


Abstract
The CFTR gene lies within an invariant topologically associated domain (TAD) demarcated by CTCF and cohesin, but shows cell-type specific control mechanisms utilizing different cis-regulatory elements (CRE) within the TAD. Within the respiratory epithelium, more than one cell type expresses CFTR and the molecular mechanisms controlling its transcription are likely divergent between them. Here, we determine how two extragenic CREs that are prominent in epithelial cells in the lung, regulate expression of the gene. We showed earlier that these CREs, located at −44 and −35 kb upstream of the promoter, have strong cell-type-selective enhancer function. They are also responsive to inflammatory mediators and to oxidative stress, consistent with a key role in CF lung disease. Here, we use CRISPR/Cas9 technology to remove these CREs from the endogenous locus in human bronchial epithelial cells. Loss of either site extinguished CFTR expression and abolished long-range interactions between these sites and the gene promoter, suggesting non-redundant enhancers. The deletions also greatly reduced promoter interactions with the 5′ TAD boundary. We show substantial recruitment of RNAPII to the −35 kb element and identify CEBPβ as a key activator of airway expression of CFTR, likely through occupancy at this CRE and the gene promoter.
INTRODUCTION
Enhancer elements, which are marked by specific modified histone proteins and recruit activating transcription factors (TFs), have pivotal roles in the regulation of gene expression. Protocols that examine the 3D organization of active chromatin (1–3) greatly increased our understanding of how enhancers, sometimes located several megabases away from the transcription start site (TSS) of their target gene, can regulate gene transcription in a temporal and cell-type specific manner by looping (4). These methods also showed that the genome is organized into compartmentalized topologically associated domains (TADs) which are maintained in part by architectural proteins, e.g. CCCTC binding factor (CTCF) and the cohesin complex (SMC1, SMC3, RAD21). Enhancers rarely loop across TAD boundaries.
While the major advances in functional genomics have come largely from genome-wide studies, there is still a critical need for detailed analysis of individual loci to generate and test new models and to provide additional precision. One of the most intensively studied loci is the cystic fibrosis conductance regulator (CFTR) gene, mutations in which cause the life-shortening inherited disorder cystic fibrosis (CF). Insights from the analysis of CFTR have repeatedly proven relevant to many individual loci across the genome (5–7). The CFTR locus is organized within a TAD flanked by CTCF and cohesin occupancy at −80.1 kb upstream and +48.9 kb downstream, of the gene. Within this TAD specific cis-regulatory elements (CREs) play critical roles in the regulation of CFTR expression, either by acting as enhancers or as structural features that facilitate the recruitment of active elements to the gene promoter (6). We identified and characterized multiple CREs (8–10) in different cell types showing that many of them are cell-type specific, acting as enhancers only in one cell type (6,8–12). Examples include (i) an intestinal enhancer element associated with a DNase I hypersensitive site (DHS) in intron 11# of the gene (DHS 11) in Caco2 cells, removal of which reduced CFTR expression by ∼80% (10) and (ii) an enhancer blocking insulator element associated with a DHS at −20.9 kb upstream of the promoter, that recruits CTCF (13) and loss of which alters chromatin architecture at the locus though has little impact on CFTR expression. There are now many such examples in other loci, where distal enhancers dictate cell-type specific regulation of gene expression by altering chromatin architecture upon recruitment of specific activating TFs (reviewed in (14)).
Though lung disease underlies most CF morbidity, the majority of cells in the post-natal airway epithelium have low levels of CFTR expression, with rare cells showing more abundant transcripts (15–19). This suggests distinct mechanisms of CFTR regulation in different cell types within the adult lung. Here we investigate the CREs associated with two DHS that are prominent at −44 and −35 kb upstream of the CFTR gene promoter in several human lung cell lines and in primary human bronchial (HBE) (10,20) and tracheal epithelial (HTE) cells (21). Though these two sites were evaluated previously using protocols available at the time, and were shown to be functionally associated (9,11), we did not evaluate their contribution in the genomic context. We showed that the site at −44 kb contained an antioxidant response element (ARE) that plays a key role in CFTR function in airway cell lines. Sulforaphane (SFN) (a natural antioxidant) caused nuclear translocation of nuclear factor, erythroid 2-like 2 (Nrf2), a TF that controls genes with AREs in their promoters, many of which are involved in response to injury. Under normoxic conditions, the −44-kb ARE is occupied by the repressor BTB and CNC homology 1, basic leucine zipper TF (BACH1) and v-Maf avian musculoaponeurotic fibrosarcoma oncogene homolog K (MAFK) heterodimers. However, following two hours of SFN treatment, Nrf2 displaces these repressive factors and activates CFTR expression (11). In contrast, the −35 kb element, which cooperates with the −44 kb site in in vitro enhancer assays, appears to be regulated at least in part by immune mediators interferon regulatory factors (IRF1 and IRF2) (9). Additionally, nuclear factor Y (NF-Y) recruitment at the −35 kb site regulates histone modifications at this element.
Here, we build upon our earlier work and apply protocols that enable functional assessment of the role of the −44 and −35 kb DHS in the genomic context in the human bronchial epithelial cell line 16HBE14o- (22). We show by CRISPR/Cas9-mediated deletions that CFTR transcription in this cell line, which exhibits high CFTR mRNA abundance (8), requires the recruitment of both of these sites. Moreover, these sites are engaged in a higher order chromatin architecture that links the 5′ TAD boundary at −80 kb, the −20.9 kb CTCF-binding insulator element, and the gene promoter and is essential for recruitment of RNA Polymerase II to the locus.
MATERIALS AND METHODS
Cell culture
Caco2 (23) and Calu3 (24) cell lines were obtained from ATCC and grown in DMEM (Dulbecco's modified Eagle's medium) with 10% FBS (fetal bovine serum). For all experiments with Caco2 cells they were harvested 48 h post-confluence, a time at which CFTR expression is close to maximum levels (25). The transformed human bronchial epithelial cell line 16HBE14o- (22) was grown in DMEM with 10% serum. All cells were grown on plastic at liquid interface.
CRISPR guide design, CRISPR/Cas9 transfection and screening
Two pairs of gRNAs flanking the −35, −44 and −20.9 kb sites were identified using the CRISPR Design Program (http://crispr.mit.edu). gBlocks from Integrated DNA Technologies (Iowa), were cloned into pSCB (Agilent) and sequenced. 16HBE14o- cells were transfected with pMJ290 (wild-type Cas9 plasmid tagged with GFP) (Addgene, #42234) and the cloned gRNAs using Lipofectamine 2000 (Life Technologies (LT), Carlsbad, CA, USA). Forty eight hours later, GFP positive cells single cells were isolated by fluorescence-activated cell sorting and seeded into 96-well plate for clonal expansion. Clones with homozygous deletions of each cis-regulatory element were confirmed by PCR of genomic DNA using primers flanking the gRNA PAM sites (specific reduction in size of product) and one flanking primer together with one located within the deletion (no product) and by Sanger sequencing. Primers are shown in Supplementary Table S1.
#This manuscript uses legacy nomenclature for the CFTR gene to be consistent with our earlier work.
Reverse transcription quantitative PCR (RT-qPCR)
Total RNA from confluent cultures was extracted with TRIzol (LT) and cDNA prepared with the TaqMan reverse transcription kit (Invitrogen). CFTR mRNA levels were assayed using a well-characterized Taqman assay (25) (Supplementary Table S2) and normalized to beta 2 microglobulin (β2M) as a housekeeping-gene control.
Enhancer RNA (eRNA) detection
Total RNA was extracted from confluent cultures with TRIzol and was DNase I treated and phenol-chloroform extracted. The treated RNA was reverse transcribed and cDNA amplified by qPCR analysis with SYBR Green reagents (Invitrogen) with primers listed in Supplementary Table S2.
Chromatin immunoprecipitation-quantitative PCR (ChIP-qPCR) and ChIP-seq
ChIP was performed by standard protocols for ChIP-qPCR and ChIP (12,26). Antibodies were specific for RNAPII (Abcam ab76123 for ChIP-qPCR or Cell Signaling Technology 14958S for ChIP-seq), CEBPβ (Santa Cruz sc-7962), H3K27Ac (Millipore 07-360), CTCF (Millipore 07-729) or rabbit IgG (Millipore 12-370). All ChIP-seq experiments were performed twice and irreproducible discovery rate (IDR) data are shown. Raw reads were processed using the AQUAS Transcription Factor and Histone ChIP-Seq processing pipeline (https://github.com/ENCODE-DCC/chip-seq-pipeline2) using the ENCODE (phase-3) guidelines on the hg19 reference genome. This includes mapping using BWA and peak calling with MACS2. Primer sequences used for qPCR are shown in Supplementary Table S2.
Luciferase assays
The pGL3B 245 (CFTR basal promoter) luciferase reporter vector (8) containing the −35 kb 350 bp core enhancer (9) was used to generate the CEBPβ mutant using the QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA). The mutagenesis primer is shown in Supplementary Table S3. pGL3B luciferase constructs were transiently cotransfected with a modified pRL Renilla luciferase control vector (Promega, Madison, WI, USA) into 16HBE14o- cells with Lipofectin (LT). Firefly and Renilla (normalizer) luciferase activities were measured 48 h after transfection.
Omni assay for transposase accessible chromatin and deep sequencing
Omni-ATAC-seq was performed on 50,000 Calu3, 16HBE14o- and Caco2 as described previously (27) with minor modifications. ATAC-seq libraries were purified with Agencourt AMPure XP magnetic beads (Beckman Coulter) with a sample to bead ratio of 1:1.2, and eluted in Buffer EB (Qiagen). Data were processed by the ENCODE-DCC/atac-seq-pipeline.
4C-seq
4C-seq libraries were generated from cultured cells as described previously (10). All 4C experiments were done a minimum of twice on the same clonal cell line (technical replicate) and each deletion event was evaluated in at least two independent clonal lines (biological replicate). NlaIII and DpnII or Csp6I were used as the primary or secondary restriction enzymes, respectively. Enzyme pairs and primer sequences used to generate 4C-seq libraries for each viewpoint are shown in Supplementary Table S4. Some of the primers contain a two nucleotide barcode at the 3′ end of P5 linker to enable multiplexing of libraries generated from same viewpoint on the same Hi-Seq 2500 flow cell for sequencing. The sequencing data were processed using the 4C-seq pipe protocol (28). All 4C-seq images were generated using default parameters of the pipeline.
Statistics
Error bars in all graphs denote standard error of the mean (SEM). Statistical analysis used the Student's unpaired t-tests in Prism software (GraphPad).
RESULTS
ATAC-seq confirms airway-selective regions of open chromatin at the CFTR locus
In earlier work we identified sites of open chromatin in airway epithelial cell lines and human tracheal epithelial (HTE) cells (8) by DNase chip and more recently in several cell lines and primary human bronchial epithelial (HBE) cells (10) by DNAse-seq. Open chromatin in airway cell lines was mapped here by assay for transposase accessible chromatin (Omni ATAC seq). Figure 1 shows regions of open chromatin across the CFTR locus by ATAC-seq in the airway cell lines Calu3 and 16HBE14o- and Caco2 colon carcinoma cells, and by DNAse-seq in HBE cells from (10). Clearly evident in all cell types are the peaks of open chromatin at −80.1 and +48.9 kb, which correspond to the TAD boundaries we identified previously (10), and at the CFTR promoter which is active in all these cell-types. Also evident in all the airway cells but not in Caco2 cells are peaks of open chromatin at −44 and −35 kb, consistent with our earlier observations with other protocols (Figure 1A). Since these two sites were shown to act as enhancers of the CFTR promoter in transient luciferase assays (9,11) we next examined the occupancy of active histone modifications (H3K27Ac) by ChIP-seq in Calu3 and 16HBE14o- cells (Figure 1A, purple and gray tracks). Both the −44 and −35 kb sites are associated with peaks of H3K27Ac (Figure 1B), suggesting they are active enhancers in CFTR-expressing airway epithelial cells. Another open chromatin site close to the −35 kb site (at −31.1 kb) was marked by H3K27Ac in 16HBE14o- but not in Calu3 cells.
Figure 1.

Identification of airway epithelial cell selective open chromatin across the CFTR locus. (A) Open chromatin mapping by Omni- ATAC-seq on Caco2 (bright blue), Calu3 (teal), 16HBE14o- (Dark blue), DNase-seq on HBE cells (light blue) and H3K27Ac occupancy by ChIP-seq on Calu3 cells (purple) and 16HBE14o- cells (gray). The location of ASZ1, CFTR and CTTNBP2 are shown at the top of the figure. Major DHS identified at the CFTR locus in all cells that express the gene and other cell-selective DHS of interest are marked below the gene track in black. This manuscript uses legacy nomenclature for the CFTR gene to be consistent with our earlier work. The −35 and −44 kb region is marked by a bar below the tracks. (B) Enlarged image of the −35 and −44 kb DHS regions.
Upstream open chromatin loops to the CFTR promoter in airway epithelial cells
The CFTR gene lies within a TAD marked by invariant CTCF sites at −80.1 and +48.9 kb and our previous analysis of chromatin architecture by 4C-seq analysis (10) also revealed several cell-type selective interactions within the TAD in intestinal, airway and epididymis epithelial cells. Here, we focus on a more detailed functional genomics analysis of airway epithelial cells in order to define key airway CREs, their recruitment to the CFTR promoter and their activating TFs. Multiple viewpoints across the CFTR locus were used for 4C-seq in 16HBE14o- and Calu3 airway epithelial cells (Figure 2) and the profiles compared with Caco2. The most notable features of the intra-TAD interactions in the airway cells are strong interactions 5′ to the gene promoter. Using a viewpoint at the CFTR promoter, more extensive interactions are seen upstream of the −20.9 kb CTCF-binding insulator element in 16HBE14o- cells than are evident in Caco2 cells. This is consistent with earlier observations on Calu3 cells. These 5′ interactions are seen more clearly with a viewpoint at the −80.1 kb 5′ TAD boundary which shows strong interactions with the −44, −35 and −20.9 kb sites in addition to the CFTR promoter. The absence of these interactions in Caco2 cells suggests that CFTR regulation in these airway cells may be dependent on recruitment of a unique set of TFs driving the looping interactions with upstream cis-regulatory elements.
Figure 2.

Airway epithelial cell chromatin architecture at the CFTR locus: 4C-seq data are shown for Caco2, 16HBE14o- and Calu3 cell lines. At the top the genomic location of CFTR and adjacent genes on chromosome 7 are shown together with known CFTR cis-regulatory elements. Below, 4C-seq data are shown for each cell line using viewpoints at the CFTR promoter (upper) and the −80.1 kb 5′ TAD boundary (lower). The 4C-seq data show the main trend of contact profile using a 5-kb window size as a black line above the domainogram. Relative interactions are normalized to the strongest point (which is set to 1) within each panel. The domainogram uses color-coded intensity values to show relative interactions with window sizes varying from 2 to 50 kb. Here, red denotes the strongest interactions and dark blue, through turquoise, to gray represent decreasing frequencies.
The −35 and −44 kb enhancers make a major contribution to CFTR expression in airway epithelial cells
Next we examined the genomic contribution of CREs within the −35 and −44 kb DHS to CFTR expression in airway epithelial cells (Figure 3A). These sites, which interact with the TAD boundary and the gene promoter, were also shown previously to act as cooperating enhancers of the CFTR promoter in the airway but not in the intestinal epithelial cells in transient luciferase assays (8,11).
Figure 3.

Deletion of the −35 or −44 kb CRE abolishes CFTR gene expression in airway cells. (A) Genomic location of the deleted CREs relative to the CFTR locus. (B) Location of gRNAs (blue) used for CRISPR/Cas9 mediated deletion of the −44 and −35 kb CREs in the 16HBE14o- cell line. (C) RT-qPCR analysis of CFTR gene expression in two Δ −35 kb clones (left) and two Δ-44 kb clones (right), compared to non-targeted WT clones from the same experiment. Results are the average of three independent experiments. Error bars represent S.E.M.
CRISPR/Cas9 technology was used to delete these elements in 16HBE14o- airway cells (Figure 3B, Supplementary Figure S1). This line has the advantage over Calu3 cells in that it is relatively amenable to single cell cloning after CRISPR/Cas9 modification. Guide RNAs targeting PAM sites that flank each CRE on the 5′ and 3′ sides were used to remove ∼1.5 kb encompassing the core of the −44 and −35 kb enhancers, as evidenced by the limits of the H3K27Ac peaks (Figure 1) and TF occupancy (29). The CRISPR/Cas9-mediated deletions were subjected to Sanger sequencing of the genomic DNA and were confirmed to be homozygous.
First, CFTR expression was assayed by RT-qPCR in 16HBE14o- clones lacking either the −35 or −44 kb CREs and was found to be almost abolished (Figure 3C). This suggests that these elements are essential in driving CFTR transcription in these cells.
Deletion of the −35 and −44 kb sites alters in intra-TAD chromatin interactions
Next, to examine the impact of deletion of the −35 and −44 kb CREs on locus architecture, we performed 4C-seq analysis. Viewpoints at the CFTR promoter and −80.1 kb 5′ TAD boundary (Figure 4), and at the −20.9 kb insulator and +48.9 kb 3′ TAD boundary (Supplementary Figure S2), were used. Two clones that were homozygous for each deletion were examined and the data for one of them is shown. In all cases, the two clones showed an equivalent profile.
Figure 4.

Deletion of the −35 or −44 kb CRE from the CFTR locus has a major impact on locus architecture. (A) 4C-seq interaction profiles of the same WT, Δ −35 kb (Clone #6) and Δ-44 kb (clone #4) 16HBE14o- clones shown in Figure 3, using viewpoints at the CFTR promoter and the −80.1 kb 5′ TAD boundary. The interpretation of the 4C-seq data is as described in the legend to Figure 2. Features of interest are shown by black or gray arrows or bars.
In both the Δ −35 kb and Δ-44 kb deletion clones, interactions of the promoter viewpoint with the −20.9 and the −80.1 kb sites are substantially reduced compared to WT cells (Figure 4 upper panels, black arrows). Similarly, with the −80.1 kb viewpoint, a clear loss of interactions is evident with all sites between this TAD boundary and the gene promoter, including the remaining −44 or −35 kb and −20.9 kb CREs (Figure 4, lower panels, black bar and arrows). In addition, interactions between −80.1 and +48.9 kb TAD boundaries are greatly reduced. In contrast, in both Δ −35 kb and Δ-44 kb deletion clones some novel interactions are seen between the −80.1 kb viewpoint and sequences 5′ to the −44 kb site (Figure 4, gray arrows).
Interactions between the −20.9 kb viewpoint are somewhat reduced in the Δ −35 kb and Δ-44 kb deletion clones, though an increase is seen with 3′ part of the locus, specifically the +6.8 and +15.6 kb sites (Supplementary Figure S2, upper panels see black arrows). Both these sites were shown previously to function as enhancer-blocking insulators, the former being associated with CTCF occupancy (13) while the latter functions independently of CTCF (30). Interactions with +48.9 kb viewpoint (Supplementary Figure S2, lower panels) were not substantially altered by deletion of the −35 kb CREs suggesting these sites do not depend upon the 3′ TAD boundary for their functions. In contrast loss of the −44 kb site somewhat enhanced interactions between the −20.9 and +48.9 kb elements.
A bifunctional role for the −20.9 kb CRE in airway epithelial cells
In classical plasmid-based enhancer-blocking insulator assays (31), the −20.9 kb element impairs the interaction of the chicken β-globin enhancer with the human γ-globin promoter driving a neor gene, when placed between these elements (30). Our observations here showing a reduction in recruitment of the −20.9 kb element to the promoter and the −80.1 kb 5′ TAD boundary in the Δ −35 kb and Δ-44 kb deletion clones encouraged us to re-evaluate the role of the −20.9 kb site in the genomic locus in airway epithelial cells. Of note, despite the activity of the strong 5′ enhancer elements, CFTR expression is low in most airway epithelial cell in comparison to other epithelia. To determine whether the −20.9 kb element functions primarily as an enhancer-blocking insulator, fine-tuning the activation of the CFTR promoter by its strong 5′ enhancers to maintain low expression levels, we used CRISPR/Cas9 to delete the site from 16HBE14o- cells (Supplementary Figure S3). If the loss of this site is accompanied by increased CFTR expression and interactions between the promoter and upstream enhancers, this would reinforce its primary insulator role. The results in Supplementary Figure S4A show that −20.9 kb del was accompanied by a significant increase in CFTR expression. However, though this deletion was associated with alterations in locus architecture (Supplementary Figure S4B), there was no substantial increase in the interactions between the 5′ enhancers and the promoter. Instead, the loss of interactions between −20.9 and the 5′ TAD boundary was accompanied by a substantial increase in interactions of the 5′ TAD boundary with the CTCF- recruiting insulator element at +6.8 kb (13), though changes in promoter interactions were more diffuse. This suggests that the −20.9 kb element has a dominant role in maintaining locus architecture and possibly a lesser role in inhibiting enhancer-promoter interactions.
The −35 kb CRE recruits RNA polymerase II in airway epithelial cells
Since removal of either the −44 or −35 kb enhancers abolished CFTR transcription, we next tested the hypothesis that these deletions altered RNA polymerase II (RNAPII) occupancy at the CFTR promoter. Using an antibody specific for RNAPII that recognizes both the promoter bound and elongating forms, we performed ChIP-qPCR with primers across the CFTR locus (Figure 5A). The mannose-6-phosphate receptor (M6PR) gene promoter provided a positive control, based on ENCODE ChIP-seq data for RNAPII subunit A (POLR2A) (29) and our RNA-seq data from 16HBE14o- cells (32). RNAPII occupancy was compared in a WT 16HBE14o- clone and a Δ −35 clone. In the WT clone (Figure 5A, black bars) primer sets at −3.4 and −2 kb upstream of the CFTR basal promoter (33) showed high RNAPII occupancy while intronic sites within the gene (DHS in introns 1, 10 and 23) showed lower levels. Of particular note was substantial RNAPII occupancy at the −35 kb region, suggesting that this enhancer element could be transcribed. In contrast RNAPII binding was reduced in the Δ −35 clone, with the greatest occupancy changes at promoter proximal sites. These data suggest the −35 kb CRE has an important role in CFTR transcription initiation in the 16HBE14o- cell line.
Figure 5.
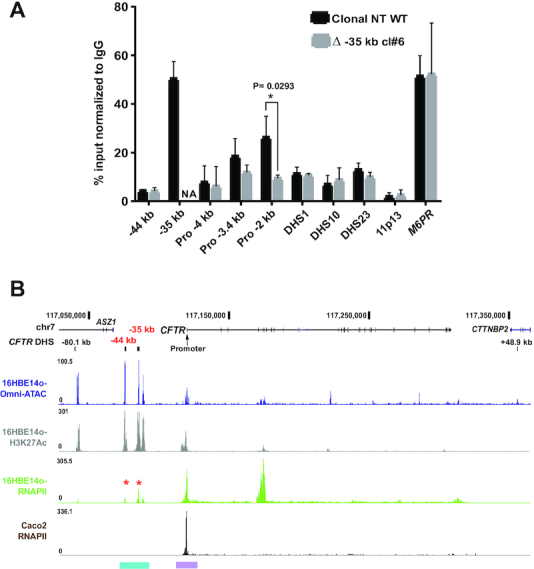
The −35 kb enhancer region binds RNAPII. (A) ChIP for RNAPII in non-targeted (NT) WT (black) and Δ −35 kb (grey) 16HBE14o- clones. RNAPII occupancy shown by qPCR using primers for CREs across the CFTR locus (−44, −35 kb, promoter regions (−4, −3.4 and −2 kb), intron 1 DHS, intron 10ab DHS, intron 23 DHS (12)). Negative and positive controls were an inactive region of chr11p13 (49), and the M6PR gene promoter, respectively. Results are n = 3 comparing a NT WT and Δ −35 kb clone #6. Error bars represent S.E.M. (B) RNAPII occupancy by ChIP-seq in 16HBE14o- (Olive green) and Caco2 (Brown) cell lines with Omni ATAC-seq and H3K27Ac ChIP-seq panels for 16HBE14o- as marked. The −44, −35 kb regions and the promoter region are highlighted in cyan and purple respectively (color scheme and UCSC browser configuration as in Figure 1).
To extend these observations we used ChIP-seq to identify genome-wide occupancy of RNAPII in both 16HBE14o- cells and the intestinal adenocarcinoma cell line Caco2 for comparison, since the latter lacks open chromatin and active histone marks at the −44 and −35 kb sites (10) (Figure 5B). The results confirmed RNAPII occupancy at the CFTR promoter and at the −35 and −44 kb sites in 16HBE14o- cells, confirming our ChIP qPCR data. In contrast, RNAPII occupancy was not seen upstream of the promoter in Caco2 cells. These results suggest that RNAPII has an important role in the promoter recruitment and utilization of the −35 and −44 kb sites in the airway cells. Though no additional novel sites of RNAPII binding were seen 5′ to the CFTR promoter in 16HBE14o- cells, a very strong peak of RNAPII was evident at an H3K4me1 enriched-site at introns 4-6 (34) (Figure 5B), which may relate to the SV40 integration site mapped close by (35) (Supplementary Figure S5).
Next we explored the possibility that the substantial recruitment of RNAPII to the −35 and the −44 kb CRE was associated with production of enhancer RNAs (eRNAs) (36). RT-qPCR was performed on total RNA from WT 16HBE14o- cells, following DNase I-treatment and reverse transcription using random hexamers. An eRNA 5′ to the lysine methyl transferase 2E (KMT2E) gene was used as a positive control for this assay due to its abundance in lung tissues (37) and parallel high levels of RNAPII occupancy (29). Although both the −44 and −35 kb regions produced low levels of non-coding (nc) RNAs, these were of low abundance compared to the KMT2E ncRNA (Supplementary Figure S6A). The asymmetric distribution of the eRNAs relative to the epigenetic features of the −44 and −35 kb CREs, as shown by H3K27Ac ChIP-seq in 16HBE14o- bronchial epithelial cells, is shown in Supplementary Figure S6B. In silico prediction algorithms found little or no coding potential associated with these low abundance eRNAs, so any functional significance remains to be determined (38).
Identification of activating transcription factors at the −35 kb CRE
The −35 kb CRE recruits both activating and repressive TFs in airway cells, though mechanistic understanding of the element is incomplete. The immune response components IRF1 (activator), IRF2 (repressor) and NF-YB were shown to modulate CFTR expression (9). Also ETS homologous factor (EHF) and Krüppel-like factor 5 (KLF5), which both repress CFTR expression, bind to the −35 kb site (39). As deletion of the −35 kb CRE nearly extinguished CFTR expression, we hypothesized that this CRE likely recruits additional critical activating factors, independently of immune challenge. Inspection of ENCODE consortium data (29), identified a peak of occupancy of CCAAT/enhancer-binding protein beta (CEBPβ) in three non-airway cell lines at core of the −35 kb CRE (9). CEBPβ is a leucine zipper domain (bZIP) containing TF that is involved in immune and inflammatory processes and interacts with the p300 coactivator. To determine if CEBPβ is recruited to the −35 kb CRE in airway cells we performed ChIP-seq with an antibody specific for this factor in Calu3 cells. Prominent peaks of CEBPβ occupancy were seen at the −35 kb site, which corresponded to a trough in H3K27Ac enrichment in Calu3 cells and at the promoter (Figure 6A, B). To determine whether recruitment of CEBPβ at the −35 kb CRE was required for its enhancer activity, we used a luciferase reporter gene assay. A 350 bp sequence encompassing the −35 kb element core was shown previously to increase CFTR promoter activity in 16HBE14o- cells by ∼40-fold (9). Mutation of the single in silico predicted CEBPβ binding site within the 350 bp enhancer fragment abolished luciferase expression (Figure 6C). This suggests that CEBPβ has a pivotal role in the enhancer activity of this site in airway epithelial cells.
Figure 6.
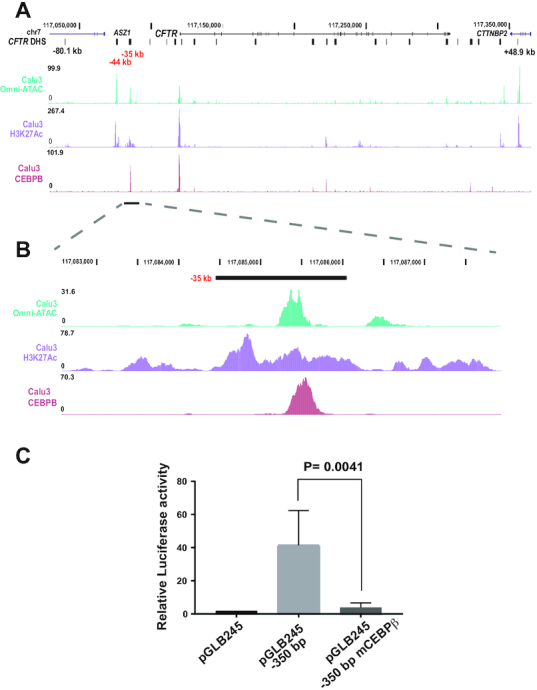
CEBPβ is recruited to the −35 kb enhancer. (A) UCSC genome browser graphic of the CFTR TAD region showing data from Calu3 cells: top, open chromatin by Omni-ATAC, middle, H3K27Ac by ChIP-seq, lower track, CEBPβ occupancy by ChIP-seq (light red). (B) Enlarged image of A) showing the −35 kb region. (C) Luciferase activity of CRE −35 kb in luciferase reporter gene assays in 16HBE14o-. Cells were transfected with pGL3B luciferase reporter constructs driven by CFTR basal promoter (pGL3B 245) alone or with the −35 kb −350 bp WT or mCEBPβ fragments at the enhancer site. pRL Renilla was the transfection control. The data are normalized to the CFTR basal promoter vector. Error bars represent S.E.M. (n = 3).
DISCUSSION
The CFTR locus has consistently provided a paradigm for the regulatory mechanisms utilized by large, multi-exon genes with complex patterns of tissue-specific regulation. These data make a valuable contribution to understanding the precise details of transcriptional control mechanisms, particularly at a time when most models of genome architecture are built upon genome wide datasets. The regulatory mechanisms for the CFTR gene depend upon the recruitment to the promoter of multiple CREs, which appear to show some cell-type selectivity (6,8,10). CREs within introns of the locus, which are particularly evident in intestinal epithelial cells have been studied most extensively to date (10,12). Here, we investigate two sites located more than 30 kb 5′ of the translational start site which are prominent in CFTR-expressing airway cells lines and in primary bronchial and tracheal epithelial cells. We focus on three aspects of these CREs that are of particular mechanistic importance: first, their role in coordinating gene expression by altering higher order chromatin structure at the CFTR locus, second their involvement in the recruitment of RNAPII and activating transcription factors to the locus, and third the fact that these CREs appear to be non-redundant.
Firstly, our 4C-seq data show that the −44 and −35 kb enhancers are in close association not only with the active CFTR promoter, but also the −80 kb TAD boundary in airway cells. The observations of promoter interactions are consistent with models whereby distal enhancers, by definition marked by the H3K27Ac and H3K4Me1 active histone marks, can promote transcription by looping onto the target gene promoter aided by the TF recruitment (reviewed in (40)). However, the enhancer association with the −80 kb TAD boundary warrants further discussion. Of interest is how the 5′ distal enhancers are physically brought close to the promoter and/or the TAD boundary, and a role for CTCF would seem plausible. We previously characterized an enhancer-blocking insulator at −20.9 kb 5′ to the promoter, which recruits both CTCF and cohesin (30). Using a 4C-seq viewpoint at this site we see that deletion of the −44 or −35 kb CREs not only diminishes interactions of −20.9 kb with the −80.1 kb TAD boundary, but also increases interactions with a 3′ CTCF-binding insulator at +6.8 kb (13) and with the 3′ TAD boundary at +48.9 kb (Supplementary Figure S2, black arrows). These results suggest that CTCF/cohesin occupancy at the −20.9 CRE may contribute to the looping of the 5′ enhancers to the CFTR promoter. Consistent with this model are CTCF ChIP-qPCR data demonstrating a significant loss of CTCF binding at the −20.9 kb element in a −35 kb 16HBE14o- deletion clone when compared to a non-targeted WT clone (Supplementary Figure S4C). However, deletion of the −20.9 kb element alone has a similar effect on locus architecture to removal of the −35 kb enhancer, with increased interactions between the 5′ TAD boundary and the +6.8 kb insulator (Supplementary Figure S4B). These results suggest that CTCF occupancy at the insulator elements may be the driving force in maintaining the required 3D conformation of the active locus. Hence, though removal of the −20.9 kb insulator element is associated with increased CFTR expression, this may not solely be due to removal of the barrier between −35 kb and the promoter. Instead it may be the result of a combination of insulator removal and the consequence of the alteration in chromatin conformation across the locus. This interpretation is consistent with our previous data in intestinal cells, where intronic rather than 5′ enhancers activate the promoter and deletion of the −20.9 kb site also alters chromatin structure at the locus. However, in intestinal cells a slight increase in CFTR expression upon deletion of the −20.9 kb site did not reach statistical significance (10), but siRNA-mediated depletion of CTCF caused a significant increase in expression of the gene (26). Presumably this is due to increased accessibility to activating transcription factors upon relaxing of the locus 3D structure.
Secondly, considering the recruitment of RNAPII to the 5′ CREs: using ChIP-seq and ChIP-qPCR we found RNAPII occupancy at both sites, with very high enrichment at the −35 kb enhancer. Moreover, the RNAPII peaks showed a bimodal distribution as is seen at promoters genome-wide, where one peak likely initiates at the active TSS and the second represents paused polymerases (41). Without additional experiments it is not possible to determine whether enhancer bound RNAPII initiates looping to the promoter or if RNAPII is recruited to the 5′ enhancers and the promoter independent of looping interactions. Long-range Lim Domain Binding 1 (LDB1) - mediated enhancer looping at the β-globin locus in erythroid cells was shown to be independent of RNAPII pre-initiation complex (PIC) formation, mediator or cohesin (42). Hence, PIC formation is not a pre-requisite for looping and specific TFs may orchestrate looping in the absence of RNAPII. Either way, it is likely that the mechanism of promoter looping of the −44 and −35 kb enhancers is in part dependent on RNAPII which connects these CREs to CTCF foci to facilitate coordinated, cell-type-selective transcription (43). Recent data also suggest that the Mediator complex may act as a functional (non-architectural) bridge between promoters and their enhancers (44).
Our earlier work on the −44 and −35 kb CREs identified a very limited repertoire of transcription factors involved in oxidative stress (−44 kb) and in the immune response (−35 kb) that were recruited to the sites (9,11). Here, we extended our findings by mining ChIP-seq data from the ENCODE consortium (29). Unfortunately, these data include very few cell lines that express CFTR, however we observed CEBPβ occupancy at the −35 kb site and RELA Proto-Oncogene, NF-κB Subunit (RELA), Activating transcription factor 2 (ATF2) and p300 at the −44 kb site. Here, we show direct binding of CEBPβ at the −35 kb site in Calu3 cells and demonstrate that disruption of the CEBPβ binding site abolishes enhancer activity of the −35 kb core in luciferase reporter gene assays. Of note we also see robust occupancy of CEBPβ at the CFTR promoter. Earlier work by others documented the role of CEBP family members (CEBPδ) in promoter-driven CFTR transcription through an inverted CCAAT element (45). More recently promoter bound, phosphorylated CEBPβ was also reported (46). Hence, it is probable that CEBPβ and RNAPII may be recruited to the upstream enhancers and the gene promoter both independently and coordinately.
Thirdly, deletion of the −44 and −35 kb enhancers individually by CRISPR/Cas9 in 16HBE14o- cells abrogates CFTR transcription suggesting that these elements are not redundant, but rather have cooperative activity that is essential for gene expression (11). These results are of particular interest in the context of enhancers for many developmental genes and gene clusters showing substantial redundancy (reviewed in (47)). For example, studies in which genome editing was used to create single and combinatorial enhancer deletions at seven loci required for mouse limb development (48) clearly showed that loss of single limb enhancers had no impact on limb morphology, while deletion of pairs of enhancers for the same gene generated clear phenotypes. Moreover, when gene expression levels were reduced at single target genes, subsequent loss of individual enhancers did impair limb development. Our observations on CFTR suggest that for a locus that is not apparently involved in a complex network of development events there may be no redundancy in cell-type specific enhancers. However, our previous work (10) supported by more recent observations presented here, demonstrate that substantial redundancy exists at sites of architectural protein occupancy at the CFTR locus. Thus loss of a key CTCF binding site either within the locus (10) or at the TAD boundary is compensated for by recruitment of adjacent structural elements to maintain normal gene expression levels.
DATA AVAILABILITY
Genome-wide data are deposited at GEO: GSE132808 and GSE140458.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr P. Faber and staff at the University of Chicago Genomics Core for all deep sequencing.
Author contributions: M.N. and A.H. conceived and designed the project, M.N., S.Y., J.L.K., H.S., A.G. and S.-H.L. designed and performed experiments, M.N. and A.H. wrote the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R01 HL094585, HL117843 to A.H., T32 GM008056 to A.P. and H.S., F31 HL146010 to H.S.]; Cystic Fibrosis Foundation [Harris 16G0, 15/17XX0, 18P0, 19G0]. Funding for open access charge: Institutional Funds.
Conflict of interest statement. The authors declare no conflict of interest. The funders had no role in the design of the study, in the collection, analyses, or interpretation of the data, in the writing of the manuscript, or in the decision to publish the results.
REFERENCES
- 1. Splinter E., de Wit E., van de Werken H.J., Klous P., de Laat W.. Determining long-range chromatin interactions for selected genomic sites using 4C-seq technology: from fixation to computation. Methods. 2012; 58:221–230. [DOI] [PubMed] [Google Scholar]
- 2. Miele A., Dekker J.. Mapping cis- and trans-chromatin interaction networks using chromosome conformation capture (3C). Methods Mol. Biol. 2009; 464:105–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Belton J.M., McCord R.P., Gibcus J.H., Naumova N., Zhan Y., Dekker J.. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods. 2012; 58:268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu Z., Tjian R.. Visualizing transcription factor dynamics in living cells. J. Cell Biol. 2018; 217:1181–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Swahn H., Harris A.. Cell-selective regulation of CFTR gene expression: relevance to gene editing therapeutics. Genes. 2019; 10:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gosalia N., Harris A.. Chromatin dynamics in the regulation of CFTR expression. Genes. 2015; 6:543–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gillen A.E., Harris A.. The role of CTCF in coordinating the expression of single gene loci. Biochem. Cell Biol. 2011; 89:489–494. [DOI] [PubMed] [Google Scholar]
- 8. Zhang Z., Ott C.J., Lewandowska M.A., Leir S.H., Harris A.. Molecular mechanisms controlling CFTR gene expression in the airway. J. Cell Mol. Med. 2012; 16:1321–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang Z., Leir S.H., Harris A.. Immune mediators regulate CFTR expression through a bifunctional airway-selective enhancer. Mol. Cell Biol. 2013; 33:2843–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang R., Kerschner J.L., Gosalia N., Neems D., Gorsic L.K., Safi A., Crawford G.E., Kosak S.T., Leir S.H., Harris A.. Differential contribution of cis-regulatory elements to higher order chromatin structure and expression of the CFTR locus. Nucleic Acids Res. 2016; 44:3082–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Z., Leir S.H., Harris A.. Oxidative stress regulates CFTR gene expression in human airway epithelial cells through a distal antioxidant response element. Am. J. Respir. Cell Mol. Biol. 2015; 52:387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ott C.J., Blackledge N.P., Kerschner J.L., Leir S.H., Crawford G.E., Cotton C.U., Harris A.. Intronic enhancers coordinate epithelial-specific looping of the active CFTR locus. PNAS. 2009; 106:19934–19939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Blackledge N.P., Ott C.J., Gillen A.E., Harris A.. An insulator element 3′ to the CFTR gene binds CTCF and reveals an active chromatin hub in primary cells. Nucleic Acids Res. 2009; 37:1086–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ong C.T., Corces V.G.. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat. Rev. Genet. 2011; 12:283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Plasschaert L.W., Zilionis R., Choo-Wing R., Savova V., Knehr J., Roma G., Klein A.M., Jaffe A.B.. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature. 2018; 560:377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Montoro D.T., Haber A.L., Biton M., Vinarsky V., Lin B., Birket S.E., Yuan F., Chen S., Leung H.M., Villoria J. et al.. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature. 2018; 560:319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Broackes-Carter F.C., Mouchel N., Gill D., Hyde S., Bassett J., Harris A.. Temporal regulation of CFTR expression during ovine lung development: implications for CF gene therapy. Hum. Mol. Genet. 2002; 11:125–131. [DOI] [PubMed] [Google Scholar]
- 18. Trezise A.E., Chambers J.A., Wardle C.J., Gould S., Harris A.. Expression of the cystic fibrosis gene in human foetal tissues. Hum. Mol. Genet. 1993; 2:213–218. [DOI] [PubMed] [Google Scholar]
- 19. Crawford I., Maloney P.C., Zeitlin P.L., Guggino W.B., Hyde S.C., Turley H., Gatter K.C., Harris A., Higgins C.F.. Immunocytochemical localization of the cystic fibrosis gene product CFTR. PNAS. 1991; 88:9262–9266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gillen A.E., Yang R., Cotton C.U., Perez A., Randell S.H., Leir S.H., Harris A.. Molecular characterization of gene regulatory networks in primary human tracheal and bronchial epithelial cells. J. Cyst. Fibros. 2018; 17:444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bischof J.M., Ott C.J., Leir S.H., Gosalia N., Song L., London D., Furey T.S., Cotton C.U., Crawford G.E., Harris A.. A genome-wide analysis of open chromatin in human tracheal epithelial cells reveals novel candidate regulatory elements for lung function. Thorax. 2012; 67:385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cozens A.L., Yezzi M.J., Kunzelmann K., Ohrui T., Chin L., Eng K., Finkbeiner W.E., Widdicombe J.H., Gruenert D.C.. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 1994; 10:38–47. [DOI] [PubMed] [Google Scholar]
- 23. Fogh J., Wright W.C., Loveless J.D.. Absence of HeLa cell contamination in 169 cell lines derived from human tumors. J. Natl. Cancer Inst. 1977; 58:209–214. [DOI] [PubMed] [Google Scholar]
- 24. Shen B.Q., Finkbeiner W.E., Wine J.J., Mrsny R.J., Widdicombe J.H.. Calu-3: a human airway epithelial cell line that shows cAMP-dependent Cl- secretion. Am. J. Physiol. 1994; 266:L493–L501. [DOI] [PubMed] [Google Scholar]
- 25. Mouchel N., Henstra S.A., McCarthy V.A., Williams S.H., Phylactides M., Harris A.. HNF1alpha is involved in tissue-specific regulation of CFTR gene expression. Biochem. J. 2004; 378:909–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gosalia N., Neems D., Kerschner J.L., Kosak S.T., Harris A.. Architectural proteins CTCF and cohesin have distinct roles in modulating the higher order structure and expression of the CFTR locus. Nucleic Acids Res. 2014; 42:9612–9622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Corces M.R., Trevino A.E., Hamilton E.G., Greenside P.G., Sinnott-Armstrong N.A., Vesuna S., Satpathy A.T., Rubin A.J., Montine K.S., Wu B. et al.. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods. 2017; 14:959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van de Werken H.J., Landan G., Holwerda S.J., Hoichman M., Klous P., Chachik R., Splinter E., Valdes-Quezada C., Oz Y., Bouwman B.A. et al.. Robust 4C-seq data analysis to screen for regulatory DNA interactions. Nat. Methods. 2012; 9:969–972. [DOI] [PubMed] [Google Scholar]
- 29. Dunham I., Kundaje A., Aldred S.F., Collins P.J., Davis C.A., Doyle F., Epstein C.B., Frietze S., Harrow J., Kaul R. et al.. The ENCODE project consortium: an integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blackledge N.P., Carter E.J., Evans J.R., Lawson V., Rowntree R.K., Harris A.. CTCF mediates insulator function at the CFTR locus. Biochem. J. 2007; 408:267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chung J.H., Whiteley M., Felsenfeld G.. A 5′ element of the chicken beta-globin domain serves as an insulator in human erythroid cells and protects against position effect in Drosophila. Cell. 1993; 74:505–514. [DOI] [PubMed] [Google Scholar]
- 32. Stolzenburg L.R., Wachtel S., Dang H., Harris A.. miR-1343 attenuates pathways of fibrosis by targeting the TGF-beta receptors. Biochem. J. 2016; 473:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Smith A.N., Barth M.L., McDowell T.L., Moulin D.S., Nuthall H.N., Hollingsworth M.A., Harris A.. A regulatory element in intron 1 of the cystic fibrosis transmembrane conductance regulator gene. J. Biol. Chem. 1996; 271:9947–9954. [DOI] [PubMed] [Google Scholar]
- 34. Wang A., Yue F., Li Y., Xie R., Harper T., Patel N.A., Muth K., Palmer J., Qiu Y., Wang J. et al.. Epigenetic priming of enhancers predicts developmental competence of hESC-derived endodermal lineage intermediates. Cell Stem Cell. 2015; 16:386–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Valley H.C., Bukis K.M., Bell A., Cheng Y., Wong E., Jordan N.J., Allaire N.E., Sivachenko A., Liang F., Bihler H. et al.. Isogenic cell models of cystic fibrosis-causing variants in natively expressing pulmonary epithelial cells. J. Cyst. Fibros. 2018; 18:476–483. [DOI] [PubMed] [Google Scholar]
- 36. De Santa F., Barozzi I., Mietton F., Ghisletti S., Polletti S., Tusi B.K., Muller H., Ragoussis J., Wei C.L., Natoli G.. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 2010; 8:e1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ren C., Liu F., Ouyang Z., An G., Zhao C., Shuai J., Cai S., Bo X., Shu W.. Functional annotation of structural ncRNAs within enhancer RNAs in the human genome: implications for human disease. Sci. Rep. 2017; 7:15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang L., Park H.J., Dasari S., Wang S., Kocher J.P., Li W.. CPAT: coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013; 41:e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mutolo M.J., Leir S.H., Fossum S.L., Browne J.A., Harris A.. A transcription factor network represses CFTR gene expression in airway epithelial cells. Biochem. J. 2018; 475:1323–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Meng H., Bartholomew B.. Emerging roles of transcriptional enhancers in chromatin looping and promoter-proximal pausing of RNA polymerase II. J. Biol. Chem. 2018; 293:13786–13794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Quinodoz M., Gobet C., Naef F., Gustafson K.B.. Characteristic bimodal profiles of RNA polymerase II at thousands of active mammalian promoters. Genome Biol. 2014; 15:R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Krivega I., Dean A.. LDB1-mediated enhancer looping can be established independent of mediator and cohesin. Nucleic Acids Res. 2017; 45:8255–8268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tang Z., Luo O.J., Li X., Zheng M., Zhu J.J., Szalaj P., Trzaskoma P., Magalska A., Wlodarczyk J., Ruszczycki B. et al.. CTCF-Mediated human 3D genome architecture reveals chromatin topology for transcription. Cell. 2015; 163:1611–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. El Khattabi L., Zhao H., Kalchschmidt J., Young N., Jung S., Van Blerkom P., Kieffer-Kwon P., Kieffer-Kwon K.R., Park S., Wang X. et al.. A pliable mediator acts as a functional rather than an architectural bridge between promoters and enhancers. Cell. 2019; 178:1145–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pittman N., Shue G., LeLeiko N.S., Walsh M.J.. Transcription of cystic fibrosis transmembrane conductance regulator requires a CCAAT-like element for both basal and cAMP-mediated regulation. J. Biol. Chem. 1995; 270:28848–28857. [DOI] [PubMed] [Google Scholar]
- 46. Viart V., Varilh J., Lopez E., Rene C., Claustres M., Taulan-Cadars M.. Phosphorylated C/EBPbeta influences a complex network involving YY1 and USF2 in lung epithelial cells. PLoS One. 2013; 8:e60211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Robson M.I., Ringel A.R., Mundlos S.. Regulatory landscaping: how enhancer-promoter communication is sculpted in 3D. Mol. Cell. 2019; 74:1110–1122. [DOI] [PubMed] [Google Scholar]
- 48. Osterwalder M., Barozzi I., Tissieres V., Fukuda-Yuzawa Y., Mannion B.J., Afzal S.Y., Lee E.A., Zhu Y., Plajzer-Frick I., Pickle C.S. et al.. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature. 2018; 554:239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stolzenburg L.R., Yang R., Kerschner J.L., Fossum S., Xu M., Hoffmann A., Lamar K.M., Ghosh S., Wachtel S., Leir S.H. et al.. Regulatory dynamics of 11p13 suggest a role for EHF in modifying CF lung disease severity. Nucleic Acids Res. 2017; 45:8773–8784. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genome-wide data are deposited at GEO: GSE132808 and GSE140458.