

Abstract
Auto-antibodies (autoAbs) can result from excessive T follicular helper (Tfh) cell activity, while (Tfr) cells negatively regulate autoAb production. Interleukin-2 knockout (IL-2 KO) mice on the BALB/c background have elevated Tfh responses, produce autoAbs and develop lethal autoimmunity. We analyzed Tfh and Tfr cells in IL-2 KO mice on the C57Bl/6 (B6) genetic background. In B6 IL-2 KO mice the spontaneous formation of Tfh cells and Germinal Center (GC) B cells was greatly enhanced along with production of anti-DNA autoAbs. IL-2 has been reported to repress Tfr cell differentiation, however Tfr cells were not increased over wildtype (WT) levels in the B6 IL-2 KO mice. To assess Tfh and Tfr cell regulation of autoAb production in IL-2 KO mice, we generated IL-2 KO mice with a T cell-specific deletion of the master Tfh cell transcription factor Bcl6. In IL-2 KO Bcl6 conditional KO (2KO-Bcl6TC) mice, Tfh cells, Tfr cells and GC B cells were ablated. In contrast to expectations, autoAb IgG titers in 2KO-Bcl6TC mice were significantly elevated over auto-Ab IgG titers in IL-2 KO mice. Specific deletion of Tfr cells with Foxp3-cre Bcl6-flox alleles in IL-2 KO mice led to early lethality, before high levels of autoAbs could develop. We found IL-2+/+ Tfr cell deficient mice produce significant levels of autoAbs. Our overall findings provide evidence that Tfh cells are dispensable for high level production of autoAbs, and also reveal a complex interplay between Tfh and Tfr cells in autoAb production and autoimmune disease.
Keywords: Autoimmunity, autoantibodies, T follicular helper cells, T follicular regulatory T cells, IL-2, regulatory T cells
Introduction
During an immune response, CD4 T helper cells can differentiate into several unique effector lineages that promote different immune responses via the secretion of distinct types of cytokines. T follicular helper (Tfh) cells are a CD4 T cell lineage whose major function is to help B cells form germinal centers (GCs) and produce high-affinity antibodies (Abs)(reviewed in (1, 2)). Tfh cells are characterized by high level of expression of the chemokine receptor Cxcr5, which binds the chemokine Cxcl13 expressed in B cell follicles. Cxcl13, acting on Cxcr5, promotes migration of Tfh cells to the B cell follicle. Tfh cells have an activated effector T cell phenotype and express elevated Icos and PD-1. Tfh cells regulate memory B cell and plasma cell development, and thus Tfh cells control both the initiation as well as the outcome of the GC B cell response (1, 2). A key cytokine produced by Tfh cells is IL-21, a factor that potently promotes B cell activation and Ab secretion (1, 2). While Tfh cells are critical for the proper production of Ab, the over-production of Tfh cells can lead to autoimmunity as Tfh cells can help B cells to produce self-reactive Abs (1–10). Thus, the proper regulation of Tfh cell differentiation is essential for normal immune function as well as preventing autoimmune disease.
The Bcl6 transcriptional repressor protein is up-regulated in Tfh cells and is considered the master regulator for Tfh cells (11–13). Forced Bcl6 expression promotes differentiation of CD4 T cells into Tfh cells, whereas Bcl6-deficient T cells cannot differentiate into Tfh cells. A conditional knockout mouse for Bcl6, that we have developed, allows direct analysis of Bcl6 function in specific T cell populations (14).
A subpopulation of follicular T cells can act as suppressors of the GC response (15–21). These cells express both FoxP3 and Bcl6 and have been termed T follicular regulatory cells or “Tfr” cells. Tfr cells have a hybrid phenotype of Tfh cells and regulatory T cells (Tregs). Like Tfh cells, Tfr cells are dependent on Bcl6 and Cxcr5 for localization to the B cell follicle. Tfr cell functions are not well understood. In many model systems, Tfr cells suppress the number of Tfh cells and GC B cells during the immune response and also regulate affinity maturation of Abs (15–21). However, Tfr cells can also promote the GC and Ab response in certain model systems (22, 23). A popular model for the function of Tfr cells is that they limit the number of Tfh cells in the GC reaction or the amount of Tfh cell help, thus preventing excessive Tfh cell help to B cells (15, 24). In the absence of Tfr cells, excessive Tfh cell activity may lead to less stringent selection of high affinity B cell clones, leading to the expansion of low affinity B cell clones. Excessive Tfh cell activity may also promote the expansion of self-reactive B cells. Indeed, Tfr cells have an important role in maintaining tolerance during the B cell response and inhibiting the development of autoAbs (10, 23, 25–30).
Many autoimmune diseases are characterized by pathogenic self-reactive or autoreactive Abs (autoAbs)(31–33). AutoAb-mediated autoimmune diseases such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), myasthenia gravis (MG) and autoimmune hemolytic anemia (AIHA) are a tremendous source of morbidity and mortality for the human population. Since Tfh cells can promote autoAb production (1-9), Tfh cells are considered as targets for developing therapies for autoAb-driven autoimmune diseases, particularly SLE. However, the extent to which Tfh cells are required for autoAb generation is not clear. Tfh cells are not strictly required to help B cells make Abs (2, 14, 34), and in some model systems, autoAbs can even be produced in a T cell-independent manner (35, 36). Understanding the interplay of Tfh and Tfr cells in the control of the autoAb response will expand our knowledge of the pathogenesis of diseases such as lupus and should lead to the development of new treatments.
Interleukin-2-deficient (IL-2 KO) mice develop a lethal autoimmune disease that varies with the genetic background of the mice. On a mixed 129/01a x C57BL/6 genetic background, IL-2 KO mice die early (at 9 weeks on average) and almost invariably develop severe colitis (37, 38). IL-2 KO mice on the BALB/c background die between 3-5 weeks from AIHA caused by the production of autoAbs to red blood cells (RBCs)(38, 39). In these mice, Tfh cells are increased in numbers as IL-2 suppresses Tfh cell differentiation by inhibiting Bcl6 transcription (40–42). Tfr cell differentiation can be inhibited by IL-2 (25), although Tfr cells have not been examined previously in the IL-2 KO mice.
In the current study, we wondered if Tfr cells were elevated in IL-2 KO mice and if Tfh and Tfr cells played specific roles in autoAb production. We observed that unexpectedly, Tfr cells were not elevated even though Tfh cells were dramatically increased in IL-2 KO mice. Furthermore, we found that in IL-2 KO mice, high levels of autoAb can be produced independent of Tfh cells, and that low levels of Tfr cells can dramatically limit autoAb production.
Materials and Methods
Mice
IL-2 KO mice on the C57Bl/6 background and MyD88 KO mice on the C57Bl/6 background were obtained from Jackson labs. CD4-cre Bcl6-flox and Foxp3-cre Bcl6-flox mice were described previously (14, 23). Crossing of strains to produce IL-2 KO CD4-cre Bcl6-flox (2KO-Bcl6TC), IL-2 KO Foxp3-cre Bcl6-flox (2KO-Bcl6FC) and IL-2 KO MyD88 double KO (IL2-MyD88) DKO mice used published genotyping protocols (14, 23), as well as genotyping protocols from Jackson labs. All IL-2 KO mice were bred from IL-2+/− parents. IL-2+/+ WT littermates were used as controls for IL-2 KO, 2KO-Bcl6TC, 2KO-Bcl6FC and IL2-MyD88 DKO mice. Both male and female mice of 5-10 weeks old were analyzed. Mice were bred under specific pathogen-free conditions at the laboratory animal facility at IUSM and were handled according to protocols approved by the IUSM Animal Use and Care Committee.
Flow cytometry reagents and cell staining for flow cytometry
Cell suspensions from spleens were prepared and filtered through a 40-μm cell strainer (Fisherbrand). Cells were washed and diluted in PBS with 1% FBS and were stained with Fc block (BioXCell) for 5 min, followed by surface staining for the indicated markers. Following labelled Abs were used: anti-CXCR5 (L138D7), anti-PD-1 (29F.1A12), anti-CD4 (RM4-5), anti-Foxp3 (MF-14), anti-CD38 (90) and anti-B220 (RA3-6B2) were obtained from BioLegend; GL7 (GL7) was purchased from BD Pharmingen. For intracellular staining, after surface markers were stained, cells were fixed and stained with anti-Foxp3, eBioscience intracellular kit was used. All samples were acquired on an LSR2 flow cytometer (Becton Dickonson) and analyzed with FlowJo software.
Anti-dsDNA titer analysis
Anti-dsDNA IgA and IgG titers were determined by ELISA, as previously reported (14). Briefly, 96-well Nunc-Immuno plates (Sigma-Aldrich) were coated overnight at 4°C with salmon sperm DNA passed through 45 μm filter (Invitrogen). Wells were blocked with 1% BSA for 1.5 hours. Diluted serum samples were applied. ELISA plates were incubated for 1 hour at 37°C for anti-dsDNA Ab detection. Peroxidase-conjugated anti-mouse IgA or anti-mouse IgG (Sigma-Aldrich) was used as secondary Ab. Anti-dsDNA ELISA plates were washed extensively after each incubation to minimize high background signals.
Autoantibody Array
Autoantibody reactivities against a penal of autoantigens were measured using an autoantigen microarray platform developed by University of Texas Southwestern Medical Center (https://microarray.swmed.edu/products/category/protein-array/). Serum samples were incubated with the autoantigen array and the autoantibodies binding to the antigens on the array were detected with laser wavelengths 532nm (cy3 labeled anti-IgG), generating Tiff images. Genepix Pro 6.0 software was used to analyze the image and generate a .GPR file (GenePix Results format). Signal-to-noise ratio (SNR= (Foreground Median-Background Median)/sd(Background)) is used as a quantitative measure of the ability to resolve true signal from background noise. A higher SNR indicates higher signal over background noise. SNR equal or bigger than 3 are considered true signal from background noise.
Pristane administration.
Mice were given a single i.p. injection of 0.5 ml Pristane (Sigma-Aldrich) at 12 weeks of age. After 4 months, mice were sacrificed. The presence of anti-dsDNA antibodies in the serum was tested by ELISA.
Statistical Analysis.
All data analysis was done using Prism Graphpad software. Unless otherwise stated, Student t test or ANOVA with Tukey post hoc analysis were used. Only significant differences (P < 0.05) are indicated in Figures.
Results
Tfh cells but not Tfr cells are increased in the absence of IL-2 in vivo.
In order to analyze the role of Tfh and Tfr cells in the IL-2 KO, we obtained IL-2 KO mice on the C57Bl/6 (B6) background, since this strain was genetically compatible with most conditional KO strains that are also on the B6 background. We found that IL-2 KO mice on the B6 background in our facility were healthier than reported for IL-2 KO mice on the BALB/c background (38, 39, 43). In our colony, most B6 IL-2 KO mice live longer than 5 weeks and have a lifespan more similar to what was seen with IL-2 KO on the mixed 129-O1a x C57Bl/6 background (37, 38). However, the B6 background IL-2 KO mice were nonetheless smaller than wild-type (WT) and IL-2+/− littermates, had a sickly appearance, had enlarged spleens (Supp. Fig 1) and were infertile. To determine if loss of IL-2 led to higher Tfh and Tfr cells in this B6 background IL-2 KO strain, we conducted flow cytometry analysis (Fig. 1). As shown in Fig. 1A–B, we observed a roughly 7-fold increase in Tfh cells in unimmunized mice IL-2 KO mice compared to unimmunized wild-type (WT) mice. Even more striking was the large increase (~15-fold) in PD-1+ populations, indicating a high level of activated CD4 T cells in the un-manipulated IL-2 KO mice. In contrast to Tfh cells, Tfr cells were not significantly increased in the IL-2 KO (Fig. 1C–D), though IL-2 has been shown to inhibit both Tfh and Tfr cell development (25, 40–42). The lack of increase in Tfr cells was not due to a general loss of Treg cells in these mice, as total numbers of Foxp3+ CD4+ T cells were not significantly different from WT (Supp. Fig. 1). This result is different from what was seen with IL-2 KO mice on the BALB/c background, which show a marked depletion of Tregs (44). The more severe phenotype of the BALB/c IL-2 KO mice may relate to the significant loss of Tregs seen in that strain. Even though overall numbers of Tregs were not significantly decreased in the B6 IL-2 KO, there was a 3-fold increase in PD-1+ Tregs (Fig. 1C–D), suggesting more Treg activation in the IL-2 KO background. Our results also show that while IL-2 can be inhibitory for Tfr cell differentiation (25), in the IL-2 KO model of autoimmunity, IL-2 is also required for Tregs to fully mature into Tfr cells.
Figure 1. Spontaneous and strong Tfh cell but not Tfr cell development in IL2 KO mice.
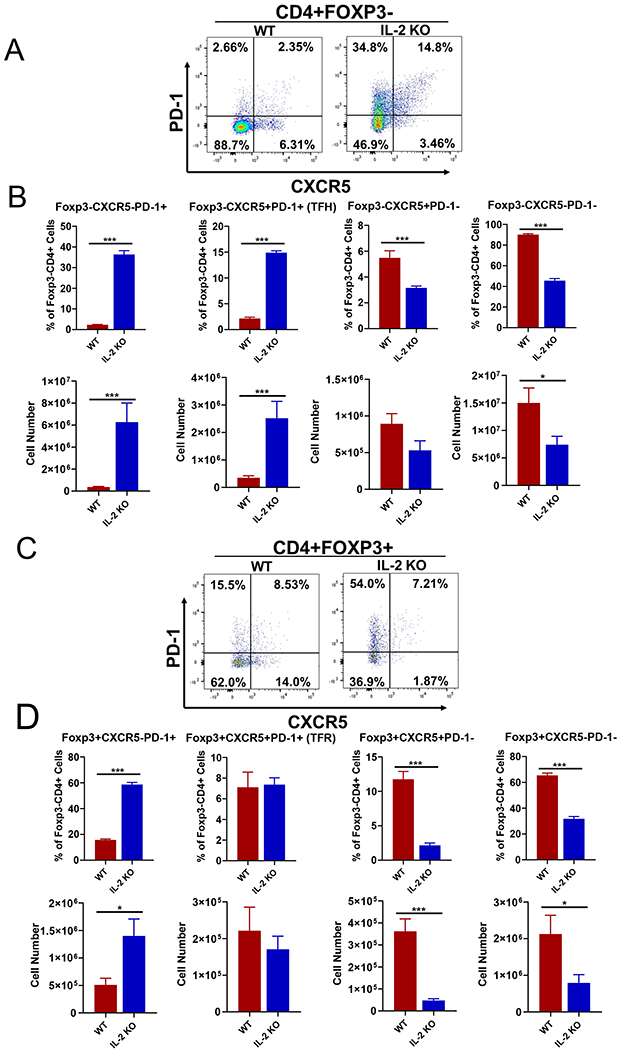
Naïve 10 weeks old wild-type (WT) and IL-2 KO mice were used to analyzed Tfh and Tfr responses. Spleens were analyzed for the indicated cell populations by flow cytometry. Representative flow cytometric dot plots for each cell staining are shown along with graphs showing average % of cells as a fraction of parental cell population and total yield of cells. (A-B) Analysis of CD4+ Foxp3-T cells as PD-1+Cxcr5-, PD-1+Cxcr5+ (Tfh) PD-1-Cxcr5+ and PD-1-Cxcr5-populations. (B) Tfh cells and non-Tfh cell populations are quantitated as a percentage of CD4+Foxp3-T cells, and absolute number per spleen. (C-D) Analysis of CD4+ Foxp3+T cells as PD-1+Cxcr5-, PD-1+Cxcr5+ (Tfr) PD-1-Cxcr5+ and PD-1-Cxcr5-populations. (D) Tfr cells are quantitated as a percentage of CD4+Foxp3+ T cells, and absolute number per spleen. P values were calculated by t test where * p < 0.05, ** p < 0.01, *** p < 0.0001. N = 4 - 6 mice, and each experiment was repeated 2 times.
Deletion of Tfh and Tfr cells in IL-2 KO mice.
To test the functional role of Tfh cells in the autoimmune disease and in autoAb production in the IL-2 KO mice, we mated the mice to the Bcl6-flox CD4-cre mouse strain (termed here Bcl6TC), which is unable to produce follicular T cells due to deletion of the Bcl6 gene in CD4 and CD8 T cells (14, 45). We therefore produced double mutant mice that we termed 2KO-Bcl6TC mice. The 2KO-Bcl6TC mice show a similar disease profile to IL-2 KO mice: both types of mice are smaller in size and both have enlarged spleens, multi-organ inflammation and similarly shortened lifespans (Supp. Figs. 1–2 and data not shown). We then examined the Tfh and GC B cell compartments in 2KO-Bcl6TC mice, and found both Tfh and GC B cells were completely ablated (Figure 2). This indicates first, that even in an IL-2-deficient environment, Bcl6 is still required for Tfh cell development. Second, these data show that Tfh cells do not correlate with overall disease in the IL-2 KO model.
Figure 2. Loss of Tfh and GCB cells in 2KO-Bcl6TC mice.
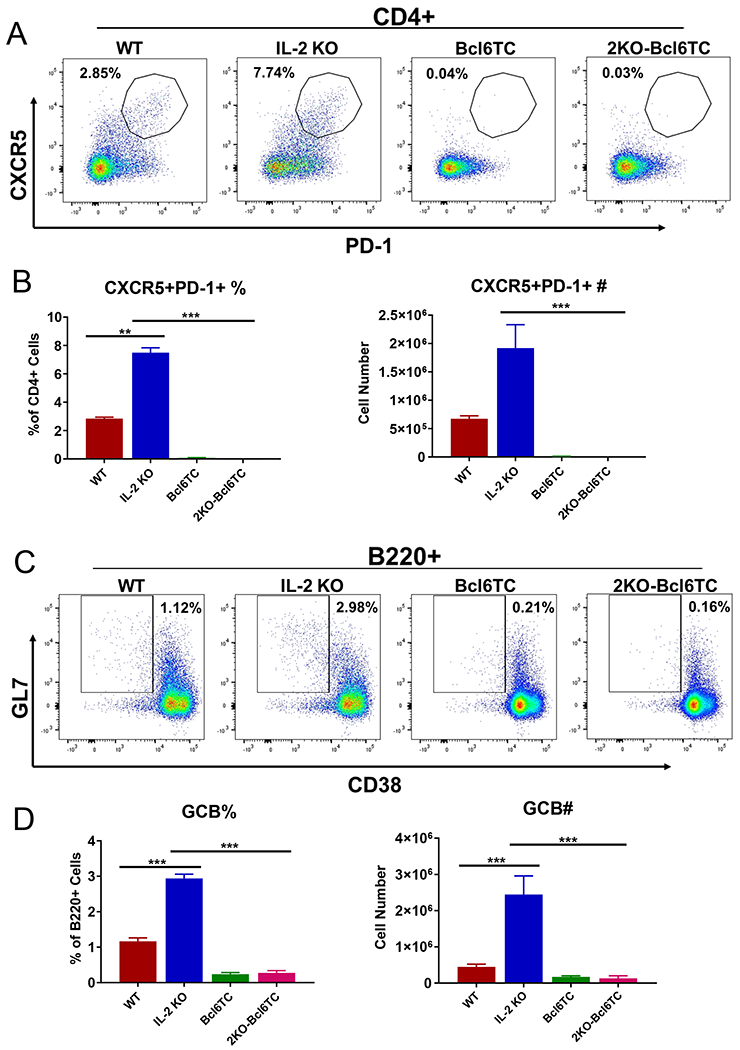
Naïve 10 week old WT, IL-2 KO, Bcl6TC (CD4-cre Bcl6-flox cKO) and 2KO-Bcl6TC (IL-2 KO and CD4-cre Bcl6-flox cKO) mice were used to analyzed germinal center responses. Spleens were analyzed for the indicated cell populations by flow cytometry. Representative flow cytometric dot plots for each cell staining are shown along with graphs showing average % of cells as a fraction of parental cell population and total yield of cells. (A) – (B) Analysis of CD4+ PD-1+Cxcr5+ follicular T cells. Follicular T cells are quantitated as a percentage of CD4+ T cells, and absolute number per spleen. (C) – (D) Analysis of B220+CD38-GL7+GCB cells. GCB cells are quantitated as a percentage of B220+ cells, and absolute number per spleen. P values were calculated by t test where * p < 0.05, ** p < 0.01, *** p < 0.0001. N = 4 - 6 mice, and each experiment was repeated 2 times.
Tfh cell-independent production of autoAbs.
We next asked whether Tfh cells were required for autoAbs in IL-2 KO mice and first measured anti-double stranded DNA Abs by ELISA (Figure 3). As expected, IL-2 KO mice had elevated levels of both IgG and IgA anti-DNA Abs compared to WT. Unexpectedly, loss of Tfh cells in 2KO-Bcl6TC mice led to significantly higher titers of both IgG (~3-fold higher) and IgA (~10-fold higher) anti-DNA Abs than seen in the IL-2 KO mice. To further analyze autoAbs in these mice, we used an auto-antigen (autoAg) microarray assay system to screen for IgG autoAbs to a panel of mouse protein antigens (Ags). Out of 94 autoAgs analyzed on the microarray, 17 autoAgs showed a significant difference in IgG reactivity between WT, IL-2 KO and 2KO-Bcl6TC groups (Fig. 4A). All 17 of these autoAgs showed a significantly higher Ab binding score for the 2KO-Bcl6TC sera over WT and IL-2 KO. Surprisingly, IL-2 KO sera showed little increase in autoAg binding over WT (Fig. 4A–B). These data showed that high levels of autoAbs could be produced in the absence of Tfh cells. To test that this result wasn’t unique to the IL-2 KO model, we used the pristane model of lupus (46, 47) to test the generation of anti-DNA Abs in WT and Bcl6TC mice. As shown in Figure 5A, WT and Bcl6TC mice both produced substantial levels of anti-DNA IgG after pristane immunization, although the production of anti-DNA IgA was lower in Bcl6TC mice. Thus, significant levels of autoreactive IgG can be produced in the absence of Tfh cells, though production of autoreactive IgA appears to require Tfh cell help.
Figure 3. Increased anti-DNA Abs in 2KO-Bcl6TC mice.
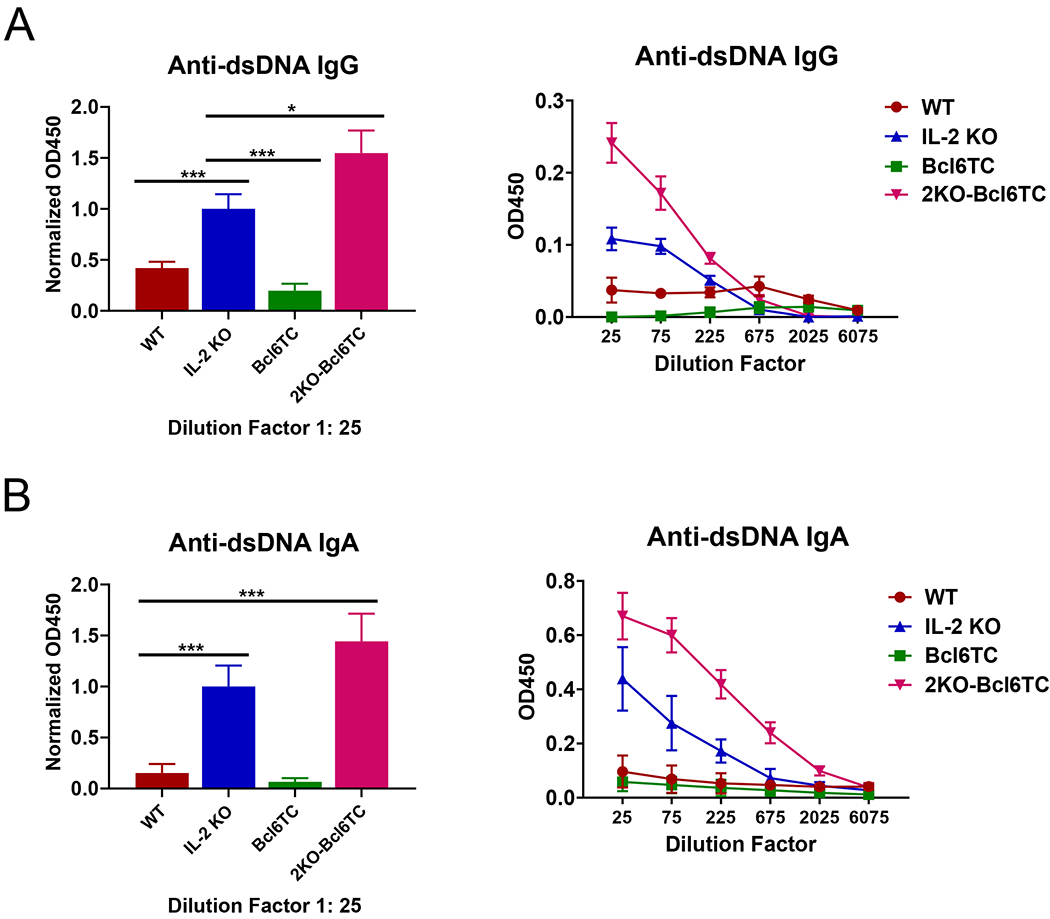
Naïve 10 weeks old WT, IL-2 KO, Bcl6TC and 2KO-Bcl6TC mice were used to analyzed serum Ab responses for reactivity to double stranded (ds) DNA. Bar graphs on left show normalized results over all experiments where OD values at 1:25 dilutions of serum are normalized relative to the average OD value for IL-2 KO serum, which is set to 1.0 for each experiment. Line graphs on right show sample ELISA results with titrations for individual sets of sera. Serum was tested for (A) anti-dsDNA IgG and (B) anti-dsDNA IgA. P values were calculated by t test where * p < 0.05, ** p < 0.01, *** p < 0.0001. N = 4 - 6 mice, and each experiment was repeated at least 3 times.
Figure 4. Increased autoAbs in 2KO-Bcl6TC mice.
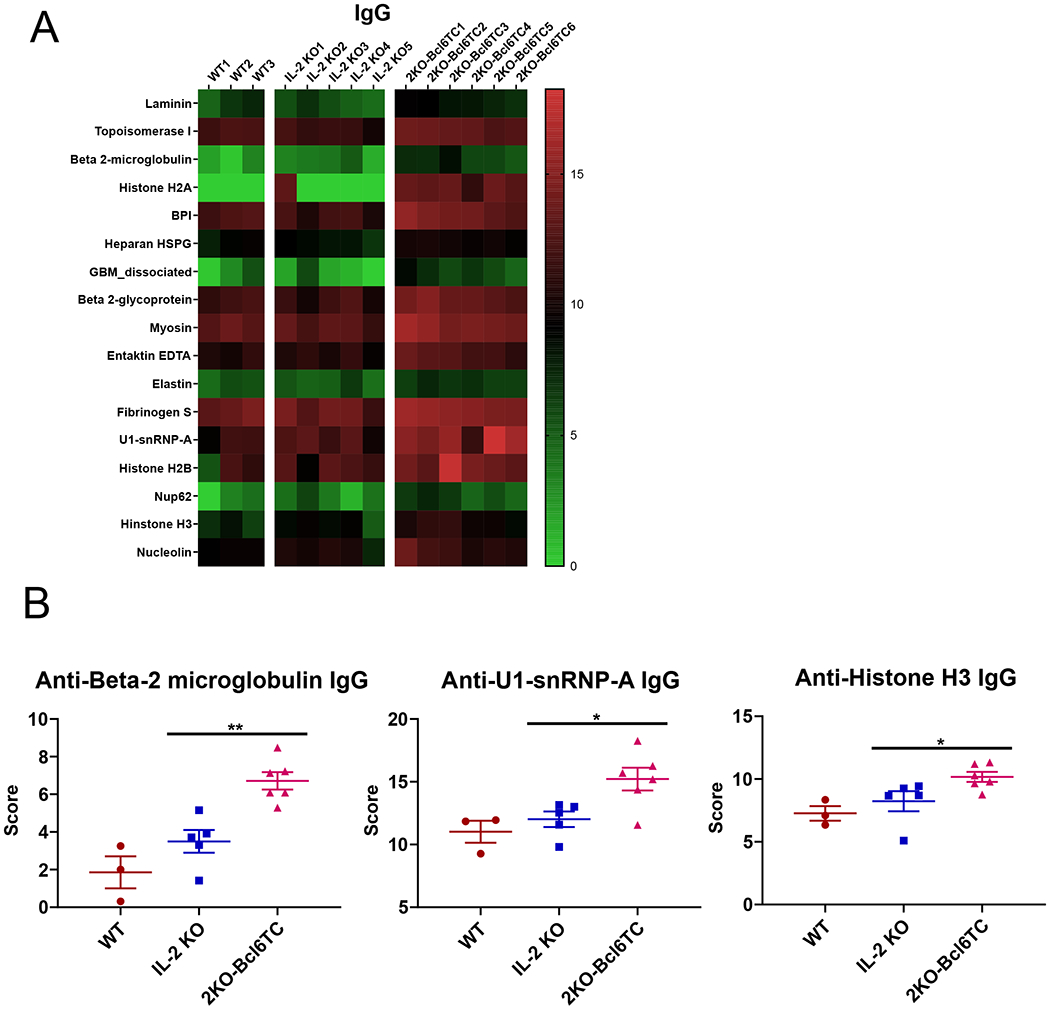
Serum from naïve 10 weeks old WT, IL-2 KO and 2KO-Bcl6TC mice was analyzed for IgG reactivity to an autoantigen (autoAg) microarray. (A) Heat map of 17 autoAgs with significantly higher reactive IgG between WT, IL-2 KO and 2KO-Bcl6TC mice based on the microarray score and multi-comparison statistical analysis. 17 autoAgs out of 94 autoAgs tested showed a significant difference in IgG reactivity between the 3 groups, and all 17 showed a higher score for the 2KO-Bcl6TC sera. (B) Graphs of selected anti-autoAg IgG scores for anti-beta-2 microglobulin, anti-U1-snRNP and anti-Histone H3 taken from the autoAg array data. P values were calculated by t test where * p < 0.05, ** p < 0.01, *** p < 0.0001. N = 4 - 6 mice.
Figure 5. Anti-DNA Abs were not affected by the deletion of germinal centers in pristane-induced lupus model.
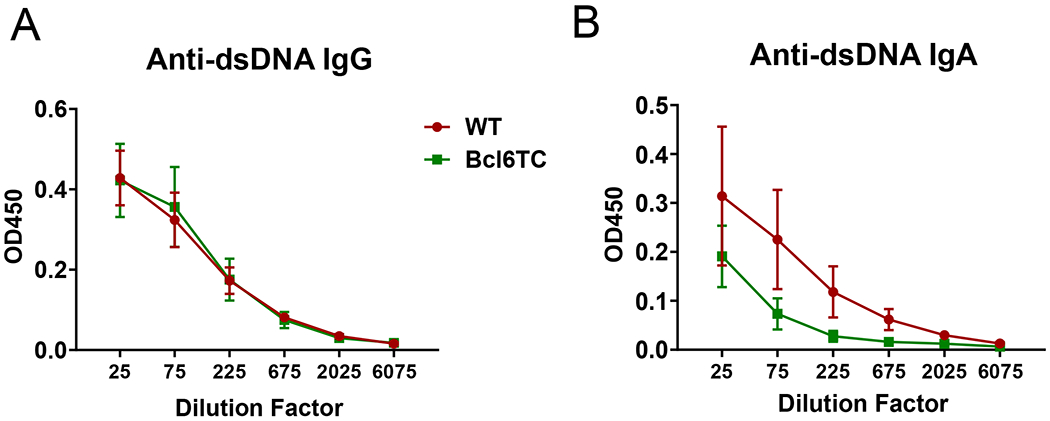
Four months after a single 0.5 mL pristane i.p. injection, serum samples were collected from control WT and Bcl6TC mice for anti-dsDNA antibody detection by ELISA. (A) Anti-dsDNA IgG and (B) anti-dsDNA IgA titers are shown. The X-axis labels are dilution factors. Data shown as mean ± SEM, n=5. Data are pooled from two independent experiments.
Regulation of autoAbs by Tfr cells.
We then wondered if the higher titers of autoAbs in the 2KO-Bcl6TC mice were due to loss of Tfr cells that would normally inhibit development of strong autoAb responses. To test this hypothesis, we mated IL-2 KO mice to Bcl6-flox Foxp3-cre mice that are specifically deficient in Tfr cells, to generate double mutant (2KO-Bcl6FC) mice also on the B6 background. In 2KO-Bcl6FC mice, we found that deletion of Bcl6 in Tregs and the loss of Tfr cells dramatically worsened the early lethality in the B6 background IL-2 KO mice. The average lifespan of 2KO-Bcl6FC mice was 3.5 weeks (Fig. 6A), and thus much shorter than the B6 IL-2 KO mice, which typically lived past 10 weeks of age. We analyzed sera from three 2KO-Bcl6FC mice that lived to 5 weeks for anti-DNA IgM and IgG levels by dsDNA ELISA (Fig. 6B–C). The 2KO-Bcl6FC mice had significant less anti-DNA IgM (Fig. 6B), but had a trend toward increased anti-DNA IgG (Fig. 6C) Abs over IL-2 KO mice. We then used the autoAg microarray assay system to screen for IgG autoAbs, but did not observe significant differences in autoAbs between in IL-2 KO and 2KO-Bcl6FC mice (data not shown). 2KO-Bcl6FC mice also did not show significantly more organ inflammation than age-matched IL-2 KO mice (data not shown). Thus, the early death of 2KO-Bcl6FC mice cannot be explained by more autoAbs or stronger inflammatory disease. At the same time, the shortened lifespan of 2KO-Bcl6FC mice likely forestalled more severe autoimmune disease as autoAbs and inflammation typically develops with increased age. To better understand the role of Tfr cells in repressing autoAb responses, we analyzed the development of autoAbs in IL-2+/+ Tfr cell-deficient Bcl6FC mice (Fig. 7). Bcl6FC mice are healthy, fertile, have a normal lifespan (23). We found that significantly increased titers of anti-DNA IgG develop in older Bcl6FC mice compared to WT controls (Fig. 7A). Additionally, aged Bcl6FC mice produced significant levels of anti-DNA anti-IgA, whereas WT mice produced undetectable levels (Fig. 7B). This finding further supports a role for Tfr cells controlling IgA production (23). We further tested autoAbs in Bcl6FC mice by autoAg microarray, and found higher levels of IgM Abs to several autoAgs Fig. (7C). These data show that in our Bcl6FC mouse model in our mouse colony, Tfr cells have a significant role in maintaining Ab self-tolerance, consistent with recent findings using different models of Tfr cell deficiency (26, 27).
Figure 6. Early death of 2KO-Bcl6FC mice and autoAb production.
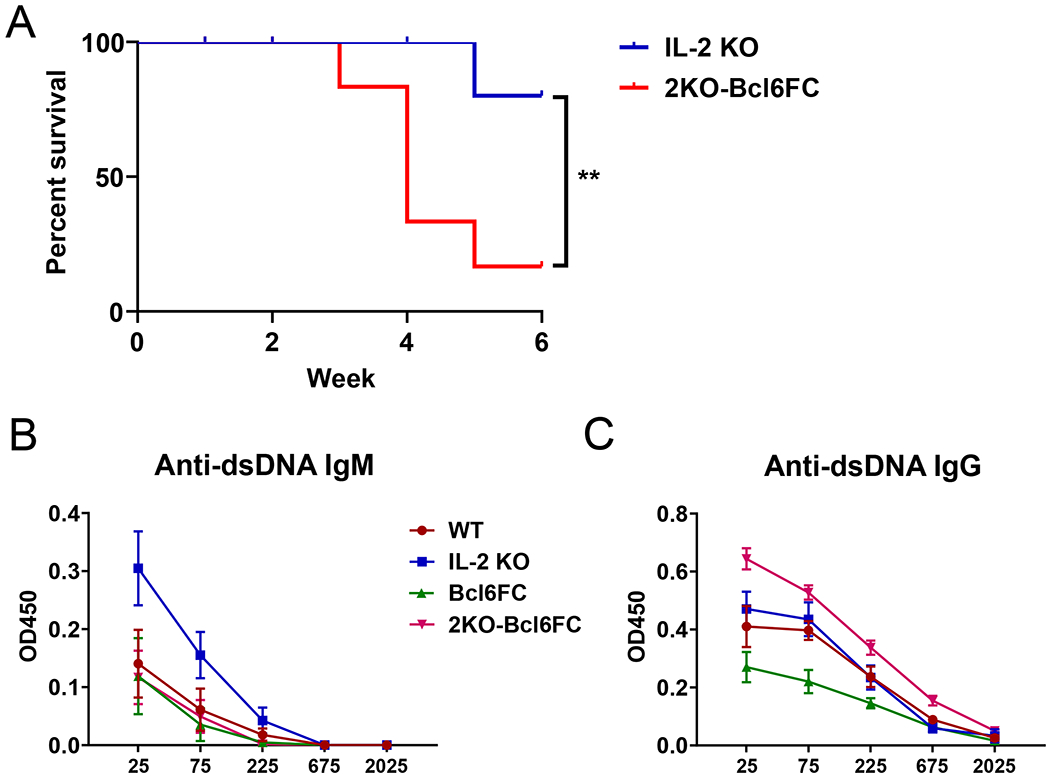
(A) Survival curve for IL-2 KO and 2KO-Bcl6FC (IL-2 KO Foxp3-cre Bcl6-flox cKO) mice over time. A Gehan-Breslow-Wilcoxon test was used to measure the statistical difference between the samples. (B-C) Serum samples were collected from 5 week old WT, IL-2 KO, Bcl6FC and 2KO-Bcl6FC mice for anti-dsDNA antibody detection by ELISA. (B) Anti-dsDNA IgM and (B) anti-dsDNA IgG titers are shown. The X-axis labels are dilution factors. Data shown as mean ± SEM, n=4 for WT, IL-2 KO, Bcl6FC and n=3 for 2KO-Bcl6FC. P values were calculated by T-test where * p < 0.05, ** p < 0.01, *** p < 0.0001.
Figure 7. Increased auto-Abs in Bcl6FC mice.
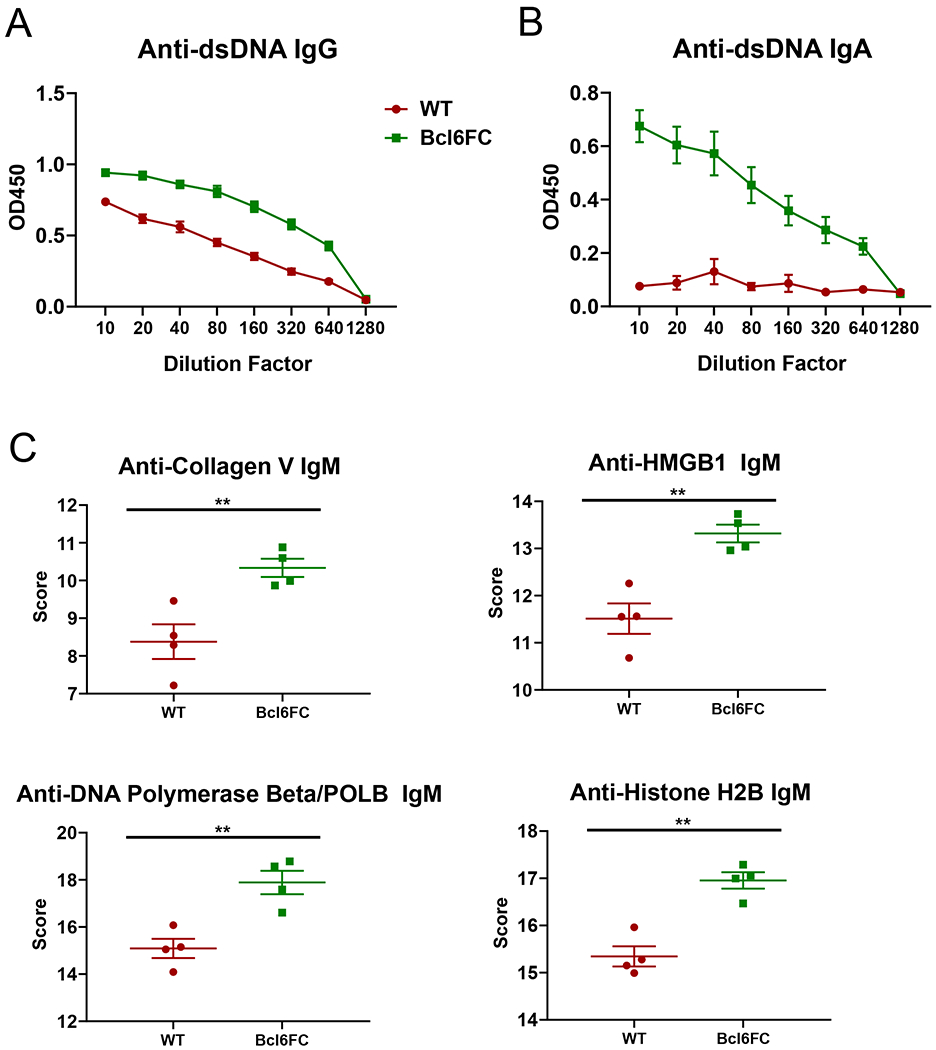
Serum from naïve WT and Bcl6FC mice was used to analyzed for anti-dsDNA Abs by ELISA and autoAg microarray. (A-B) Serum from 20 week old mice was tested for (A) anti-dsDNA IgG and (B) anti-dsDNA IgA. P values were calculated by t test where * p < 0.05, ** p < 0.01, *** p < 0.0001. N = 6 mice, experiment was repeated at 2 times. (C) Serum from 5 week old mice was tested for anti-autoAg IgG by microarray. 38 autoAgs out of 118 autoAgs tested showed a significant difference in IgG reactivity between WT and Bcl6FC, and all 38 showed a higher score for the Bcl6FC sera. All Graphs show selected anti-autoAg IgG scores for anti-collagen V, anti-Hmgb1, anti-DNA polymerase beta and anti-Histone H2B taken from the autoAg array data. P values were calculated by t test where ** p < 0.01. N = 4 mice.
Discussion
In this study, we have used the IL-2 KO mouse model of autoimmunity to probe the roles of Tfh cells and Tfr cells in the development of autoAbs. Tfh cells are required for the germinal center reaction and the production of high affinity antibodies to antigen, and excessive development of Tfh cells can promote autoAb production. Tfr cell functions are less well defined due to conflicting data on whether Tfr cells have a suppressive or helper function in the GC (20). However, there is general consensus that Tfr cells repress the development of autoAbs (23, 25–30). In our work here analyzing the IL-2 KO model of autoimmunity, we found unexpectedly that Tfh cells were not required for the production of high levels of autoAbs, and further that Tfr cells appear to potently inhibit the magnitude of autoAb responses.
Previous work on the IL-2 KO model showed strong differences depending on the genetic background. On a mixed 129/01a x C57BL/6 background, IL-2 KO mice die at an early age, most likely from severe colitis (37, 38). AutoAbs specific for colon cells were detected in this mixed background IL-2 KO (37). In BALB/c IL-2 KO mice, high levels of autoAbs to RBCs lead to severe AIHA and much earlier death than on a mixed background (38, 39). In BALB/c IL-2 KO mice, conventional CD4+ Tfh cells are elevated, as well as an unusual CD8+ Tfh-like population with B cell helper activity (39). Both CD4+ and CD8+ cells contribute to autoAb production and autoimmune disease in the BALB/c IL-2 KO model (39). Our B6 IL-2 KO mice live longer than what has been reported for BALB/c IL-2 KO mice, and to a similar age as reported for IL-2 KO mice on a mixed background. We did not observe significant development of colitis in our B6 background IL-2 KO mice, but did find significant inflammatory infiltration in the lung and liver and kidney (Supp. Fig. 2). Tfh cells were not analyzed in the mixed IL-2 KO mice but we observed that Tfh cells were elevated similarly to what was reported for BALB/c IL-2 KO mice. In terms of autoAb production, B6 IL-2 KO produced very little if any autoAbs to self-proteins but developed significant levels of anti-double stranded DNA Abs. We did not assess anti-RBC autoAbs directly, however we did observe apparent anemia (decreased hematocrit) in the B6 IL-2 KO mice (data not shown). Overall, the B6 background IL-2 KO mice had a somewhat different phenotype from both the mixed background and BALB/c background mice, although there are many similarities (Supp. Table 1). How many of these differences is due to the pure B6 background or from the conditions of different mouse colonies and related effects such as elimination of specific pathogens and microbiota is not clear.
Our original hypothesis for this study was that the increased Tfh cells were critical for promoting the autoimmune disease in the IL-2 KO mice by driving formation of autoAbs. However, when we deleted Tfh cells from the B6 IL-2 KO background, the 2KO-Bcl6TC mice had a similar disease phenotype as the B6 IL-2 KO strain (Supp. Fig. 1–2, Supp. Table 1). Note that both CD4+ Tfh cells and CD8+ Tfh-like cells are dependent on Bcl6 for development and are deleted in Bcl6TC cKO mice (14, 45). Even more unexpected than the similar disease phenotype of IL-2 KO mice and 2KO-Bcl6TC mice was that despite loss of Tfh cells, 2KO-Bcl6TC mice had higher levels of not only anti-DNA IgG autoAbs but also IgG autoAbs to self-proteins. Among other things, these data show that Tfh cells are not required for the robust production of IgG autoAbs in B6 IL-2 KO mice. Our data contrasts with data from the BALB/c background, where depletion of either CD4 or CD8 T cells led to lower total Ig, decreased anti-RBC Abs and improved overall survival of the mice (39). However, it seems likely that non-Tfh cells drive autoAb production in both the B6 and BALB/c IL-2 KO models.
The strongly increased levels of autoAbs in 2KO-Bcl6TC mice showed that a population of T cells that would normally suppress autoAb production was removed by the loss of T cell-specific Bcl6. The major candidates for such cells are Tfh cells and Tfr cells. While in principle, Tfh cells could play a suppressive role in driving autoAb generation, perhaps by diverting autoreactive B cells into the GC reaction and mutating them away from auto-reactivity, such a mechanism is speculative. A more likely explanation is that loss of Tfr cells in 2KO-Bcl6TC mice led to a loss of suppression of auto-reactive B cells. We tested this idea by generating 2KO-Bcl6FC mice, creating an IL-2 KO background where Tfr cells are deleted without affecting Tfh cells. 2KO-Bcl6FC mice had significantly worse disease than 2KO-Bcl6TC mice, with markedly stunted growth and 80% lethality by 4 weeks of age. However, 2KO-Bcl6FC mice did not have increased levels of autoAbs nor did they have increased organ inflammation compared to IL-2 KO mice, indicating a more complex pathogenesis in these mice. BALB/c IL-2 KO mice have a loss of Treg function as well as a loss of Treg cell numbers that correlates with autoimmune disease and that can be restored by IL-2 treatment (44). However, we did not observe decreased Treg cells in the B6 IL-2 KO or IL2-Bcl6TC mice, and the normal numbers of Tregs in these mice correlated with less severe disease than in BALB/c IL-2 KO mice. In the 2KO-Bcl6FC mice, we hypothesize that loss of Bcl6 expression coupled with loss of IL-2 signals led to a disabled, depleted or destabilized the overall Treg cell population. A loss of Tregs or Treg function coupled with normal T effector cells can promote severe inflammatory disease and/or autoimmune disease. We did not detect stronger inflammatory disease or autoAb production in the 2KO-Bcl6FC mice, although the early death and stunted growth of these mice made pathology difficult to elucidate.
While deletion of Tfh cells did not inhibit the production of anti-DNA autoAbs in the IL-2 KO mice, we did observe that deletion of MyD88 in the IL-2 KO mice did block anti-DNA autoAbs (Supp. Fig. 3). IL-2-MyD88 double KO mice also appeared to be healthier than IL-2 KO mice and developed to a normal size (data not shown). MyD88 is a downstream signaling protein for TLR molecules, and MyD88-dependent TLR signals are known to promote inflammation and amplify autoimmunity (48). Our data indicate that innate signals, likely nucleic acids released from dead cells (49), as well as commensal-derived TLR-ligands (50), help to promote the formation of autoimmune disease and autoAbs in the IL-2 KO mice. Strikingly, TFR cells were increased in IL-2-MyD88 double KO mice over WT levels (Supp. Fig. 3), suggesting that in a less pro-inflammatory environment, IL-2 is indeed inhibitory for Tfr cell generation. In other words, the loss of Tfr cells observed in IL-2 KO mice maybe due to increased destabilization of the Treg population from inflammatory cytokines.
In summary, our studies on the IL-2 KO model of autoimmunity have generated new insights into the roles of Tfh and Tfr cells in controlling autoAbs formation and in the role of IL-2 in regulating Tfr cell differentiation. Further work is needed to understand more completely how Tfr cells control autoimmunity and the function of Tfr cells in IL-2 KO mice.
Supplementary Material
Acknowledgements
This work was supported by NIAID grant R01 AI132771 to A.L.D.. M.M.X. was supported by a Careers in Immunology Fellowship from American Association of Immunologists. Core facility usage was supported by IU Simon Cancer Center Support Grant P30 CA082709 and U54 DK106846. Support provided by the Herman B Wells Center was in part from the Riley Children’s Foundation.
Footnotes
Disclosures
The authors declare no conflict of interest with this work.
References
- 1.Crotty S 2011. Follicular helper CD4 T cells (TFH). Annu Rev Immunol 29: 621–663. [DOI] [PubMed] [Google Scholar]
- 2.Vinuesa CG, Linterman MA, Yu D, and MacLennan IC. 2016. Follicular Helper T Cells. Annu Rev Immunol 34: 335–368. [DOI] [PubMed] [Google Scholar]
- 3.Odegard JM, Marks BR, DiPlacido LD, Poholek AC, Kono DH, Dong C, Flavell RA, and Craft J. 2008. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J Exp Med 205: 2873–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, Yu D, Domaschenz H, Whittle B, Lambe T, Roberts IS, Copley RR, Bell JI, Cornall RJ, and Goodnow CC. 2005. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature 435: 452–458. [DOI] [PubMed] [Google Scholar]
- 5.Linterman MA, Rigby RJ, Wong RK, Yu D, Brink R, Cannons JL, Schwartzberg PL, Cook MC, Walters GD, and Vinuesa CG. 2009. Follicular helper T cells are required for systemic autoimmunity. J Exp Med 206: 561–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu D, and Vinuesa CG. 2010. Multiple checkpoints keep follicular helper T cells under control to prevent autoimmunity. Cell Mol Immunol 7: 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.King C, Tangye SG, and Mackay CR. 2008. T follicular helper (TFH) cells in normal and dysregulated immune responses. Annu Rev Immunol 26: 741–766. [DOI] [PubMed] [Google Scholar]
- 8.Crotty S 2014. T follicular helper cell differentiation, function, and roles in disease. Immunity 41: 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mountz JD, Hsu HC, and Ballesteros-Tato A. 2019. Dysregulation of T Follicular Helper Cells in Lupus. J Immunol 202: 1649–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu Y, Zou L, and Liu YC. 2016. T follicular helper cells, T follicular regulatory cells and autoimmunity. Int Immunol 28: 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, Dent AL, Craft J, and Crotty S. 2009. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 325: 1006–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, Wang YH, and Dong C. 2009. Bcl6 mediates the development of T follicular helper cells. Science 325: 1001–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, Srivastava M, Linterman M, Zheng L, Simpson N, Ellyard JI, Parish IA, Ma CS, Li QJ, Parish CR, Mackay CR, and Vinuesa CG. 2009. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity 31: 457–468. [DOI] [PubMed] [Google Scholar]
- 14.Hollister K, Wu H, Kusam S, Clegg N, Mondal A, Sawant DV, and Dent A. 2013. Insights into the Role of Bcl6 in Follicular Helper T Cells Using a New Conditional Mutant Mouse Model. J Immunol 191: 3705–3711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, Srivastava M, Divekar DP, Beaton L, Hogan JJ, Fagarasan S, Liston A, Smith KG, and Vinuesa CG. 2011. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med 17: 975–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alexander CM, Tygrett LT, Boyden AW, Wolniak KL, Legge KL, and Waldschmidt TJ. 2011. T regulatory cells participate in the control of germinal centre reactions. Immunology 133: 452–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wollenberg I, Agua-Doce A, Hernandez A, Almeida C, Oliveira VG, Faro J, and Graca L. 2011. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J Immunol 187: 4553–4560. [DOI] [PubMed] [Google Scholar]
- 18.Sage PT, and Sharpe AH. 2016. T follicular regulatory cells. Immunol Rev 271: 246–259. [DOI] [PubMed] [Google Scholar]
- 19.Sage PT, Francisco LM, Carman CV, and Sharpe AH. 2013. The receptor PD-1 controls follicular regulatory T cells in the lymph nodes and blood. Nat Immunol 14: 152–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie MM, and Dent AL. 2018. Unexpected Help: Follicular Regulatory T Cells in the Germinal Center. Front Immunol 9: 1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, Wang YH, Lim H, Reynolds JM, Zhou XH, Fan HM, Liu ZM, Neelapu SS, and Dong C. 2011. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med 17: 983–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laidlaw BJ, Lu Y, Amezquita RA, Weinstein JS, Vander Heiden JA, Gupta NT, Kleinstein SH, Kaech SM, and Craft J. 2017. Interleukin-10 from CD4(+) follicular regulatory T cells promotes the germinal center response. Sci Immunol 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu H, Chen Y, Liu H, Xu LL, Teuscher P, Wang S, Lu S, and Dent AL. 2016. Follicular regulatory T cells repress cytokine production by follicular helper T cells and optimize IgG responses in mice. Eur J Immunol 46: 1152–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pratama A, and Vinuesa CG. 2014. Control of TFH cell numbers: why and how? Immunology and cell biology 92: 40–48. [DOI] [PubMed] [Google Scholar]
- 25.Botta D, Fuller MJ, Marquez-Lago TT, Bachus H, Bradley JE, Weinmann AS, Zajac AJ, Randall TD, Lund FE, Leon B, and Ballesteros-Tato A. 2017. Dynamic regulation of T follicular regulatory cell responses by interleukin 2 during influenza infection. Nat Immunol 18: 1249–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fu W, Liu X, Lin X, Feng H, Sun L, Li S, Chen H, Tang H, Lu L, Jin W, and Dong C. 2018. Deficiency in T follicular regulatory cells promotes autoimmunity. J Exp Med 215: 815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vaeth M, Muller G, Stauss D, Dietz L, Klein-Hessling S, Serfling E, Lipp M, Berberich I, and Berberich-Siebelt F. 2014. Follicular regulatory T cells control humoral autoimmunity via NFAT2-regulated CXCR5 expression. J Exp Med 211: 545–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fazilleau N, and Aloulou M. 2018. Several Follicular Regulatory T Cell Subsets With Distinct Phenotype and Function Emerge During Germinal Center Reactions. Front Immunol 9: 1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maceiras AR, Almeida SCP, Mariotti-Ferrandiz E, Chaara W, Jebbawi F, Six A, Hori S, Klatzmann D, Faro J, and Graca L. 2017. T follicular helper and T follicular regulatory cells have different TCR specificity. Nat Commun 8: 15067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sage PT, Schildberg FA, Sobel RA, Kuchroo VK, Freeman GJ, and Sharpe AH. 2018. Dendritic Cell PD-L1 Limits Autoimmunity and Follicular T Cell Differentiation and Function. J Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lleo A, Invernizzi P, Gao B, Podda M, and Gershwin ME. 2010. Definition of human autoimmunity--autoantibodies versus autoimmune disease. Autoimmun Rev 9: A259–266. [DOI] [PubMed] [Google Scholar]
- 32.Tan EM 2012. Autoantibodies, autoimmune disease, and the birth of immune diagnostics. J Clin Invest 122: 3835–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Naparstek Y, and Plotz PH. 1993. The role of autoantibodies in autoimmune disease. Annu Rev Immunol 11: 79–104. [DOI] [PubMed] [Google Scholar]
- 34.Kotov JA, and Jenkins MK. 2019. Cutting Edge: T Cell-Dependent Plasmablasts Form in the Absence of Single Differentiated CD4(+) T Cell Subsets. J Immunol 202: 401–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Groom JR, Fletcher CA, Walters SN, Grey ST, Watt SV, Sweet MJ, Smyth MJ, Mackay CR, and Mackay F. 2007. BAFF and MyD88 signals promote a lupuslike disease independent of T cells. J Exp Med 204: 1959–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herlands RA, Christensen SR, Sweet RA, Hershberg U, and Shlomchik MJ. 2008. T cell-independent and toll-like receptor-dependent antigen-driven activation of autoreactive B cells. Immunity 29: 249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, and Horak I. 1993. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell 75: 253–261. [DOI] [PubMed] [Google Scholar]
- 38.Sadlack B, Lohler J, Schorle H, Klebb G, Haber H, Sickel E, Noelle RJ, and Horak I. 1995. Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. Eur J Immunol 25: 3053–3059. [DOI] [PubMed] [Google Scholar]
- 39.Valentine KM, Davini D, Lawrence TJ, Mullins GN, Manansala M, Al-Kuhlani M, Pinney JM, Davis JK, Beaudin AE, Sindi SS, Gravano DM, and Hoyer KK. 2018. CD8 Follicular T Cells Promote B Cell Antibody Class Switch in Autoimmune Disease. J Immunol 201: 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnston RJ, Choi YS, Diamond JA, Yang JA, and Crotty S. 2012. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med 209: 243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nurieva RI, Podd A, Chen Y, Alekseev AM, Yu M, Qi X, Huang H, Wen R, Wang J, Li HS, Watowich SS, Qi H, Dong C, and Wang D. 2012. STAT5 protein negatively regulates T follicular helper (Tfh) cell generation and function. J Biol Chem 287: 11234–11239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ballesteros-Tato A, Leon B, Graf BA, Moquin A, Adams PS, Lund FE, and Randall TD. 2012. Interleukin-2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity 36: 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Isakson SH, Katzman SD, and Hoyer KK. 2012. Spontaneous autoimmunity in the absence of IL-2 is driven by uncontrolled dendritic cells. J Immunol 189: 1585–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barron L, Dooms H, Hoyer KK, Kuswanto W, Hofmann J, O’Gorman WE, and Abbas AK. 2010. Cutting edge: mechanisms of IL-2-dependent maintenance of functional regulatory T cells. J Immunol 185: 6426–6430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leong YA, Chen Y, Ong HS, Wu D, Man K, Deleage C, Minnich M, Meckiff BJ, Wei Y, Hou Z, Zotos D, Fenix KA, Atnerkar A, Preston S, Chipman JG, Beilman GJ, Allison CC, Sun L, Wang P, Xu J, Toe JG, Lu HK, Tao Y, Palendira U, Dent AL, Landay AL, Pellegrini M, Comerford I, McColl SR, Schacker TW, Long HM, Estes JD, Busslinger M, Belz GT, Lewin SR, Kallies A, and Yu D. 2016. CXCR5(+) follicular cytotoxic T cells control viral infection in B cell follicles. Nat Immunol 17: 1187–1196. [DOI] [PubMed] [Google Scholar]
- 46.Batten M, Ramamoorthi N, Kljavin NM, Ma CS, Cox JH, Dengler HS, Danilenko DM, Caplazi P, Wong M, Fulcher DA, Cook MC, King C, Tangye SG, de Sauvage FJ, and Ghilardi N. 2010. IL-27 supports germinal center function by enhancing IL-21 production and the function of T follicular helper cells. J Exp Med 207: 2895–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Calvani N, Caricchio R, Tucci M, Sobel ES, Silvestris F, Tartaglia P, and Richards HB. 2005. Induction of apoptosis by the hydrocarbon oil pristane: implications for pristane-induced lupus. J Immunol 175: 4777–4782. [DOI] [PubMed] [Google Scholar]
- 48.Liu Y, Yin H, Zhao M, and Lu Q. 2014. TLR2 and TLR4 in autoimmune diseases: a comprehensive review. Clin Rev Allergy Immunol 47: 136–147. [DOI] [PubMed] [Google Scholar]
- 49.Nundel K, Green NM, Shaffer AL, Moody KL, Busto P, Eilat D, Miyake K, Oropallo MA, Cancro MP, and Marshak-Rothstein A. 2015. Cell-intrinsic expression of TLR9 in autoreactive B cells constrains BCR/TLR7-dependent responses. J Immunol 194: 2504–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lartigue A, Colliou N, Calbo S, Francois A, Jacquot S, Arnoult C, Tron F, Gilbert D, and Musette P. 2009. Critical role of TLR2 and TLR4 in autoantibody production and glomerulonephritis in lpr mutation-induced mouse lupus. J Immunol 183: 6207–6216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.