

ABSTRACT
Motivation
Clustered regularly interspaced short palindromic repeats (CRISPR) technologies allow for facile genomic modification in a site-specific manner. A key step in this process is the in silico design of single guide RNAs to efficiently and specifically target a site of interest. To this end, it is necessary to enumerate all potential off-target sites within a given genome that could be inadvertently altered by nuclease-mediated cleavage. Currently available software for this task is limited by computational efficiency, variant support or annotation, and assessment of the functional impact of potential off-target effects.
Results
To overcome these limitations, we have developed CRISPRitz, a suite of software tools to support the design and analysis of CRISPR/CRISPR-associated (Cas) experiments. Using efficient data structures combined with parallel computation, we offer a rapid, reliable, and exhaustive search mechanism to enumerate a comprehensive list of putative off-target sites. As proof-of-principle, we performed a head-to-head comparison with other available tools on several datasets. This analysis highlighted the unique features and superior computational performance of CRISPRitz including support for genomic searching with DNA/RNA bulges and mismatches of arbitrary size as specified by the user as well as consideration of genetic variants (variant-aware). In addition, graphical reports are offered for coding and non-coding regions that annotate the potential impact of putative off-target sites that lie within regions of functional genomic annotation (e.g. insulator and chromatin accessible sites from the ENCyclopedia Of DNA Elements [ENCODE] project).
Availability and implementation
The software is freely available at: https://github.com/pinellolab/CRISPRitzhttps://github.com/InfOmics/CRISPRitz.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Clustered regularly interspaced short palindromic repeats (CRISPR) genome editing has revolutionized the ability to modify a genome of interest in a targeted and programable way (Cong et al., 2013; Mali et al., 2013). The initially described CRISPR system for eukaryotic genome editing involves a single guide RNA (hereafter referred to as a guide or sgRNA) to direct Streptococcus pyogenes-derived Cas9 (SpCas9) protein for site-specific genomic cleavage upstream of a protospacer adjacent motif (PAM), which is NGG for SpCas9. The Cas9-mediated double strand break is repaired by endogenous repair pathways including non-homologous end joining (NHEJ), microhomology-mediated end joining (MMEJ) and homology-directed repair (HDR). NHEJ/MMEJ often result in the introduction of insertions/deletions (indels) while exploitation of the HDR pathway allows for precise integration of customized sequence by providing a donor repair template (Komor et al., 2017). Since the initial description of eukaryotic genome editing by SpCas9, the CRISPR toolbox has been greatly expanded to include a variety of novel- and modified-nucleases with distinct PAM sequences (e.g. Cas12a, Cas9 derived from different species, and modified-Cas nucleases) (Komor et al., 2017). Although designed for site-specific cleavage, CRISPR nuclease-mediated cleavage may occur at other genomic sites, termed off-target sites. Off-target cleavage(s) commonly occur at sites of sequence homology to the on-target site; however, the rules governing off-target cleavage are incompletely understood. In general, mismatches may lead to a reduction in cleavage activity or have no effect at all depending on the specific base change and the relative position as described in Doench et al. (2014, 2016). Off-target cleavage(s) can produce unintended cellular effects that can confound analysis of a phenotype of interest. In particular, off-target cleavage(s) can impose cellular stress/toxicity and/or disrupt functional genomic regions (i.e. disruption of tumor suppressor genes). Several genome-wide assays have been developed to profile off-target effects in an unbiased manner (Frock et al., 2015; Kim et al., 2015; Tsai et al., 2015, 2017; Yan et al., 2017).
A common task during guide design is to scan the genome of interest for homologous sequences up to k mismatches (usually up to 4–7; Hsu et al., 2013) and/or with RNA/DNA bulges (1–5 bp; Lin et al., 2014), since these sequences may correspond to potential off-target sites. Of note, it is necessary to take bulges into account for off-target analysis because sgRNAs with bulges (up to 5) have been demonstrated to have cleavage activity (Lin et al., 2014). Many available tools for in silico off-target site prediction have employed strategies involving scanning for k mismatches, considering their proximity to the PAM site, and with or without support for DNA/RNA bulges (Bae et al., 2014; Doench et al., 2016, 2014; Haeussler et al., 2016; Hsu et al., 2013; Labun et al., 2016; Lin and Wong, 2018; Listgarten et al., 2018; Moreno-Mateos et al., 2015).
Short-read aligners, such as Burrows-Wheeler Aligner, can be applied to scan a reference genome with up to a predetermined number of mismatches (Li and Durbin, 2009). However, short-read aligners are not straightforward to use (see Supplementary File S1 Section S1 and Supplementary Fig. S1A). For these reasons, a variety of tools have been recently proposed to perform this task efficiently. For example, Cas-OFFinder scans a reference genome to identify all PAM-restricted genomic sites followed by base-by-base comparison with all identified PAM-restricted sites with the on-target site including quantification of the number of mismatches and DNA/RNA bulges (Bae et al., 2014). The output list of potential off-target sites includes all sites up to a maximum number of mismatches as set by the user. Another approach utilized by FlashFry and Off-Spotter software (McKenna and Shendure, 2018; Pliatsika and Rigoutsos, 2015) involves the creation of a reference genome index for PAM-restricted sites. With this index, the two software can efficiently identify all potential off-target sites including all sites up to a user-set number of mismatches. Of note, these software do not consider DNA/RNA bulges (Lin et al., 2014) in their analysis. Prior studies have utilized variant-aware off-target site identification using databases such as the 1000 Genomes Project and Exome Aggregation Consortium (Lessard et al., 2017; Scott and Zhang, 2017). Variant-aware sgRNA design is important for clinical applications of CRISPR genome editing and has been previously utilized in a CRISPR pooled screening format to minimize false positive and false negatives (e.g. mediated by PAM creation or disruption) due to variants (Canver et al., 2017, 2018; Lessard et al., 2017; Scott and Zhang, 2017); highlighting the need for tools that can efficiently search accounting for personal genetic variants. However, the tools presented in these studies are limited by long execution times and lack of support for RNA/DNA bulges. Cas-OFFinder is the only tool that considers bulges to the best of our knowledge at the time of this article. However, the bulge-related computation is incomplete (e.g. does not take into account possible ‘jumps’ in the analyzed sequence, see Supplementary Section S2) and also requires long computation time, as shown in Figure 5. It is computationally intensive to compute analysis with more than two bulges due to this prolonged computation time although previous work has shown cleavage activity with greater than two RNA bulges (Lin et al., 2014).
Fig. 5.

Running time comparison between CRISPRitz, Cas-OFFinder, FlashFry and OFF-Spotter. (A) Performance by varying the number of analyzed guides with a mismatch threshold set to 5. (B) Running time to search for 1000 guides with an increasing mismatch threshold. (C) Performance by varying the number of analyzed guides with thresholds set to 5, 1, 1, for number of mismatches, DNA bulges and RNA bulges threshold, respectively. (D) Running time (in log scale) for searching 1000 guides with an increasing number of mismatches and a fixed number of DNA and RNA bulges (1, 1)
Taken together, the currently available tools for in silico off-target site identification commonly suffer from long execution times, lack of support for RNA/DNA bulges, may fail to report all possible regions, and/or lack support for variants (variant-unaware) (see Supplementary File S1 Section S2 and Supplementary Fig. S1B). In addition, tools available at present offer limited data visualization capabilities to aid the user understanding of a given guides possible effect on functionally annotated non-coding regions, on the positional distribution of mismatches among the off-targets or on the impact of genetic variants on off-target in silico prediction, see Supplementary File S1 Section S8 and Supplementary Table S6).
To address these issues, we present CRISPRitz, a suite of tools for rapid, comprehensive and variant-aware CRISPR off-target site identification. Specifically, the CRISPRitz suite offers optimized data structures to naturally encode genetic variants and an arbitrary number of mismatches as well as RNA/DNA bulges. It also incorporates functional annotations and visualization features to assess the potential impact of putative off-target sites for both coding and non-coding regions. A detailed comparison of CRISPRitz with similar tools and an analysis of its performance is presented in Section 3.1.
2 Materials and methods
2.1 CRISPRitz overview
CRISPRitz is a suite of tools to rapidly enumerate and annotate putative off-target sequences and to assess their potential impact on the functional genome. Figure1 shows CRISPRitz functionalities, required and optional inputs, and generated output for each tool. CRISPRitz has three required inputs: (i) PAM sequence, (ii) a list of guides and (iii) a reference genome (in FASTA format). A collection of variants (in VCF format) and/or genomic annotations (in BED format) can be included as optional inputs. CRISPRitz performs the off-target site search by supporting a user-specified number of mismatches and/or DNA/RNA bulges. Importantly, the parallelism capability of multi-core architectures is utilized for all the computational-intensive tasks, such as search and index-genome, in order to minimize execution time. The following subsections present the implementation details and the data structure of each tool and the datasets used for performance evaluation and testing.
Fig. 1.

Overview of CRISPRitz. Starting from a reference genome and a set of genetic variants, the add-variants tool builds a new reference genome that incorporates population or personal variants (see Section 2.3). To perform searches with bulges in addition to mismatches it is necessary to create an index for the reference genome through the index-genome tool. This tool scans the genome and collects all the candidate targets for any PAM sequence given in input. The output is a compressed representation of candidate targets found on chromosomes (see Section 2.4). Targets and off-targets are found by the search tool, which takes in input the reference genome file or the previously created genome index with variants and a list of sgRNAs, the mismatches threshold [mandatory], and the bulges threshold [optional]. To understand the functional impact of the ongoing CRISPR experiment on a genome, starting from an input file of functional annotations in BED format, the annotate-results tool lists the number of guide matches that fall in exons, introns, promoters, CTCF and DNase I regions on the genome (see Section 2.6). Finally, generate-report (see Section 2.6) implements a graphical visualization through radar charts and motif logos of any guide behavior in a specific condition (i.e. number of mismatches and/or bulges)
2.2 PAM search
This operation is implemented using the Aho-Corasick string matching algorithm (Aho and Corasick, 1975), an efficient solution widely used to find all occurrences of any finite number of patterns in a reference string. This step is necessary for two search operations: one based on an index genome (with support for mismatches and bulges) and for a simplified search with only mismatches and not requiring the genome index.
The algorithm is based on deterministic automata that efficiently represent all the sequences corresponding to a given PAM. These automata can be used to scan the entire genome in linear time and enumerate all the sites compatible with the user-specified PAM (see Fig. 2 and Supplementary File S1 Section S3).
Fig. 2.
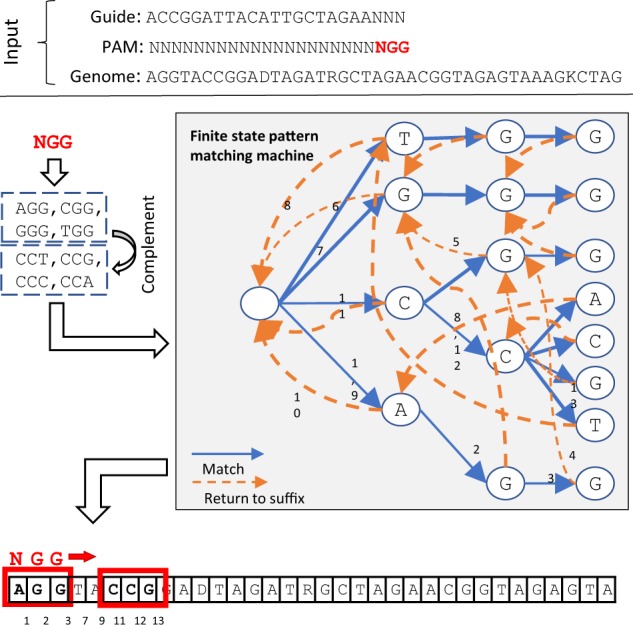
The PAM search. Searching for the PAM NGG in the genome starts by matching the base A at position 0 in the genome with the root children T, G, C and A of the pattern matching machine. This example illustrates the first 11 transitions of the automata that correspond to the identification of three candidate targets (0, 5 and 6). For more details see Supplementary File S1 Section S2
2.3 Encoding genetic variants in a reference genome
If a collection of variants is supplied as an optional input, the add-variants tool adopts the IUPAC notation to represent genetic variants via ambiguous DNA characters in the reference genome. As an example, if the nucleotide is G in a given position of the reference genome and the variant is A, the tool encodes the two alternatives by using the ambiguous nucleotide R, which corresponds to the IUPAC code for G or A.
The required inputs for the add-variants tool include (i) a reference genome and (ii) list of variants included in VCF file(s) format. The tool will output two versions of the reference genome with the first one containing single nucleotide polymorphisms (SNPs) and the second one including both SNPs and indels. Both genomes are coded with the IUPAC notation (Johnson, 2010) and four-bit encoding (see the table in Fig. 3).
Fig. 3.
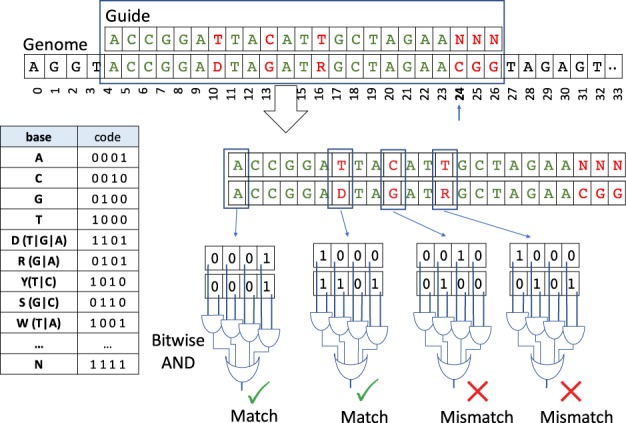
The mismatch-aware guide matching strategy. Characters are encoded using four-bit notation, as illustrated in the table on the left. For a given genomic position, this step compares the characters of the guide with the characters of the genome using bitwise operations. In the example the guide matching starts from index 24
2.4 Genome indexing for rapid bulge search
The two required inputs prior to genome indexing include (i) PAM sequence and (ii) a reference genome (in FASTA format). CRISPRitz then identifies and compiles all PAM-restricted sites (hereafter referred to as candidate off-target sites) within the provided genome into a genome index data structure (i.e. one genome index for each input PAM). This allows for a reduction in execution time for all subsequent searching with bulges (see Section 2.5.2). The index-genome tool starts by searching for all occurrences of the user-specified PAM in the genome, as explained in Section 2.2.
For each identified PAM sequence, the genomic sequence adjacent to the PAM with length equal to the input guide sequence is extracted and represents a candidate off-target site. The adjacent sequence is upstream or downstream as specified by the user to accommodate CRISPR nucleases available at the time of this article (e.g. Cas9 and Cas12a). All identified candidate off-target sequences are collected, sorted following a lexicographical order and encoded using the four-bit notation as shown in the table presented in Figure 3. index-genome is based on a ternary search tree (TST) data structure (Bentley and Sedgewick, 1998), which is optimized for approximate string search, such as utilized for text auto-completion or spell checking (see Supplementary File S1 Section S4).
2.5 Search for candidate off-target sites
The search tool searches for candidate off-target sites with mismatches only (Section 2.5.1) or with both mismatches and bulges (Section 2.5.2). It is necessary to use an index genome when considering both mismatches and bulges because the bulge-related search is computational expensive, and the index genome is fundamental to obtain a robust reduction in computational time by pre-computing an efficient data structure to perform searches.
2.5.1 Efficient search with mismatches
The search operation is computationally intensive task largely due to the required number of base-to-base comparisons (see Fig. 3). However, CRISPRitz implements this task through a four-bit-based encoding to represent each nucleotide of the IUPAC code to allow for efficient bitwise operations (see table in Fig. 3).
The inputs for analysis include: (i) the arrays of indices generated by the PAM search, (ii) the input list of guides, (iii) the user-specified maximum number of tolerated mismatches (α) and (iv) a reference genome/enriched genome (reference genome with variants). If a given guide from the input guide list matches to a region of the genome without mismatches, the index (or the indices) represents the starting position of one (or more) on-target site(s), otherwise the reported position is referred to as a candidate off-target site.
2.5.2 Efficient search with mismatches and RNA/DNA bulges
CRISPRitz also implements a search algorithm that handles a user-specified number of mismatches and optimized for the identification of DNA/RNA bulges. This algorithm requires the construction of an index genome as presented in Section 2.4. The usage of an index genome requires only operations to search for a sequence of length k in a TST with n candidate off-target sites.
This search is implemented with a function that recursively visits the TST. For all the candidate off-target sites, the algorithm begins from the TST root and visits all TST branches by checking if the nucleotide comparison corresponds to a match, a mismatch, or a bulge (DNA/RNA). When the user allows for bulges, the CRISPRitz can return duplicate results. For example, the results may represent the same target with a different number of mismatches and/or bulges or in different positions. In the mismatch-bulge search type, the visit on the branch can reach the leaf if the bulge threshold is sufficient to visit all the nodes in the branch. This may happen since the DNA bulges are treated like supplementary characters on the guide that can match with supplementary characters on the candidate off-target site. The mismatch-bulge search type stops when a branch of the TST is visited completely (i.e. when the leaf is reached), when the TST is visited partially and any threshold of mismatches or bulges has been exceeded, or when the visit has reached a number of nodes equal to the guide length (see Fig. 4). A more detailed description of this approach is presented in Supplementary File S1 Section S5.
Fig. 4.
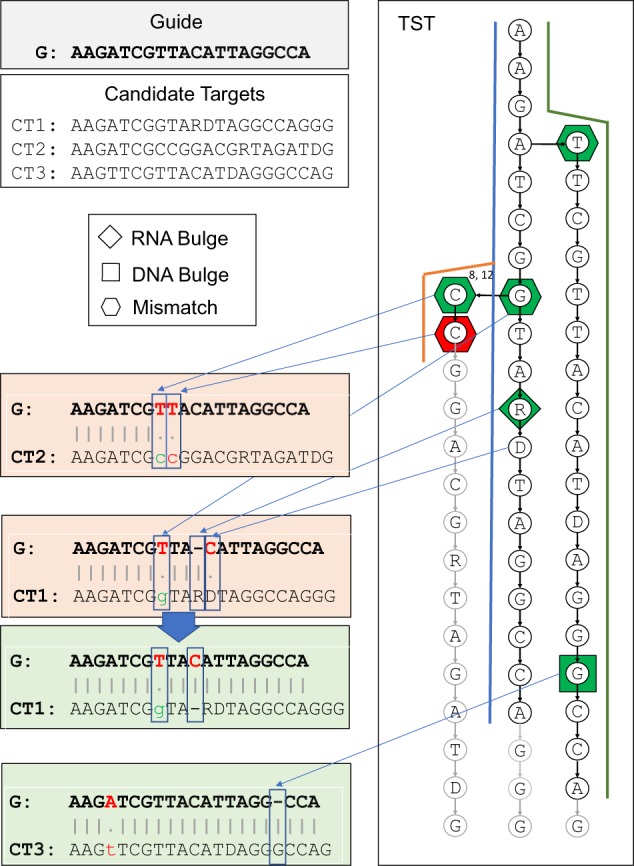
Example of guide matching by considering up to one mismatch and up to one RNA/DNA bulge. The search starts by visiting the left-most path of the tree, which represents the candidate sequence CT2. After the second mismatch (T versus C), the algorithm verifies that no bulges are allowed and stops the visit over the CT2 path. Back to the previous branch G, which in the example represents the first mismatch, it continues over the CT1 path. It verifies that the second mismatch cannot be considered as a DNA bulge but it can be considered as RNA bulge. It concludes the CT1 path visit to the leaf, thus identifying CT1 as an off-target with one mismatch and one RNA bulge. Similarly, the algorithm jumps back to the previous branch A and reaches the CT3 leaf, thus identifying CT3 as an off-target with one mismatch and one DNA bulge
2.5.3 Scoring candidate off-targets
Evaluation of the activity of a given guide RNA requires assessment of on-target activity as well as the probability of cleavage at genomic at sites distinct from the intended on-target region. Several scoring methods have been developed to quantify on- and off-target cleavage potential (Doench et al., 2014, 2016). CRISPRitz integrates two previously published scores: On-Target Efficacy Scoring (Doench et al., 2016) and cutting frequency determination (CFD) (Doench et al., 2016). The On-Target Efficacy Scoring is based on Azimuth machine-learning model (Doench et al., 2016) trained with more than 4000 sgRNAs targeting different positions in the protein coding regions of 17 genes. The CFD is used to quantitatively assess the off-target potential for a given guide. The CFD is based on a position weight matrix that calculates the probability of binding between each nucleotide of the sgRNA and each nucleotide of the DNA sequence. CRISPRitz executes the scoring functions for all identified targets after the completion of the search.
2.6 CRISPRitz output: search results, annotations and graphical report
The search tool generates three output files:
A list of matching DNA sequences with genomic coordinates, number of mismatches, DNA/RNA bulges and scores (CFD and On-Target Efficacy Scoring).
An overall mismatch and bulge profile for each guide considering all the putative off-target sites. The profile consists of a matrix in which the rows are the guides and the columns represent the number of their on-target(s) and off-target sites with respect to the mismatch values up to the user-specified threshold.
Motif matrices based on all the off-targets. One matrix is created per guide and per mismatch count. Rows correspond to nucleotides, columns to base-pair, and each entry represents the number of occurrences of a given nucleotide (row) in a given base-pair (column) for all the sites enumerated.
CRISPRitz also includes two additional tools, annotate-results that annotates the candidate off-target sites, and generate-report that creates a summary graphical report for each guide from the input guide list, annotate-results annotates all identified off-target sites for each guide using a set of predefined functional genomic annotations or using custom annotations provided by the user (in BED format). The predefined functional genomic annotations include gene body (promoter, exon, and intron) as well as DNase I hypersensitive (DHS) and CCCTC-binding factor (CTCF) sites obtained from the ENCyclopedia Of DNA Elements (ENCODE) project (Consortium et al., 2004). The DHS annotation file was obtained by considering 62 tracks derived from different ENCODE cell types. Only the peaks passing QC by the ENCODE pipeline were included and merged into a single file containing their union. CTCF site annotation was created in a similar fashion using 308 available ENCODE tracks. We have also evaluated CTCF annotations based on putative binding sites based on PWM motif models from the JASPAR database (Khan et al., 2017; (see Supplementary File S1 Section S6 and Supplementary Figs S2–S5).
Based on the annotated results, generate-report creates a graphical report to aid in the assessment of the potential functional impact of the off-target sites for a given guide (see Fig. 6A and B and Supplementary File S1 Section S6 and Supplementary Figs S2–S6). Specifically, a radar chart is created for each mismatch threshold. Each axis of the radar chart corresponds to a functional annotation and the plotted value represents the similarity (in terms of found on-/off-target sites) to the examined guide as compared with its own guide set (so the user can assess an individual guide’s behavior as compared with all other guides from the input guide list) or to a previously analyzed guide library (e.g. the Gecko Library v2; Sanjana et al., 2014, see Section 2.7). The area in the radar chart allows the user to quickly evaluate the off-target potential for a given guide. A small area on the radar chart corresponds to a guide with reduced candidate off-target sites (the exact number is displayed for each annotation) whereas a large area corresponds to guides with increased candidate off-target sites in multiple functional regions. generate-report also produces mismatch profiles where each position corresponds to a bar that represents the number of observed mismatches for each nucleotide normalized on the maximum number of mismatches.
Fig. 6.

Visual representation of CRISPRitz results. (A) and (B) show the behavior of two guides from the CCR5 set (hg19 reference genome, with up to 4 mismatches and no bulges). (A) and (B) were created by comparing results from the CCR5 dataset with the previous computed results based on the Gecko Library v2, as explained in Section 2.6. The area shown in radar chart is constructed joining the points on the ‘y’ axis of each annotation. This area helps the user to obtain an overview on the behavior of a single guide of the analyzed dataset as compared with a reference set of guides. A small area represents a guide with a poor activity in terms of off-targets; on the contrary, a big area represents a guide with a rich activity. (C) Bar plot to show the relative increase in the count of off-targets when accounting for genetic variants from the 1000 Genomes project
If genetic variants and enriched genomes are used during the search operation, generate-report can also plot a bar plot showing the percentage gain for each annotation and the additional sites as compared with the reference genome using the annotation file created by annotate-results (see Fig. 6C).
2.7 Reference genomes and validation datasets
For all validation testing, we used both the hg19 reference genome and a modified version, enriched genome, which incorporates genetic variants (SNPs and indels) from the 1000 Genome Project (Consortium et al., 2015) through the four-bit IUPAC encoding presented in Section 2.3. Of note, CRISPRitz supports any reference genome for which a FASTA file is available. We tested the performance and functionality of CRISPRitz by considering the following datasets:
Random guides from the hg19 reference genome. To test the scalability of CRISPRitz as compared with other tools, we sampled different number of guides (i.e. 1, 10, 100 and 1000) from the human reference genome among more than 300 million positions compatible with the NGG PAM. The guides were randomly selected without any filter.
Therapeutic guides. This set is composed of 124 guides targeting the CCR5 gene derived from (Lessard et al., 2017).
Gecko Library v2. Genome-wide CRISPR library used for knock-out screens (Sanjana et al., 2014) containing 111 671 guides with six sgRNA per gene target and 2000 non-targeting control guides designed without any perfect genomic matches.
3 Results and discussion
We evaluated the computational performance of CRISPRitz using benchmark datasets (see Section 3.1). In addition, we evaluated its application in solving two commonly encountered situations related to genome editing experiments (see Section 3.2): (i) Systematically assessing off-target potential from large CRISPR libraries (e.g. genome-wide sgRNA libraries); (ii) Evaluating and selecting guides targeting a therapeutically relevant locus with consideration for personalized genetic variants and potential off-target sites in functional genomic regions. These two applications also highlight key features of CRISPRitz and the utility of the proposed output graphical report.
3.1 Performance evaluation and comparison with similar tools
To evaluate the performance of CRISPRitz, we used a general dataset of guides randomly sampled from the human reference genome hg19 using the NGG PAM (see Section 2.7). The tests were performed on a machine equipped with an Intel(R) Xeon(R) CPU E5-2650 v4, clocked at 2200 MHz and 64 GBs RAM, and the Ubuntu operating system (version 16.04).
We performed a head-to-head comparison of CRISPRitz with Cas-OFFinder (Bae et al., 2014), FlashFry (McKenna and Shendure, 2018) and OFF-Spotter (Pliatsika and Rigoutsos, 2015) as shown in Figure 5A and B. In Figure 5C and D, we compared CRISPRitz only with Cas-OFFinder since it was the only available tool (at the time of writing this article), that allowed searches with both mismatches and DNA/RNA bulges. Both CRISPRitz and Cas-OFFinder can take advantage of multi-core architectures. CRISPRitz was implemented using the well-known OpenMP API (Dagum and Menon, 1998), while Cas-OFFinder is implemented using OpenCL API (Munshi, 2009).
First, performance testing without DNA/RNA bulges was performed using a different number of guides with up to five mismatches. FlashFry and OFF-Spotter are faster than CRISPRitz and Cas-OFFinder, ranging from a speed-up of 30–70× comparing CRISPRitz and Flashfry and a ranging from 7 to 25× when comparing CRISPRitz to OFF-Spotter. By using CRISPRitz, we observed an ∼2-fold or greater reduction in execution time as compared with Cas-OFFinder (see Fig. 5A). Notably, the execution time slightly increase for CRISPRitz and Cas-OFFinder tools with respect to the number of input guides; however we observed a significant difference between Cas-OFFinder and CRISPRitz with 1000 guides (Cas-OFFinder performed the search in 10 000 s with respect to the 3400 s used by CRISPRitz).
We evaluated the impact of the number of mismatches on the execution time using a fixed number of guides (e.g. 1000 in Fig. 5B). This analysis has shown that Cas-OFFinder, FlashFry and CRISPRitz execution time increases similarly with the increase in the number of mismatches compared with OFF-Spotter which execution time increases of and passing from three to four and from four to five mismatches, respectively. Of note, the performance testing in Figure 5A and B was run by using two CPU cores for CRISPRitz and Cas-OFFinder and one core for FlashFry and OFF-Spotter, because the two tools lack a parallel implementation.
Next, using the same hardware, we tested the performance of CRISPRitz and Cas-OFFinder when DNA/RNA bulges were also considered in the analysis. The same guides were tested as in Figure 5A and B with up to five mismatches but allowing also one DNA and one RNA bulge. Although the execution times are similar for one guide, we observed a 4-fold reduction in execution time for CRISPRitz with respect to Cas-OFFinder when the number of guides increased (Fig. 5C). Furthermore, the effect of the number of mismatches was evaluated with a fixed number of guides (n = 1000) with the number of DNA and RNA bulges set to one. The difference in execution times was greatest (up to 74-fold reduction) when considering three mismatches (Fig. 5D). However, the magnitude of execution time reduction decreased to ∼4-fold with an increased number of mismatches. This is because the number of visited branches of the tree (and of the execution time as a consequence) by CRISPRitz increases due to increasing the mismatch and bulge thresholds.
Importantly, CRISPRitz showed robust scalability by varying the number of CPU cores (from 2 to 8), the mismatch threshold from 3 to 5 and the predicted off-target activity per guide (see Supplementary File S1 Section S7, Supplementary Fig. S7 and Supplementary Table S1). The results highlight that CRISPRitz performance scales approximately linearly over the number of CPU cores (1.8).
Finally, we compared the features and the running time of CRISPRitz with additional four software; CRISPOR (Haeussler et al., 2016), CHOPCHOP (Montague et al., 2014), CRISPRseek (Zhu et al., 2014) and CRISPRtool (Lessard et al., 2017), on searching with mismatches and bulges on the reference genome and genome with variants (see Supplementary File S1 Section S8 and Supplementary Tables S1–S6). CRISPRitz is the only software able to perform a search taking into account genetic variants as well as mismatches and bulges while still maintaining computational efficiency. Furthermore, in a comparison performed with only mismatches allowed in the search, the fastest tool was FlashFry, followed by Off-Spotter, CHOPCHOP and CRISPRitz. Of note, the speed-up range between FlashFry and OFF-Spotter was from 2- to 4-fold (100 s by FlashFry as compared with >400 s by Off-Spotter; Supplementary Table S2). In a comparison performed with mismatches allowed in the search and using a variant genome (only supported by CRISPRitz and CRISPRtool), CRISPRitz was the faster tool with a speed-up range of 8.5- to 35-fold (3000 s by CRISPRitz as compared >100 000 s taken by CRISPRtool) (Supplementary Table S3). In a comparison performed using mismatches and bulges allowed in the search (only supported by CRISPRitz and Cas-OFFinder), we determined that CRISPRitz was the faster tool with a speed-up range from 4- to 75-fold (50 000 s by CRISPRitz as compared with >200 000 seconds by Cas-OFFinder; Supplementary Table S4). In a comparison performed using mismatches, bulges and a variant genome in the search, only CRISPRitz supported this combination of features with no available alternatives at the time of this article (Supplementary Table S5; refer to Supplementary Section S8 for further information on all comparison testing). Taken together, when using only mismatches FlashFry is the fastest tool; however, when bulges or genetic variants are included CRISPRitz offers a significant speedup over all the available tools. Importantly, CRISPRitz is the only tool that allows a complete enumeration of target and off-target sites when accounting simultaneously for mismatches, bulges and genetic variants.
3.2 CRISPRitz high-throughput and variant-aware enumeration of potential off-target sites on the functional genome
In some cases, it may be necessary to identify potential off-target sites for a large number of guides, such as for genome-wide CRISPR libraries. In addition, it is often important to evaluate for off-target sites in multiple genomes, such as when performing experiments in multiple cell lines and/or evaluating the effect of personalized variants in individuals (Canver et al., 2018) given that these variants can affect the number of on- and off-target sites or lead to false positive/negative if not accounted for. Furthermore, once off-target sites are identified, it is useful to inform potential risk in the setting of off-target cleavage through genomic annotation, such as off-target sites in coding sequence or off-targets in non-coding sequence affecting function sequences (e.g. CTCF sites). Therefore, CRISPRitz was designed to support genetic variants as well as provide intuitive enrichment analysis using a set of genomic annotations for a variety of functional regions, as discussed in Section 2.3.
To showcase these features, we analyzed the GeCKO Library v2 (see Section 2.7) genome-wide library, using up to five mismatches (and no bulges). For this analysis we constructed a single enriched genome including 84.4 million genetic variants obtained from 2504 individuals across 26 populations from the 1000 Genome Project Phase 3 (Consortium et al., 2015). CRISPRitz analysis of this dataset with a variant-aware genome and also the reference hg19 genome resulted in a total time of 3500 min (2.4 days), for both analyses. Of note, differences were observed between the CRISPRitz results when using the hg19 reference genome as compared with the enriched genome with variants, which is consistent with previous work (we obtain an average increase of 50–60% in all the analyzed genetic regions) (Lessard et al., 2017; Scott and Zhang, 2017). It is worth noting, that although this analysis was performed using all the variants present in at least one individual, with CRISPRitz a similar analysis can be performed using variants from a single individual to create a personalized, enriched genome. The results obtained from this analysis were used as a template for comparison with subsequent analysis of the guides targeting CCR5 (see Fig. 6).
Next, we evaluated the guides targeting CCR5 coding sequence, which is a therapeutic target for patients with human immunodeficiency virus (HIV) infection. Using the hg19 and the enriched genome described in the previous section derived from the 1000 Genomes Project database, we observed an increase of 18–24% in putative off-target sites when accounting for SNPs (Fig. 6C), which is consistent with previous works (Lessard et al., 2017; Scott and Zhang, 2017). In addition the visualization of the functional annotation of identified off-target sites can aid in the identification of specific and non-specific guides as shown in Figure 6A and B. We also perform an analysis on the CCR5 guides, including bulges in the search, to show that also the inclusion of bulges is fundamental to obtain an accurate analysis (see Supplementary Section S6 and Supplementary Fig. S6). The bulges inclusion led to a dramatic increase in the total number of possible off-target sites, from 36 000 with four mismatches and no bulges to 2.5 million with inclusion of 1 DNA and 1 RNA bulge.
Multiple flavors of high-fidelity Cas9 (e.g. SpCas9-HF1/eSpCas9/HypaCas9/evoCas9) are available that exhibit reduced off-target activity while maintaining on-target editing efficiency (Casini et al., 2018; Chen et al., 2017; Kleinstiver et al., 2016; Slaymaker et al., 2016; Vakulskas et al., 2018). High-fidelity nucleases still maintain probability of cleavage at putative off-target sites albeit a lower probability as compared with standard reagents. Moreover, use of a high-fidelity Cas9 does not preclude off-target analysis as these nucleases can still mediate off-target cleavage, particularly if a sgRNA is utilized with significant off-target potential. CRISPRitz analysis may be able to aid nuclease selection prior to initiation of wet-lab experiments as high-fidelity nucleases may be preferred for sgRNAs with increased off-target potential.
In summary, we offer CRISPRitz as a rapid, high-throughput, and variant-aware tool for off-target site identification for CRISPR genome editing, which includes visualization capabilities to allow for functional annotation of identified genomic sites in coding and/or non-coding regions. A detailed documentation covering CRISPRitz installation, usage and a walk-trough example is available as Supplementary File S2 and online at GitHub (see code availability).
Supplementary Material
Acknowledgements
We would like to thank Giuseppe Ritrovato for the first prototype of the TST data structure. Jonathan Y. Hsu, Karl Petri and other members of the Pinello and the Joung labs for their helpful discussions and/or software testing. We also would like to thank you the authors of Off-spotter and CHOPCHOP for the kind support received to test their software. In addition we would like to thank the Centro Piattaforme Tecnologiche (CPT) located in the University of Verona that provide us with all the hardware necessary to perform all the tests.
Funding
L.P. was supported by a National Human Genome Research Institute (NHGRI) Career Development Award [R00HG008399] and Genomic Innovator Award [R35HG010717] and a Centers for Excellence in Genomic Science (CEGS) Award [RM1HG009490].
Conflict of Interest. none declared.
References
- Aho A.V., Corasick M.J. (1975) Efficient string matching: an aid to bibliographic search. Commun. ACM, 18, 333–340. [Google Scholar]
- Bae S. et al. (2014) Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics, 30, 1473–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley J., Sedgewick B. (1998) Ternary search trees. Dr. Dobb’s J., 23, [Google Scholar]
- Canver M. et al. (2017) Variant-aware saturating mutagenesis using multiple Cas9 nucleases identifies regulatory elements at trait-associated loci. Nat. Genet., 49, 625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canver M.C. et al. (2018) Impact of genetic variation on CRISPR-Cas targeting. CRISPR J., 1, 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casini A. et al. (2018) A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat. Biotechnol., 36, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.S. et al. (2017) Enhanced proofreading governs CRISPR–Cas9 targeting accuracy. Nature, 550, 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L. et al. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science, 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium G.P. et al. (2015) A global reference for human genetic variation. Nature, 526, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium E.P. et al. (2004) The ENCODE (ENCyclopedia Of DNA Elements) Project. Science, 306, 636–640. [DOI] [PubMed] [Google Scholar]
- Dagum L., Menon R. (1998) OpenMP: an industry standard API for shared-memory programming. IEEE Comput. Sci. Eng., 5, 46–55. [Google Scholar]
- Doench J.G. et al. (2014) Rational design of highly active sgRNAs for CRISPR-Cas9–mediated gene inactivation. Nat. Biotechnol., 32, 1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench J.G. et al. (2016) Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol., 34, 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frock R.L. et al. (2015) Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol., 33, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler M. et al. (2016) Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol., 17, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P.D. et al. (2013) DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol., 31, 827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson A.D. (2010) An extended IUPAC nomenclature code for polymorphic nucleic acids. Bioinformatics, 26, 1386–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A. et al. (2017) JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res., 46, D260–D266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. et al. (2015) Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Methods, 12, 237. [DOI] [PubMed] [Google Scholar]
- Kleinstiver B.P. et al. (2016) High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature, 529, 490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor A.C. et al. (2017) CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell, 168, 20–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labun K. et al. (2016) CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res., 44, W272–W276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard S. et al. (2017) Human genetic variation alters CRISPR-Cas9 on-and off-targeting specificity at therapeutically implicated loci. Proc. Natl. Acad. Sci. USA, 201714640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. (2009) Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J., Wong K.-C. (2018) Off-target predictions in CRISPR-Cas9 gene editing using deep learning. Bioinformatics, 34, i656–i663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. et al. (2014) CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res., 42, 7473–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listgarten J. et al. (2018) Prediction of off-target activities for the end-to-end design of CRISPR guide RNAs. Nat. Biomed. Eng., 2, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P. et al. (2013) RNA-guided human genome engineering via Cas9. Science, 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A., Shendure J. (2018) FlashFry: a fast and flexible tool for large-scale CRISPR target design. BMC Biol., 16, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montague T.G. et al. (2014) CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res., 42, W401–W407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Mateos M.A. et al. (2015) CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods, 12, 982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munshi A. (2009). The OpenCL specification In: 2009 IEEE Hot Chips 21 Symposium (HCS), pp. 1–314. IEEE, Stanford, CA. [Google Scholar]
- Pliatsika V., Rigoutsos I. (2015) “Off-spotter”: very fast and exhaustive enumeration of genomic lookalikes for designing CRISPR/Cas guide RNAs. Biol. Direct, 10, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana N.E. et al. (2014) Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods, 11, 783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott D.A., Zhang F. (2017) Implications of human genetic variation in CRISPR-based therapeutic genome editing. Nat. Med., 23, 1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaymaker I.M. et al. (2016) Rationally engineered Cas9 nucleases with improved specificity. Science, 351, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai S.Q. et al. (2015) Guide-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol., 33, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai S.Q. et al. (2017) Circle-seq: a highly sensitive in vitro screen for genome-wide CRISPR–Cas9 nuclease off-targets. Nat. Methods, 14, 607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakulskas C.A. et al. (2018) A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med., 24, 1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W.X. et al. (2017) Bliss is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat. Commun., 8, 15058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L.J. et al. (2014) CRISPRseek: a bioconductor package to identify target-specific guide RNAs for CRISPR-Cas9 genome-editing systems. PLoS One, 9, e108424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.