

Abstract
The chapter will review early and more recent seminal contributions to the discovery and characterization of heparanase and non-anticoagulant heparins inhibiting its peculiar enzymatic activity. Indeed, heparanase displays a unique versatility in degrading heparan sulfate chains of several proteoglycans expressed in all mammalian cells. This endo-β-D-glucuronidase is overexpressed in cancer, inflammation, diabetes, atherosclerosis, nephropathies and other pathologies. Starting from known low- or non-anticoagulant heparins, the search for heparanase inhibitors evolved focusing on structure-activity relationship studies and taking advantage of new chemical-physical analytical methods which have allowed characterization and sequencing of polysaccharide chains. New methods to screen heparanase inhibitors and to evaluate their mechanism of action and in vivo activity in experimental models prompted their development. New non-anticoagulant heparin derivatives endowed with anti-heparanase activity are reported. Some leads are under clinical evaluation in the oncology field (e.g., acute myeloid leukemia, multiple myeloma, pancreatic carcinoma) and in other pathological conditions (e.g., sickle cell disease, malaria, labor arrest).
Keywords: Heparanase, Heparanase inhibitors, Non-anticoagulant heparin, Heparin derivatives, Cancer therapy
Introduction
Heparan sulfate proteoglycan (HSPG) physiological functions, fundamental for development, homeostasis and signaling, are dependent on the integrity of heparan sulfate (HS). Heparanase is an endo-β-D-glucuronidase which cleaves chains of HS present at both cell surfaces and the extracellular matrix (ECM). Early on, its deregulation appeared to be involved in tumor cell growth, migration and metastasis, and later on in other pathologies such as inflammation, diabetes, atherosclerosis, and nephropathy [1–4].
Heparin, exclusively produced by mast cells, is a highly sulfated form of HS. Early studies evidenced the HS higher charged congener unfractionated heparin (UFH) as an efficient heparanase inhibitor as well as substrate [5, 6]. Then, investigations were mainly oriented to identify non-anticoagulant heparin derivatives to overcome the UFH anticoagulation and bleeding side effects as well as to improve its pharmacokinetics and bioavailability. This chapter describes how heparin-derivative heparanase inhibitor research has evolved referring to landmark [7] and more recent contributions focusing on structure-activity relationship. Perspectives for the development of new agents of this class as potential drugs in cancer and other pathologies are also illustrated.
Heparan Sulfate
Heparanase targets are the HS chains covalently linked to core proteins of the ubiquitous and multifunctional HSPGs, present at cell surfaces and ECM and involved in cell signaling, survival, proliferation, migration, and invasion. The HSPG biological functions are mostly carried out through electrostatic binding/interaction of the negatively charged HS chains with a multitude of proteins including chemokines and cytokines, growth factors and their receptors as well as enzymes. HS is expressed in almost all cells of mammalian species as linear polyanionic chains, differing in size (20 up to 100 kDa) and sulfated domain distribution among species and tissues of the same species. The composition of natural physiological HS chains can be altered in several pathological conditions [8–11]. Heparin and HS-like glycosaminoglycans (GAGs) have been isolated from avian intestinal mucosa [12], terrestrial invertebrates [13, 14], marine crustacean and mollusk species [15–17] as well as bacterial and virus species [12]. In the Golgi compartment of all animal cells and mast cells of connective tissue, biosynthesis of HS and heparin chains, respectively are completed through common steps leading to the formation of a specific tetrasaccharidic linkage region (D-GlcA β-1-3-O-D-Gal β-1-3-O-D-Gal1-β-3-O-D-Gal-1-β-4-O-D-xylopyranosyl-1-α) to join L-serine of the HSPG core protein. The following elongation steps lead to a common linear high molecular weight (HMW) homogeneous GAG, N-acetyl-heparosan, constituted by 4-O-β-D-glucopyranosyl 1–4-O-N-acetyl- α D-glucosamine.
A sequence of incomplete enzymatic reactions induces modifications and heterogeneity in terms of composition and size distinct for HS and heparin chains. The highly sulfated domains (NS), constituted by sequences of the trisulfated disaccharide 4-O-L-IdoA2S 1α-4-O-D-GlcNS6S are prevalent in heparin chains, while in the N-acetylated domains (NA), the disaccharide 4-O-α-D-GlcA-1β-4-O-D-GlcNAc) is more abundant in HS chains. NS and NA domains in both HS and heparin are disseminated among mixed transition regions (NS/NA) [9–18].
Experimental studies showed that the heparin chains can interact with a great number of proteins, classified as heparin binding proteins (HBPs) . Taking into consideration the ubiquitous presence of HSPGs and their multifunctional interactions in physiological media with a great variety of proteins, these should be better classified as HS binding proteins (HSBPs) [10]. However, functional studies are hampered by the fact that samples of pure HS are very expensive and difficult to isolate. The first HS rich preparation was isolated in 1948 as by-product of heparin manufacturing process and named “heparin monosulfate” for its low sulfate content [19]. On the other hand, the presently available pharmaceutical heparin is mainly derived from the porcine intestinal mucosa extracts which contain other linear GAGs, [dermatan- (DeS) and chondroitin-sulfate (ChS)] which are difficult to separate from HS [12]. In fact, DeS and ChS are present as minor components of an antithrombotic drug (Danaparoid sodium) containing 80% of low molecular weight (LMW) HS, and known in the EC market as Orgaran [20].
Heparanase: Discovery and Characterization
The term “heparanase” appeared for the first time in a paper of 1978 [21] to define a “heparitinase”-like endoglucuronidase, present in guinea-pigs basophil leukocytes, able to cleave GAGs resistant to chondroitinase ABC, such as HS but not heparin. However, three years earlier in 1975, a heparanase-like endoglucuronidase from murine mast cells was found able to cleave macromolecular heparin to functional heparin [22] and in 1983 lymphoma cells were found able to degrade HS chains of HSPGs of subendothelial ECM [23]. Finally, in 1984 other authors reported for the first time in the title of their paper the term heparanase to design an endoglucuronidase, able to degrade HS, produced by the highly invasive lung metastatic murine B16-BL6 melanoma cells [24]. Endoglucuronidase activities were previously described in other mammalian tissues and cells such as rat liver tissues [25], human skin fibroblasts and placenta [26], human platelets [27] and activated T lymphocytes [28]. A partially purified human platelet enzyme was found able to cleave both HS and heparin [29]. In the following years, the increasing interest in heparanase function and role in several pathological contexts has been documented by an ever-growing number of reports summarized and commented in [3] and in Vlodavsky et al., Chap. 10.1007/978-3-030-34521-1_1 in this volume. Since 1984 heparanase-like activity was detected in several human normal and malignant cells and tissues. However, the lack of selective activity assays along with the low concentration and instability of crude enzyme have hindered heparanase isolation and characterization until 1999 when five teams have independently reported the cloning and functional expression of the human heparanase gene in mammalian and insect cells [30–34] (Vlodavsky et al., Ilan et al., Gaskin et al., Pinhal et al., Chaps. 10.1007/978-3-030-34521-1_1, 10.1007/978-3-030-34521-1_7, 10.1007/978-3-030-34521-1_9 and 10.1007/978-3-030-34521-1_36 in this volume). Heparanase, at present the only known mammalian endo β-D-glucuronidase, is physiologically expressed primarily in platelets, activated white blood cells and placenta. The heparanase human gene encodes for a pre-proenzyme which undergoes removal of the N-terminal signal peptide in the endoplasmic reticulum to give rise to the pro-enzyme of 65 kDa. Cleavage of a 6 kDa linker peptide by cathepsins in the lysosomes leads to an active enzyme constituted of two subunits of 50 and 8 kDa that are not covalently linked, as also confirmed by X-ray crystal structures [35–39] and (Vlodavsky et al., Gaskin et al., Chaps. 10.1007/978-3-030-34521-1_1 and 10.1007/978-3-030-34521-1_7 in this volume).
Modeling investigations on heparanase interactions with HSPGs, HS/heparin, and related oligosaccharides have also been reported along with molecular model of human heparanase proposing the binding mode of HS oligosaccharide to catalytic amino acids [40]. Early studies identified the minimal HS sequences recognized by heparanase in a GlcA flanked by two NS,6S D-GlcN units, the second one can be also 3-O-sulfated [41, 42] (Fig. 20.1a).
Fig. 20.1.

(a) minimal heparanase recognized sequence: R = H; SO3−; R1 and R2 uronic acids of Hep/HS chain. (b) Antithrombin binding site: R = SO3−; NAc R1 and R2 = uronic acids of Hep/HS chain; R3 = SO3; Fondaparinux: R = SO3−; R1 = H; R2 = Me; R3 = SO3−
The disaccharide sequence GlcAβ1–4-GlcNS,3,6S [42], is a constituent of the pentasaccharide heparin-antithrombin binding region (ATBR) and of the synthetic mimic α-methyl glycoside of N-sulfated pentasaccharide Fondaparinux (Fig. 20.1b) clinically used as antithrombotic agent [43]. As a good heparanase substrate, it has been included as a component of heparanase activity assay kit particularly useful for kinetic analysis and screening of enzyme inhibitors [44, 45]. The heparanase cleavage site was described to be also dependent on the sulfation pattern of the neighboring sequences [46]. The same team identified the most suitable cleavage site in the pentasaccharide GlcNAc6S-GlcA-GlcNS-Ido2S-GlcNS6S [46]. Other common cleavage sites have been identified by the analytical profiling of heparanase digests of HS of different origin and sulfation degree [47].
HS cleavage is affected by heparanase expression levels and proteolytic activation but can also be influenced by the activity of other HS biosynthetic and modifying enzymes. Notably, the multiple substrate recognition allows heparanase to degrade HS chains independently of cell specificity and environment [9]. Hence, heparanase localization and activating processes are relevant in determining its biological function in a variety of healthy and malignant cells and tissues [35]. Moreover, heparanase upregulation has been described in several malignancies and pathological conditions including acute and chronic inflammation, fibrosis, amyloidosis, diabetes and related nephropathies, osteoarthritis, atherosclerosis and other vessel wall pathologies [3, 6] and (Vlodavsky et al., Ilan et al., Elkin; Simeonovic et al., Masola et al. Li and Zhang, Chaps. 10.1007/978-3-030-34521-1_1, 10.1007/978-3-030-34521-1_9, 10.1007/978-3-030-34521-1_17, 10.1007/978-3-030-34521-1_24, 10.1007/978-3-030-34521-1_25 and 10.1007/978-3-030-34521-1_27; in this volume).
The first evidence of heparanase activity in the murine melanoma B16-BL6 [24] and T-lymphoma [23] experimental models, were provided by Nakajima et al. [24] and Vlodavsky et al. [23] associating the in vivo metastatic potential of these cells with HS degradation. Excellent reviews [2, 3, 6, 7, 11] reported findings on heparanase overexpression in several malignancies and functional studies in cancer models highlighting its causal relevant role in sustaining tumor growth and progression.
Of note, heparanase overexpression was shown to accelerate HSPG turnover along with upregulation of their HS N- and O-sulfation degree suggesting a functional correlation between the endo-β-D-glucuronidase expression level and HS oversulfation [48, 49]. Interestingly, the N-unsubstituted D-glucosamine (GlcNH3+) in the disaccharide GlcA-β1–4-GlcNH3+6S, very uncommon (0.7 to 4% of total GlcN) in natural HS chains [50], was quite abundant in HS of mammary carcinoma cells [51, 52]. More recently, the same disaccharide was also found in a significant amount in HS chains of highly invasive breast cancer cell lines expressing heparanase. These findings suggest a possible role of GlcNH3+6S in imparting heparanase degradation resistance to HS chains in these malignant cells. Indeed, the synthetic tetrasaccharide (TD 4–143,1): GlcAβ1–4-O-GlcNH3+6S-1α-4-O-GlcAβ1–4-O-GlcNH3+6S α-octyl glycoside (Fig. 20.2 a) inhibited heparanase and suppressed cancer cell invasion in vitro [53].
Fig. 20.2.

Structures of two synthetic oligosaccharide heparanase inhibitors: (a) TD 4–143,1 [53]; (b) pseudopentasaccharide [ED 80061] [53]
Also, the synthetic pseudopentasaccharide [ED 80061] (Fig. 20.2b), bearing at the reducing end a 2-deoxy-1 N-imido D-glucuronic acid moiety, was shown to be a potent heparanase inhibitor (IC50 11 nM) with antimetastatic activity in the B16-F10 and MAT 13702 experimental models [54].
Overall, these findings provide useful information concerning the heparanase-HS interaction and structural determinants to be exploited for the design of efficient heparanase inhibitors devoid of side effects.
Heparosan-Related Heparanase Inhibitors
Natural and Semi-Synthetic Derivatives
Invertebrate and bacterial N-acetyl heparosan derivatives endowed with a peculiar structural chemo-diversity have provided the opportunity to perform in-depth SAR study and to define structural determinants responsible for different biological activities.
A heterogeneous HS (Mw ~ 27 KDa), mainly constituted of N-acetyl heparosan sequences (GlcA-GlcNAc)n was isolated from viscera of the bivalve mollusk . NMR analysis indicated that the major disaccharide 4-O-D-GlcAβ1–4-D-GlcNAc showed a low sulfation degree due to partial and random 2- and/or 3-O-sulfation of D-GlcA along with partial N- and 6-O sulfation of GlcNAc (Fig. 20.3). Endowed with heparanase and P-selectin inhibitory activity and a low anticoagulant activity (five-fold lower than porcine heparin), the mollusk HS showed anti-metastatic and anti-inflammatory effects in vivo without bleeding effect [55].
Fig. 20.3.

Major disaccharide units of HS. D-GlcA:glucuronic acid; D-GlcN: glucosamine
A capsular polymeric (Mw 35–49 kDa) GAG of the Escherichia coli strain K5 showed the same structure of the HS/heparin natural biosynthetic precursor N-acetyl heparosan constituted by a regular sequence of [GlcAβ1–4-GlcNAc α1–4]n [56]. This discovery was extremely useful in the search for new anticoagulant and antithrombotic heparins endowed with better pharmacokinetic and fewer side effects and analogously, in the identification of non-anticoagulant congeners to be evaluated in other therapeutic fields. The progress in the knowledge of the HS/heparin biosynthetic pathway [57, 58] has opened the way for chemo-enzymatic synthesis of polymers called “bioheparin” [59] and “bioengineered heparins” [60]. These novel approaches were stimulated, at the end of the nineties, by the “mad-cow crisis”, which urged the search for new animal sources of heparin or semisynthetic derivatives to compensate the withdrawal of bovine heparin from the market. Looking for heparin-like GAGs, several N-deacetylated N-sulfated sulfoamino heparosans were firstly obtained and then subjected to O-sulfation at the 6-O position of GlcNS and 2-O, 3-O sulfation of GlcA [59]. A number of semisynthetic O-sulfated sulfamino heparosans (SAHSs), differing in degree and pattern of O-sulfation as well as molecular size, were tested in the mouse B16-BL6 melanoma model. Among these compounds, the two high Mw SAHS-2 (Mw 25,7 kDa) and SAHS-4 (Mw 22.7 kDa) and a low molecular weight derivative SAHS-5 (Mw 3.2 KDa), showed a remarkable anti-metastatic activity, with the sole SAHS-4 displaying a modest anticoagulant activity [61]. Highly N,O-sulfated heparosans were found to bind FGF-2 and inhibit FGF-2-induced endothelial cell proliferation and angiogenesis likely interfering with the formation of FGF-2/FGFR/HS complexes [62–64]. Various species of O-sulfated N-acetyl heparosan (OSK5) (Fig. 20.4) were reported to bind FGF-1, −2 and − 8 with different FGF signaling antagonist activity influenced by the type of FGF and FGFR expressed and by the cellular context [65]. This class of derivatives (OSK5), along with new preparations of O-sulfated sulfamino heparosans (NSOSK5), were also tested as heparanase inhibitors in a translational project “Heparanase” supported by the EC, which recognized the enzyme as a potential therapeutic target for cancer. New powerful analytical tools, such as 2-D NMR spectroscopy, have allowed a better characterization of the component profile and sequence of heparosan derivatives and complex GAGs [66]. Focusing on their biological activities, the most representative are the HMW derivatives NSOS-K5 and OS-K5 (11–15 kDa) which displayed a stronger heparanase inhibitory in vitro in comparison with the corresponding ultra LMWH (2–3 kDa) [67].
Fig. 20.4.

Predominant disaccharide units of capsular polysaccharide from E. coli K5 (K5PS) (a), its sulfated variants (b, c) and typical heparin trisulfated disaccharide (d)
The anticoagulant activity of LMW NS,OS and OS derivatives was found negligible and lower than that of HMW NS,OS congener. The HMW OS-K5 and NS,OS-K5 preparations were shown to inhibit metastatic dissemination of human breast cancer MDA-MB-231 cells [65]. Interestingly, the same K5 derivatives, endowed with heparanase inhibitory activity, inhibited HIV replication in T cells and macrophages, likely preventing the virus attachment to the host cells [63], an event that implicates interaction of the virus with cell surface HS (Agelidis and Shukla, Chap. 10.1007/978-3-030-34521-1_32 in this volume). A very HMW (35 kDa) NS,OS-K5, endowed with moderate anticoagulant activity, was reported to inhibit in vivo bone osteolysis and tumor growth of the highly metastatic human breast cancer MDA-MB-231 (SA) cell line [68].
The project “ bioheparin” by chemoenzymatic processes led to derivatives endowed with modest or low anticoagulant activity, tested as potential antiangiogenic, antiviral, and anti-inflammatory agents [69]. The other project of chemoenzymatic synthesis of GAGs starting from N-acetyl heparosan led to a “bioengineered heparin” [70] and to some intermediate oligosaccharides which have allowed to define the substrate specificity of heparanase [46, 70–72].
Heparin Derivatives
Early studies showed an effective heparanase inhibition by UFH, even if some of UFH sequences can be recognized and cleaved by heparanase [4, 5, 41]. However, its unwanted anticoagulant activity hampered its safe use as an inhibitor of heparanase. The elimination or at least the reduction of the anticoagulant activity can be obtained through different types of chemical modifications of the structure of heparin such as oversulfation, partial desulfation, reduction of Mw or selective modifications of the residues at the antithrombin binding site representing the major determinant of anticoagulant activity. Among heparins endowed with heparanase inhibitory activity, N-acetylated and O-desulfated species of non-anticoagulant heparins were the first to be tested and found active in vivo in the metastatic B16 melanoma model whereas carboxy reduced heparin was almost inactive [4, 5]. Moreover, UFH fragments, consisting of at least 16 units, were found active both in vitro and in vivo [5].
LMWs, Ultra LMWHs and Derivatives
The clinical use of LMWHs in oncology has been approved for preventing venous thromboembolism as well as for their better pharmacokinetic and pharmacodynamic properties compared with UFH [73]. Results of randomized studies concerning the benefits of UFH and LMWHs in cancer patients were published since the early 1980s without a clear conclusion on their real impact on cancer patients’ response to therapy. Heparanase and selectins are inhibited by LMWHs, albeit with a somewhat lower efficacy than UFH. Taking into consideration the heterogeneity of the starting UFH chains in term of size, beyond the anticoagulant activity, their depolymerization can offer other compositional and interaction differences affecting the pharmacological properties of LMWH. For example, early in vivo evaluation of antimetastatic effects evidenced a significantly higher activity of dalteparin (Fragmin) in comparison with that of nadroparin or enoxaparin [5]. Size fractionation of tinzaparin allowed separation of HMW fractions whose components, not present in the other two LMWHs, are endowed with high selectin inhibitory activity but low antiXa activity [74]. Preclinical in vivo evaluation in tumor xenografts models evidenced that tinzaparin was able to sensitize cis-platin resistant ovarian cancer [75]. A non-anticoagulant ultra LMWH (2.5 kDa), obtained by hydrogen peroxide catalyzed radical heparin hydrolysis assisted by ultrasonic waves, exhibited anti-heparanase activity intermediate compared with those of tinzaparin (Mw 7 kDa), dalteparin (6.3 kDa) and enoxaparin (5.5 kDa) [76]. A glycopolymer constituted by a N-sulfated poly2-aminoethyl methacrylate carrying the heparin disaccharide ΔU2S-GlcNS,6S, was reported to inhibit heparanase, B16 melanoma cell migration, and adhesion to platelets and microvascular endothelial cells [77]. Of note, the old orally active Sulodexide, constituted by LMWH and DeS in a 80:20 mixture, isolated from porcine intestinal mucosa, has been used since 1974 as antithrombotic drug, displayed low anticoagulant activity and bleeding effects [78]. More recent studies evidenced that Sulodexide provided benefits in patients with diabetic nephropathies through inhibiting heparanase [79].
Supersulfated Heparins
A supersulfated LMWH (ssLMWH) , prepared by controlled depolymerization of UFH and endowed with low anticoagulant activity was demonstrated to inhibit heparanase, proinflammatory molecules such as leukocyte elastase, cathepsin G and hepcidin [80–82]. Tested in vivo in the metastatic B16 melanoma model, it exhibited antimetastatic activity similar to that of UFH [61]. Recently, a remarkable antitumor activity of ssLMWH has been demonstrated both in vitro and in vivo in synovial sarcoma experimental models [83]. Inhibition of synovial sarcoma cell growth and invasion was associated with downregulation of the activity of receptor tyrosine kinases of the EGFR, PDGFR and IGFIR families and heparanase inhibition. The combination of ssLMWH with an inhibitor of IGF receptors, synergistically inhibited cell proliferation and motility and promoted apoptosis. In vivo ssLMWH synergized with the receptor tyrosine kinase inhibitor to suppress orthotopic synovial sarcoma growth and spontaneous lung metastatic dissemination [83]. However, it is necessary to consider the risk that derivatives with a high degree of sulfation, may stimulate other competitive biological mechanism or cause unwanted reactions such as the activation of prekallikrein as observed with oversulfated chondroitin sulfate, known as OSCS [84, 85].
O-Desulfated Heparins
The presence of 2-O and 6-O sulfation , with at least one of the two positions retaining a high sulfation degree, was found to be essential for inhibition of heparanase. As shown in Fig. 20.5, effective heparanase inhibition was exhibited by fully 2-O-desulfated heparin whereas two intermediates bearing modified 2-O-desulfated IdoA units, namely 2,3 epoxy L-uronic acid and GalA, were practically inactive [86, 87].
Fig. 20.5.

Heparanase inhibitory activity of heparin (Hep) and desulfated heparin derivatives: (a) heparin (Hep), 6-O desulfated heparin (6OdeSH), 2,3-O desulfated heparin (2O-deSH), 2,3-O desulfated heparin with change of configuration (L,GalA), N-acetyl heparin (NAH); (b) Inhibition of heparanase by N-acetyl heparins with different acetylation degree and corresponding 25% glycol-split derivatives
Basic conditions needed for 2-O-desulfation can cleave also the 3-O-sulfate of GlcN, fundamental for the AT binding thus, producing a further decrease in the anticoagulant activity of the 2,3-O-desulfated heparin (2OdesH / ODSH) . Along with a high potency in inhibiting heparanase, 2OdesH showed in vivo antitumor activity in CaPan-2 pancreatic adenocarcinoma xenografts and antimetastatic activity in the B16-F10 mouse melanoma experimental model [88, 89]. Recently, 2OdesH has been described to block the release of the inflammation mediator, high mobility group box 1, by inhibition of p300 acetyltransferase activity [90]. A 2OdesH is currently under clinical investigation as CX01 in combination treatment of acute myeloid leukemia [Sect. 20.7.5].
6-O-desulfated heparin (6OdesH) also showed selectin inhibitory activity [86, 87] along with low anticoagulant activity. Interestingly, its LMW congener inhibited the aggregation of Plasmodium falciparum-infected red blood cells with uninfected erythrocytes to form rosettes [91].
Regarding other glycoconjugates and their applications, a 6-O-desulfated nadroparin conjugate with deoxycholic acid was orally active and able to suppress neovascularization and bone destruction in murine arthritis experimental models [92].
N-Acyl-N-Desulfated Heparins
Heparin N-desulfation can be modulated from 10 up to 100% and the products used as intermediates to obtain N-acyl heparins. When the remaining N-sulfation degree is low, compounds were non-anticoagulant and generally endowed with low heparanase inhibitory activity (Fig. 20.5b). In vivo antimetastatic activity was reported for fully N-acetyl [4, 5], N-hexanoyl [5], low and ultra low N-succinyl heparins [93]. Beside of being almost non-anticoagulant, the advantage of N-acetyl derivatives over UFH is their incapacity of releasing active bFGF from cells and ECM [94], an event that promotes tumor growth and angiogenesis [95].
A SAR study on N-acetyl heparins with N-acetylation degree ranging from 29 up to 100% showed a drastic decrease of heparanase inhibitory activity with degree of N-acetylation higher than 50%. These findings suggest that the interaction with the enzyme needs at least one N-sulfated glucosamine per tetrasaccharide [87]. Surprisingly, the formation of flexible joints inside the chain obtained by periodate oxidation of the non-sulfate uronic acid residues showed increased anti-heparanase activity (Fig. 20.5, 5b). This result has highlighted the glycol split (gs) derivative of fully N-desulfo-N acetyl heparin (G4000, 100NA-ROH, SST0001) for the development of a potential drug (Roneparstat) which will be discussed in more detail in the next section. In a follow-up study, N-acetyl heparins, ranging from 39 up to 100% N-acetylation were tested as P- and L-selectin inhibitors in comparison with UFH. The 58% N-acetylated heparin displayed a good selectin inhibitory activity as well as anti-metastatic activity when tested in vivo in murine MC38 colon carcinoma and B16 melanoma experimental models [95].
Glycol-Split Heparins: Semisynthesis and Activities
Controlled depolymerization of UFH by periodate oxidation which cleaves the linkage between the hydroxylated C(2)-C(3) of non-sulfated hexuronic acid gives oxyheparin (oxyH), susceptible to be reduced with NaBH4 leading to reduced oxyheparin (ROH) . This approach incorporates the gs-residues while preserving the sulfation pattern and degree with a low reduction of Mw [96]. Structural characterization of ROHs obtained from UFH from different animal sources are reported by Alekseeva et al. [97]. They displayed a significant reduction of anticoagulant activity mainly related to the periodate oxidation of the GlcA residues linked to tri-O-sulfated GlcN of the ATBR, whose integrity is essential for the anticoagulant activity of heparin [98, 99]. Early investigations suggested that, other than temperature and pH values, neighboring residues could influence and differentiate the periodate oxidation rate of IdoA versus GlcA [100, 101]. Other studies evidenced that mild acid hydrolysis of ROH gave oligomers bearing at the non-reducing end of non-anticoagulant LMWH, the N,3,6 trisulfated glucosamine residues [102]. In a recent kinetics study of enoxaparin periodate oxidation, NMR-HSQC showed that the complete IdoA oxidation occurred in 2 hr. while GlcA was only partially oxidized after 8 hr. This difference may be explained by the higher conformational flexibility of gs IdoA suitable for the periodate cyclic complex intermediates whose formation could be partially hindered by the GlcA neighboring residues [103]. A number of non-anticoagulant ROH were tested in a variety of therapeutic areas where UFH was active. The residual anticoagulant activity of ROH as well as of N- and O-desulfated heparins may result from interactions outside the ATBR, mediated by heparin cofactor II and the release of vascular tissue factor pathway inhibitor [104]. Investigators of Glycomed (Alameda) and the University of Boston observed severe bleeding in mice harboring human pancreatic adenocarcinoma Ca Pan-2 xenografts and murine B16-F10 melanoma administered with s.c. ROH, an effect likely due to antiplatelet and heparin cofactor II activity [88]. A modified preparation of ROH (Mw 11 kDa), with a cofactor II activity comparable to that of heparin, but lower (10–15%) anti-Xa activity was developed by Glycomed as an adjuvant in cardiovascular intervention to prevent vascular restenosis [105].
A non-anticoagulant oxy-heparin fragment carrying a hydrophobic polystyrene chain (NAC-HCPS) exhibited in vitro and in vivo antiangiogenic and antimetastatic activities in murine B16 melanoma and Lewis lung cancer (3LL) models. NAC-HCPS also inhibited 3LL tumor growth and vascularization, likely by means of inhibiting endothelial cell proliferation stimulated by VEGF165, FGF-2, or HGF [106]. ROH prepared according to the method of Casu et al. [96] inhibited P-selectin-mediated cell adhesion of human colon carcinoma cells to immobilized platelets [107]. ROH preparation, designated as low anticoagulant heparin (LAC), showed good tolerability without bleeding complication when given s.c., i.p. and i.v. to mice at dosages able to inhibit tumor cell dissemination in several murine metastatic models. LAC activity was associated with inhibition of cancer cell adhesion and extravasation in lung capillary by competing with cell-surface HS interaction [108].
The seminal report of Folkman et al. 1983, disclosing the inhibition of angiogenesis and tumor growth by heparin and its fragments, was the start-up for investigating the molecular mechanisms underlying tumor neovascularization (neoangiogenesis) [109]. Heparin chains bind with high-affinity FGFs and in particular, FGF-2, recognized as one of the major angiogenesis promoting factors. The heparin minimum FGF binding fragment was identified in the pentasulfated trisaccharide GlcNS6S-Ido2S-GlcNS6S (PST) followed by IdoA2S (Fig. 20.6 (2) mainly present in the high sulfated region of UFHs [110]. With the aim of generating 2-O-sulfation gaps along heparin chains, alkaline treatment of UFH led to heparin derivatives bearing epoxy uronic acid units that were then hydrolyzed to L-galacturonic acid units. These heparin derivatives were converted by periodate oxidation/NaBH4 reduction to gs-derivatives. Graded 2-O-desulfation led to a heparin derivative characterized by a 1:1 ratio IdoA2S: (GlcA+IdoA) residues. The following periodate oxidation/NaBH4 reduction gave a heparin chains bearing 50% gs uronic acids which showed a low anticoagulant activity due to glycol-splitting of GlcA essential for the binding to ATBR. The gs-residues generating flexible joints along the chains improved the FGF-2 antagonist and angiostatic effect as well as anti-heparanase activity [111, 112].
Fig. 20.6.

Prevalent sequences in regular regions of heparin and chemically modified heparins. (1) heparin; (2) 50% 2-O-desulfated heparin (prevalently PST.U sequences), ST1514; (3) 50% 2-O-desulfated and glycol-split LMW heparin (prevalently PST.sU sequences); ST2184. R = SO3− or Ac; TDS trisulfated disaccharide; PTS pentasulfated trisaccharide
Derivate 2 in Fig. 20.6 named ST1514 (Mw 11 kDa), was further investigated as along with its LMW derivatives 3 ST2184 (Mw 5.8 kDa). ST2184 displayed in vitro antiangiogenic and VEGF165 antagonist activity by interfering with the binding of the growth factor to its receptors [112]. ST1514 and ST2184 inhibited in vivo metastatic lung dissemination of B16-BL6 melanoma cells in mice. ST2184 was also reported to reduce angiogenesis in human MeVo melanoma xenografts and to potentiate the antitumor activity of a camptothecin derivative [113]. In addition, ST1514, being a potent heparanase inhibitor, was able to reduce wound vascular density and inflammation in heparanase overexpressing transgenic mouse model of wound healing and delayed-type hypersensitivity [114, 115].
To evaluate new potential heparin applications, N-acetyl and gs-heparin and their LMW derivatives were assessed as HS competitors and anti-inflammatory agents in chronic airway diseases caused by Pseudomonas aeruginosa. Indeed, HS has been recognized as cellular receptor for Pseudomonas aeruginosa and binding site for its flagella. HS competitions by heparin derivatives can reduce bacterial burden acting as adjuvants with clinically used antibiotics. N-acetyl heparin (C23, Mw 17.2 kDa), its LMW derivative (8 kDa), ROH (C3gs20, Mw 16,5) and three LMW derivatives (8,12.6, 9.6 kDa, respectively) were tested in a mouse model of chronic Pseudomonas aeruginosa air way inflammation. Only the HMW products were able to reduce the inflammatory response, an effect mediated by reduction of cyto−/chemo-kine levels and of neutrophil elastase activity, and by inhibition of neutrophil recruitment [116] that correlated with anti-heparanase activity.
To dissect structural determinants for effective heparanase inhibition, a library of non-anticoagulant heparins was prepared by graded or fully O-desulfation, N-acetylation of N-desulfated of UFH. Periodate oxidation and borohydride reduction were also applied to give the corresponding gs ROHs. Preliminary tests showed that some derivatives from this library, including the gs derivative of fully N-desulfo-N acetyl heparin (100NA-ROH), were effective in inhibiting lung metastasis from B16-BL6 mouse melanoma cells [117]. Further experiments showed that both the heparanase and selectin inhibitors 58NAH (58% N-acetyl heparin) and ROH were able to inhibit lung metastasis formation in MC38 colon carcinoma mouse model expressing selectin ligands [72].
The choice of fully N-acetyl ROH as the lead compound was based on its inability to release FGF-2 from ECM, its low anticoagulant activity, its remarkable inhibition of heparanase enzymatic activity and its in vivo antimetastatic activity in the B16-BL6 melanoma model. Conversely, it was found inactive in vivo on the MC38 colon carcinoma model correlating with a lack of P- and L-selectin inhibitory activity [72]. Likewise, the low anticoagulant and weak heparanase inhibitor NAH was inactive as a selectin inhibitor. Nevertheless, it displayed a higher antimetastatic activity than UFH, in the MC38 colon carcinoma model. These findings suggested that its antimetastatic effects could be independent of inhibition of coagulation, heparanase, and selectins [72]. Indeed, other studies evidenced an anti-inflammatory activity of NAH which inhibited the function of inflammatory mediators such as human neutrophil elastase, IL-8 and TNFα [118–120]. Two non-anticoagulant LMW-ROH (8 and 10 kDa) obtained by heparinase I depolymerization of UFH followed by glycol-splitting, showed in vivo antimetastatic activity in the B16-F10 metastatic model. Conversely, only the 8 kDa LMW-ROH was able to inhibit spontaneous lung dissemination when B16-F10 cells were inoculated s.c. without affecting the primary tumor growth [74].
The increase of heparanase inhibitory activity produced by glycol splitting of UFH, LMWH and ultra LMWH [76, 87] was confirmed by assessing the effect of the heparin-related synthetic trisaccharide 4-OMeGlcNS6S-GlcA-α1,6 anhydro GlcNS. Strikingly, its gs derivative showed an increase of one order of magnitude in inhibiting the enzyme (IC50 = 30 μg/ml versus IC50 = 2 μg/ml for the gs derivative). Both trisaccharides were used for molecular modeling studies validated by NOESY-NMR data, the first evidencing the gs-GlcA conformation [122].
Of note, in comparison with the previously described LMW-ROHs, the structural peculiarities of 100NA-ROH reside in a semisynthetic process based on reactions which preserve the UFH natural 2,3,6-O sulfation as well as the Mw range. The structural characterization showed the presence of both gs-GlcA within the ATBR sequence and the gs-uronic acid residues mainly interspersed within 6-O-N-sulfated disaccharides. Indeed, the reduced (~30%) overall sulfation degree lowered protein unspecific interactions and the chain higher flexibility, induced by the gs-residues, conferred to 100NA-ROH an enhanced heparanase inhibitory activity and a more selective proteins interaction [123]. A recent study reported kinetic analysis and modeling of the heparanase-inhibiting mechanism of Roneparstat. Dose-inhibition kinetics confirmed its high potency in inhibiting heparanase enzymatic activity (IC50 = 3 nM) and suggested different interaction features implicating a complex binding mechanism, involving one or multiple 100NA-ROH molecules depending on concentration ratios. Analysis of docking solutions indicated that a single chain of the inhibitor (e.g., Roneparstat) could interact with both heparin-binding domains of the enzyme or two different sequences of Roneparstat can interact with each of the heparin-binding domains, depending on the inhibitor/enzyme binding stoichiometry [124].
New Glycol-Split Non-anticoagulant Heparin as Heparanase Inhibitors
A follow-up translational project entitled “Novel heparanase inhibitors for cancer therapy” proposed and developed by teams of the Ronzoni Institute (Milan, Italy), the Technion University (Israel Institute of Technology, Haifa, Israel) and the University of Alabama at Birmingham (USA), was supported by the National Institutes of Health (NIH). This project was mainly devoted to optimizing ROHs inhibiting heparanase and multiple myeloma growth in experimental models [125]. Some compounds that emerged in these studies are described below.
N-Desulfated ROHs
Periodate oxidation of GlcN residues of N-deacetylated heparin to obtain non anticoagulant heparin was reported in a patent [126]. For the preparation of new heparanase inhibitors, periodate oxidation of N-desulfated heparins was performed in aqueous neutral media to split C2-C3 linkages of both N-desulfo GlcN units and of non-sulfated uronic acid residues. A previous study has reported a graded N-desulfation of UFH (from 20 up to 100%) by known modification methods [87, 127]. The N-desulfated gs-compounds were obtained by a final borohydride reduction [128]. Physical and biological properties of three representatives of this new class of ROHs are shown in Table 20.1.
Table 20.1.
Comparison of physical and biological properties of RO-N desulfated heparins
| Code | description | % gs/monomers | Mw kDa | Anti-Heparanase activity IC50 ng/mL | In vivo % CAG tumor inhibition∗ |
|---|---|---|---|---|---|
| G8340 | RO-N-desulfated heparin | 62 | 8.4 | 20 | 75 |
| G8438 | RO-N-desulfated heparin | 44 | 6.8 | 60 | n.d. |
| G9578 | RO-N-desulfated heparin | 47 | 6.3 | 75 | 63 |
| G4000 | Roneparstat | 25 | 16.0 | 3 | 62 |
∗in vivo human CAG multiple myeloma growth inhibition after 14 days treatment with drugs administered at 60 mg/Kg/day by subcutaneous continuous delivery [128]
The significant reduction of Mw values can be explained by the instability induced by depolymerization due to the formation of two adjacent gs residues generated by nonsulfated uronic acids and N-desulfated glucosamines naturally present in UFH. When compared with Roneparstat, the newly generated compounds exhibited a somewhat lower but still significant heparanase inhibitory effect, which can be explained by their lower Mw. Regardless, the new compounds displayed a comparable antimyeloma activity. As previously observed with substitution of N-sulfate groups with nonpolar N-acetyl groups [87], the addition to gs-uronic acid residues of further flexible joints, randomly generated from the gs-glucosamines, maintained the heparanase inhibitory activity, suggesting that the unmodified sequences can still bind and inhibit the enzyme.
Dicarboxylated Oxy-Heparins (DCoxyHs)
Periodate oxidation of UFH nonsulfated uronic acid residues led to oxy-heparins (oxyHs), characterized by the split of the C2-C3 linkage and the formation of two aldehyde groups which were further oxidized to carboxy groups yielding dicarboxylated oxy heparins (DCoxHs). A number of oxy-heparins obtained by periodate oxidation of UFH or of its derivatives, such as partially or fully 2-O-desulfated fully-acetyl-N-desulfated and partially N-desulfated heparins [87], were used as intermediates. The oxidation of aldehyde to carboxyl was performed using sodium chlorite (NaClO2) in aqueous media, pH 4, 0 °C for 24 h or at a neutral pH in the presence of oxidation catalysts [129]. The data in Table 20.2 show that the majority of the reported DCoxHs exhibited high efficacy in inhibiting in vitro heparanase and CAG multiple myeloma growth in vivo, independently of the Mw. It is noteworthy that the random presence of about 40% of dicarboxylated gs-uronic acid residues, instead of both 2-O-sulfated and non-sulfated uronic acid, along the heparin sequences, did not affect the interaction with the enzyme or its inhibition. Accordingly, these modifications did not significantly change the in vivo tumor growth inhibition in comparison with Roneparstat.
Table 20.2.
ROHs and DCoxHs . Comparison of physical and biological properties
| code | description | %RO/UA | Mw KDa | Anti-Heparanase activity IC50 ng/mL | In vivo % CAG tumor inhibition |
|---|---|---|---|---|---|
| G8223 | RO heparin | 25 | 17 | 2–8 | n.d. |
| G8249 | 50%RO heparin | 50 | 9.8 | 18 | 60 |
| G4000 | Roneparstat | 25 | 16.0 | 3 | 62 |
| %DC/UA | |||||
| G10810 | DCoxy heparin | 15.0 | 10 | 50 | |
| G9685 | DCoxy heparin50%2Odes | 38 | 11.7 | 10 | 68 |
| G8767 | DCoxy heparin50%2Odes | 40 | 9.1 | n.d | 52 |
| G7927 | DCoxy heparin50%2Odes | 47 | 6.4 | 10 | n.d. |
| G8733 | DCoxy heparin100%2Odes | 14 | 5.5 | n.d. | 53 |
| G10847 | DCoxy heparin100%2Odes | 63 | 5.5 | n.d. | 25 |
∗UA = uronic acid
The interaction with heparanase and its inhibition by a tricarboxylate moiety was first demonstrated with the natural enzyme inhibitor trachyspic acid, a metabolite of Talaromyces trachyspermus [130], and was later confirmed by its synthetic (+) enantiomer (Fig. 20.7) [131].
Fig. 20.7.
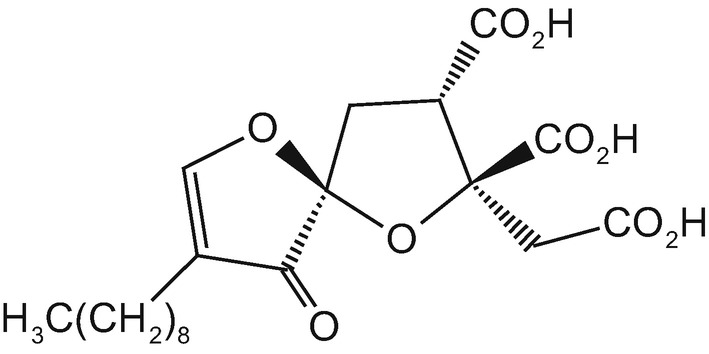
Structure of trachyspic acid
New Biotin-Conjugated N-Acetyl-Glycol Split Heparins
Recently, different classes of biotinylated N-acetyl gs-heparins were obtained by several approaches. Taking advantage of diverse reactive functions of oxy-N-acetyl heparins, intermediate spacers with different size have been used for coupling and keeping the biotin moiety at different distances from the heparin chains. The advantages of the biotin coupling are related to its chemical moiety and its easy detection by chemico-physical methods useful for pharmacokinetic studies. As biotin receptors are overexpressed in several cancer cell lines and solid tumors, biotin-conjugation not only improves bioavailability but can also contribute to tumor targeting and drug delivery [132]. All the biotin conjugates prepared showed heparanase inhibiting IC50 in the nM range similar to Roneparstat [133]. Likewise, these compounds displayed a similar efficacy comparable to that of Roneparstat in inhibiting CAG myeloma tumor growth and metastatic dissemination of B16-F10 melanoma cells.
Clinical Candidates and New Applications
Heparin Derivatives and Oligomers Interacting with Viral Envelope
The role of heparanase enzymatic activity in supporting viral infection has recently emerged, suggesting new potential applications of heparanase inhibitors [134] (Angelidis and Shukla, Chap. 10.1007/978-3-030-34521-1_32 in this volume). As reported by Skidmore [135], UFH inhibited the interaction of dengue, Herpes simplex, yellow fever and T lymphocyte viruses with HS, known to favor viral entry. UFH and heparosan derivatives, including N-acetyl and de-O-sulfated derivatives were assayed upon H5N1 virus infection. 2-O-desulfated and ssLMWH showed an anti-viral activity comparable to that of UFH whereas N-acetylation was detrimental [91]. In a recent study investigating the interaction of GAGs with the Zika virus envelop protein (Zikave), porcine intestinal UFH was shown to bind Zikave more efficiently than other GAGs (ChS, DeS). Among the UFH oligomers, the binding with Zikave was inhibited starting from the 18-mer [136]. A number of derivatives, including heparin and N-acetyl heparin and their 2-O, 6-O, 2,6 di O desulfated derivatives, were assayed for effect on H5N1 influenza virus invasion comprising a H5 pseudo typed HIV system. In comparison with UHF, 2-O-desulfation increased the activity, supersulfation led to a comparable activity, and N-desulfation-N-acetylation exhibited somewhat lower activity [135].
Sevuparin (DF F01) and Tafoxiparin (DFX232)
Early studies evidenced that strains of Plasmodium falciparum associated with severe forms of malaria use HS as a host adhesion receptor. Taking into consideration the structural and functional analogies between HS and UFH, the latter was used to treat severe malaria with overall positive outcomes. However, UFH administration was discontinued due to severe intracranial bleeding [137]. It was demonstrated that inhibition of interactions between parasite and erythrocytes can be achieved by heparin fragments sizing more than 3.5 kDa (dodecasaccharide) bearing natural N,6-O and 2-O sulfation [138]. A nonanticoagulant LMW ROH, Sevuparin (Mw 7.4 kDa), obtained by mild acid hydrolysis of ROH, showed the same activity of UFH both in vitro and in vivo in severe malaria models [139]. Clinical trials are evaluating the adjuvant activity of Sevuparin both in malaria patients and subjects with sickle cell disease, an inherited form of anemia [140, 141]. Another LMW ROH, Tafoxiparin (Mw 6.0 kDa) obtained by mild alkaline β-elimination, was found to disrupt rosettes, especially in the majority of fresh blood isolated from children with complicated malaria. Tafoxiparin represents potential adjuvant agent in the treatment of severe malaria [139]. As previous in vitro studies have demonstrated that Tafoxiparin increased both myometrial smooth muscle cell contractility and IL-8 activity in cervical fibroblasts [142], it is currently being evaluated in Phase II trial in pregnant women with slow progressing labor or labor arrest. In addition, with humanitarian approval, two pediatric patients suffering from Gorham-Stout syndrome, a rare bone disorder characterized by progressive bone loss and lymphatic vessel leakage, were successfully treated with Tafoxiparin [143].
Necuparanib (M 402)
A rationally designed LMW ROH (M 402, Mw 5.5–6.0 kDa) obtained by nitrous acid controlled depolymerization of UFH followed by glycol splitting, showed a reduced anticoagulant activity. Through its high-affinity binding, it displayed the ability to interfere with the function of several HS-binding proteins, such as chemokines, pro-angiogenic factors, P-selectin and heparanase (IC50 = 5 μg/mL). It showed an efficient in vivo antitumor, antiangiogenic, and antimetastatic activity in preclinical models [144, 145]. It underwent clinical evaluation in breast and pancreatic cancer, but the subsequent Phase 2 trial was discontinued after interim futility analysis for insufficient efficacy [146].
Roneparstat (G4000, 100NA-ROH, SST0001)
Preclinical studies evidenced the pleiotropic effects of this N-acetyl ROH heparin including interference with heparanase-syndecan-1 axis relevant in multiple myeloma development, [125, 147, 148]. Roneparstat was also shown to interfere with the function of several HS-binding proteins other than heparanase. Indeed, other studies indicated its ability to interfere with receptor tyrosine kinase signaling and heparanase-induced expression of genes associated with aggressive tumor phenotypes [149, 150]. This NA-ROH (Mw 16 kDa) demonstrated a remarkable antitumor, antiangiogenic, immunomodulatory and antimetastatic activity in several preclinical models of both hematological (e.g., multiple myeloma, lymphoma) and solid tumors (e.g., sarcomas, pancreatic and breast carcinoma). It was safely administered in mice in prolonged treatment schedules both alone and in combination with other antitumor agents [151–159]. An excellent safety profile was also emerged by recent results of Phase I clinical trial in advanced relapsed/refractory multiple myeloma [160] (Noseda and Barbieri, Chap. 10.1007/978-3-030-34521-1_21 in this volume). A recent study has assessed the role of heparanase in developing renal fibrosis arising in transplanted organ as a consequence of ischemia/reperfusion damage. In vivo tests showed that active doses of Roneparstat were well tolerated in animal models. A recent study evidenced that Roneparstat, by inhibiting heparanase, almost restored renal function, plasma creatinine and albuminuria. These results opened the way to further investigations on the potential efficacy of Roneparstat in reducing acute kidney injury and preventing chronic pro-fibrotic damage induced by ischemia/reperfusion injury [161].
CX-O1 (ODSH)
The low anticoagulant CX-01 retaining most of the anti-inflammatory properties of heparin is under clinical evaluation as adjuvant in acute myeloid leukemia and in refractory myelodysplastic syndrome [https://clinicaltrials.gov/ct2/show/NCT02995655]. It has recently received Orphan Drug and Fast Track Designations from the FDA for the treatment of acute myeloid leukemia [162]. It has been shown to interfere with CLC12/CXCR4 axis, inhibiting leukemia stem cell homing in the marrow stromal niches by competitive interaction with HS. Moreover, a randomized phase II trial in untreated metastatic pancreatic cancer has assessed that the combination of CX-01 with gemcitabine/nab-paclitaxel appears beneficial in term of disease control [163]. Worthy of note, local treatment with CX-01 , significantly reduced neutrophil elastase in cystic fibrosis patients in combination with dornase [164].
Concluding Remarks
UFH remains the main source of semisynthetic efficient heparanase inhibitors. Non-anticoagulant heparin derivatives endowed with heparanase inhibitory activity reported in this chapter, retain part of the pleiotropic pharmacological effects of the starting heparin. This class of heparin derivatives interferes with the emerging role of heparanase in inflammatory diseases and other pathologies. All show good safety and tolerability along with hints of efficacy on several pathologies. On the other hand, the anticancer agents under clinical trials, namely Roneparstat and CX-01, given their HMW and low sulfation degrees, compositionally better mimic HS. These peculiarities allow these compounds to better interfere and inhibit the interactions between HSPGs and heparanase overexpressed by tumor cells and present in their environment. Given the need for long-term treatments, the development of orally active agents of this class represents an attractive research field.
Acknowledgments
The authors thank Giuliana Cassinelli for her helpful comments and critical revision of the manuscript. We also apologize for not citing several relevant articles due to space limits.
Contributor Information
Israel Vlodavsky, Email: vlodavsk@mail.huji.ac.il.
Ralph D. Sanderson, Email: ralphsanderson@uabmc.edu
Neta Ilan, Email: netailan@technion.ac.il.
Annamaria Naggi, Email: naggi@ronzoni.it.
References
- 1.Bernfield M, Götte M, Park PW, et al. Functions of cell surface heparan sulfate proteoglycans. Annual Review of Biochemistry. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 2.Hammond E, Khurana A, Shridhar V, et al. The role of heparanase and sulfatases in the modification of heparan sulfate proteoglycans within the tumor microenvironment and opportunities for novel cancer therapeutics. Frontiers in Oncology. 2014;4:195. doi: 10.3389/fonc.2014.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rivara S, Milazzo FM, Giannini G. Heparanase: A rainbow pharmacological target associated to multiple pathologies including rare diseases. Future Medicinal Chemistry. 2016;8:647–680. doi: 10.4155/fmc-2016-0012. [DOI] [PubMed] [Google Scholar]
- 4.Irimura T, Nakajima M, Nicolson GL. Chemically modified heparins as inhibitors of heparan sulfate specific endo-. Beta-glucuronidase (heparanase) of metastatic melanoma cells. Biochemistry. 1986;25:5322–5328. doi: 10.1021/bi00366a050. [DOI] [PubMed] [Google Scholar]
- 5.Bitan M, Mohsen M, Levi E, et al. Structural requirements for inhibition of melanoma lung colonization by heparanase inhibiting species of heparin. Israel Journal of Medical Sciences. 1995;31:106–118. [PubMed] [Google Scholar]
- 6.Vlodavsky I, Ilan N, Naggi A, et al. Heparanase: Structure, biological functions, and inhibition by heparin-derived mimetics of heparan sulfate. Current Pharmaceutical Design. 2007;13:2057–2073. doi: 10.2174/138161207781039742. [DOI] [PubMed] [Google Scholar]
- 7.Vlodavsky I, Singh P, Boyango I, et al. Heparanase: From basic research to therapeutic applications in cancer and inflammation. Drug Resistance Updates. 2016;29:54–75. doi: 10.1016/j.drup.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gallagher JT. Heparan sulphate: A heparin in miniature. In: Lever R, Mulloy B, Page CP, editors. Heparin-a century of Progress. Berlin, Heidelberg: Springer; 2012. pp. 347–360. [Google Scholar]
- 9.Lindahl U, Kjellén L. Pathophysiology of heparan sulphate: Many diseases, few drugs. Journal of Internal Medicine. 2013;273:555–571. doi: 10.1111/joim.12061. [DOI] [PubMed] [Google Scholar]
- 10.Xu D, Esko JD. Demystifying heparan sulfate–protein interactions. Annual Review of Biochemistry. 2014;83:129–157. doi: 10.1146/annurev-biochem-060713-035314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lanzi C, Zaffaroni N, Cassinelli G. Targeting heparan sulfate proteoglycans and their modifying enzymes to enhance anticancer chemotherapy efficacy and overcome drug resistance. Current Medicinal Chemistry. 2017;24:2860–2886. doi: 10.2174/0929867324666170216114248. [DOI] [PubMed] [Google Scholar]
- 12.van der Meer JY, Kellenbach E, van den Bos LJ. From farm to pharma: An overview of industrial heparin manufacturing methods. Molecules. 2017;22:1025. doi: 10.3390/molecules22061025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee, Y. S., Yang, H. O., Shin, K. H., et al. (2003). Suppression of tumor growth by a new glycosaminoglycan isolated from the African giant snail Achatina fulica. European Journal of Pharmacology, 465, 191–198. [DOI] [PubMed]
- 14.Lanzi C, Cassinelli G. Heparan sulfate mimetics in cancer therapy: The challenge to define structural determinants and the relevance of targets for optimal activity. Molecules. 2018;23:2915. doi: 10.3390/molecules23112915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colliec-Jouault S, Bavington C, Delbarre-Ladrat C. Heparin-like entities from marine organisms. In: Lever R, Mulloy B, Page CP, editors. Heparin-a century of Progress. Berlin, Heidelberg: Springer; 2012. pp. 423–449. [DOI] [PubMed] [Google Scholar]
- 16.Pavão MS. Glycosaminoglycans analogs from marine invertebrates: Structure, biological effects, and potential as new therapeutics. Frontiers in Cellular and Infection Microbiology. 2014;4:123. doi: 10.3389/fcimb.2014.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gomes, A. M., Kozlowski, E. O., Pomin, V. H., et al. (2010). Unique extracellular matrix heparan sulfate from the bivalve Nodipecten nodosus (linnaeus, 1758) safely inhibits arterial thrombosis after photo chemically induced endothelial lesion. The Journal of Biological Chemistry, 285(10), 7312–7323. [DOI] [PMC free article] [PubMed]
- 18.Casu B, Naggi A, Torri G. Re-visiting the structure of heparin. Carbohydrate Research. 2015;403:60–68. doi: 10.1016/j.carres.2014.06.023. [DOI] [PubMed] [Google Scholar]
- 19.Jorpes JE, Gardell S. On heparin monosulfuric acid. The Journal of Biological Chemistry. 1948;176(1):267–276. [PubMed] [Google Scholar]
- 20.Gardini C, Urso E, Guerrini M, et al. Characterization of Danaparoid complex extractive drug by an orthogonal analytical approach. Molecules. 2017;22(7):1116. doi: 10.3390/molecules22071116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orenstein NS, Galli SJ, Dvorak AM, et al. Sulfated glycosaminoglycans of Guinea pig basophilic leukocytes. The Journal of Immunology. 1978;121(2):586–592. [PubMed] [Google Scholar]
- 22.Ogren SIREN, Lindahl U. Cleavage of macromolecular heparin by an enzyme from mouse mastocytoma. The Journal of Biological Chemistry. 1975;250(7):2690–2697. [PubMed] [Google Scholar]
- 23.Vlodavsky I, Fuks Z, Bar-Ner M, Ariav Y, Schirrmacher V. Lymphoma cell-mediated degradation of sulfated proteoglycans in the subendothelial extracellular matrix: Relationship to tumor cell metastasis. Cancer research. 1983;43:2704–2711. [PubMed] [Google Scholar]
- 24.Nakajima M, Irimura T, Di Ferrante N, Nicolson G L (1984) Metastatic melanoma cell heparanase. Characterization of heparan sulfate degradation fragments produced by B16 melanoma endoglucuronidase. The Journal of Biological Chemistry 259(4):2283–2290. [PubMed]
- 25.Höök M, Wasteson Å, Oldberg Å. A heparan sulfate-degrading endoglycosidase from rat liver tissue. Biochemical and Biophysical Research Communications. 1975;67(4):1422–1428. doi: 10.1016/0006-291X(75)90185-0. [DOI] [PubMed] [Google Scholar]
- 26.Klein U, Kresse H, von Figura K. Evidence for degradation of heparan sulfate by endoglycosidases: Glucosamine and hexuronic acid are reducing terminals of intracellular heparan sulfate from human skin fibroblasts. Biochemical and Biophysical Research Communications. 1976;69(1):158–166. doi: 10.1016/S0006-291X(76)80286-0. [DOI] [PubMed] [Google Scholar]
- 27.Wasteson A, Glimelius B, Busch C, et al. Effect of a platelet endoglycosidase on cell surface associated heparan sulphate of human cultured endothelial and glial cells. Thrombosis Research. 1977;11(3):309–321. doi: 10.1016/0049-3848(77)90184-0. [DOI] [PubMed] [Google Scholar]
- 28.Naparstek Y, Cohen IR, Fuks Z, Vlodavsky I. Activated T lymphocytes produce a matrix-degrading heparan sulphate endoglycosidase. Nature. 1984;310(5974):241. doi: 10.1038/310241a0. [DOI] [PubMed] [Google Scholar]
- 29.Oohira A, Wight TN, McPherson, et al. Biochemical and ultrastructural studies of proteoheparan sulfates synthesized by PYS-2, a basement membrane-producing cell line. The Journal of Cell Biology. 1982;92(2):357–367. doi: 10.1083/jcb.92.2.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vlodavsky I, Friedmann Y, Elkin M, et al. Mammalian heparanase: Gene cloning, expression and function in tumor progression and metastasis. Nature Medicine. 1999;5(7):793. doi: 10.1038/10518. [DOI] [PubMed] [Google Scholar]
- 31.Hulett MD, Freeman C, Hamdorf BJ, et al. Cloning of mammalian heparanase, an important enzyme in tumor invasion and metastasis. Nature Medicine. 1999;5(7):803. doi: 10.1038/10525. [DOI] [PubMed] [Google Scholar]
- 32.Kussie PH, Hulmes JD, Ludwig DL, et al. Cloning and functional expression of a human heparanase gene. Biochemical and Biophysical Research Communications. 1999;261(1):183–187. doi: 10.1006/bbrc.1999.0962. [DOI] [PubMed] [Google Scholar]
- 33.Fairbanks MB, Mildner AM, Leone JW, et al. Processing of the human heparanase precursor and evidence that the active enzyme is a heterodimer. The Journal of Biological Chemistry. 1999;274(42):29587–29590. doi: 10.1074/jbc.274.42.29587. [DOI] [PubMed] [Google Scholar]
- 34.Toyoshima M, Nakajima M. Human heparanase purification, characterization, cloning, and expression. The Journal of Biological Chemistry. 1999;274(34):24153–24160. doi: 10.1074/jbc.274.34.24153. [DOI] [PubMed] [Google Scholar]
- 35.Zetser A, Levy-Adam F, Kaplan V, et al. Processing and activation of latent heparanase occurs in lysosomes. Journal of Cell Science. 2004;117(11):2249–2258. doi: 10.1242/jcs.01068. [DOI] [PubMed] [Google Scholar]
- 36.Fux L, Feibish N, Cohen-Kaplan V, et al. Structure-function approach identifies a COOH-terminal domain that mediates heparanase signaling. Cancer Research. 2009;69(5):1758–1767. doi: 10.1158/0008-5472.CAN-08-1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu L, Viola CM, Brzozowski AM, et al. Structural characterization of human heparanase reveals insights into substrate recognition. Nature Structural & Molecular Biology. 2015;22(12):1016. doi: 10.1038/nsmb.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levy-Adam F, Feld S, Suss-Toby E, et al. Heparanase facilitates cell adhesion and spreading by clustering of cell surface heparan sulfate proteoglycans. PLoS One. 2008;3(6):e2319. doi: 10.1371/journal.pone.0002319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sapay N, Cabannes É, Petitou M, et al. Molecular model of human heparanase with proposed binding mode of a heparan sulfate oligosaccharide and catalytic amino acids. Biopolymers. 2012;97(1):21–34. doi: 10.1002/bip.21696. [DOI] [PubMed] [Google Scholar]
- 40.Gandhi NS, Freeman C, Parish CR, et al. Computational analyses of the catalytic and heparin-binding sites and their interactions with glycosaminoglycans in glycoside hydrolase family 79 endo-β-D-glucuronidase (heparanase) Glycobiology. 2011;22(1):35–55. doi: 10.1093/glycob/cwr095. [DOI] [PubMed] [Google Scholar]
- 41.Pikas DS, Li JP, Vlodavsky I, et al. Substrate specificity of heparanases from human hepatoma and platelets. The Journal of Biological Chemistry. 1998;273(30):18770–18777. doi: 10.1074/jbc.273.30.18770. [DOI] [PubMed] [Google Scholar]
- 42.Okada Y, Yamada S, Toyoshima M, et al. Structural recognition by recombinant human heparanase that plays critical roles in tumor metastasis hierarchical sulfate groups with differential effects and the essential target disulfated trisaccharide sequence. The Journal of Biological Chemistry. 2002;277(45):42488–42495. doi: 10.1074/jbc.M206510200. [DOI] [PubMed] [Google Scholar]
- 43.Petitou M, Duchaussoy P, Lederman I, et al. Synthesis of heparin fragments: A methyl α-pentaoside with high affinity for antithrombin III. Carbohydrate Research. 1987;167:67–75. doi: 10.1016/0008-6215(87)80268-9. [DOI] [PubMed] [Google Scholar]
- 44.Alban S, Schiemann S. Novel heparanase activity assay based on a fluorescence sensor technology. Planta Medica. 2011;77(12):SL13. [Google Scholar]
- 45.Melo CM, Tersariol ILS, Nader HB, et al. Development of new methods for determining the heparanase enzymatic activity. Carbohydrate Research. 2015;412:66–70. doi: 10.1016/j.carres.2015.04.020. [DOI] [PubMed] [Google Scholar]
- 46.Peterson SB, Liu J. Multi-faceted substrate specificity of heparanase. Matrix Biology. 2013;32(5):223–227. doi: 10.1016/j.matbio.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 47.Mao Y, Huang Y, Buczek-Thomas JA, et al. A liquid chromatography-mass spectrometry based approach to characterize the substrate specificity of mammalian Heparanase. The Journal of Biological Chemistry. 2014;289(49):34141–34151. doi: 10.1074/jbc.M114.589630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Escovar Galvis ML, Jia J, Zhang X, et al. Transgenic or tumor-induced expression of heparanase upregulates sulfation of heparan sulfate. Nature Chemical Biology. 2007;3(12):773. doi: 10.1038/nchembio.2007.41. [DOI] [PubMed] [Google Scholar]
- 49.Sembajwe, L. F., Katta, K., & Grønning, M. (2018). The exostosin family of glycosyltransferases: mRNA expression profiles and heparan sulphate structure in human breast carcinoma cell lines. Bioscience Reports, 38(4). 10.1042/BSR20180770. [DOI] [PMC free article] [PubMed]
- 50.Westling C, Lindahl U. Location of N-unsubstituted glucosamine residues in heparan sulfate. The Journal of Biological Chemistry. 2002;277(51):49247–49255. doi: 10.1074/jbc.M209139200. [DOI] [PubMed] [Google Scholar]
- 51.Fujii M, Yusa A, Yokoyama Y. Cytoplasmic expression of the JM403 antigen GlcA-GlcNH 3+ on heparan sulfate glycosaminoglycan in mammary carcinomas-a novel proliferative biomarker for breast cancers with high malignancy. Glycoconjugate Journal. 2010;27(7–9):661–672. doi: 10.1007/s10719-010-9311-4. [DOI] [PubMed] [Google Scholar]
- 52.Weyers A, Yang B, Yoon DS, et al. A structural analysis of glycosaminoglycans from lethal and nonlethal breast cancer tissues: Toward a novel class of theragnostics for personalized medicine in oncology? OMICS. 2012;16(3):79–89. doi: 10.1089/omi.2011.0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nadanaka S, Purunomo E, Takeda N, et al. Heparan sulfate containing unsubstituted glucosamine residues: Biosynthesis and heparanase inhibitory activity. The Journal of Biological Chemistry. 2014;289(22):15231–15243. doi: 10.1074/jbc.M113.545343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Driguez PA, Petitou M. Patent application no 11/625,994. 2007. Azasugar derivatives, Heparanase inhibitors, method for preparing same, compositions containing same, use thereof U.S. [Google Scholar]
- 55.Gomes, A. M., Kozlowski, E. O., Borsig, L., et al. (2014). Antitumor properties of a new non-anticoagulant heparin analog from the mollusk Nodipecten nodosus: Effect on P-selectin, heparanase, metastasis and cellular recruitment. Glycobiology, 25(4), 386–393. [DOI] [PubMed]
- 56.Vann, W. F., Schmidt, M. A., Jann, B., & Jann, K. (1981). The structure of the capsular polysaccharide (K5 Antigenn) of urinary-tract-infective Escherichia coli 010: K5: H4: A polymer similar to Desulfo-heparin. European Journal of Biochemistry, 116(2), 359–364. [DOI] [PubMed]
- 57.Jacobsson I, Lindahl U, Jensen JW, et al. Biosynthesis of heparin. Substrate specificity of heparosan N-sulfate D-glucuronosyl 5-epimerase. The Journal of Biological Chemistry. 1984;259(2):1056–1063. [PubMed] [Google Scholar]
- 58.Lindahl ULF, Kusche M, Lidholt K, Oscarsson LG. Biosynthesis of heparin and heparan sulfate. Annals of the New York Academy of Sciences. 1989;556(1):36–50. doi: 10.1111/j.1749-6632.1989.tb22488.x. [DOI] [PubMed] [Google Scholar]
- 59.Casu, B., Grazioli, G., Razi, N., et al. (1994). Heparin-like compounds prepared by chemical modification of capsular polysaccharide from E. coli K5. Carbohydrate Research, 263(2), 271–284. [DOI] [PubMed]
- 60.Linhardt RJ, Toida T. Heparin oligosaccharides: New analogues development and applications. In: Witczak ZJ, Nieforth KA, editors. Carbohydrates in drug design. New York: Marcel Dekker Inc; 1997. pp. 277–341. [Google Scholar]
- 61.Poggi A, Rossi C, Casella N, et al. Inhibition of B16-BL6 melanoma lung colonies by semisynthetic sulfaminoheparosan sulfates from E. coli K5 polysaccharide. Seminars in Thrombosis and Hemostasis. 2002;28(4):383–392. doi: 10.1055/s-2002-34308. [DOI] [PubMed] [Google Scholar]
- 62.Leali, D., Belleri, M., Urbinati, C., et al. (2001). Fibroblast growth factor-2 antagonist activity and angiostatic capacity of sulfated Escherichia coli K5 polysaccharide derivatives. The Journal of Biological Chemistry, 276(41), 37900–37908. [DOI] [PubMed]
- 63.Rusnati M, Oreste P, Zoppetti G, Presta M. Biotechnological engineering of heparin/heparan sulphate: A novel area of multi-target drug discovery. Current Pharmaceutical Design. 2005;11(19):2489. doi: 10.2174/1381612054367553. [DOI] [PubMed] [Google Scholar]
- 64.Li P, Sheng J, Liu Y, et al. Heparosan-derived Heparan Sulfate/heparin-like compounds: One kind of potential therapeutic agents. Medicinal Research Reviews. 2013;33(3):665–692. doi: 10.1002/med.21263. [DOI] [PubMed] [Google Scholar]
- 65.Borgenström, M., Jalkanen, M., & Salmivirta, M. (2003). Sulfated derivatives of Escherichia coli K5 polysaccharides as modulators of fibroblast growth factor signaling. The Journal of Biological Chemistry, 278(50), 49882–49889. [DOI] [PubMed]
- 66.Guerrini M, Naggi A, Guglieri S, et al. Complex glycosaminoglycans: Profiling substitution patterns by two-dimensional nuclear magnetic resonance spectroscopy. Analytical Biochemistry. 2005;337(1):35–47. doi: 10.1016/j.ab.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 67.Borgenström M, Wärri A, Hiilesvuo K, et al. O-sulfated bacterial polysaccharides with low anticoagulant activity inhibit metastasis. Seminars in Thrombosis and Hemostasis. 2007;33(5):547–556. doi: 10.1055/s-2007-982087. [DOI] [PubMed] [Google Scholar]
- 68.Pollari S, Käkönen RS, Mohammad KS. Heparin-like polysaccharides reduce osteolytic bone destruction and tumor growth in a mouse model of breast cancer bone metastasis. Molecular Cancer Research. 2012;10(5):597–604. doi: 10.1158/1541-7786.MCR-11-0482. [DOI] [PubMed] [Google Scholar]
- 69.Naggi, A., Torri, G., Casu, B., et al. (2001). Toward a biotechnological heparin through combined chemical and enzymatic modification of the Escherichia coli K5 polysaccharide. Seminars in Thrombosis and Hemostasis, 27(5), 437–444. [DOI] [PubMed]
- 70.Wang Z, Yang B, Zhang Z, et al. Control of the heparosan N-deacetylation leads to an improved bioengineered heparin. Applied Microbiology and Biotechnology. 2011;91(1):91–99. doi: 10.1007/s00253-011-3231-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Teng, L., Fu, H., Wang, M., et al. (2015). Immunomodulatory activity of heparan sulfate mimetics from Escherichia coli K5 capsular polysaccharide in vitro. Carbohydrate Polymers, 115, 643–650. [DOI] [PubMed]
- 72.Stevenson JL, Choi SH, Varki A. Differential metastasis inhibition by clinically relevant levels of heparins—Correlation with selectin inhibition, not antithrombotic activity.Clin. Cancer Research. 2005;11(19):7003–7011. doi: 10.1158/1078-0432.CCR-05-1131. [DOI] [PubMed] [Google Scholar]
- 73.Lyman GH, Bohlke K, Khorana AA, et al. Venous thromboembolism prophylaxis and treatment in patients with cancer: American Society of Clinical Oncology clinical practice guideline update 2014. JCO. 2015;33(6):654. doi: 10.1200/JCO.2014.59.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kragh M, Loechel F. Non-anti-coagulant heparins: A promising approach for prevention of tumor metastasis. International Journal of Oncology. 2005;27(4):1159–1167. [PubMed] [Google Scholar]
- 75.Muelle, T., Pfankuchen, D., Wantoch von Rekowski, K., et al. (2017). The impact of the low molecular weight heparin Tinzaparin on the sensitization of Cisplatin-resistant ovarian cancers-preclinical in vivo evaluation in Xenograft tumor models. Molecules, 22(5), 728. [DOI] [PMC free article] [PubMed]
- 76.Achour O, Poupard N, Bridiau N, et al. Anti-heparanase activity of ultra-low-molecular-weight heparin produced by physicochemical depolymerization. Carbohydrate Polymers. 2016;135:316–323. doi: 10.1016/j.carbpol.2015.08.041. [DOI] [PubMed] [Google Scholar]
- 77.Cai Z, Teng L, Zhou J. Design and synthesis of a native heparin disaccharide grafted poly-2-aminoethyl methacrylate glycopolymer for inhibition of melanoma cell metastasis. International Journal of Biological Macromolecules. 2019;126:612–619. doi: 10.1016/j.ijbiomac.2018.11.255. [DOI] [PubMed] [Google Scholar]
- 78.Lauver DA, Lucchesi BR. Sulodexide: A renewed interest in this glycosaminoglycan. Cardiovascular Drug Reviews. 2006;24(3–4):214–226. doi: 10.1111/j.1527-3466.2006.00214.x. [DOI] [PubMed] [Google Scholar]
- 79.Masola V, Onisto M, Zaza G, et al. A new mechanism of action of sulodexide in diabetic nephropathy: Inhibits heparanase-1 and prevents FGF-2-induced renal epithelial-mesenchymal transition. Journal of Translational Medicine. 2012;10(1):213. doi: 10.1186/1479-5876-10-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Naggi, A., Torri, G., & Casu, B. (1987). “Supersulfated” heparin fragments, a new type of low-molecular weight heparin. Physico-chemical and pharmacological properties. Biochem Pharmacol, 36(12), 1895–1900. [DOI] [PubMed]
- 81.Sissi C, Naggi A, Torri G, et al. Effects of sulfation on antithrombin-thrombin/factor Xa interactions in semisynthetic low molecular weight heparins. Seminars in Thrombosis and Hemostasis. 2001;27(5):483–488. doi: 10.1055/s-2001-17959. [DOI] [PubMed] [Google Scholar]
- 82.Asperti M, Naggi A, Esposito E, et al. High sulfation and a high molecular weight are important for anti-hepcidin activity of heparin. Frontiers in Pharmacology. 2016;6:316. doi: 10.3389/fphar.2015.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cassinelli G, Dal Bo L, Favini E, et al. Supersulfated low-molecular weight heparin synergizes with IGF1R/IR inhibitor to suppress synovial sarcoma growth and metastases. Cancer Letters. 2018;415:187–197. doi: 10.1016/j.canlet.2017.12.009. [DOI] [PubMed] [Google Scholar]
- 84.Guerrini M, Beccati D, Shriver Z, et al. Oversulfated chondroitin sulfate is a contaminant in heparin associated with adverse clinical events. Nature biotech. 2008;26(6):669. doi: 10.1038/nbt1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kishimoto TK, Viswanathan K, Ganguly T, et al. Contaminated heparin associated with adverse clinical events and activation of the contact system. New England J of Med. 2008;358(23):2457–2467. doi: 10.1056/NEJMoa0803200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Casu B, Guerrini M, Guglieri S, et al. Undersulfated and glycol-split heparins endowed with antiangiogenic activity. Journal of Medicinal Chemistry. 2004;47(4):838–848. doi: 10.1021/jm030893g. [DOI] [PubMed] [Google Scholar]
- 87.Naggi A, Casu B, Perez M, et al. Modulation of the heparanase-inhibiting activity of heparin through selective desulfation, graded N-acetylation, and glycol-splitting. The Journal of Biological Chemistry. 2005;280(13):12103–12113. doi: 10.1074/jbc.M414217200. [DOI] [PubMed] [Google Scholar]
- 88.Lapierre F, Holme K, Lam L, et al. Chemical modifications of heparin that diminish its anticoagulant but preserve its heparanase-inhibitory, angiostatic, anti-tumor and anti-metastatic properties. Glycobiology. 1996;6(3):355–366. doi: 10.1093/glycob/6.3.355. [DOI] [PubMed] [Google Scholar]
- 89.Rao NV, Argyle B, Xu X, et al. Low anticoagulant heparin targets multiple sites of inflammation, suppresses heparin-induced thrombocytopenia, and inhibits interaction of RAGE with its ligands. American Journal of Physiology. Cell Physiology. 2010;299(1):C97–C110. doi: 10.1152/ajpcell.00009.2010. [DOI] [PubMed] [Google Scholar]
- 90.Zheng S, Kummarapurugu AB, Afosah DK, et al. 2-O, 3-O desulfated heparin blocks high mobility group box 1 release by inhibition of p300 acetyltransferase activity. Am J Respir Cell Mol. 2017;56(1):90–98. doi: 10.1165/rcmb.2016-0069OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Skidmore, M. A., Dumax-Vorzet, A. F., Guimond, S. E., et al. (2008). Disruption of rosetting in Plasmodium falciparum malaria with chemically modified heparin and low molecular weight derivatives possessing reduced anticoagulant and other serine protease inhibition activities. Journal of Medicinal Chemistry, 51(5), 1453–1458. [DOI] [PubMed]
- 92.Hwang SR, Seo DH, Al-Hilal TA, et al. Orally active desulfated low molecular weight heparin and deoxycholic acid conjugate, 6ODS-LHbD, suppresses neovascularization and bone destruction in arthritis. Journal of Controlled Release. 2012;163(3):374–384. doi: 10.1016/j.jconrel.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 93.Sciumbata T, Caretto P, Pirovano P, Pozzi P, et al. Treatment with modified heparins inhibits experimental metastasis formation and leads, in some animals, to long-term survival. Invasion & Metastasis. 1996;16(3):132–143. [PubMed] [Google Scholar]
- 94.Vlodavsky I, Korner G, Ishai-Michaeli R, et al. Extracellular matrix-resident growth factors and enzymes: Possible involvement in tumor metastasis and angiogenesis. Cancer Metastasis Reviews. 1990;9(3):203–226. doi: 10.1007/BF00046361. [DOI] [PubMed] [Google Scholar]
- 95.Hostettler N, Naggi A, Torri G, et al. P-selectin-and heparanase-dependent antimetastatic activity of non-anticoagulant heparins. The FASEB Journal. 2007;21(13):3562–3572. doi: 10.1096/fj.07-8450com. [DOI] [PubMed] [Google Scholar]
- 96.Casu B, Diamantini G, Fedeli G, et al. Retention of antilipemic activity by periodate-oxidized non-anticoagulant heparins. Arzneimittel-Forschung. 1986;36(4):637–642. [PubMed] [Google Scholar]
- 97.Alekseeva A, Casu B, Torri G, et al. Profiling glycol-split heparins by high-performance liquid chromatography/mass spectrometry analysis of their heparinase-generated oligosaccharides. Analytical Biochemistry. 2013;434(1):112–122. doi: 10.1016/j.ab.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Choay J, Lormeau JC, Petitou M, et al. Anti-Xa active heparin oligosaccharides. Thrombosis Research. 1980;18(3):573–578. doi: 10.1016/0049-3848(80)90356-4. [DOI] [PubMed] [Google Scholar]
- 99.Casu B, Oreste P, Torri G, et al. The structure of heparin oligosaccharide fragments with high anti-(factor Xa) activity containing the minimal antithrombin III-binding sequence chemical and 13C nuclear-magnetic-resonance studies. The Biochemical Journal. 1981;197(3):599–609. doi: 10.1042/bj1970599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fransso LÅ, Malmström A, Sjöberg I, et al. Periodate oxidation and alkaline degradation of heparin-related glycans. Carbohydrate Research. 1980;80(1):131–145. doi: 10.1016/S0008-6215(00)85321-5. [DOI] [Google Scholar]
- 101.Conrad, H. E., & Guo, Y. (1992). Structural analysis of periodate-oxidized heparin. In D. A. Lane, I. Björk, & U. Lindahl (Eds.), Heparin and related polysaccharides, Springer (pp. 31–36). Boston: MA. [DOI] [PubMed]
- 102.Islam T, Butler M, Sikkander SA, et al. Further evidence that periodate cleavage of heparin occurs primarily through the antithrombin binding site. Carbohydrate Research. 2002;337(21–23):2239–2243. doi: 10.1016/S0008-6215(02)00229-X. [DOI] [PubMed] [Google Scholar]
- 103.Alekseeva A, Elli S, Cosentino C, et al. Susceptibility of enoxaparin reducing end amino sugars to periodate oxidation. Carbohydrate Research. 2014;400:33–43. doi: 10.1016/j.carres.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Casu B, Vlodavsky I, Sanderson RD. Non-anticoagulant heparins and inhibition of cancer. Pathophysiology of Haemostasis and Thrombosis. 2007;36(3–4):195–203. doi: 10.1159/000175157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sobel M, Bird KE, Tyler-Cross R, et al. Heparins designed to specifically inhibit platelet interactions with von Willebrand factor. Circulation. 1996;93(5):992–999. doi: 10.1161/01.CIR.93.5.992. [DOI] [PubMed] [Google Scholar]
- 106.Ono K, Ishihara M, Ishikawa K, et al. Periodate-treated, non-anticoagulant heparin-carrying polystyrene (NAC-HCPS) affects angiogenesis and inhibits subcutaneous induced tumour growth and metastasis to the lung. British Journal of Cancer. 2002;86(11):1803. doi: 10.1038/sj.bjc.6600307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wei M, Tai G, Gao Y, et al. Modified heparin inhibits P-selectin-mediated cell adhesion of human colon carcinoma cells to immobilized platelets under dynamic flow conditions. The Journal of Biological Chemistry. 2004;279(28):29202–29210. doi: 10.1074/jbc.M312951200. [DOI] [PubMed] [Google Scholar]
- 108.Yoshitomi Y, Nakanishi H, Kusano Y, et al. Inhibition of experimental lung metastases of Lewis lung carcinoma cells by chemically modified heparin with reduced anticoagulant activity. Cancer Letters. 2004;207(2):165–174. doi: 10.1016/j.canlet.2003.11.037. [DOI] [PubMed] [Google Scholar]
- 109.Folkman J, Langer R, Linhardt RJ, et al. Angiogenesis inhibition and tumor regression caused by heparin or a heparin fragment in the presence of cortisone. Science. 1983;221(4612):719–725. doi: 10.1126/science.6192498. [DOI] [PubMed] [Google Scholar]
- 110.Ornitz DM, Herr AB, Nilsson M, et al. FGF binding and FGF receptor activation by synthetic heparan-derived di-and trisaccharides. Science. 1995;268(5209):432–436. doi: 10.1126/science.7536345. [DOI] [PubMed] [Google Scholar]
- 111.Casu B, Guerrini M, Naggi A, et al. Short heparin sequences spaced by glycol-split uronate residues are antagonists of fibroblast growth factor 2 and angiogenesis inhibitors. Biochemistry. 2002;41(33):10519–10528. doi: 10.1021/bi020118n. [DOI] [PubMed] [Google Scholar]
- 112.Pisano C, Aulicino C, Vesci L, et al. Undersulfated, low-molecular-weight glycol-split heparin as an antiangiogenic VEGF antagonist. Glycobiology. 2004;15(2):1C–6C. doi: 10.1093/glycob/cwi007. [DOI] [PubMed] [Google Scholar]
- 113.Pisano C, Foderà R, Marcellin, et al (2002) Antiangiogenic and antitumoral activity of novel heparin derivatives. European Journal of Cancer 38: S73-S73).
- 114.Zcharia E, Zilka R, Yaar A, et al. Heparanase accelerates wound angiogenesis and wound healing in mouse and rat models. FASEB. 2005;19(2):211–221. doi: 10.1096/fj.04-1970com. [DOI] [PubMed] [Google Scholar]
- 115.Edovitsky E, Lerner I, Zcharia E, et al. Role of endothelial heparanase in delayed-type hypersensitivity. Blood. 2006;107(9):3609–3616. doi: 10.1182/blood-2005-08-3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lorè, N., Veraldi, N., Riva, C., et al. (2018). Synthesized Heparan Sulfate competitors attenuate Pseudomonas aeruginosa lung infection. International Journal of Molecular Sciences, 19(1), 207. [DOI] [PMC free article] [PubMed]
- 117.Vlodavsky I, Zcharia E, Goldshmidt O, et al. Involvement of heparanase in tumor progression and normal differentiation. Pathophysiol Haemos Thromb. 2003;33(1):59–61. doi: 10.1159/000073296. [DOI] [PubMed] [Google Scholar]
- 118.Huang L, Kerns RJ. Diversity-oriented chemical modification of heparin: Identification of charge-reduced N-acyl heparin derivatives having increased selectivity for heparin-binding proteins. Bioorganic & Medicinal Chemistry. 2006;14(7):2300–2313. doi: 10.1016/j.bmc.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 119.Lider O, Baharav E, Mekori YA, et al. Suppression of experimental autoimmune diseases and prolongation of allograft survival by treatment of animals with low doses of heparins. The Journal of Clinical Investigation. 1989;83(3):752–756. doi: 10.1172/JCI113953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Black, S. C., Gralinski, M. R., Friedrichs, G. S., et al. (1995). Cardioprotective effects of heparin or N-acetylheparin in an in vivo model of myocardial ischaemic and reperfusion injury. Cardiovascular Research, 29(5), 629–636. [PubMed]
- 121.Veraldi N, Hughes AJ, Rudd TR, et al. Heparin derivatives for the targeting of multiple activities in the inflammatory response. Carbohydrate Polymers. 2015;117:400–407. doi: 10.1016/j.carbpol.2014.09.079. [DOI] [PubMed] [Google Scholar]
- 122.Ni M, Elli S, Naggi A, et al. Investigating glycol-split-heparin-derived inhibitors of heparanase: A study of synthetic trisaccharides. Molecules. 2016;21(11):1602. doi: 10.3390/molecules21111602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Alekseeva A, Mazzini G, Giannini G, et al. Structural features of heparanase-inhibiting non-anticoagulant heparin derivative Roneparstat. Carbohydrate Polymers. 2017;156:470–480. doi: 10.1016/j.carbpol.2016.09.032. [DOI] [PubMed] [Google Scholar]
- 124.Pala D, Rivara S, Mor M, et al. Kinetic analysis and molecular modeling of the inhibition mechanism of roneparstat (SST0001) on human heparanase. Glycobiology. 2016;26(6):640–654. doi: 10.1093/glycob/cww003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yang Y, MacLeod V, Dai Y, et al. The syndecan-1 heparan sulfate proteoglycan is a viable target for myeloma therapy. Blood. 2007;110(6):2041–2048. doi: 10.1182/blood-2007-04-082495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Conrad H E, Yuchuan G Non-anticoagulant heparin derivatives. U.S. Patent 5,280,016 18 Jan 1994.
- 127.Nagasawa K, Inoue Y, Kamata T. Solvolytic desulfation of glycosaminoglycuronan sulfates with dimethyl sulfoxide containing water or methanol. Carbohydrate Research. 1977;58(1):47–55. doi: 10.1016/S0008-6215(00)83402-3. [DOI] [PubMed] [Google Scholar]
- 128.Torri, G., & Naggi, A. (2016). Derivatives of N-desulfated glucosaminoglycans and use as drugs. U.S. Patent Application, 15(/032), 248.
- 129.Torri, G., & Naggi A. (2017). Carboxylated derivatives of Glucosaminoglycans and use as drugs. U.S. Patent Application, 15(/034), 555.
- 130.Shiozawa, H., Takahashi, M., Takatsu, T., et al. (1995). Trachyspic acid, a new metabolite produced by Talaromyces trachyspermus, that inhibits tumor cell heparanase: Taxonomy of the producing strain, fermentation, isolation, structural elucidation, and biological activity. The Journal of Antibiotics, 48(5), 357–362. [DOI] [PubMed]
- 131.Zammit SC, Ferro V, Hammond E, Rizzacasa MA. Enantiospecific synthesis of the heparanase inhibitor (+)-trachyspic acid and stereoisomers from a common precursor. Organic & Biomolecular Chemistry. 2007;5(17):2826–2834. doi: 10.1039/b708594j. [DOI] [PubMed] [Google Scholar]
- 132.Leamon CP, Reddy JA. Folate-targeted chemotherapy. Advanced Drug Delivery Reviews. 2004;56(8):1127–1141. doi: 10.1016/j.addr.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 133.Giannini, G., Naggi, A., & Esposito, E. (2018). Biotin-conjugated N-acetyl glycol split heparin WO2018188990 (A1) 2018-10-18. [DOI] [PubMed]
- 134.Thakkar N, Yadavalli T, Jaishankar D. Emerging roles of heparanase in viral pathogenesis. Pathogens. 2017;6(3):43. doi: 10.3390/pathogens6030043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Skidmore MA, Kajaste-Rudnitski A, Wells NM, et al. Inhibition of influenza H5N1 invasion by modified heparin derivatives. Med Chem Comm. 2015;6(4):640–646. doi: 10.1039/C4MD00516C. [DOI] [Google Scholar]
- 136.Kim SY, Zhao J, Liu X, et al. Interaction of Zika virus envelope protein with glycosaminoglycans. Biochemistry. 2017;56(8):1151–1162. doi: 10.1021/acs.biochem.6b01056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Vogt AM, Pettersson F, Moll K, et al. Release of sequestered malaria parasites upon injection of a glycosaminoglycan. PLoS Pathogens. 2006;2(9):e100. doi: 10.1371/journal.ppat.0020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Barragan, A., Spillmann, D., Kremsner, P. G., et al. (1999). Plasmodium falciparum: Molecular background to strain-specific rosette disruption by glycosaminoglycans and sulfated glycoconjugates. Experimental Parasitology, 91(2), 133–143. [DOI] [PubMed]
- 139.Leitgeb, A. M., Blomqvist, K., Cho-Ngwa, F., et al. (2011). Low anticoagulant heparin disrupts Plasmodium falciparum rosettes in fresh clinical isolates. Am J Trop Med, 84(3), 390–396. [DOI] [PMC free article] [PubMed]
- 140.Batchvarova M, Shan S, Zennadi R, et al. Sevuparin reduces adhesion of both sickle red cells and leukocytes to endothelial cells in vitro and inhibits vaso-occlusion in vivo. Blood. 2013;122(21):182. doi: 10.1182/blood.V122.21.182.182. [DOI] [Google Scholar]
- 141.Leitgeb, A. M., Charunwatthana, P., Rueangveerayut, R., et al. (2017). Inhibition of merozoite invasion and transient de-sequestration by sevuparin in humans with Plasmodium falciparum malaria. PLoS One, 12(12), e0188754. [DOI] [PMC free article] [PubMed]
- 142.Ekman-Ordeberg G, Hellgren M, Åkerud A, et al. Low molecular weight heparin stimulates myometrial contractility and cervical remodeling in vitro. Acta Obstetricia et Gynecologica Scandinavica. 2009;88(9):984–989. doi: 10.1080/00016340903176818. [DOI] [PubMed] [Google Scholar]
- 143.Brodszki N, Länsberg JK, Dictor M, et al. A novel treatment approach for paediatric Gorham–stout syndrome with chylothorax. Acta Paediatrica. 2011;100(11):1448–1453. doi: 10.1111/j.1651-2227.2011.02361.x. [DOI] [PubMed] [Google Scholar]
- 144.Zhou H, Roy S, Cochran E, et al. M402, a novel heparan sulfate mimetic, targets multiple pathways implicated in tumor progression and metastasis. PLoS One. 2011;6(6):e21106. doi: 10.1371/journal.pone.0021106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: Linking inflammation and cancer. Journal of Immunology. 2009;182(8):4499–4506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.O’Reilly EM, Roach J, Miller P, et al. Safety, pharmacokinetics, pharmacodynamics, and antitumor activity of Necuparanib combined with nab-paclitaxel and gemcitabine in patients with metastatic pancreatic Cancer: Phase I results. The Oncologist. 2017;22(12):1429–e139. doi: 10.1634/theoncologist.2017-0472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Purushothaman A, Hurst DR, Pisano C, et al. Heparanase-mediated loss of nuclear syndecan-1 enhances histone acetyltransferase (HAT) activity to promote expression of genes that drive an aggressive tumor phenotype. The Journal of Biological Chemistry. 2011;286(35):30377–30383. doi: 10.1074/jbc.M111.254789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ramani VC, Zhan F, He J, et al. Targeting heparanase overcomes chemoresistance and diminishes relapse in myeloma. Oncotarget. 2016;7(2):1598. doi: 10.18632/oncotarget.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jung O, Trapp-Stamborski V, Purushothaman A, et al. Heparanase-induced shedding of syndecan-1/CD138 in myeloma and endothelial cells activates VEGFR2 and an invasive phenotype: Prevention by novel synstatins. Oncogene. 2016;5(2):e202. doi: 10.1038/oncsis.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cassinelli G, Lanzi C, Tortoreto M, et al. Antitumor efficacy of the heparanase inhibitor SST0001 alone and in combination with antiangiogenic agents in the treatment of human pediatric sarcoma models. Biochemical Pharmacology. 2013;85(10):1424–1432. doi: 10.1016/j.bcp.2013.02.023. [DOI] [PubMed] [Google Scholar]
- 151.Ritchie JP, Ramani VC, Ren Y, et al. SST0001, a chemically modified heparin, inhibits myeloma growth and angiogenesis via disruption of the heparanase/syndecan-1 axis. Clinical Cancer Research. 2011;17(6):1382–1393. doi: 10.1158/1078-0432.CCR-10-2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ramani VC, Vlodavsky I, Ng M, et al. Chemotherapy induces expression and release of heparanase leading to changes associated with an aggressive tumor phenotype. Matrix Biology. 2016;55:22–34. doi: 10.1016/j.matbio.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hao M, Franqui-Machin R, Xu H, et al. NEK2 induces osteoclast differentiation and bone destruction via heparanase in multiple myeloma. Leukemia. 2017;31(7):1648. doi: 10.1038/leu.2017.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Meirovitz A, Hermano E, Lerne I, et al. Role of heparanase in radiation-enhanced invasiveness of pancreatic carcinoma. Cancer Research. 2011;71(7):2772–2780. doi: 10.1158/0008-5472.CAN-10-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Shafat I, Ben-Arush MW, Issakov J, et al. Pre-clinical and clinical significance of heparanase in Ewing’s sarcoma. Journal of Cellular and Molecular Medicine. 2011;15(9):1857–1864. doi: 10.1111/j.1582-4934.2010.01190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cassinelli G, Favini E, Dal Bo L, et al. Antitumor efficacy of the heparan sulfate mimic roneparstat (SST0001) against sarcoma models involves multi-target inhibition of receptor tyrosine kinases. Oncotarget. 2016;7(30):47848. doi: 10.18632/oncotarget.10292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Rossini A, Zunino F, Ruggiero G, et al. Microenvironment modulation and enhancement of antilymphoma therapy by the heparanase inhibitor roneparstat. Hematological Oncology. 2018;36(1):360. doi: 10.1002/hon.2466. [DOI] [PubMed] [Google Scholar]
- 158.Zhang L, Ngo JA, Wetzel MD, Marchetti D. Heparanase mediates a novel mechanism in lapatinib-resistant brain metastatic breast cancer. Neoplasia. 2015;17(1):101–113. doi: 10.1016/j.neo.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Barbieri, P., Paoletti, D., Giannini, G., et al. (2017). Roneparstat and heparanase inhibition: A new toll for cancer treatment. J Pharmacol Clin Toxicol, 5(2), 1071–1074.
- 160.Galli M, Magen H, Einsele H, et al. Roneparstat (SST0001), an innovative heparanase (HPSE) inhibitor for multiple myeloma (MM) therapy: First in man study. Blood. 2015;126(23):3246. doi: 10.1182/blood.V126.23.3246.3246. [DOI] [Google Scholar]
- 161.Masola V, Bellin G, Vischini G, et al. Inhibition of heparanase protects against chronic kidney dysfunction following ischemia/reperfusion injury. Oncotarget. 2018;9(90):36185. doi: 10.18632/oncotarget.26324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Huselton, E., Cashen, A. F., & Jacoby, M. (2018). CX-01, an inhibitor of CXCL12/CXCR4 axis and of platelet factor 4 (PF4), with azacitidine (AZA) in patients with hypomethylating agent (HMA) refractory AML and MDS. J Clin Oncl, 36(15), 7027–7027.
- 163.Sigal D., Marcus S. G., Rosen P. J., et al. (2013). Association of 2-O, 3-O desulfated heparin (ODSH) plus combination gemcitabine (G)/nab-paclitaxel (A) with preliminary benefit in untreated metastatic pancreatic cancer. Journal of Clinical Oncology,31(34), abst. 2348
- 164.Sagel SD, Chmiel JF, Konstan MW. Sputum biomarkers of inflammation in cystic fibrosis lung disease. Proceedings of the American Thoracic Society. 2007;4(4):406–417. doi: 10.1513/pats.200703-044BR. [DOI] [PMC free article] [PubMed] [Google Scholar]