the assay (between 0.4–0.6 mg/ml for the rat brain membranes, and 50–100 μg/ml for the recombinant APN), the specific binding of [125I]RB 129 was between 80–98% (not shown).
3.2. Kinetic studies
The binding kinetics of [125I]RB 129 to rat brain membranes appeared to follow a simple bimolecular reaction with rates of both association and dissociation being monophasic after transformation of the specific binding data (Fig. 1). Specific [125I]RB 129 (100 pM) binding reached equilibrium after 80 min incubation at 35°C, and remained constant for a further 200 min.
Figure 1.

Kinetic analysis of specific [125I]RB 129 (100 pM) binding to rat brain membranes at 35°C. A: Time course association of [125I]RB 129 binding. Inset: Transformation of association data appropriate for pseudo‐first‐order conditions. B: Time course of dissociation of [125I]RB 129 binding initiated with 1 μM unlabeled RB 129. Inset: Transformation of specific binding data according to the first‐order dissociation rate equation.
Association data were transformed according to the pseudo‐first‐order rate equation on the basis that only ∼10% of added ligand was bound at equilibrium. The data were resolved into a single‐exponential function from which the association rate constant, k +1, was calculated as 94.7×106 M−1 min−1.
The rate of dissociation of [125I]RB 129 was also resolved into a single‐exponential function when the specific binding data were transformed according to the first‐order rate reaction, from which the dissociation rate constant (k −1) was calculated as 0.022 min−1. The kinetically derived dissociation constant (k d=k −1/k +1) was 0.23 nM.
3.3. Saturation binding analysis
In both rat brain membranes and recombinant APN, [125I]RB 129 demonstrated saturable, high affinity binding to an apparent single population of non‐interacting sites when the non‐specific binding was defined as the amount of radioligand remaining in the presence of 1 μM unlabeled RB 129 (Fig. 2). The equilibrium dissociation constant (K d) and maximal capacity of binding sites (B max) obtained from the specific binding data were 3.4±0.3 nM and 722±88 fmol/mg of protein, respectively, in the rat brain homogenates, and 0.9±0.2 nM and 650±67 fmol/mg of protein, respectively, in the recombinant APN wild‐type. No specific binding was measured on the E350A mutated APN (Fig. 2). The presence of the mutated enzyme was verified by Western blot (data not shown).
Figure 2.

Saturation analysis and Scatchard transformation (inset) of [125I]RB 129 binding to rat brain membranes (A) and recombinant APN (• represents the specific binding of [125I]RB 129 to the wild‐type, and □ specific binding to the mutant E350D) expressed in COS‐7 cells (B). Non‐specific binding to each tissue was defined with 1 μM unlabeled RB 129.
3.4. Competition studies
All unlabeled inhibitors (compound 1, RB 129, RB 38A, Phe‐thiol) produced a concentration‐dependent inhibition of specific [125I]RB 129 binding to membranes prepared from both rat brain and transfected COS‐7 cells. Moreover, a rather good correlation between the efficiency of these compounds to inhibit [125I]RB 129 binding and their inhibitory potency towards APN, determined in previous enzymatic experiments, was obtained, except for the hydroxamate RB 38A and the Phe‐thiol inhibitors in the rat brain membranes [17, 21, 22, 24] (Table 1). The competition curves were monophasic, and the slope values were always not significantly different from unity (Fig. 3).
Table Table 1.
K i values (nM) of inhibitors determined in binding studies on rat brain membranes and recombinant APN expressed in COS‐7 cells or determined by enzymatic assays
Figure 3.
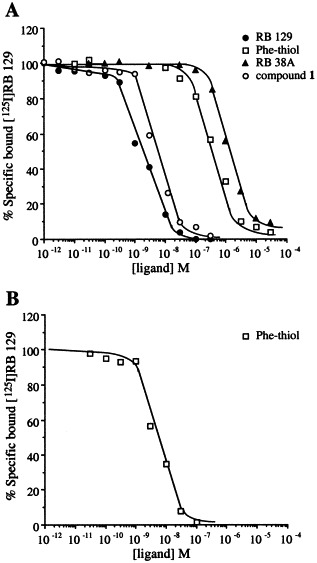
Competition between [125I]RB 129 and increasing concentrations of compound 1, RB 129, RB 38A, Phe‐thiol for APN in rat brain membranes (A) and COS‐7 cells in which recombinant wild‐type APN has been transfected (B).
3.5. Autoradiography of rat intestinal APN
Fig. 4 shows the distribution of APN labeled with [125I]RB 129. Staining of intestine was absent in the controls which were incubated in presence of the same concentration of radioiodinated inhibitor (1 nM) and an excess of cold ligand. Specific binding of [125I]RB 129 was observed in the circular layer of the intestine.
Figure 4.

Radioautography of rat intestine showing the specific binding of 1 nM [125I]RB 129 on APN localized in the intestinal membranes. A complete absence of binding was observed in presence of 1 μM unlabeled RB 129.
4. Discussion
RB 129 is the first highly potent and selective APN inhibitors which has been described [17]. In this study we have characterized the binding of its radioiodinated form, [125I]RB 129, using either rat brain membranes or recombinant APN expressed in COS‐7 cells. The binding was tissue concentration dependent, reversible and saturable. Saturation analysis indicated that [125I]RB 129 binds with high affinity to a homogeneous population of non‐interacting sites in rat brain membranes and COS‐7 cells transfected with APN wild‐type. Thus, Scatchard plots were linear, and Hill plots had slopes which were not significantly different from unity. From rat brain homogenates [125I]RB 129 bound to a single class of binding sites with a K d value of 3.4 nM and a B max of 722 fmol/mg of protein. Specific binding was high, being almost 95% at the K d value. In binding studies using recombinant APN expressed in COS‐7 cells [125I]RB 129 had a K d of 0.9 nM and again a high specific binding (∼92% at the K d value). Recently, E350 has been characterized as a critical residue involved in the N‐terminal amine binding site of APN [19]. The mutation E350A led to a non‐measurable specific binding. This result was expected, as E350 is essential to stabilize aminophosphinic inhibitors in the S1 subsite, and the binding data are in good agreement with the enzymatic assay demonstrating that this mutated APN shows an activity at least 10 000‐fold lower than that of the wild‐type APN [19]. This reinforces the argument that E350, found highly conserved in zinc metallopeptidase by sequence comparison, interacts with the amino group of substrates and inhibitors and participates to the exopeptidase specificity of aminopeptidases.
Strikingly, displacement of the radiolabeled APN inhibitor from rat brain membranes by increasing concentration of RB 38A or Phe‐thiol inhibitors yielded K i values of 1618 nM and 203 nM, respectively, which were very different to the K i values obtained in enzymatic studies in which pure porcine APN is used (K i=120 nM and 12.1 nM, respectively) [21, 22, 23]. These more than 10‐ and 15‐fold lower affinities of both inhibitors observed in binding studies are very likely due to the lower selectivity of RB 38A and Phe‐thiol inhibitors for APN as compared to the aminophosphinic inhibitors in regard to the various other aminopeptidases present in rat brain membranes. This is strongly supported by the results obtained with the recombinant APN expressed in COS‐7 cells, which could be considered as a partially purified enzyme preparation. Thus, Phe‐thiol inhibitor inhibited the [125I]RB 129 binding on COS‐7 cell membranes transfected with APN with K i value of 7.7 nM which parallels that found in enzymatic assay (K i=12.1 nM).
The iodinated APN inhibitor [125I]RB 129 was synthesized (i) to determine the level of membrane‐bound APN in different brain regions after various pharmacological treatments [25, 26, 27, 28]; (ii) to perform a localization of the enzyme in the central nervous system and at the periphery of different species. The use of such an inhibitor for autoradiography has advantages over enzymatic assay which is very imprecise or immunological methods for which quantitation could be hampered by differences in the tertiary structure and epitope presentation of the enzyme in various tissues, as shown in the case of angiotensin converting enzyme [29, 30]. Furthermore, although the primary sequences of APN from different species are highly conserved [4, 31, 32, 33, 34], it cannot be excluded that the enzyme appears to be differentially glycosylated not only in different species but also in different tissues of the same species, as already observed for neprilysin, another zinc metallopeptidase (NEP) [35], which might affect its recognition. Moreover, the use of 125I needs only short exposure times to obtain autoradiograms, and due to its high specific activity offers the possibility of a precise visualization of the enzyme in various tissues. As a preliminary experiment, we have visualized APN in the rat intestine with [125I]RB 129 and found a distribution at the level of the brush border in good agreement with previous studies reporting immunolabeling of APN in intestinal membranes, where its physiological function appears to be the hydrolysis of dietary peptides [36, 37].
In conclusion, the highly potent and selective APN inhibitor [125I]RB 129, is an excellent ligand for binding studies that could be easily automatized for a high throughput screening, and autoradiography, and could allow the direct detection of nanogram quantities of the enzyme from tissue extracts, as already shown with [125I]RB 104, a potent inhibitor of NEP [21, 22]. Furthermore, this radiolabeled compound could be modified to generate new dual inhibitors of NEP and APN, which both are involved in enkephalin degradation, offering the possibility to follow its bioavailability after different routes of administration, an important factor considering the potential clinical application of such molecules (review in [9]). In vivo displacement could also be performed to study the physiological modulation of enkephalin pathways by others neuromodulators.
Acknowledgments
This work was supported by grants from Synthelabo Recherche, and European Community (BMH4 CT98 2267).
Noble Florence,Luciani Nathalie,Da Nascimento Sophie,Laï-Kuen René,Bischoff Laurent,Chen Huixiong,Fournié-Zaluski Marie-Claude and Roques Bernard P.(2000), Binding properties of a highly potent and selective iodinated aminopeptidase N inhibitor appropriate for radioautography, FEBS Letters, 467, doi: 10.1016/S0014-5793(99)01645-2
References
- 1. Maroux S., Louvard D., Baratti J., Biochim. Biophys. Acta, 321, (1973), 282– 295. [DOI] [PubMed] [Google Scholar]
- 2. Sanderink G.J., Artur Y., Siest G., J. Clin. Chem. Clin. Biochem., 26, (1988), 795– 807. [DOI] [PubMed] [Google Scholar]
- 3. Look A.T., Ashmun R.A., Shapiro L.H., Peiper S.C., J. Clin. Invest., 83, (1989), 1299– 1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Delmas B., Gelfi J., Kut E., Sjostrom H., Noren O., Laude H., J. Virol., 68, (1994), 5216– 5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yeager C.L., Ashmun R.A., Williams R.K., Cardellichio C.B., Shapiro L.H., Look A.T., Holmes K.V., Nature, 357, (1992), 420– 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Waksman G., Bouboutou R., Devin J., Bourgoin S., Cesselin F., Hamon M., Fournié-Zaluski M.C., Roques B.P., Eur. J. Pharmacol., 117, (1985), 233– 243. [DOI] [PubMed] [Google Scholar]
- 7. Montiel J.L., Cornille F., Roques B.P., Noble F., J. Neurochem., 68, (1997), 354– 361. [DOI] [PubMed] [Google Scholar]
- 8. Zini S., Fournié-Zaluski M.C., Chauvel E., Roques B.P., Corvol P., Llorens-Cortes C., Proc. Natl. Acad. Sci. USA, 93, (1996), 11968– 11973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Roques B.P., Noble F., Daugé V., Fournié-Zaluski M.C., Beaumont A., Pharmacol. Rev., 45, (1993), 87– 146. [PubMed] [Google Scholar]
- 10. Hersh L.B., Aboukhair N., Watson S., Peptides, 8, (1987), 523– 532. [DOI] [PubMed] [Google Scholar]
- 11. Turner, A.J., Hooper, N.M. and Kenny, A.J. (1987) in: Mammalian Ectoenzymes (Kenny, A.J. and Turner, A.J., Eds.), pp. 211–248, Elsevier Science Publisher, Amsterdam.
- 12. Solhonne B., Gros C., Pollard H., Schwartz J.C., Neuroscience, 22, (1987), 225– 232. [DOI] [PubMed] [Google Scholar]
- 13. Zajac, J.M., Gacel, G., Dodey, P., Fournié-Zaluski, M.C., Roques, B.P., Roy, J., Morgat, J.L., Chaillet, P., Collado, H., Costentin, J., Petit, F. and Rossignol, P. (1983) in: Phencyclidine and Related Arylcyclohexylamines: Present and Future Applications (Kamenka, J.M., Domino, E.F. and Geneste, P., Eds.), pp. 143–152, NPP Books, Ann Arbor.
- 14. Matthews B.W., Acc. Chem. Res., 21, (1988), 333– 340. [Google Scholar]
- 15. Sales N., Dutriez I., Mazière B., Ottaviani M., Roques B.P., Regul. Pept., 33, (1991), 209– 222. [DOI] [PubMed] [Google Scholar]
- 16. Chen H., Noble F., Coric P., Fournié-Zaluski M.C., Roques B.P., Proc. Natl. Acad. Sci. USA, 95, (1998), 12028– 12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen H., Roques B.P., Fournié-Zaluski M.C., Bioorg. Med. Chem. Lett., 9, (1999), 1511– 1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen, H., Bischoff, L., Fournié-Zaluski, M.C. and Roques, B.P. (1999) J. Lab. Comp. Radiopharm., in press.
- 19. Luciani N., Marie-Claire C., Ruffet E., Beaumont A., Roques B.P., Fournié-Zaluski M.C., Biochemistry, 37, (1998), 686– 692. [DOI] [PubMed] [Google Scholar]
- 20. Roques B.P., Fournié-Zaluski M.C., Soroca E., Lecomte J.M., Malfroy B., Llorens C., Schwartz J.C., Nature, 288, (1980), 286– 288. [DOI] [PubMed] [Google Scholar]
- 21. Fournié-Zaluski M.C., Coric P., Turcaud S., Bruetschy L., Lucas E., Noble F., Roques B.P., J. Med. Chem., 35, (1992), 1259– 1266. [DOI] [PubMed] [Google Scholar]
- 22. Fournié-Zaluski M.C., Soleilhac J.M., Turcaud S., Laï-Kuen R., Crine P., Beaumont A., Roques B.P., Proc. Natl. Acad. Sci. USA, 89, (1992), 6388– 6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schmidt C., Peyroux J., Noble F., Fournié-Zaluski M.C., Roques B.P., Eur. J. Pharmacol., 192, (1991), 253– 262. [DOI] [PubMed] [Google Scholar]
- 24. Bouboutou R., Waksman G., Devin J., Fournié-Zaluski M.C., Roques B.P., Life Sci., 35, (1984), 1023– 1030. [DOI] [PubMed] [Google Scholar]
- 25. van Hal P.T., Hopstaken-Broos J.P., Prins A., Favaloro E.J., Huijbens R.J., Hilvering C., Figdor C.G., Hoogsteden H.C., J. Immunol., 153, (1994), 2718– 2728. [PubMed] [Google Scholar]
- 26. Vahdat L., Maslak P., Miller W.H., Eardley A., Heller G., Scheinberg D.A., Warrell R.P., Blood, 84, (1994), 3843– 3849. [PubMed] [Google Scholar]
- 27. Kehlen A., Gohring B., Langner J., Riemann D., Clin. Exp. Immunol., 111, (1998), 435– 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Phillips A.J., Tomasec P., Wang E.C., Wilkinson G.W., Borysiewicz L.K., Virology, 250, (1998), 350– 358. [DOI] [PubMed] [Google Scholar]
- 29. Jackson B., Cubela R., Johnston C.I., Aust. J. Exp. Biol. Med. Sci., 64, (1986), 149– 155. [DOI] [PubMed] [Google Scholar]
- 30. Strittmatter S.M., Snyder S.H., Neuroscience, 21, (1987), 407– 420. [DOI] [PubMed] [Google Scholar]
- 31. Olsen J., Cowell G.M., Konigshofer E., Danielsen E.M., Moller J., Laustsen L., Hansen O.C., Welinder K.G., Engberg J., Hunziker W., Spiess M., Sjöström M., Noren O., FEBS Lett., 238, (1988), 307– 314. [DOI] [PubMed] [Google Scholar]
- 32. Malfroy B., Kado-Fong H., Gros C., Giros B., Schwartz J.C., Hellmiss R., Biochem. Biophys. Res. Commun., 161, (1989), 236– 241. [DOI] [PubMed] [Google Scholar]
- 33. Watt V.M., Yip C.C., J. Biol. Chem., 264, (1989), 5480– 5487. [PubMed] [Google Scholar]
- 34. Yang X.F., Milhiet P.E., Gaudoux F., Crine P., Boileau G., Biochem. Cell Biol., 1993, (1993), 278– 287. [DOI] [PubMed] [Google Scholar]
- 35. Relton J.M., Gee N.S., Matsas R., Turner A.J., Kenny A.J., Biochem. J., 1983, (1983), 519– 523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sjöström N., Noren O., Jeppesen L., Staun M., Svensson B., Christiansen L., J. Biochem., 88, (1978), 503– 511. [DOI] [PubMed] [Google Scholar]
- 37. Terashima H., Wong H., Kobayashi R., Bunnett N.W., Gastroenterology, 102, (1992), 1867– 1876. [DOI] [PubMed] [Google Scholar]