

Abstract
Extraction of chemicals from biota leads to co-extraction of lipids. When dosing such extracts into in vitro bioassays, co-dosed lipids act as an additional phase that can reduce the bioavailability of the chemicals and the apparent sensitivity of the assay. Equilibrium partitioning between medium, cells, and co-dosed lipids was described with an existing equilibrium partitioning model for cell-based bioassays extended by an additional lipid phase. We experimentally investigated the influence of co-dosed lipids on the effects elicited by four test chemicals of different hydrophobicity in two bioassays, indicative of the aryl hydrocarbon receptor and oxidative stress response (AREc32). The partitioning model explained the effect of the test chemicals in the presence of spiked triolein within a factor of 0.33–5.83 between the measured and predicted effect concentration (EC). We applied the model to marine mammal blubber extracted with silicone. Extracts dosed in the AREc32 bioassay showed a linear increase of apparent EC with increasing lipid fraction. The partitioning model was used to interpret the role of the co-extracted lipid. A quantitative lipid correction of bioassay results in the presence of co-dosed lipids was possible for known compounds and defined mixtures, while we could only estimate a range for mixtures of unknown chemicals.
Introduction
Bioanalytical tools are widely used to assess the burden of chemical mixtures, among others in biota extracts, including mussel,1,2 fish,3,4 and marine mammals.5,6 In vitro cell-based reporter gene bioassays are highly sensitive, target specific modes of actions, allow high-throughput applications, and can be used for the effect characterization of the chemical mixture present in an environmental sample.7 However, the bioavailability of chemicals in the bioassay system can be influenced by various processes, such as binding to medium constituents (lipids and proteins),8,9 sorption to the well plate,10,11 and, in the case of biota samples, binding to co-extracted lipids2,12 that form droplets on the medium surface. Co-dosed lipids add an additional partitioning phase to the bioassay system and can thus reduce the bioavailability of chemicals.
The preparation of biota samples for bioanalytical testing is commonly done using either exhaustive solvent extraction3,6,12 or passive equilibrium sampling with polymers, like the silicone polydimethylsiloxane (PDMS).5,13 In the case of solvent extraction, all the lipids present in the sample are extracted along with the chemicals. Hence, extensive clean-up procedures to eliminate the lipid residues, for example, treatment with sulfuric acid,1,14,15 gel permeation chromatography,12 sorbent gels,12,14,16 and acetonitrile extraction, followed by freeze-filtration6 are required. However, clean-up procedures may alter the chemical mixture composition of the extract and could introduce blank contamination. This can be largely circumvented using extraction with PDMS, which reduces the amount of co-extracted lipids substantially while conserving the chemical composition of the mixture.17−19 PDMS is suitable for nonpolar, hydrophobic organic chemicals with virtually constant partition constants between PDMS and different matrices,20 including biota,5,21−24 for a wide range of chemicals. Sampling the tissue of varying lipid fractions with PDMS led to a rather uniform uptake of between 0.55 and 1.2%22 lipid into the PDMS, which presumably is an absorption process.25 Therefore, the lipid fraction in PDMS may be considered independent of the sampled tissue and will be transferred into the solvent extract. The lipid fraction that is then co-dosed with the chemical mixture into a bioassay will vary depending on the potency of the sample, which determines how much extract needs to be dosed to elicit the desired response. Former studies observed a reduction of the sensitivity2,12 in in vitro cell-based bioassays when extracts of biological materials containing lipids were dosed. Jin et al.5 investigated the kinetic effect of blubber extracts in the AhR-CAFLUX assay (chemically activated fluorescence expression) by comparing the time course of the receptor activation of the reference compound 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) with and without the presence of lipids. The authors observed similar effects of TCDD with and without blubber extracts after an exposure time of 72 h but measured an enhanced activation of the receptor after 24 h in the presence of co-extracted lipids, hypothesizing that colloidal lipids can facilitate the uptake into cells.
The main objectives of this study were to develop a model to describe the partitioning of chemicals in in vitro bioassays and to systematically assess the influence of co-dosed lipids in these systems. We hypothesized that lipids act as an additional partitioning phase that reduces the bioavailability of the dosed chemicals and thus the apparent sensitivity of the bioassay. We screened the influence of different dosed volumes of the spiked model lipid triolein, a synthetic triglyceride, in two cell-based in vitro bioassays: AREc32 for the Nrf2-mediated activation of the antioxidant response element (ARE) and AhR-CALUX (chemically activated luciferase expression) for the activation of the aryl hydrocarbon receptor (AhR), which previously have been applied for testing blubber extracts.5,13 Triolein was spiked with two bioactive test chemicals for each assay that differ in hydrophobicity. Hence, a difference in partitioning from the lipid phase to the bioassay medium and cells was expected. We extended an existing mass balance model26 by the lipid phase to describe the bioassay system. As both cell lines AREc32 and AhR are cultured in the same assay medium, the test conditions were the same. We validated the model with our experimental data and applied it to bioanalytical data of lipid-containing extracts of the marine mammal blubber from the PDMS-based passive equilibrium sampling. We identified conditions when a correction of bioassay results in the presence of co-dosed lipids is necessary and provide recommendations on how to apply this correction in practice.
Theory
Partitioning Model for Lipid-Containing Cell-Based In Vitro Bioassays
The equilibrium distribution of chemicals in in vitro cell-based systems has been described as the partitioning between water, medium components, and cells.26,27 Here, we simplified the bioassay partitioning system and accounted only for two compartments, cells and medium. Each of these compartments is composed of lipid, protein, and water, with water representing all nonsorptive materials. Co-dosed lipids from biota extracts constitute a third compartment (Figure SI1). Binding to the plastic of the well plates is expected to be negligible under the test conditions.10 Losses to the air were not expected because the medium/air partition constant Kmedium/air of the tested chemicals was above the volatility cut-off of Kmedium/air > 104.28 The fractions of chemicals in the co-dosed lipid, flipid, and in the cells, fcells, were calculated with the mass balance eqs 1 and 2.
| 1 |
| 2 |
The volumes of co-dosed lipids, medium, and cells, Vlipid, Vmedium, and Vcells, sum up to the total volume in the cell assay system, Vtotal. The fraction of Vlipid per Vtotal was termed “volume fraction”, Vflipid (eq 3).
| 3 |
Partition constants K between the lipid and cells, Klipid/cell, and between the medium and cells, Kmedium/cell, were derived from partition constants K between lipid/water, Klipid/w, medium/water, Kmedium/w, and cell/water, Kcell/w (eqs 4 and 5).
| 4 |
| 5 |
Triolein served as a surrogate for co-dosed lipids, and experimental triolein/water partition constants, Ktriolein/w, were used as a proxy for Klipid/w. Triolein is a synthetic triglyceride with known physicochemical properties. Natural oils would pose the disadvantage of unknown purity and chemical partition constants.
The cellular effect concentration, ECcell, can be calculated from the nominal effect concentration, EC, the fraction of chemical in the cell fcell, Vcell, and Vtotal (eq 6).26
| 6 |
We assume that ECcell is constant28 and independent of Vflipid, that is, ECcell with lipid in the system (ECcell,x% lipid) is equal to ECcell without lipid in the system (ECcell,0% lipid). Therefore, the EC with lipids in the system (ECx% lipid) can be predicted from the measured EC without lipids in the system (EC0% lipid) for any Vflipid (eq 7).
| 7 |
Materials and Methods
Chemicals and Materials
The test chemicals for the AREc32 assay were benzo[a]pyrene (Sigma-Aldrich, #50-32-8, ≥96%) and dichlorvos (Dr. Ehrenstorfer, #62-73-7, 97.6%), and for the AhR-CALUX, they were 3,3′,4,4′,5-pentachlorobiphenyl (PCB 126, Dr. Ehrenstorfer, #57465-28-8, 94.5%) and β-naphthoflavone (Sigma-Aldrich, #6051-87-2, ≥98%) (chemical structures in Figure SI2). The reference compound for the AhR-CALUX assay was 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, Dr. Ehrenstorfer). The solvents used were ethyl acetate (Merck, SupraSolv, GC–MS), methanol (Merck, SupraSolv, for GC ECD FID), and dimethyl sulfoxide (DMSO, Applichem, ≥99.5%). Triolein was purchased from Sigma-Aldrich (≥99%). PDMS sheets (SSP-M823) with a thickness of 1 mm, supplied by Specialty Silicone Products (Ballston Spa, USA), were Soxhlet-extracted with ethyl acetate for at least 16 h prior to use.
Cell Lines
In this study, we used two in vitro reporter gene bioassays measuring the Nrf2-dependent oxidative stress response with the AREc32 cell line29 obtained by the courtesy of C. Roland Wolf, Cancer Research UK, and the AhR activation with the AhR-CALUX cell line30 obtained by the courtesy of Michael Denison, University of California, Davis, USA. For both cell lines, the same assay medium was used (90% Dulbecco’s modified Eagle’s medium with Glutamax and 10% fetal bovine serum (FBS), supplemented with penicillin (100 U mL–1) and streptomycin (100 μg mL–1), Thermo Fisher Scientific #31966021, #10099-141, #15140-035). The protein and lipid contents of the medium and the cells31 are given in Section SI1 and Table SI1. Culturing conditions and methods are described in previous studies.28,32 Two bioactive chemicals were tested per bioassay. Briefly, in preparation of the bioassay experiments, 24 h prior to the dosing of the chemicals, 30 μL of cell suspension with a defined number of cells (AREc32: 2500/well, AhR-CALUX: 3250/well) were plated with a multi-mode Dispenser (Biotek) in a 384-well polystyrene microtiter plate with a clear bottom (AREc32 #3764, AhR-CALUX BioCoat #356663, Corning), termed “cell assay plate”. As experience had shown that the cell number stays constant for 24 h since the cells need to adhere and adapt to the new conditions,33 the seeded cell number was considered as the starting cell number for the experiment. To calculate the mean cell number, the average of the difference between the seeded cell number and the cell number after 24 h exposure to the test chemicals was used, which means 48 h after seeding (Table SI1).28
Sample Preparation and Dosing Procedure for Spiked Triolein
Triolein was spiked with each of the test chemicals (benzo[a]pyrene, dichlorvos, PCB 126, β-naphthoflavone) individually in a serial dilution of the single chemical. It was then dosed into the wells of the bioassay at a constant triolein volume but with decreasing concentration of the test chemical. Two main experimental set-ups were tested to show the importance of the pre-equilibration of the spiked chemical with triolein and the medium components. In this context, a non-equilibrated set-up (termed “0 h”) and a 24 h-equilibrated set-up (termed “24 h”) were tested. An overview of the dosing procedures is shown in Figure SI3.
For the “0 h” experiments, spiked triolein was pipetted directly into 40 μL of the medium with cells in a 384-well cell assay plate with a digital analytical syringe (eVol, SGE Analytical Science). The volume of triolein was constant in each well for a given Vflipid, and the Vflipid ranged between 0.5 and 4% in the “0 h” experiments. For the “24 h” experiments, spiked triolein was pipetted with the digital analytical syringe to the 70 μL cell assay medium in glass vials. The triolein–medium mix was pre-equilibrated in closed vials for 24 h at room temperature in the dark on an orbital shaker (IKA) at 140 rpm. After 24 h of pre-equilibration, the complete volume in the glass vials was transferred to a glass-coated 384-well plate (Thermo Fischer) for dosing. In preparation of dosing, the medium in the 384-well cell assay plate was removed with a microplate washer (BioTek Instruments), leaving 10 μL assay medium, including cells. 70 μL of each well in the glass-coated 384-well dosing plate was then transferred to the cell assay plate with a multichannel pipette (Eppendorf). Vflipid in the “24 h” experiments ranged between 0.25 and 4%.
Each experiment was performed in three independent replicates. The first experiment was done with a serial 1:2 dilution of the test chemical in a constant Vftriolein to identify the concentration range of interest. For the second and third experiments, a linear dilution series of the chemical was dosed.
Sample Preparation and Dosing Procedure for Environmental Samples
Biota samples were processed using silicone-based equilibrium sampling of homogenized blubber tissue from marine mammals with PDMS sheets.5,22 The samples originated from eight individuals of dugongs (Dugong dugon, obtained from Caroline Gaus, the University of Queensland, Queensland, Australia), two harbor porpoises (Phocoena phocoena), one ringed seal (Phoca hispida), and one gray seal (Halichoerus grypus) originating from the waters of Schleswig-Holstein, Germany. All marine mammals were stranded and found dead. The dugongs were found near the beaches of Queensland in Australia. The harbor porpoises and the ringed seal were found in the area of the North Sea in Germany and the gray seal in the Baltic Sea in Germany. Blubber from dugong was homogenized with a cryogenic grinding mill (Retsch), and the other blubber samples were homogenized with a blender (Büchi). The homogenized tissue samples were stored at −80 °C.
2.5–8.7 g homogenized dugong blubber tissue and 1.6–2.3 g PDMS were brought in contact by sandwiching PDMS sheets and blubber and wrapping in an aluminum foil. The homogenized tissues of the harbor porpoises and ringed and gray seals were an oil-like liquid and the PDMS sheets (5.0–5.6 g) were placed vertically in a 125 mL jar in approximately 100 g homogenized blubber, allowing the sample to fill the room between the PDMS sheets. The mass-to-volume ratio satisfied negligible depletion conditions of the blubber tissue (<5% depletion by partitioning to PDMS).5,22 For this calculation, an average K between the lipid and PDMS, Klipid/PDMS, of 30 LPDMS Llipid–15 was used. The thermodynamic equilibrium between the blubber and PDMS was typically achieved in less than 24 h with 1 mm PDMS sheets according to previous work,5,22 and hence, PDMS and blubber were equilibrated for 24 h at 4 °C. After cleaning the PDMS surface thoroughly with lint-free tissues, the sheets were extracted in 10 mL ethyl acetate per 1 g of PDMS for at least 2 h. The extraction was repeated once. The combined solvent was blown down to dryness, and the extract was re-suspended in 2 mL methanol (dugong samples) and 2 mL ethyl acetate (all other samples).
In preparation of the dosing into the bioassay, a defined volume of the sample extract was blown down to dryness and re-suspended in the medium in a dosing vial. The mass of co-dosed lipids in the bioassay system was determined gravimetrically by weighing the preweighed dosing vial with a micro-analytical balance (Mettler Toledo). It was assumed that the weight gain after the blow-down of the solvent is exclusively from lipids, neglecting other co-extracted matrix and the chemicals themselves. As for single chemicals, dosing was performed in two set-ups. The blubber extracts (i) were dosed in a direct set-up without any pre-equilibration (termed “0 h”) and (ii) were pre-equilibrated with the medium for 24 h at room temperature in the dark on an orbital shaker (IKA) at 140 rpm in dosing vials prior to the dosing procedure (termed “24 h”). The dosing procedure was described previously by Neale at al.34 Briefly, two 96-well dilution plates were prepared with a Hamilton robot (Hamilton Microlab Star) by diluting the sample with the medium either in a serial 1:2 dilution or in a linear dilution. For dosing, 10 μL of the diluted sample in the dilution plate was transferred into the 384-well cell plate containing 30 μL of the medium and the cells.
This experiment was performed two times in independent replicates. The first dosing was done with a serial 1:2 dilution of the extracts to identify the concentration range of interest. For the second dosing, a linear dilution series of the extracts was dosed.
Detection of Cytotoxicity and Reporter Gene Activation
The cell viability was measured before dosing and again after 24 h exposure to the dosing extract with an IncuCyte S3 live cell-imaging system (Essen BioScience). The growth of the cells and the cytotoxicity of the extracts were derived from the measured cell confluency in the 384-well cell plates (number of cells given in Table SI1). The nominal inhibitory concentration for 10% reduction of cell viability (IC10) was derived from the concentration–inhibition curves, which were linear up to 40% inhibition.35
After monitoring the confluency, the cell plates were prepared for the detection of the reporter gene activation. The AREc32 and AhR-CALUX cell lines have a luciferase reporter gene that was detected after cell lysis by the quantification of the produced luciferase with the substrate luciferin complemented by adenosine triphosphate.32 The effect concentration, ECIR1.5, triggering an induction ratio (IR) of 1.5 in the AREc32 assay was derived from the linear concentration–induction curves up to IR 4, under the condition that these concentrations were below the IC10. The EC10 in the AhR-CALUX assay was derived analogously for 10% response relative to the maximum effect elicited by the potent agonist TCDD below 40% and below IC10.35 The linear regressions were performed with GraphPad Prism 8.2.1.
Results and Discussion
Effect of the Test Chemicals in the Presence of Constant Triolein Volume Fractions
Linear concentration–response curves of all chemicals in the presence of a certain Vftriolein (Figures SI4–13) were well repeatable, and therefore, a common ECx% triolein was derived from triplicate measurements35 (Table SI2).
Dosing-spiked triolein to the cells resulted in a higher ECx% triolein than without triolein (EC0% triolein) for all test chemicals, and ECx% triolein increased with the increasing Vftriolein (Figure 1) for both assays. A higher EC corresponds to an apparently lower potency of the tested chemical, likely resulting from a reduced bioavailability of the test chemical in the bioassay system.
Figure 1.

Comparison between “0 h” samples (red triangles) and “24 h” samples (blue diamonds) in dependency of the Vftriolein: ECIR1.5 in the AREc32 assay for (A) benzo[a]pyrene and (B) dichlorvos and EC10 in the AhR assay for (C) PCB 126 and (D) β-naphthoflavone. The filled symbols represent the measurements without triolein (EC0% triolein).
The dosing of the extract without pre-equilibration (“0 h”) led to a 5.1–9.5 times higher ECIR1.5 in comparison to the 24 h pre-equilibrated samples with benzo[a]pyrene (Figure 1A), 3.4–6.2 times higher EC IR1.5 with dichlorvos (Figure 1B), and 1.9–16.5 times higher EC10 with PCB 126 (Figure 1C). However, the higher EC was not proportional to the increasing Vftriolein. The higher EC at the “0 h” experiment could have resulted from the time needed to reach equilibrium partitioning between triolein and the medium, which would have delayed cellular uptake, which is typically established in standard set-ups within a few hours.33 In contrast, dosing β-naphthoflavone with the “0 h” approach led to a 1.4–3.7 times lower EC10 in comparison with the “24 h” samples (Figure 1D). The uptake into the cells took 1.4 h (time to reach 95% of equilibrium) for benzo[a]pyrene in a previous study using the medium supplemented with 10% FBS.33 In the current set-up with the chemicals in triolein dosed to the medium and cells for the “0 h” experiment, the chemicals need to partition from triolein to the medium before cellular uptake is possible. Though it is important to mix the dosing solution with the assay medium in the cell plate properly during dosing,36 the time to reach the equilibrium between all compartments in the well cannot be accelerated. If this partitioning is slow and is happening during the 24 h of the cell-exposure experiment, the biologically effective dose in the cells is likely to be much smaller than in the absence of the additional triolein phase. In the “24 h” experiment, triolein was pre-equilibrated with the medium for 24 h while shaking to establish an equilibrium between triolein and the medium prior to dosing of the cells, so that the biologically effective dose in the cells can be reached as fast as in the absence of triolein.
If the tested chemicals are not stable under the test conditions, the additional 24 h equilibration with the medium might have led to degradation or reactions with medium components, which could have occurred for β-naphthoflavone (Figure 1D). Since the differences were small and since chemicals present in the blubber and other lipid-rich tissues are typically rather persistent, we did not further pursue this issue. The three other test chemicals appeared to be stable in the triolein-containing medium, as supported by very similar EC10,0% triolein for the “0 h” and “24 h” experiments.
We also noticed that dichlorvos spiked to triolein showed higher cytotoxic effects in the “0 h” experiment than in the “24 h” experiment. Since the IC10 was at concentrations where the IR had often not yet reached the threshold of IR 1.5, this cytotoxic effect masked the induction (Table SI2). As the spiked triolein was not mixed, the inhomogeneous distribution of dichlorvos could have triggered the higher cytotoxicity of the cells.36
Validation of the Three-Phase Partitioning Model for the Reduction of Bioavailability
The experimentally determined ECs from the “24 h” experiments were compared to the predicted values for all test chemicals in Figure 2. The mean EC0% triolein of the “0 h” experiments was used to predict the ECx% triolein of the “24 h” samples that had attained equilibrium to assure that stability issues as encountered with β-naphthoflavone did not impact the model. More information on the partition constants used as the model input is given in Section SI2 and Table SI3.
Figure 2.

Comparison between experimental EC of the “24 h” experiments and EC predicted with eq 7: ECIR1.5 for the AREc32 assay for (A) benzo[a]pyrene and (B) dichlorvos and EC10 for the AhR-CALUX assay for (C) PCB 126 and (D) β-naphthoflavone. The filled symbols represent the measurements without triolein (EC0% triolein).
Benzo[a]pyrene and dichlorvos were tested in the AREc32 assay because they are known to activate the Nrf2-dependent oxidative stress response.13,37Vftriolein ranged from 0.25 to 4% in both experiments, resulting in 89–98% for the more hydrophobic benzo[a]pyrene and 15−75% for the less hydrophobic dichlorvos. For this range of Vftriolein, the ECIR1.5,x% triolein was increased by a factor of 9–25 compared to ECIR1.5,0% triolein (Figure 1A) but only by a factor of 2–3 for dichlorvos (Figure 1B). The ratio between modeled and experimental ECIR1.5,x% triolein ranged from 0.33−1.9 for benzo[a]pyrene (Figure 2A) and from 0.48−2.2 for dichlorvos (Figure 2B).
The two AhR-activating chemicals, PCB 12638 and β-naphthoflavone,39 were tested in the AhR-CALUX assay at the same Vftriolein in the range 0.25–4%. The ftriolein ranged between 81 and 99% for the more hydrophobic PCB 126 and from 28 to 87% for the less hydrophobic β-naphthoflavone. Testing spiked triolein with PCB 126 in AhR-CALUX showed a reduction by a factor of 10–400 of the apparent sensitivity in the bioassay with the tested range of Vftriolein (Figure 1C). The model underestimated the effect of triolein but could predict the EC10,x% triolein within a factor between 1.5−5.8 (Figure 2C). EC10,0% triolein for β-naphthoflavone were 2–9 times lower than EC10,x% triolein (Figure 1D). EC10,x% triolein was predicted within a factor of 1.0–2.3 (Figure 2D).
ECIR1.5,x% triolein for benzo[a]pyrene and dichlorvos and EC10,x% triolein for PCB 126 and β-naphthoflavone could be fairly precisely predicted with the three-phase model, indicating that the expansion of the previously published multimedia partition models for cell-based bioassays27,40 by the additional lipid compartment appears valid. The model was highly sensitive to the partition constants used for the calculations, especially to KBSA/w and Ktriolein/w, as demonstrated by the sensitivity analysis described in Section SI3 and Figure SI14.
Blubber Extracts of Marine Mammals Sampled with PDMS
To test the three-phase partitioning model with environmental samples, PDMS extracts of marine mammal blubber were examined in the AREc32 bioassay. As described above, the PDMS-sampled extracts were either dosed without pre-equilibration (“0 h”) or pre-equilibrated for 24 h prior to dosing (“24 h”) into the AREc32 assay. The comparison between “0 h” and “24 h” dosing showed that for extracts with a lower Vflipid the apparent sensitivity of the bioassay can be improved with a 24 h pre-equilibration, as these extracts showed on average a 1.4 lower ECIR1.5 (Figures SI15–18 and Table SI5). Since the experiments with single chemicals described above also showed that the apparent sensitivity of the assay was increased by 24 h pre-equilibration, we discuss only the “24 h”-dosing.
Vflipid in the “24 h” extracts ranged between 0.01 and 0.06% at the ECIR1.5 (Table SI5 and Figure 3). For highly potent samples with a low ECIR1.5, a low volume of the total extract was dosed, resulting in a low Vflipid in the cell assay well. For less-potent samples with higher ECIR1.5, more extract had to be dosed to achieve an IR of 1.5, and consequently, the Vflipid at the ECIR1.5 was higher. The correlation of Vflipid to ECIR1.5 in Figure 3 had r2 = 0.61 and root-mean-square error (RMSE) = 0.016 but was driven by one high value at Vflipid of 0.06%. Therefore, we added the ECIR1.5 from the “0 h” experiments and additional samples from previous experiments (unpublished) with 24 additional dugong blubber samples with Vflipid from 0.007 to 0.1% in Figure SI 19, resulting in a more robust linear correlation of Vflipid to ECIR1.5 with r2 = 0.71 and RMSE = 0.026. The regression lines are not shown in Figures 3 and SI19 because the database is too weak to derive a model. This correlation can be explained by the same amount of lipid extracted in each experiment but a dosing of higher extract volumes (and hence Vflipid) of low-potency samples. The effect of lipids appeared independent of the source of the blubber, which was derived from dugong, ringed seal, or gray seal, but species-specific effects remain to be investigated in future studies.
Figure 3.
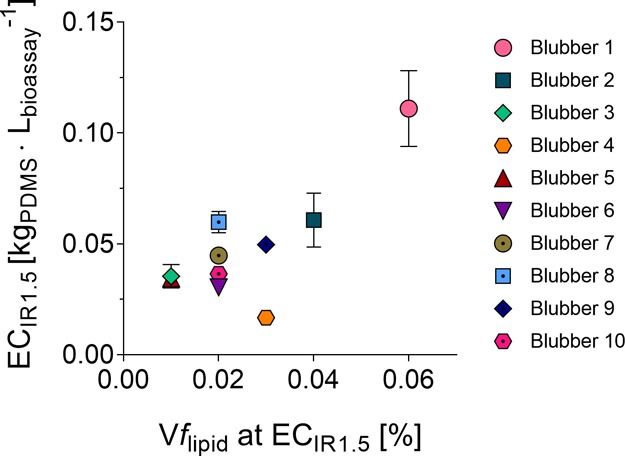
Relationship between ECIR1.5 and Vflipid measured in the AREc32 assay (“24 h” dosing). Bars represent the standard error (in some cases smaller than the symbols). PDMS extracts from blubber tissue are plotted using different symbols: blubber 1–8 from dugong, blubber 9 from ringed seal, and blubber 10 from gray seal.
The chemical mixtures present in the PDMS-based blubber extracts are of unknown composition and mainly consist of hydrophobic chemicals. While log Klipid/w is linearly correlated with log Kow, log Kmedium/w and log Kcell/w are slightly offset from that correlation for less-hydrophobic chemicals because of the contribution of sorption to proteins and water in the medium and cells (Figure 4A). In contrast, log Kmedium/cell and log Klipid/cell are fairly constant at log Kow > 3 (Figure 4A). Hence, over the hydrophobicity range of chemicals likely to occur in blubber,42,43 the concentration in the lipid is proportional to the concentration in the cells. Correspondingly, the composition of the mixtures remains intact from the lipid to cell as it is the case for mixtures dosed into the medium without an additional lipid phase. This specification is important because each component of the mixture contributes to the effect approximately in proportion to its fraction in the mixture independent of the presence of the lipid phase.
Figure 4.

(A) Partition constants log Klipid/water, log Kcell/water, log Kmedium/water, log Klipid/cell, and log Kmedium/cell as a function of log Kow. (B) Relationship between flipid and Vflipid for log Kow between 0 and 9. The shaded area highlights Vflipid at the EC of lipid-containing “24 h” extracts observed in the experiments in this study (0.01–0.06% in the AREc32 assay). (C) Lipid correction for the “24 h” blubber extracts (eq 7). Color code according to Figure 3. The span of the bars shows the range of the lipid correction for chemicals with log Kow of 3–9. The dotted line indicates the deviation by a factor of 3 from the 1:1 line.
The additional lipid phase reduces the fraction of chemicals available for cellular uptake and hence the apparent sensitivity of the assay. As shown in Figure 4B, the lipid phase usually is the main sorptive phase, especially for hydrophobic chemicals and higher Vflipid. Even for a low Vflipid of 0.01%, the flipid for chemicals with log Kow between 3 and 9 ranges from 2−26%. For Vflipid of 0.06%, flipid increases to 12–68% for the same log Kow range. flipid is only moderately dependent on the hydrophobicity for chemicals, with log Kow between 3 and 9 (Figure 4B). On this understanding, we estimated a range of lipid-corrected ECx% lipid in Figure 4C. The lipid correction was performed for the “24 h” PDMS-based blubber extracts for log Kow range between 3 and 9. The span of lipid-corrected ECIR1.5,0% lipid and the distance to the experimentally determined value is dependent on Vflipid. Although the lipid correction covered 6 orders of magnitude of Kow, the corrected span of ECIR1.5,0% lipid ranged up only to a factor of 3. Up to Vflipid of 0.27%, the deviation between non-corrected EC to the lipid-corrected value ECIR1.5,0% lipid is expected to be less than 1 order of magnitude.
Lipid correction to a precise EC0% lipid for samples with an unknown chemical composition has limitations because the reduction of the bioavailability is dependent on the individual partitioning behavior of each chemical of the mixture. In contrast, precise lipid correction is possible for single compounds with a known partitioning behavior. It is essential to consider experimental differences in the lipid content when linking bioanalytical results with analytical results by the so-called “iceberg” mixture modeling.37 With the iceberg model, the combination of chemical analysis and mixture effect profiling can explain how much of the total effect can be explained by the fraction of known chemicals.37 With the application of the lipid correction, the accuracy of the iceberg modeling can be improved.
Recommendations
When dosing samples with co-extracted lipids to cell-based bioassays, the lipid content should be determined before dosing to be able to correct for the reduced bioavailability in the presence of co-dosed lipids. If working with PDMS extracts from passive equilibrium sampling, the amount of co-extracted lipid can be derived from the weight gain of the PDMS during sampling. The lipid-containing extract should be transferred into the dosing vial in a solvent that can dissolve lipids, such as ethyl acetate, to ensure quantitative dosing, but the solvent should be blown down prior to adding the medium to avoid solvent effects.36
The model was developed for AhR CALUX and AREc32 because previous studies showed the activity of blubber samples in these assays.5,13 Provided there is enough information on the lipid and protein content of cells and the medium, the partitioning model can be adapted to any other cell line or bioassay set-up.
To increase the apparent sensitivity of the assay and to facilitate the application of the three-phase partitioning model, we suggest a 24 h pre-equilibration of the extract, ideally blown down to dryness to avoid disturbances by solvents, with assay medium prior to dosing to assure the equilibration of extracted chemicals between the co-extracted lipids and the medium. This approach is possible only for chemicals that are stable under bioassay test conditions, but most chemicals in the blubber are rather persistent and hence likely to be stable during the 48 h of pre-equilibration and incubation in the bioassay.
It is possible to quantitatively correct for the lipid effect on the activity of single chemicals. However, if unresolved complex mixtures are extracted from the tissue, the correction yields only a range of lipid-corrected EC values due to the dependence of the partitioning of the hydrophobicity of the individual components, which are not known. We recommend to test only up to Vflipid 0.27%, where the range of the lipid-corrected EC is within a factor of 10 for the undefined mixtures in blubber extracts. In such cases, it seems acceptable to omit the lipid correction, especially if similar tissues are compared and no comparison with chemical analysis is attempted.
Acknowledgments
The authors thank Jörg Watzke for technical help; Fabian Fischer for help with the modeling; Rita Schlichting, Jenny John, Lisa Glauch, and Niklas Wojtysiak for assistance with the bioassays; Caroline Gaus for the blubber samples; and Anne Jäger and Anna Sophia Martin for assistance with the biota extraction.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.est.9b07850.
Schematic illustration of the three-phase system, water, protein, and lipid contents of the used medium and the different cell lines; workflow of the dosing procedure; concentration-effect curves for all test chemicals and tested blubber extracts in all assays and supplementary analyses; additional information on the partition constants for the three-phase partitioning model; and sensitivity analysis of the model (PDF)
Author Contributions
The article was written through contributions of all authors. All authors have given approval to the final version of the article.
This project received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 715173, CHEMO-RISK). We gratefully acknowledge access to the CITEPro platform (Chemicals in the Terrestrial Environment Profiler) funded by the Helmholtz Association.
The authors declare no competing financial interest.
Supplementary Material
References
- Khim J. S.; Villeneuve D. L.; Kannan K.; Hu W. Y.; Giesy J. P.; Kang S. G.; Song K. J.; Koh C. H. Instrumental and bioanalytical measures of persistent organochlorines in blue mussel (Mytilus edulis) from Korean coastal waters. Arch. Environ. Contam. Toxicol. 2000, 39, 360–368. 10.1007/s002440010116. [DOI] [PubMed] [Google Scholar]
- Bayen S.; Gong Y.; Chin H. S.; Lee H. K.; Leong Y. E.; Obbard J. P. Androgenic and estrogenic response of green mussel extracts from Singapore’s coastal environment using a human cell-based bioassay. Environ. Health Perspect. 2004, 112, 1467–1471. 10.1289/ehp.6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtman C. J.; Van Oostveen A. M.; Brouwer A.; Lamoree M. H.; Legler J. Identification of estrogenic compounds in fish bile using bioassay-directed fractionation. Environ. Sci. Technol. 2004, 38, 6415–6423. 10.1021/es049750p. [DOI] [PubMed] [Google Scholar]
- Rostkowski P.; Horwood J.; Shears J. A.; Lange A.; Oladapo F. O.; Besselink H. T.; Tyler C. R.; Hill E. M. Bioassay-directed identification of novel antiandrogenic compounds in bile of fish exposed to wastewater effluents. Environ. Sci. Technol. 2011, 45, 10660–10667. 10.1021/es202966c. [DOI] [PubMed] [Google Scholar]
- Jin L.; Gaus C.; van Mourik L.; Escher B. I. Applicability of passive sampling to bioanalytical screening of bioaccumulative chemicals in marine wildlife. Environ. Sci. Technol. 2013, 47, 7982–7988. 10.1021/es401014b. [DOI] [PubMed] [Google Scholar]
- Desforges J.-P.; Levin M.; Jasperse L.; De Guise S.; Eulaers I.; Letcher R. J.; Acquarone M.; Nordøy E.; Folkow L. P.; Hammer Jensen T.; Grøndahl C.; Bertelsen M. F.; St. Leger J.; Almunia J.; Sonne C.; Dietz R. Effects of Polar Bear and Killer Whale Derived Contaminant Cocktails on Marine Mammal Immunity. Environ. Sci. Technol. 2017, 51, 11431–11439. 10.1021/acs.est.7b03532. [DOI] [PubMed] [Google Scholar]
- Escher B.; Leusch F.. Bioanalytical Tools in Water Quality Assessment; IWA publishing, 2011. [Google Scholar]
- Clemedson C.; Dierickx P. J.; Sjöström M. The prediction of human acute systemic toxicity by the EDIT/MEIC in vitro test battery: the importance of protein binding and of partitioning into lipids. Altern. Lab. Anim. 2003, 31, 245–256. 10.1177/026119290303100306. [DOI] [PubMed] [Google Scholar]
- Gülden M.; Mörchel S.; Tahan S.; Seibert H. Impact of protein binding on the availability and cytotoxic potency of organochlorine pesticides and chlorophenols in vitro. Toxicology 2002, 175, 201–213. 10.1016/s0300-483x(02)00085-9. [DOI] [PubMed] [Google Scholar]
- Fischer F. C.; Cirpka O. A.; Goss K.-U.; Henneberger L.; Escher B. I. Application of Experimental Polystyrene Partition Constants and Diffusion Coefficients to Predict the Sorption of Neutral Organic Chemicals to Multiwell Plates in in Vivo and in Vitro Bioassays. Environ. Sci. Technol. 2018, 52, 13511–13522. 10.1021/acs.est.8b04246. [DOI] [PubMed] [Google Scholar]
- Kramer N. I.; Di Consiglio E.; Blaauboer B. J.; Testai E. Biokinetics in repeated-dosing in vitro drug toxicity studies. Toxicol. In Vitro 2015, 30, 217–224. 10.1016/j.tiv.2015.09.005. [DOI] [PubMed] [Google Scholar]
- Simon E.; Lamoree M. H.; Hamers T.; Weiss J. M.; Balaam J.; de Boer J.; Leonards P. E. G. Testing endocrine disruption in biota samples: a method to remove interfering lipids and natural hormones. Environ. Sci. Technol. 2010, 44, 8322–8329. 10.1021/es101912z. [DOI] [PubMed] [Google Scholar]
- Jin L.; Gaus C.; Escher B. I. Adaptive stress response pathways induced by environmental mixtures of bioaccumulative chemicals in dugongs. Environ. Sci. Technol. 2015, 49, 6963–6973. 10.1021/acs.est.5b00947. [DOI] [PubMed] [Google Scholar]
- Suzuki G.; Tue N. M.; van der Linden S.; Brouwer A.; van der Burg B.; van Velzen M.; Lamoree M.; Someya M.; Takahashi S.; Isobe T.; Tajima Y.; Yamada T. K.; Takigami H.; Tanabe S. Identification of major dioxin-like compounds and androgen receptor antagonist in acid-treated tissue extracts of high trophic-level animals. Environ. Sci. Technol. 2011, 45, 10203–10211. 10.1021/es2024274. [DOI] [PubMed] [Google Scholar]
- Wong H. L.; Giesy J. P.; Siu W. H. L.; Lam P. K. S. Estrogenic and dioxin-like activities and cytotoxicity of sediments and biota from Hong Kong mudflats. Arch. Environ. Contam. Toxicol. 2005, 48, 575–586. 10.1007/s00244-004-0166-1. [DOI] [PubMed] [Google Scholar]
- Simon E.; Bytingsvik J.; Jonker W.; Leonards P. E. G.; de Boer J.; Jenssen B. M.; Lie E.; Aars J.; Hamers T.; Lamoree M. H. Blood plasma sample preparation method for the assessment of thyroid hormone-disrupting potency in effect-directed analysis. Environ. Sci. Technol. 2011, 45, 7936–7944. 10.1021/es2016389. [DOI] [PubMed] [Google Scholar]
- Jahnke A.; McLachlan M. S.; Mayer P. Equilibrium sampling: partitioning of organochlorine compounds from lipids into polydimethylsiloxane. Chemosphere 2008, 73, 1575–1581. 10.1016/j.chemosphere.2008.09.017. [DOI] [PubMed] [Google Scholar]
- Baltussen E.; Cramers C.; Sandra P. Sorptive sample preparation—a review. Anal. Bioanal. Chem. 2002, 373, 3–22. 10.1007/s00216-002-1266-2. [DOI] [PubMed] [Google Scholar]
- Jahnke A.; Mayer P.; Schäfer S.; Witt G.; Haase N.; Escher B. I. Strategies for Transferring Mixtures of Organic Contaminants from Aquatic Environments into Bioassays. Environ. Sci. Technol. 2016, 50, 5424–5431. 10.1021/acs.est.5b04687. [DOI] [PubMed] [Google Scholar]
- Jahnke A.; Mayer P. Do complex matrices modify the sorptive properties of polydimethylsiloxane (PDMS) for non-polar organic chemicals?. J. Chromatogr. A 2010, 1217, 4765–4770. 10.1016/j.chroma.2010.05.046. [DOI] [PubMed] [Google Scholar]
- Ossiander L.; Reichenberg F.; McLachlan M. S.; Mayer P. Immersed solid phase microextraction to measure chemical activity of lipophilic organic contaminants in fatty tissue samples. Chemosphere 2008, 71, 1502–1510. 10.1016/j.chemosphere.2007.11.060. [DOI] [PubMed] [Google Scholar]
- Jahnke A.; Mayer P.; Broman D.; McLachlan M. S. Possibilities and limitations of equilibrium sampling using polydimethylsiloxane in fish tissue. Chemosphere 2009, 77, 764–770. 10.1016/j.chemosphere.2009.08.025. [DOI] [PubMed] [Google Scholar]
- Rusina T. P.; Carlsson P.; Vrana B.; Smedes F. Equilibrium Passive Sampling of POP in Lipid-Rich and Lean Fish Tissue: Quality Control Using Performance Reference Compounds. Environ. Sci. Technol. 2017, 51, 11250–11257. 10.1021/acs.est.7b03113. [DOI] [PubMed] [Google Scholar]
- Rojo-Nieto E.; Muz M.; Koschorreck J.; Rüdel H.; Jahnke A. Passive equilibrium sampling of hydrophobic organic compounds in homogenised fish tissues of low lipid content. Chemosphere 2019, 220, 501–504. 10.1016/j.chemosphere.2018.12.134. [DOI] [PubMed] [Google Scholar]
- Smedes F.; Rusina T. P.; Beeltje H.; Mayer P. Partitioning of hydrophobic organic contaminants between polymer and lipids for two silicones and low density polyethylene. Chemosphere 2017, 186, 948–957. 10.1016/j.chemosphere.2017.08.044. [DOI] [PubMed] [Google Scholar]
- Fischer F. C.; Henneberger L.; König M.; Bittermann K.; Linden L.; Goss K.-U.; Escher B. I. Modeling Exposure in the Tox21 in Vitro Bioassays. Chem. Res. Toxicol. 2017, 30, 1197–1208. 10.1021/acs.chemrestox.7b00023. [DOI] [PubMed] [Google Scholar]
- Armitage J. M.; Wania F.; Arnot J. A. Application of mass balance models and the chemical activity concept to facilitate the use of in vitro toxicity data for risk assessment. Environ. Sci. Technol. 2014, 48, 9770–9779. 10.1021/es501955g. [DOI] [PubMed] [Google Scholar]
- Escher B. I.; Glauch L.; König M.; Mayer P.; Schlichting R. Baseline Toxicity and Volatility Cutoff in Reporter Gene Assays Used for High-Throughput Screening. Chem. Res. Toxicol. 2019, 32, 1646–1655. 10.1021/acs.chemrestox.9b00182. [DOI] [PubMed] [Google Scholar]
- Wang X. J.; Hayes J. D.; Wolf C. R. Generation of a stable antioxidant response element-driven reporter gene cell line and its use to show redox-dependent activation of nrf2 by cancer chemotherapeutic agents. Cancer Res. 2006, 66, 10983–10994. 10.1158/0008-5472.can-06-2298. [DOI] [PubMed] [Google Scholar]
- Brennan J. C.; He G.; Tsutsumi T.; Zhao J.; Wirth E.; Fulton M. H.; Denison M. S. Development of Species-Specific Ah Receptor-Responsive Third Generation CALUX Cell Lines with Enhanced Responsiveness and Improved Detection Limits. Environ. Sci. Technol. 2015, 49, 11903–11912. 10.1021/acs.est.5b02906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneberger L.; Mühlenbrink M.; König M.; Schlichting R.; Fischer F. C.; Escher B. I. Quantification of freely dissolved effect concentrations in in vitro cell-based bioassays. Arch. Toxicol. 2019, 93, 2295–2305. 10.1007/s00204-019-02498-3. [DOI] [PubMed] [Google Scholar]
- Escher B. I.; Dutt M.; Maylin E.; Tang J. Y. M.; Toze S.; Wolf C. R.; Lang M. Water quality assessment using the AREc32 reporter gene assay indicative of the oxidative stress response pathway. J. Environ. Monit. 2012, 14, 2877–2885. 10.1039/c2em30506b. [DOI] [PubMed] [Google Scholar]
- Fischer F. C.; Abele C.; Droge S. T. J.; Henneberger L.; König M.; Schlichting R.; Scholz S.; Escher B. I. Cellular Uptake Kinetics of Neutral and Charged Chemicals in in Vitro Assays Measured by Fluorescence Microscopy. Chem. Res. Toxicol. 2018, 31, 646–657. 10.1021/acs.chemrestox.8b00019. [DOI] [PubMed] [Google Scholar]
- Neale P. A.; Altenburger R.; Aït-Aïssa S.; Brion F.; Busch W.; de Aragão Umbuzeiro G.; Denison M. S.; Du Pasquier D.; Hilscherová K.; Hollert H.; Morales D. A.; Novák J.; Schlichting R.; Seiler T.-B.; Serra H.; Shao Y.; Tindall A. J.; Tollefsen K. E.; Williams T. D.; Escher B. I. Development of a bioanalytical test battery for water quality monitoring: Fingerprinting identified micropollutants and their contribution to effects in surface water. Water Res. 2017, 123, 734–750. 10.1016/j.watres.2017.07.016. [DOI] [PubMed] [Google Scholar]
- Escher B. I.; Neale P. A.; Villeneuve D. L. The advantages of linear concentration-response curves for in vitro bioassays with environmental samples. Environ. Toxicol. Chem. 2018, 37, 2273–2280. 10.1002/etc.4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanneberger K.; Rico-Rico A.; Kramer N. I.; Busser F. J. M.; Hermens J. L. M.; Schirmer K. Effects of Solvents and Dosing Procedure on Chemical Toxicity in Cell-Basedin VitroAssays. Environ. Sci. Technol. 2010, 44, 4775–4781. 10.1021/es100045y. [DOI] [PubMed] [Google Scholar]
- Escher B. I.; Aït-Aïssa S.; Behnisch P. A.; Brack W.; Brion F.; Brouwer A.; Buchinger S.; Crawford S. E.; Du Pasquier D.; Hamers T.; Hettwer K.; Hilscherová K.; Hollert H.; Kase R.; Kienle C.; Tindall A. J.; Tuerk J.; van der Oost R.; Vermeirssen E.; Neale P. A. Effect-based trigger values for in vitro and in vivo bioassays performed on surface water extracts supporting the environmental quality standards (EQS) of the European Water Framework Directive. Sci. Total Environ. 2018, 628-629, 748–765. 10.1016/j.scitotenv.2018.01.340. [DOI] [PubMed] [Google Scholar]
- Ghorbanzadeh M.; van Ede K. I.; Larsson M.; van Duursen M. B. M.; Poellinger L.; Lücke-Johansson S.; Machala M.; Pěnčíková K.; Vondráček J.; van den Berg M.; Denison M. S.; Ringsted T.; Andersson P. L. In vitro and in silico derived relative effect potencies of ah-receptor-mediated effects by PCDD/Fs and PCBs in rat, mouse, and guinea pig CALUX cell lines. Chem. Res. Toxicol. 2014, 27, 1120–1132. 10.1021/tx5001255. [DOI] [PubMed] [Google Scholar]
- Seidel S. D.; Li V.; Winter G. M.; Rogers W. J.; Martinez E. I.; Denison M. S. Ah receptor-based chemical screening bioassays: application and limitations for the detection of Ah receptor agonists. Toxicol. Sci. 2000, 55, 107–115. 10.1093/toxsci/55.1.107. [DOI] [PubMed] [Google Scholar]
- Henneberger L.; Mühlenbrink M.; Heinrich D. J.; Teixeira A.; Nicol B.; Escher B. I. Experimental Validation of Mass Balance Models for in Vitro Cell-Based Bioassays. Environ. Sci. Technol. 2020, 54, 1120–1127. 10.1021/acs.est.9b06144. [DOI] [PubMed] [Google Scholar]
- Tanabe S.; Iwata H.; Tatsukawa R. Global Contamination by Persistent Organochlorines and Their Ecotoxicological Impact on Marine Mammals. Environ. Sci. Technol. 1994, 154, 163–177. 10.1016/0048-9697(94)90086-8. [DOI] [PubMed] [Google Scholar]
- Ross P. S. Marine Mammals as Sentinels in Ecological Risk Assessment. Hum. Ecol. Risk Assess. 2000, 6, 29–46. 10.1080/10807030091124437. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.