

Abstract
Purpose:
Although most children with medulloblastoma are cured of their disease, Sonic Hedgehog (SHH) subgroup medulloblastoma driven by TRP53 mutations is essentially lethal. Casein kinase 1a (CK1α) phosphorylates and destabilizes GLI transcription factors, thereby inhibiting the key effectors of SHH signaling. We therefore tested a second-generation CK1α activator against TRP53-mutant, MYCN-amplified medulloblastoma.
Experimental Design:
The ability of this CK1α activator to block SHH signaling was determined in vitro using GLI reporter cells, granular precursor primary cultures, and PATCHED1 (PTCH1)-mutant sphere cultures. While in vivo efficacy was tested using 2 different medulloblastoma mouse models: PTCH1 and ND2:SMOA1. Finally, the clinical relevance of CK1α activators was demonstrated using a TRP53-mutant, MYCN-amplified patient-derived xenograft.
Results:
SSTC3 inhibited SHH activity in vitro, acting downstream of the vismodegib target SMOOTHENED (SMO), and reduced the viability of sphere cultures derived from SHH medulloblastoma. SSTC3 accumulated in the brain, inhibited growth of SHH medulloblastoma tumors, and blocked metastases in a genetically engineered vismodegib-resistant mouse model of SHH medulloblastoma. Importantly, SSTC3 attenuated growth and metastasis of orthotopic patient-derived TRP53-mutant, MYCN-amplified, SHH subgroup medulloblastoma xenografts, increasing overall survival.
Conclusions:
Using a newly described small-molecule, SSTC3, we show that CK1α activators could address a significant unmet clinical need for patients with SMO inhibitor–resistant medulloblastoma, including those harboring mutations in TRP53.
Introduction
Medulloblastoma is the most common malignant pediatric brain tumor, for which the standard of care is surgery followed by chemotherapy and/or radiation (1). Although the overall cure rate in medulloblastoma is high, genomic stratification has demonstrated that certain subgroups have extremely poor prognosis (2). In addition, even patients with medulloblastoma that survive have severe cognitive and quality of life issues as a result of the devastating effect of current therapies, particularly radiation, on the developing brain (1, 3). Therefore genomic stratification is being used to guide development of novel subgroup–specific targeted therapies with improved efficacy and side-effect profiles (4–6). One-third of patients with medulloblastoma harbor tumors associated with constitutive Sonic Hedgehog activity (SHH subgroup; refs. 5–7). Although the survival of most patients with medulloblastoma is over 70%, a subset of SHH subgroup patients characterized by TRP53 mutations display de novo resistance and all typically die despite therapy (8–13), representing the highest risk form of SHH medulloblastoma.
SHH signaling is initiated upon binding to the receptor PATCHED1 (PTCH1), resulting in derepression of the 7-transmembrane protein SMOOTHENED (SMO; ref. 14). Alternatively, SMO is also activated by loss-of-function mutations in PTCH1, which is the major molecular driver for SHH subgroup medulloblastoma (15). Ultimately, SMO regulates a signaling pathway that stabilizes and activates the GLI family of transcription factors (16), triggering a growth-promoting pathway. A number of small-molecule SMO inhibitors have been evaluated in the clinic. One of these inhibitors, vismodegib, was FDA-approved for treatment of advanced basal cell carcinoma (BCC; ref. 17). Given its efficacy against BCC driven by SHH signaling, vismodegib was rapidly repurposed in clinical trials for patients with medulloblastoma (18). Vismodegib also exhibited efficacy against patients with SHH subgroup medulloblastoma. These patients’ cancer was predominantly driven by mutations in PTCH1 (19), which acts upstream of SMO (14). However, rapid tumor recurrence has already been observed in patients with medulloblastoma, likely driven by mutations in or downstream of SMO (20). These examples with the SMO inhibitor vismodegib illustrate the clinical potential of attenuating SHH signaling in patients with medulloblastoma, as well as the importance of developing and clinically evaluating SHH inhibitors that act downstream of SMO.
We previously described a first-in-class CK1α activator, pyrvinium, which acts as a potent inhibitor of SHH signaling downstream of SMO, via the phosphorylation and destabilization of GLI1 and GLI2 (21). Although we showed that pyrvinium was able to reduce the growth of SHH subgroup medulloblastoma in vivo, its inability to accumulate in serum and cross the blood–brain barrier (BBB) limited efficacy studies to medulloblastoma implanted in the flanks of mice and dosed by local administration of pyrvinium. We recently described a second-generation CK1α activator with significantly improved pharmacokinetics, SSTC3 (22). We now show the ability of SSTC3 to inhibit SHH signaling in vitro and in vivo. We take advantage of the improved pharmacokinetics of SSTC3, demonstrating its ability to cross the BBB, attenuate the growth and metastases of SHH subgroup medulloblastoma mouse models, and prolong survival. Most importantly, we demonstrate the efficacy of SSTC3 against an orthotopically implanted, patient-derived xenograft (PDX) of SHH medulloblastoma with a TRP53 mutation and MYCN amplification, one of the most clinically challenging forms of medulloblastoma (8, 12, 13, 23).
Materials and Methods
Cell and molecular biology
NIH-3T3 and HEK293T cells were purchased from ATCC and cultured as recommended by ATCC. The efficacy of SSTC was validated using LIGHT2 cells, a cell line derived from NIH-3T3 cells stably expressing a GLI-dependent firefly luciferase reporter gene and a constitutive Renilla luciferase reporter gene, in presence of 0.1% FCS. GLI-driven firefly luciferase activity was normalized to the Renilla luciferase control, and luciferase activity determined using a Dual Luciferase Kit (Promega; ref. 24). SUFU −/− MEFs were a gift of Dr. Toftgard (Karolinska Institute, Stockholm, Sweden; ref. 25). Wild-type (WT) and GLI null MEFs were gifts of Dr. Bushman (University of Wisconsin-Madison, Madison, WI; ref. 26), and were all cultured in DMEM and 10% FBS. Medulloblastoma sphere cultures (MSC) were obtained by digesting medulloblastoma tissue for 10 minutes using Accutase (Invitrogen) at 37°C and sequentially selecting single-cell suspensions using 100-μm and 70-μm cell strainers (BD Biosciences). The resulting cell suspensions were grown ex vivo in DMEM/F12, 2% B27, and penicillin–streptomycin (Invitrogen), and allowed to form spheres for up to 10 days. MSCs were maintained in culture for a maximum of 10 passages (27). For analysis by indirect immunofluorescence, the MSCs were plated on chamber slides (Millipore) previously coated with poly-L-ornithine/laminin (Sigma). The isolation and culture of granular precursor cells (GPC) from P4–6 C57BL/6 or ND2:SMOA1 (C57BL/6-Tg(NEUROD2-SMO A1)199JOLS/J) mice was performed using the Papain Dissociation System (Worthington Biochemical Corporation). Cells were plated on poly-L-lysine–coated plates in Neurobasal-A, 1% glutamax, 2% B27, and 250 μmol/L KCl (28). Plasmids expressing GLI1, a gift of Dr. Oro (Stanford University, Stanford, CA), WT SMO (Addgene), or the SMO D473M2 double mutant (Addgene), were transfected into cells using Lipofectamine 2000 (Invitrogen). All lentiviral shRNA constructs were in the pLKO.1 vector (Dharmacon), and the subsequent lentivirus packaged and transduced into cells as described previously (29). shRNA-expressing LIGHT2 and MSC were selected using 10 μg/mL and 300 ng/mL puromycin, respectively.
For gene expression analyses, total RNA was TRIzol (Invitrogen)-extracted, converted into cDNA (Applied Biosystems), and analyzed using qRT-PCR) and TaqMan Probes (Invitrogen). GAPDH expression was used for normalization. RIPA buffer (Thermo Fisher Scientific) was used for protein extraction and levels of the indicated proteins determined by immunoblotting using antibodies purchased from Cell Signaling Technology (GLI1, GAPDH, and HSP90), Santa Cruz Biotechnology (CK1α), R&D Systems (GLI2), BD Biosciences (phosphoserine/threonine), or Calbiochem (α-TUBULIN). GLI-binding beads, and nonspecific beads, were prepared by conjugating biotinylated double-stranded oligonucleotides containing a defined GLI-binding site or a nonspecific sequence to Pierce streptavidin magnetic beads, as described previously (24). Protein levels were quantified using Image Studio Software (LI-COR Biosciences). Cell proliferation was determined using a bromodeoxyuridine (BrdU) Assay, in which BrdU (10 μmol/L; BioLegend) was added to cells 2 hours prior to fixation and staining (Cell Signaling Technology; ref. 29). Reduction of 3-(4,5-dimethyl-2-thiazolyl) 2,5-diphenyl-2H-tetrazolium bromide (MTT) to formazan was used to determine cell viability (30). SSTC3 abundance in plasma and brain tissue was determined following protein precipitation, methanol extraction, analyses by LC/MS-MS, and comparison with a corresponding calibration curve. After separation on a C18 reverse phase HPLC column (Agilent) using an acetonitrile-water gradient system, peaks were analyzed by mass spectrometry using ESI ionization in multiple reaction monitoring mode. LC/MS-MS was performed using an Agilent 6410 mass spectrometer coupled with an Agilent 1200 HPLC and a CTC PAL chilled autosampler, controlled by MassHunter Software (Agilent).
Mouse work
All mouse work was conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee at the University of Miami. PTCH1 (PTCH1TM1MPS/J(25)) or ND2: SMOA1 (C57BL/6-Tg(NEUROD2-SMO A1)199JOLS/J) mice (Jackson Laboratory) were mated to generate breeding colonies. Spontaneous tumors from PTCH1+/− mice were expanded and maintained in 6-week-old male CD1-Foxn1nu mice as allografts (Charles River Laboratories). A similar protocol was used to expand a PDX with a TRP53 germline missense mutation (A138P) and MYCN amplification (determined by FISH) from a patient with medulloblastoma from St Jude Children Research Hospital (Memphis, TN; TB-14–7196). Primary PDX medulloblastoma tissue was initially implanted in the cortices of NSG mice, and then subsequently amplified by cerebellar implantation in CD1-Foxn1nu mice. For orthotopic implantation, 1 × 104 PTCH1+/− mouse medulloblastoma cells or 1 × 106 TB-14–7196 human medulloblastoma cells were resuspended in 3 μL serum-free media and implanted into the cerebella of 6-week-old male CD1-Foxn1nu mice. Coordinates used were 2-mm down lambda, 2-mm right of the midline suture, and 2-mm deep (31). SSTC3 (StemSynergy Therapeutics Inc.) was diluted in DMSO to a final volume of 50 μL and administrated via intra-peritoneal injection. For IHC analyses, mouse tissues were fixed in 10% formalin for 48 hours prior to processing and staining with the indicated antibodies, following the manufacturer’s recommendations (Cell Signaling Technology). A TACS TdT In Situ DAB Kit (R&D Systems) for terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling was used to detect DNA fragmentation, following the manufacturer’s recommendations. Tumor area was quantified using hematoxylin and eosin (H&E)–stained slides imaged using an Olympus IX7I microscope, and measured using CellSens Software (Olympus). Metastatic tumor spread was assessed using a blinded visual score of H&E-stained tissues from at least 6 independent mice. Tumor lesions bigger than 50 mm2 in brain tissue outside of the posterior fossa or in the leptomeningeal space were quantified using similar software. In symptom-free survival experiments, mice were sacrificed upon developing signs of medulloblastoma (hunching, circling, hemi-neglect, or ataxia) or 9 months after treatment withdrawal for ND2:SMOA1 mice or 90 days for TB-14–7196 PDX. Presence of tumors was confimed by H&E staining.
In vivo tumor detection in ND2:SMOA1 mice was determined after intravenous injection of AngioSense 680 EX and 750 EX (Perkin Elmer). For luciferase imaging of TB-14–7196 tumors, resected tumor cells were centrifuged in the presence of luciferase-expressing lentivirus at 2,000 rpm for 2 hours at 4°C prior to implantation. D-Luciferin (Perkin Elmer) was administrated intra-peritoneally to each mouse (150 mg/kg) 10 minutes before imaging. All tumor imaging was performed using a Caliper/Xenogen IVIS SPECTRUM. Luciferase intensity was determined by measuring luminescence signal in the brain or in the spinal cord (after covering the primary tumor location) using Living Image Advanced In Vivo Imaging Software (Perkin Elmer). Increases in the size of TB-14–7196 tumors were determined by dividing the tumor luminescence signal in the brain at day 35 (posttreatment) by that at day 15 (pretreatment).
Statistical analysis
Results shown from in vitro analyses represent the mean of at least 3 independent experiments ± SEM. For BrdU staining 4 fields per condition, from 3 independent experiments, were quantified. For IHC quantification, the results shown represent the mean and SEM of at least 4 fields from 3 different mice. For in vivo analyses, the RNA expression and protein results shown represent the mean and SEM of at least 4 mice per experimental condition. For tumor size studies, the results shown represent the mean and SEM of at least 6 mice per experimental condition. Significance for 2-sample analyses was determined using an unpaired Student t test. In multiple group comparisons, significance was determined using a 1-way ANOVA followed by a post hoc Student–Newman–Keuls analysis. For survival analyses, at least 10 mice per experimental condition were used and significance calculated using a log-rank (Mantel–Cox) test. Statistical significance was reached when P < 0.05. The significance of mice showing distant metastasis, determined by IVIS imaging, was calculated using a χ2 test, and was considered statistically significant when α < 0.05.
Results
The second-generation CK1α activator SSTC3 attenuated the activity of a commonly used SHH-dependent reporter gene assay (EC50 = 68 nmol/L), relative to its inactive structural analogue SSTC111 (Fig. 1A). SSTC3 was also able to reduce the expression of a known SHH target gene (GLI1) in a time- and dose-dependent manner (Fig. 1B), and this attenuation of GLI1 expression occurred in a CK1α-dependent manner (Fig. 1C; Supplementary Fig. S1A). Consistent with the positive role that CK1α also plays in SMO activation (32), knockdown of CK1α reduced the overall levels of SMO agonist (SAG)-induced reporter gene activation. To further determine the ability of SSTC3 to attenuate SHH signaling, we also utilized primary GPC whose proliferation is dependent on SHH activity. SSTC3 attenuated the SAG-driven proliferation of GPC (Fig. 1D and E) in an on-target manner, as it also attenuated the expression of SHH target genes (Fig. 1F). Consistent with SSTC3 acting downstream of SMO, it was able to attenuate SHH activity driven by 2 clinically relevant, vismodegib-resistant drivers of medulloblastoma: loss of SUFU and a constitutive active form of SMO engineered to be vismodegib-resistant (SMOM2: D473; Fig. 1G and H). As the stability and activity of GLI1 and GLI2 are regulated by CK1α (33, 34), we determined whether SSTC3 was acting on GLIs to regulate SHH signaling. SSTC3 attenuated SHH signaling in WT, GLI1−/−, and GLI2−/− MEFs. However, it had little effect on GLI1/2−/− MEFs, consistent with SSTC3 acting on both GLI1 and GLI2 (Fig. 1I and J).
Figure 1.

SSTC3 acts downstream of SMO to attenuate SHH activity. A, LIGHT2 cells were treated with 1 μg/mL recombinant SHH for 24 hours, followed by addition of the indicated concentrations of SSTC3 or SSTC111 for an additional 48 hours. Firefly luciferase activity was then determined and normalized to Renilla luciferase activity. B, NIH-3T3 cells were incubated with the SAG (100 nmol/L) and the indicated concentrations of SSTC3. The expression of GLI1 was determined at the indicated timepoints and normalized to that of the housekeeping gene GAPDH. Data were normalized to a vehicle control. C, LIGHT2 cells were transduced with lentivirus expressing the indicated shRNA to generate stable polyclonal cell lines. These cells were treated with SAG (100 nmol/L) and SSTC3 (200 nmol/L) for 48 hours. The expression of GLI1 was determined and normalized to that of GAPDH. D, GPCs were incubated in the presence of DMSO, SAG (100 nmol/L), or SAG (100 nmol/L) and SSTC3 (200 nmol/L), for 24 hours prior to quantification of BrdU incorporation. E, Representative images from similarly treated GPC are shown. F, GPC were treated with vehicle, vismodegib (100 nmol/L), or SAG (100 nmol/L) and SSTC3 (200 nmol/L), and expression of the indicated genes determined 6 hours later. Data were normalized to a SAG-treated vehicle control. G, SUFU−/− MEFs were treated for 48 hours with vehicle, the indicated concentrations of SSTC3, or vismodegib (200 nmol/L). The expression of GLI1 was then determined and normalized to that of GAPDH. Data were normalized to a vehicle control. H, LIGHT2 cells expressing WT SMO, the vismodegib-resistant oncogenic SMO mutant M2-D473, or GLI1, were treated with vehicle, vismodegib (200 nmol/L), or SSTC3 (200 nmol/L). Firefly luciferase activity was then determined and normalized to Renilla luciferase activity. Data were normalized to a SMO WT vehicle control. I, The indicated MEFs were treated with SAG (100 nmol/L) and SSTC3 (200 nmol/L) for 24 hours, and the levels of PTCH1 expression determined and normalized to that of housekeeping gene GAPDH by qRT-PCR. Data were normalized to a SAG-treated vehicle control. J, The indicated MEFs were treated with SAG (100 nmol/L) and SSTC3 (200 nmol/L) for 24 hours, and the levels of GLI1 expression determined and normalized to that of housekeeping gene GAPDH by qRT-PCR. Data were normalized to a SAG-treated vehicle control.
A number of groups have isolated and characterized MSCs from mouse SHH subgroup medulloblastoma (35–40), the majority of which are insensitive to SMO antagonists. Segal and colleagues recently described a novel culturing system that allows MSCs to retain their sensitivity to SMO antagonists (27). We isolated sphere cultures from a mouse PTCH1-mutant–driven medulloblastoma using these culturing conditions and validated them, showing that their viability and expression of 2 SHH target genes was sensitive to low doses of vismodegib (EC50 = ~ 50 nmol/L; Fig. 2A; Supplementary Fig. S2A). SMO knockdown also reduced the viability of the MSC and reduced the expression of GLI1 (Fig. 2B; Supplementary Fig. S2B), relative to a control shRNA, further confirming activation of SHH signaling in these sphere cultures. We next treated these characterized MSCs with SSTC3 or SSTC111 and showed that SSTC3 attenuated their viability in a potent, dose-dependent manner relative to SST111 treatment (Fig. 2C). The decrease in MSC viability observed upon SSTC3 treatment is likely due to decreased proliferation, as we observed decreased incorporation of BrdU upon 24 hours SSTC3 exposure (Supplementary Fig. S2C). SSTC3 also attenuated the expression of SHH target genes in these MSCs (Fig. 2D), and reduced GLI1 and GLI2 protein levels (Fig. 2E; Supplementary Fig. S2D). Consistent with SSTC3 acting on-target to reduce medulloblastoma growth, CK1α shRNA–transduced cells were more resistant to SSTC3 than those transduced with control shRNA lentivirus (Fig. 2F and G).
Figure 2.

SSTC3 inhibits the growth of SHH subgroup medulloblastoma. A, MSCs were isolated from a PTCH1+/− medulloblastoma. The MSCs were incubated for 3 days with the indicated concentrations of vismodegib and cell viability determined using a MTT reduction assay. Data were normalized to a vehicle control. B, PTCH1-mutant MSCs were transduced with the indicated shRNA and cell viability determined 5 days later using a MTT assay. Data were normalized to control shRNA (Scramble)-transduced cells. C, PTCH1-mutant MSC were treated with the indicated concentrations of SSTC3 or SSTC111, and cell viability determined 3 days later. Data were normalized to a vehicle control. D, Expression of the indicated genes in similarly treated cells was determined 24 hours after treatment. Data were normalized to a vehicle control. E, MSCs were treated with SSTC3 (400 nmol/L) or vehicle control at the indicated time points. The levels of the indicated proteins were then determined by immunoblotting. Representative blots are shown. F, MSC were stably transduced with the indicated shRNA and CK1α expression determined by qRT-PCR. Data were normalized to control shRNA (Scramble)-transduced cells. G, MSCs were stably transduced with the indicated shRNA and then treated with SSTC3 (2,000 nmol/L) or vehicle control. Cell viability was determined 3 days later using a MTT assay. Data were normalized to a vehicle control. H, SSTC3 distribution was determined in mouse plasma and brain tissue at the indicated times (N = 3). I, Mice carrying orthotopically implanted PTCH1-mutant medulloblastoma tumors were treated every other day with SSTC3 (10 mg/kg i.p. in DMSO) or vehicle for 30 days. The residual tumors from these mice were immunostained for the proliferation biomarker PCNA and the numbers of PCNA+ cells per field in 4 fields from 3 independent mice quantified. J, Representative images of PCNA-immunostained cells are shown. K, These residual tumors were immunostained for the apoptosis biomarker Cleaved CASPASE-3 and the numbers of Cleaved CASPASE-3+ cells per field in 4 fields from 3 independent mice quantified. L, Representative images of Cleaved CASPASE-3 immunostaining are shown.
One of the challenges of treating brain tumors with drugs is penetration across the BBB. We show that SSTC3 is BBB penetrant, enriching in the brain to levels comparable with those found in serum (Fig. 2H). This is in stark contrast to pyrvinium, whose levels in the serum are essentially undetectable (41). Taking advantage of this enrichment of SSTC3 in the brain, established mice with orthotopic PTCH1 medulloblastoma implants were treated with SSTC3 for 30 days. The SSTC3-exposed residual tumors were significantly smaller than vehicle-treated tumors (Supplementary Fig. S2E and S2F). Furthermore, tumor cells displayed reduced proliferation (Fig. 2I and J; Supplementary Fig. S2G) and increased apoptosis (Fig. 2K and L; Supplementary Fig. S2G) in vivo. Consistent with SSTC3 acting on-target in vivo, SSTC3-exposed tissue exhibited reduced SHH target gene expression (Supplementary Fig. S2H) and decreased levels of GLI1 and GLI2 protein (Supplementary Fig. S2I and S2J). Thus, SSTC3 is BBB penetrant and exhibits efficacy against a PTCH1-mutant–driven orthotopic mouse model for medulloblastoma.
To validate efficacy in a model in which the BBB was never manipulated mechanically, we next tested SSTC3 in a genetically engineered mouse model (GEMM) of medulloblastoma driven by SMOA1, a vismodegib relative insensitive oncogenic SMO mutant (42–45). ND2:SMOA1-mutant GPC showed relative resistance to vismodegib compared with WT GPCs, while SSTC3 exhibited comparable efficacy on both types of GPCs (Fig. 3A), validating the relative insensitivity of SMOA1 to vismodegib. We next treated a cohort of 8-week-old ND2:SMOA1 mice with SSTC3 or vehicle for 1 month and determined tumor burden by IVIS imaging (Fig. 3B; Supplementary Fig. S3A), or pathologically (Fig. 3C and D). In both cases, SSTC3 exhibited dramatic effects on medulloblastoma growth. Importantly, the number of metastases in cerebrum and leptomeninges, which are commonly observed in this GEMM (43), was also significantly reduced in response to SSTC3 (Fig. 3E; Supplementary Fig. S3B). Furthermore, SSTC3-treated tumors exhibited decreased proliferation (Fig. 3F and G; Supplementary Fig. S3C) and increased apoptosis (Fig. 3H and I; Supplementary Fig. S3C–S3F). Consistent with SSTC3 attenuating tumor growth in an on-target manner, medulloblastoma tissue exposed to SSTC3 exhibited decreased expression of SHH target genes (Fig. 3J) and GLI2 protein levels (Fig. 3K; Supplementary Fig. S3G). We next treated a cohort of 8-week-old ND2:SMOA1 mice for 1 month with SSTC3 and monitored survival. Even with this limited time of exposure, mice treated with SSTC3 showed a significant increase in survival (Fig. 3L), with 40% of the SSTC3 mice showing no signs of tumor burden 9 months after treatment withdrawal. The observation that more than the half of the SSTC3-treated cohort died despite the treatment suggests that 1-month dosing may not be sufficient to provide complete tumor regression.
Figure 3.

SSTC3 increases the symptom-free survival of a GEMM of SHH subgroup medulloblastoma. A, SAG (100 nmol/L)-induced WT- or ND2:SMOA1-derived GPC were treated with the indicated concentrations of vismodegib or SSTC3. GPC proliferation was subsequently determined by BrdU incorporation 24 hours later. Data were normalized to a vehicle control. B, Two-month-old ND2:SMOA1 mice were treated with vehicle or SSTC3 (20 mg/kg i.p. in DMSO) daily for 1 month. Representative IVIS images from vehicle or SSTC3-treated mice are shown. C, Similar mice were treated every other day with SSTC3 (10 mg/kg i.p. in DMSO) for a month. Representative images of brains and H&E staining from WT vehicle, ND2:SMOA1 vehicle, or SSTC3-treated mice are shown. D, The area of tumor from the vehicle and SSTC3-treated mice was determined (N = 6). E, Metastatic lesions were quantified from similarly treated mice (N = 6). F, Tumors from these mice were immunostained for the proliferation biomarker PCNA and the numbers of positive cells per field in 4 fields from 3 independent mice quantified. G, Representative images of PCNA immunostaining are shown. H, Orthotopic tumors were immunostained for the apoptosis biomarker Cleaved CASPASE-3 and the numbers of positive cells per field in 4 fields from 3 independent mice quantified. I, Representative images of Cleaved CASPASE-3 immunostaining are shown. J, Four-month-old ND2:SMOA1 mice were treated with vehicle or SSTC3 (10 mg/kg i.p. in DMSO) for 2 consecutive days. The mice were then sacrificed and their cerebella harvested 6 hours after the last injection. The expression of the indicated genes was then determined and normalized to the expression of GAPDH. Data was normalized to a vehicle control (N = 5). K, The levels of indicated proteins in similarly treated mice were determined by immunoblotting from a cohort of vehicle (1–5) or SSTC3 (6–10)-treated mice. L, Two-month-old ND2:SMOA1 mice were treated with SSTC3 (10 mg/kg i.p. in DMSO) every other day for 1 month and medulloblastoma symptom-free survival monitored for 9 additional months (N = 10).
In a recent clinical trial, vismodegib exhibited poor efficacy against TRP53-mutant patients with SHH subgroup medulloblastoma (19), likely due to GLI activation (46) or amplification (23, 47) driving canonical SHH signaling downstream of SMO (14). As SSTC3 acts downstream of SMO, we hypothesized that it would exhibit efficacy against this most clinically challenging form of medulloblastoma. Mice carrying such a PDX, TB-14–7196, developed signs of tumor burden within 5 weeks. Similar to the histology reported for primary TRP53-mutant, MYCN-amplified, SHH subgroup medulloblastoma samples (10), these PDXs displayed large-cell histology that was highly invasive (Supplementary Fig. S4A). Three weeks after tumor implantation, we treated this cohort of mice with vehicle or SSTC3 for 2 days and then harvested medulloblastoma tissue. SSTC3 attenuated the expression of a subset of human-specific GLI-driven target genes in these PDXs relative to vehicle-treated mice (Fig. 4A). Moreover, the levels of GLI proteins were reduced in tumor tissues from SSTC3-treated mice. In addition, immunoblotting of these extracts with phospho-serine/threonine–specific antibodies showed increased phosphorylation of GLI2, relative to total GLI2 levels, in SSTC3-treated tumor tissues (Fig. 4B). Although, we cannot rule out that the proteins identified as phospho-GLI1 and phospho-GLI2 actually correspond to GLI-binding proteins with similar molecular sizes, whose phosphorylation is increased in response to SSTC3. All together, these results suggest that SSTC3 can reduce GLI signaling in human SHH subgroup medulloblastoma tissue known to be vismodegib-resistant in the clinic (19). Cells derived from this PDX were implanted orthotopically into the cerebella of immunocompromised mice and chronically treated with vehicle or SSTC3. Tumors from SSTC3-treated mice were significantly smaller than vehicle-treated ones (Fig. 4C and D). In addition, we implanted mice with TB-14–7196 PDX cells infected with a luciferase-expressing virus. Fifteen days after implantation, mice were treated with vehicle or SSTC3 for 20 consecutive days and tumor growth monitored afterwards. Similarly, SSTC3-treated tumors were smaller than vehicle-treated ones (Fig. 4E; Supplementary Fig. S4B). Importantly, a reduced number of metastatic lesions were observed for both distant (spinal metastasis; Fig. 4F; Supplementary Fig. S4C) and cranial metastasis (located out of the posterior fossa; Fig. 4G and H) upon SSTC3 treatment. Consistent with this reduction in tumor size and spread, tumor tissue from mice exposed to SSTC3 showed reduced proliferation histologically (Fig. 4I and J; Supplementary Fig. S4D) and increased levels of apoptotic biomarkers relative to vehicle-treated mice (Fig. 4K and L; Supplementary Fig. S4D–S4G). A larger cohort of mice was also treated with SSTC3 for 30 days and then monitored for survival. SSTC3-treated mice exhibited a significant increase in survival over vehicle-treated mice (35 days median survival for the vehicle treated mice vs. 57 days for the SSTC3 ones; Fig. 4M). Thus, SSTC3 exhibits significant efficacy in reducing the growth and metastases of TRP53-mutant SHH medulloblastoma PDX.
Figure 4.
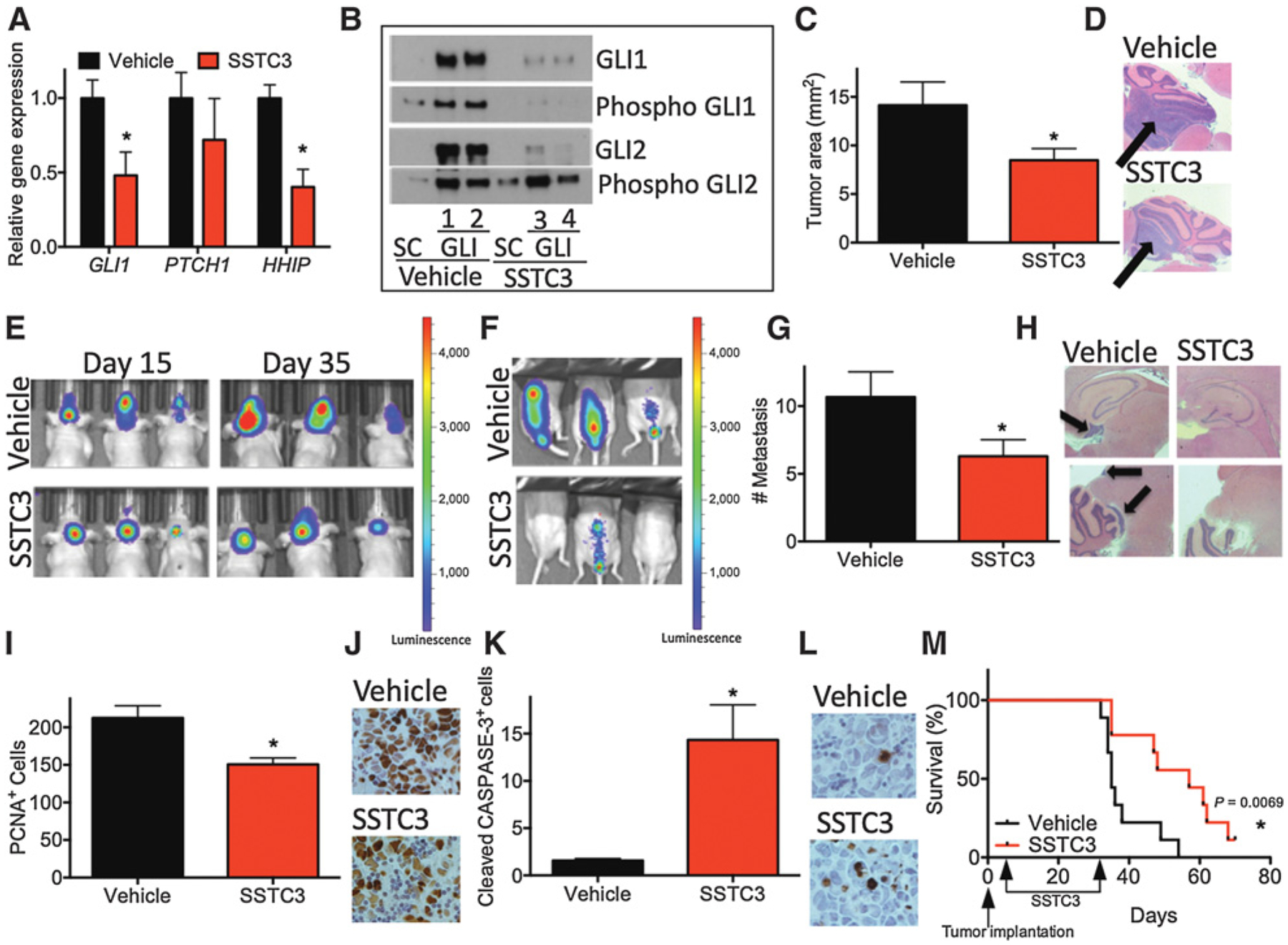
SSTC3 inhibits the growth of a patient-derived TRP53-mutant, MYCN-amplified medulloblastoma model. A, Mice carrying orthotopically implanted SHH subgroup medulloblastoma PDXs (TB-14–7196) for 3 weeks were treated for 2 consecutive days with vehicle or SSTC3 (10 mg/kg i.p in DMSO). Residual tumor tissue was harvested 6 hours after the last injection and expression of the indicated genes determined and normalized to that of GAPDH. Data were normalized to a vehicle control (N = 10). B, Mice carrying subcutaneous medulloblastoma PDX, TB-14–7196, were similarly treated with vehicle (1,2) or SSTC3 (3,4). GLI proteins were enriched using a DNA pull-down assay, containing either GLI binding site or scramble (SC) oligonucleotide beads. The GLI affinity-bead enriched proteins were subsequently immunoblotted with antibodies to GLI1, GLI2, or phospho-serine/threonine. Phospho-GLI1 and Phospho-GLI2 were identified on the basis of their similar molecular size to GLI1 or GLI2, respectively. A representative blot is shown. C, Mice carrying an orthotopically implanted medulloblastoma PDX, TB-14–7196, were treated with vehicle or SSTC3 (10 mg/kg i.p. in DMSO) every other day for 30 days. The size of the tumors from these mice was then determined (N = 10). D, Representative H&E staining of tumors from these mice. E, Mice carrying an orthotopically implanted medulloblastoma PDX (TB-14–7196) for 15 days, and engineered to express luciferase, were treated daily with vehicle or SSTC3 (10 mg/kg i.p. in DMSO) for an additional 20 days. Tumor growth was determined by IVIS imaging on day 15 (before dosing) and 35 (after dosing). Representative IVIS imaging is shown. F, Distant metastasis in similarly treated mice was detected by IVIS imaging on day 35 after implantation. Representative IVIS imaging is shown. G, The number of cranial lesions were quantified in similar mice after 30 days treatment (N = 10). H, Representative images of cerebrum and leptomeningeal tumor spread in these mice. I, Residual tumors from these mice were immunostained for the proliferation biomarker PCNA and the number of positive cells per field, in 4 fields from 3 independent mice, quantified. J, Representative images of PCNA immunostaining are shown. K, Residual tumors from these mice were immunostained for the apoptosis biomarker Cleaved CASPASE-3 and the numbers of positive cells per field, in 4 fields from 3 independent mice, quantified. L, Representative images of Cleaved CASPASE-3 immunostaining are shown. M, Mice carrying an orthotopically implanted medulloblastoma PDX, TB-14–7196, were treated with vehicle or SSTC3 (10 mg/kg i.p in DMSO) every other day for 30 days and medulloblastoma symptom-free survival monitored (N = 10).
Discussion
We show that a novel small-molecule CK1α activator, SSTC3, attenuates GLI signaling in a CK1α-dependent manner. SSTC3 inhibits SHH signaling downstream of SMO in vitro and in vivo, bypassing mechanisms of SMO inhibitor resistance commonly observed in the clinic (45, 48, 49). Furthermore, unlike the first-in-class CK1α activator pyrvinium (21), SSTC3 enriches in serum and crosses the BBB to attenuate SHH signaling, reduce tumor growth, and increase the survival of 2 distinct mouse models of SHH subgroup medulloblastoma. These mouse models of medulloblastoma exhibit numerous pathologic markers of leptomeningeal metastases (43), which is uniformly lethal in patients with medulloblastoma (11, 50). Significantly, SSTC3 was also able to reduce the emergence of these pathologic markers of metastasis. Thus, CK1α activators represent a BBB permeable, small-molecule GLI inhibitor with the potential to target patients with SHH subgroup medulloblastoma, including those that exhibit leptomeningeal metastasis.
We have previously shown that a subset of SHH subgroup medulloblastoma–initiating cells are actually WNT-dependent, and that inhibition of WNT signaling attenuates medulloblastoma growth (31). We have also shown that CK1α activators can function as WNT inhibitors in vitro and in vivo (22, 51). However, we did not observe significant activation of WNT signaling in the PTCH1 sphere cultures or mouse medulloblastoma models. Thus, we did not observe significant changes in WNT target gene expression upon SSTC3 exposure, consistent with SSTC3 acting as a GLI inhibitor in these mouse models of medulloblastoma (Supplementary Fig. S5A and S5B). However, consistent with our previous mouse PTCH1; TRP53-mutant medulloblastoma model, TRP53-mutant PDXs (TB-14–7196) exhibited significant WNT target gene expression and this expression could be reduced upon exposure to SSTC3 (Supplementary Fig. S5C). Thus, although unlikely, additional contributions to SSTC3’s efficacy against SHH-subgroup medulloblastoma may also result from attenuating WNT signaling.
Two SMO inhibitors, vismodegib and sonidegib, are currently FDA approved for use in patients with advanced BCC (17, 52), and a number of others are in various stages of clinical trials (53). Although exhibiting dramatic initial efficacy, BCCs typically relapse after treatment due to vismodegib resistance (54, 55). Deep sequencing of relapsed BCC tissue identified mutations in SMO as a common mechanism of resistance, and also identified mutations in genes that act downstream of SMO-GLI amplification and loss-of-function mutations in SUFU (45, 48, 49). The promising results observed in patients with advanced BCC resulted in the rapid repurposing of vismodegib for patients with medulloblastoma (18). Vismodegib exhibited significant efficacy in patients with SHH subgroup medulloblastoma, although rapid tumor recurrence due to resistance was also commonly observed (20). These mechanisms of vismodegib resistance underscore the clinical need for novel targeted therapies that act downstream of SMO, especially those that act on GLI proteins themselves. Consistent with transcription factors being difficult to target in the clinic, only a handful of such inhibitors have been described previously (56). One of these, arsenic trioxide, binds directly to the zinc finger region of GLI proteins to inhibit their activity (57). As arsenic trioxide is already FDA-approved for a subtype of leukemia (58), clinical trials repurposing it as a GLI inhibitor have already being performed for patients with advanced BCC (NCT01791894). Unfortunately, the limited potency and BBB permeability of arsenic trioxide (59) is likely to limit clinical utility for patients with medulloblastoma. The family of bromodomain and extraterminal domain inhibitors has also been shown to attenuate GLI activity, binding to and inhibiting the transcription of GLI1 and GLI2 (60–62). One of these small molecules, OTX015, has been shown to permeate the BBB (63) and is currently being evaluated in clinical trials for glioblastoma (NCT02296476).
Improved treatment options for patients with SHH subgroup medulloblastoma remains a significant unmet need in pediatric brain tumor therapy. While some progress has been made in identifying such agents, their clinical efficacy has been limited by early resistance (vismodegib) or lack of ability to cross the BBB (arsenic trioxide). Here, we have demonstrated the efficacy of SSTC3 downstream of known vismodegib resistance mechanisms, as well its ability to penetrate the BBB. Importantly, our work shows that SSTC3 exhibits efficacy against a patient-derived TRP53-mutant, MYCN-amplified SHH medulloblastoma, a form of SHH subgroup medulloblastoma that remains clinically refractory (23). TRP53 mutations are present in approximately 30% of childhood SHH medulloblastoma, both as somatic and germline mutations (Li-Fraumeni syndrome), and are considered the most important risk factor for patients with SHH subgroup medulloblastoma (12, 13, 23, 64). Clinical trials of vismodegib in patients with medulloblastoma showed no benefit for this subset of SHH subgroup patients (19), likely due to the downstream amplifications of GLI and MYCN commonly associated with TRP53 mutation in medulloblastoma (46, 47). Thus, CK1α agonists exhibit significant promise for treating this subset of patients with SHH subgroup medulloblastoma, who have few other therapeutic options.
Supplementary Material
Translational Relevance.
Despite the favorable outcome for most patients with Sonic Hedgehog (SHH) subgroup medulloblastoma, those that harbor mutations in TRP53 still have a dismal outcome. Thus, alternative treatment approaches are critically needed to manage this type of patient. Here, we present a novel brain barrier–permeable casein kinase 1a (CK1α) agonist, SSTC3, with the ability to block SHH-driven medulloblastoma growth and metastasis in 2 vismodegib-resistant medulloblastoma models, including those that are TRP53 mutant, MYCN amplified. Moreover, CK1α activation in this latter medulloblastoma model increased overall survival and decreased metastatic leptomeningeal spread, which is uniformly lethal in the clinic. Thus, CK1α activators represent a powerful and innovative approach for treating this most lethal form of patients with SHH subgroup medulloblastoma.
Acknowledgments
We would like to thank members of the Robbins, Capobianco, and Ayad laboratories, as well as Dr. M. Nadji (University of Miami, Pathology Department, Miami, FL) for providing their insights during discussions regarding this manuscript. We would like to also thank the Department of Surgery Tissue and Pathology core for tumor sample processing, and the Sylvester Cancer Modeling Shared Resource for assisting in IVIS imaging. This work was supported by Alex Lemonade Stand Foundation (M1201547, to D.J. Robbins), B*Cured (to D.J. Robbins), Live Like Bella Pediatric Cancer Research Initiative (8LA03, to D.J. Robbins), Childhood Brain Tumor Foundation (to J. Rodriguez-Blanco), FICYT (POST10-27 to J. Rodriguez-Blanco), The Ross K. MacNeill (to W.A. Weiss), Samuel Waxman Cancer Research Foundations (to W.A. Weiss), and funds from the Sylvester Comprehensive Cancer Center (to J. Rodriguez-Blanco and D.J. Robbins).
Footnotes
Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
D.J. Robbins is an employee of and has ownership interests (including patents) at StemSynergy Therapeutics, Inc. D. Orton has ownership interests (including patents) in StemSynergy Therapeutics, Inc. and reports receiving commercial research grants from NIH. M.F. Roussel is an employee of University of Tennessee. W.A. Weiss has ownership interests (including patents) in StemSynergy Therapeutics, Inc. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Packer RJ, Cogen P, Vezina G, Rorke LB. Medulloblastoma: clinical and biologic aspects. Neuro Oncol 1999;1:232–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuzan-Fischer CM, Guerreiro Stucklin AS, Taylor MD. Advances in genomics explain medulloblastoma behavior at the bedside. Neurosurgery 2017;64:21–6. [DOI] [PubMed] [Google Scholar]
- 3.MacDonald TJ, Rood BR, Santi MR, Vezina G, Bingaman K, Cogen PH, et al. Advances in the diagnosis, molecular genetics, and treatment of pediatric embryonal CNS tumors. Oncologist 2003;8:174–86. [DOI] [PubMed] [Google Scholar]
- 4.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007;114:97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 2012;123:465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Louis DN, Perry A, Burger P, Ellison DW, Reifenberger G, von Deimling A, et al. International Society Of Neuropathology–Haarlem consensus guide-lines for nervous system tumor classification and grading. Brain Pathol 2014;24:429–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017;547:311–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B, et al. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell 2017;31:737–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neumann JE, Swartling FJ, Schuller U. Medulloblastoma: experimental models and reality. Acta Neuropathol 2017;134:679–89. [DOI] [PubMed] [Google Scholar]
- 10.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 2016;131:803–20. [DOI] [PubMed] [Google Scholar]
- 11.Garzia L, Kijima N, Morrissy AS, De Antonellis P, Guerreiro-Stucklin A, Holgado BL, et al. A hematogenous route for medulloblastoma leptomeningeal metastases. Cell 2018;172:1050–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwalbe EC, Lindsey JC, Nakjang S, Crosier S, Smith AJ, Hicks D, et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: a cohort study. Lancet Oncol 2017;18:958–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhukova N, Ramaswamy V, Remke M, Pfaff E, Shih DJ, Martin DC, et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol 2013;31:2927–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robbins DJ, Fei DL, Riobo NA. The Hedgehog signal transduction network. Sci Signal 2012;5:re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kool M, Jones DT, Jäager N, Northcott PA, Pugh TJ, Hovestadt V, et al. Genome sequencing of SHH medulloblastoma predicts geno-type-related response to smoothened inhibition. Cancer Cell 2014; 25:393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hui CC, Angers S. Gli proteins in development and disease. Annu Rev Cell Dev Biol 2011;27:513–37. [DOI] [PubMed] [Google Scholar]
- 17.Dubey AK, Dubey S, Handu SS, Qazi MA. Vismodegib: the first drug approved for advanced and metastatic basal cell carcinoma. J Postgrad Med 2013;59:48–50. [DOI] [PubMed] [Google Scholar]
- 18.Gajjar A, Stewart CF, Ellison DW, Kaste S, Kun LE, Packer RJ, et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: a pediatric brain tumor consortium study. Clin Cancer Res 2013; 19:6305–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robinson GW, Orr BA, Wu G, Gururangan S, Lin T, Qaddoumi I, et al. Vismodegib exerts targeted efficacy against recurrent sonic hedgehog-subgroup medulloblastoma: results from Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J Clin Oncol 2015;33: 2646–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med 2009;361:1173–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li B, Fei DL, Flaveny CA, Dahmane N, Baubet V, Wang Z, et al. Pyrvinium attenuates Hedgehog signaling downstream of smoothened. Cancer Res 2014;74:4811–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li B, Orton D, Neitzel LR, Astudillo L, Shen C, Long J, et al. Differential abundance of CK1αlpha provides selectivity for pharmacological CK1alpha activators to target WNT-dependent tumors. Sci Signal 2017; 10:eaak9916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramaswamy V, Nor C, Taylor MD. p53 and meduloblastoma. Cold Spring Harb Perspect Med 2015;6:a026278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fei DL, Li H, Kozul CD, Black KE, Singh S, Gosse JA, et al. Activation of Hedgehog signaling by the environmental toxicant arsenic may contribute to the etiology of arsenic-induced tumors. Cancer Res 2010; 70:1981–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Svard J, Heby-Henricson K, Persson-Lek M, Rozell B, Lauth M, Bergström A, et al. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev Cell 2006;10:187–97. [DOI] [PubMed] [Google Scholar]
- 26.Lipinski RJ, Bijlsma MF, Gipp JJ, Podhaizer DJ, Bushman W. Establishment and characterization of immortalized Gli-null mouse embryonic fibroblast cell lines. BMC Cell Biol 2008;9:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao X, Ponomaryov T, Ornell KJ, Zhou P, Dabral SK, Pak E, et al. RAS/MAPK activation drives resistance to Smo inhibition, metastasis, and tumor evolution in Shh pathway-dependent tumors. Cancer Res 2015; 75:3623–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee HY, Greene LA, Mason CA, Manzini MC. Isolation and culture of post-natal mouse cerebellar granule neuron progenitor cells and neurons. J Vis Exp 2009;23:990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodriguez-Blanco J, Schilling NS, Tokhunts R, Giambelli C, Long J, Liang Fei D, et al. The hedgehog processing pathway is required for NSCLC growth and survival. Oncogene 2013;32:2335–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodriguez-Blanco J, Martín V, Herrera F, García-Santos G, Antolín I, Rodriguez C. Intracellular signaling pathways involved in post-mitotic dopaminergic PC12 cell death induced by 6-hydroxydopamine. J Neurochem 2008;107:127–40. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez-Blanco J, Pednekar L, Penas C, Li B, Martin V, Long J, et al. Inhibition of WNT signaling attenuates self-renewal of SHH-subgroup medulloblastoma. Oncogene 2017;36:6306–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Y, Sasai N, Ma G, Yue T, Jia J, Briscoe J, et al. Sonic Hedgehog dependent phosphorylation by CK1alpha and GRK2 is required for ciliary accumulation and activation of smoothened. PLoS Biol 2011;9: e1001083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pan Y, Bai CB, Joyner AL, Wang B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol Cell Biol 2006;26:3365–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan Y, Wang C, Wang B. Phosphorylation of Gli2 by protein kinase A is required for Gli2 processing and degradation and the Sonic Hedgehog-regulated mouse development. Dev Biol 2009;326:177–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ward RJ, Lee L, Graham K, Satkunendran T, Yoshikawa K, Ling E, et al. Multipotent CD15+ cancer stem cells in patched-1-deficient mouse medulloblastoma. Cancer Res 2009;69:4682–90. [DOI] [PubMed] [Google Scholar]
- 36.Huang X, Ketova T, Litingtung Y, Chiang C. Isolation, enrichment, and maintenance of medulloblastoma stem cells. J Vis Exp 2010;43:2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res 2003;63:5821–8. [PubMed] [Google Scholar]
- 38.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature 2004;432: 396–401. [DOI] [PubMed] [Google Scholar]
- 39.Vanner RJ, Remke M, Gallo M, Selvadurai HJ, Coutinho F, Lee L, et al. Quiescent sox2+ cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell 2014; 26:33–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corno D, Pala M, Cominelli M, Cipelletti B, Leto K, Croci L, et al. Gene signatures associated with mouse postnatal hindbrain neural stem cells and medulloblastoma cancer stem cells identify novel molecular mediators and predict human medulloblastoma molecular classification. Cancer Discov 2012;2:554–68. [DOI] [PubMed] [Google Scholar]
- 41.Smith TC, Kinkel AW, Gryczko CM, Goulet JR. Absorption of pyrvinium pamoate. Clin Pharmacol Ther 1976;19:802–6. [DOI] [PubMed] [Google Scholar]
- 42.Hatton BA, Villavicencio EH, Pritchard J, LeBlanc M, Hansen S, Ulrich M, et al. Notch signaling is not essential in sonic hedgehog-activated medulloblastoma. Oncogene 2010;29:3865–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hatton BA, Villavicencio EH, Tsuchiya KD, Pritchard JI, Ditzler S, Pullar B, et al. The Smo/Smo model: hedgehog-induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res 2008;68: 1768–76. [DOI] [PubMed] [Google Scholar]
- 44.Yang J, Huang W, Tan W. Solasonine, a natural glycoalkaloid compound, inhibits Gli-mediated transcriptional activity. Molecules 2016;21:1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pricl S, Cortelazzi B, Dal Col V, Marson D, Laurini E, Fermeglia M, et al. Smoothened (SMO) receptor mutations dictate resistance to vismodegib in basal cell carcinoma. Mol Oncol 2015;9:389–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aberger F, Ruiz IAA. Context-dependent signal integration by the GLI code: the oncogenic load, pathways, modifiers and implications for cancer therapy. Semin Cell Dev Biol 2014;33:93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rausch T, Jones DT, Zapatka M, Stütz AM, Zichner T, Weischenfeldt J, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012;148:59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Atwood SX, Sarin KY, Whitson RJ, Li JR, Kim G, Rezaee M, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell 2015;27:342–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sharpe HJ, Pau G, Dijkgraaf GJ, Basset-Seguin N, Modrusan Z, Januario T, et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 2015;27:327–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramaswamy V, Taylor MD. Medulloblastoma: from myth to molecular. J Clin Oncol 2017;35:2355–63. [DOI] [PubMed] [Google Scholar]
- 51.Li B, Flaveny CA, Giambelli C, Fei DL, Han L, Hang BI, et al. Repurposing the FDA-approved pinworm drug pyrvinium as a novel chemotherapeutic agent for intestinal polyposis. PLoS One 2014;9:e101969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jain S, Song R, Xie J. Sonidegib: mechanism of action, pharmacology, and clinical utility for advanced basal cell carcinomas. Onco Targets Ther 2017;10:1645–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rimkus TK, Carpenter RL, Qasem S, Chan M, Lo HW. Targeting the sonic hedgehog signaling pathway: review of smoothened and GLI inhibitors. Cancers 2016;8:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dong X, Wang C, Chen Z, Zhao W. Overcoming the resistance mechanisms of Smoothened inhibitors. Drug Discov Today 2018;23:704–10. [DOI] [PubMed] [Google Scholar]
- 55.Atwood SX, Whitson RJ, Oro AE. Advanced treatment for basal cell carcinomas. Cold Spring Harb Perspect Med 2014;4:a013581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bhagwat AS, Vakoc CR. Targeting transcription factors in cancer. Trends Cancer 2015;1:53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim J, Lee JJ, Kim J, Gardner D, Beachy PA. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc Natl Acad Sci U S A 2010; 107:13432–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Antman KH. Introduction: the history of arsenic trioxide in cancer therapy. Oncologist 2001;6:1–2. [DOI] [PubMed] [Google Scholar]
- 59.Au WY, Tam S, Fong BM, Kwong YL. Determinants of cerebrospinal fluid arsenic concentration in patients with acute promyelocytic leukemia on oral arsenic trioxide therapy. Blood 2008;112:3587–90. [DOI] [PubMed] [Google Scholar]
- 60.Zhao Y, Yang CY, Wang S. The making of I-BET762, a BET bromodomain inhibitor now in clinical development. J Med Chem 2013;56: 7498–500. [DOI] [PubMed] [Google Scholar]
- 61.Tang Y, Gholamin S, Schubert S, Willardson MI, Lee A, Bandopadhayay P, et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat Med 2014;20: 732–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Long J, Li B, Rodriguez-Blanco J, Pastori C, Volmar CH, Wahlestedt C, et al. The BET bromodomain inhibitor I-BET151 acts downstream of smoothened protein to abrogate the growth of hedgehog protein-driven cancers. J Biol Chem 2014;289:35494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Berenguer-Daize C, Astorgues-Xerri L, Odore E, Cayol M, Cvitkovic E, Noel K, et al. OTX015 (MK-8628), a novel BET inhibitor, displays in vitro and in vivo antitumor effects alone and in combination with conventional therapies in glioblastoma models. Int J Cancer 2016;139:2047–55. [DOI] [PubMed] [Google Scholar]
- 64.Tabori U, Baskin B, Shago M, Alon N, Taylor MD, Ray PN, et al. Universal poor survival in children with medulloblastoma harboring somatic TP53 mutations. J Clin Oncol 2010;28:1345–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.