

INTRODUCTION:
Epigenetic modifications have been implicated to mediate several complications of diabetes mellitus (DM), especially nephropathy and retinopathy. Our aim was to ascertain whether epigenetic alterations in whole blood discriminate among patients with DM with normal, delayed, and rapid gastric emptying (GE).
METHODS:
Using the ChIP-seq (chromatin immunoprecipitation combined with next-generation sequencing) assays, we compared the genome-wide enrichment of 3 histone modifications (i.e., H3K4me3, H3K9ac, and H3K27ac) in buffy coats from 20 diabetic patients with gastrointestinal symptoms and normal (n = 6), delayed (n = 8), or rapid (n = 6) GE.
RESULTS:
Between patients with DM with delayed vs normal GE, there were 108 and 54 genes that were differentially bound (false discovery rate < 0.05) with H3K27ac and H3K9ac, respectively; 100 genes were differentially bound with H3K9ac in patients with rapid vs normal GE. The differentially bound genes with H3K27ac were functionally linked to the type 2 immune response, particularly Th2 cell activation and function (e.g., CCR3, CRLF2, CXCR4, IL5RA, and IL1RL1) and glucose homeostasis (FBP-1, PDE4A, and CMKLR1). For H3K9ac, the differentially occupied genes were related to T-cell development and function (e.g., ICOS and CCR3) and innate immunity (RELB, CD300LB, and CLEC2D). Compared with normal GE, rapid GE had differential H3K9ac peaks at the promoter site of diverse immunity-related genes (e.g., TNFRSF25 and CXCR4) and genes related to insulin resistance and glucose metabolism. Motif analysis disclosed enrichment of binding sites for transcription factors relevant to the pathogenesis and complications of DM.
DISCUSSION:
GE disturbances in DM are associated with epigenetic alterations that pertain to dysimmunity, glucose metabolism, and other complications of DM.
INTRODUCTION
Up to 50% of patients with moderately controlled type 1 and type 2 diabetes mellitus (DM) have delayed gastric emptying (GE) (1–4). Full-thickness gastric biopsies reveal loss and damage to the interstitial cells of Cajal (ICC), loss of neurons and nerve fibers, and an immune infiltrate characterized predominantly by CD68 cells, a marker for macrophages, in patients with diabetic gastroparesis (5). Likewise, in streptozotocin-induced DM, classically activated macrophages produce cytokines, which damage the ICC and delay GE (6). Except for an association between longer poly-GT repeats in the heme oxygenase-1 gene and nausea, there are no known genes, loci, or single-nucleotide polymorphisms associated with diabetic gastroparesis (7).
In the Diabetes Control and Complications Trial (DCCT), intensive insulin therapy reduced the risk and rate of progression of complications (i.e., nephropathy, retinopathy, and neuropathy) vs conventional insulin regimens in patients with type 1 DM (8). Nearly 30 years later, these benefits persist despite the loss of glycemic separation over time between the intensive and conventional groups (9–11). This metabolic memory is at least partly explained by epigenetic changes that are induced by the metabolic imbalance in DM, alter the expression of genes without affecting the DNA sequence, and are evident in the blood and peripheral blood mononuclear cells (12–16). These epigenetic changes include increased acetylation of the lysine 9 residue on histone H3 protein (i.e., H3K9ac) of the promoter regions of several genes in monocytes, which causes active transcription (14) of several proinflammatory genes, including the nuclear factor kB inflammatory pathway. Other in vivo, ex vivo, and in vitro studies also demonstrate that DM complications are associated with alterations in histone post-translational modifications not only in target organs but also in immune cells (17–22). However, the epigenome in diabetic gastroenteropathy (DGE) has not been evaluated.
Among 74 randomly selected patients in the EDIC cohort, 50% had normal GE, 48% had delayed GE, and 2% had rapid GE (2). Delayed GE was associated with early and prolonged hyperglycemia before the DCCT began and, to a lesser extent, with the glycated hemoglobin (HbA1c) averaged over the DCCT and EDIC. Intrigued by the possibility that epigenetic mechanisms might explain GE disturbances, this study compared the epigenome among patients with DGE with normal, delayed, and rapid GE.
METHODS
Participants
These studies were approved by the Mayo Clinic Institutional Review Board. Blood samples for epigenetic analyses were collected in 29 patients with DGE who were undergoing a clinically indicated assessment of GE and had consented to participate in research designed to evaluate the relationship between GE and upper gastrointestinal symptoms. Twenty-one patients were recruited from a completed study (23) and 8 patients were recruited from an ongoing study. Of these 29 patients, epigenetic analyses were successfully completed in 20 patients with DM and upper gastrointestinal symptoms (8 women: mean [SD], age 47 [17] years and body mass index 30 [7] kg/m2) who are included in this report. The major exclusion criteria were severe nausea or vomiting, which may affect the ability of participants to complete the GE study, medications that affect gastric motility (e.g., narcotics, glucagon-like peptide-1 [GLP-1] agonists, or prokinetic agents), severe systemic diseases that may interfere with the study, previous gastric, major intestinal, or colonic surgery, or previous abdominal radiotherapy.
Gastrointestinal symptoms
Gastrointestinal symptoms were evaluated with the Rome III symptom criteria (24) and the Patient Assessment of Upper Gastrointestinal Disorders—Symptom Severity (PAGI-SYM) questionnaire of symptoms over the past 2 weeks (25).
Gastric emptying
GE of solids was evaluated with scintigraphy (i.e., 296 kcal meal) (23). Normal ranges were defined based on the 95th and fifth percentile values in men (1 hour: 4.7%–40%, 2 hours: 28.4%–82%, and 4 hours: 77%–100%) and women (1 hour: 4.3%–31.4%, 2 hours: 25%–71%, and 4 hours: 76.2%–100%). GE lower than the fifth percentile values at 2 or 4 hours and higher than the 95th percentile values at 1 or 2 hours was considered to be delayed and rapid, respectively (26).
Chromatin immunoprecipitation experiments
Buffy coat samples obtained from the participants were preserved in an antifreezing buffer (90% fetal bovine serum and 10% dimethyl sulfoxide) at −80 °C in a freezer. The samples were thawed and cross-linked with 1% formaldehyde, followed by quenching with 125 mM glycine at room temperature. Fixed cells were subjected to chromatin preparation, immunoprecipitation, and library preparation as described (27). The following antibodies were used in the experiment: anti-H3K4me3 (Epigenomics Development Laboratory [EDL], Lot1), anti-H3K9ac (EDL, Lot1), and anti-H3K27ac (CST, Cat. No. 8173, Lot1). The libraries were sequenced to 51 base pairs from both ends on an Illumina HiSeq 2000 or 4000 instrument in the Mayo Clinic Center for Individualized Medicine Medical Genomics Facility. Details of the ChIP-seq experiment are described in Supplementary Digital Content 1, http://links.lww.com/CTG/A201.
Bioinformatics
The ChIP-seq data were analyzed with the HiChIP pipeline (28). Briefly, the paired-end reads were mapped to the hg19 genome reference with the Burrows–Wheeler Alignment tool (29); peaks were identified with the Model-based Analysis of ChIP-Seq (MACS2) software package at an false discovery rate (FDR) ≤1% and fold change (FC) ≥2 over the input (30), and differential binding analysis was performed using the DiffBind R package (31). Supplementary Digital Content 1, http://links.lww.com/CTG/A201, describes the analysis in more detail.
Pathway analysis
Because H3K9ac preferentially marks active gene promoters, genes whose transcription start sites are within ±2.5 kbp of differentially enriched peaks (FDR < 0.05) were used for the pathway analysis using the Ingenuity Pathway Analysis software. The active enhancers, marked by H3K27ac, can affect the gene transcription independent of their orientation or distance (32). Hence, genes closest to all differentially enriched sites (FDR < 0.05) for H3K27ac were considered for the pathway analysis using the Ingenuity Pathway Analysis.
Motif analysis
The MEME suite (version 5.0.5) software was used for motif analysis. Enrichment of known transcription factor (TF) motifs (described in the motif database “JASPAR CORE [2018] vertebrates”) in the differentially bound sequences was assessed by analysis of motif enrichment (33) using default parameters to identify potentially relevant TFs.
Statistical analysis
The comparison of epidemiological and clinical features among the study groups was performed with the Kruskal–Wallis rank sum test for continuous variables and the Fisher exact test for categorical variables. The statistical analyses were performed with JMP Pro 14.
RESULTS
Clinical features
Of 20 patients, 8 had type 1 DM (Table 1). The mean duration of DM was 17 ± 13 years (mean ± SD), and the mean glycated HbA1c was 8.1 ± 1.7%, which suggests moderately controlled glycemia. Twelve patients (60%) had microvascular complications, including peripheral neuropathy (11 patients), retinopathy (5 patients), and nephropathy (8 patients). One patient also had cardiovagal, adrenergic, and postganglionic sudomotor failure. Fifteen of 20 patients with DM were treated with insulin alone, 4 were treated with oral hypoglycemic agents, and 1 patient was treated with insulin and metformin. Six patients had normal GE, 8 had delayed GE, and 6 had rapid GE. GE was associated with age (P = 0.02) (Table 1). The other features were not significantly associated with the type of GE.
Table 1.
Demographic and clinical features of patients with diabetes mellitus (N = 20)

Among 19 patients who completed the ROME III questionnaire, 16 patients had ROME III criteria for upper gastrointestinal (GI) symptoms (i.e., functional dyspepsia in 12 patients, nausea/vomiting and/or rumination in 3 patients, and functional abdominal pain in 1 patient). Three patients had upper GI symptoms but did not satisfy the Rome III criteria. Of these, 2 had a score of 2 or greater, suggestive of moderate symptoms or worse on at least one subscale of the PAGI-SYM questionnaire, and 1 had milder symptoms (i.e., highest score: 1–1.99). Of the 19 patients who had an upper endoscopy, 10 had a normal study. The remaining patients had esophagitis and/or gastritis (6 patients), benign gastric polyps (2 patient), and reactive gastropathy (1 patient).
ChIP-seq analysis
In 19, 16, and 17 patients, the data for genome-wide profiling of H3K27ac, H3K9ac, and H3K4me3, respectively, matched or exceeded quality control thresholds (Supplementary Digital Content 2, http://links.lww.com/CTG/A202, Table 1). The genome-wide distribution across genomic regions (promoter regions were defined by HOMER as −1 kb to +100 bp from transcription start sites) and binding profiles for individual histone marks in different GE phenotypes are shown in Supplementary Digital Contents 3–5, http://links.lww.com/CTG/A203, http://links.lww.com/CTG/A204, and http://links.lww.com/CTG/A205. For each mark, the genome-wide patterns of distribution of these histone marks were similar to the previous studies in human and other mammalian cells (34–36) and among the GE groups in this study. For example, for H3K9ac, 26% of peaks in patients with delayed GE and 28% each in normal and rapid GE were located at the promoter region of the genome.
Alteration of H3K27ac marks
There were 140 H3K27ac differentially bound sites between DM patients with normal and delayed GE. The unbiased hierarchical clustering based on the binding affinity at these sites segregated between these groups (Figure 1, Supplementary Digital Content 6, http://links.lww.com/CTG/A206). These differential peaks were located at 108 genes of which 95 genes had higher and 13 had lower binding with H3K27ac in patients with delayed GE compared with normal GE. The pathway analysis inferred that these genes were related to activation and function of T helper cells and degradation of sorbitol (Table 2). The differentially occupied genes include chemokines (i.e., CXCR4 and CCR3 [log2FC = 2.7]), cytokines and their receptors (i.e., IL-34 [3.7], IL5RA [3.5], and IL1RL1 [3.3]), other inflammatory genes (i.e., PDE4A [2.8], TLR5 [−2.5], PTGDR2 [3.4], PRDM1 [3.0], and CRLF2 [2.8]), and genes related to glucose homeostasis (i.e., FBP1 [2.3], PDE4A [2.8], SORD [2.7], and CMKLR1 [2.2]) (Figure 2 and Table 3). By contrast, the comparison of H3K27ac peaks between patients with rapid and normal GE only identified 9 differentially bound sites, with lower binding in 8 sites, among patients with rapid GE than normal GE (Supplementary Digital Content 7, http://links.lww.com/CTG/A207).
Figure 1.

Differential binding of H3K27ac between DM patients with delayed and normal GE. The affinity heat map (a) shows 140 individual sites (rows) that are differentially bound with H3K27ac between patients with normal and delayed GE. The inset shows the color scale for the binding affinity. Observe that the samples from patients with DM with delayed and normal GE are clustered separately. In the volcano plot (b), only a small fraction of all peaks are differentially bound with H3K27ac between patients with DM with delayed and normal GE. Red and green symbols represent significant differentially bound peaks (FDR < 0.05) in which the log2 (Fold change) was negative and positive in patients with delayed vs normal GE, respectively. Gray symbols represent nonsignificant differential peaks. DM, diabetes mellitus; FDR, false discovery rate; GE, gastric emptying.
Table 2.
Ingenuity pathway analysis for the differentially bound genes between GE categories
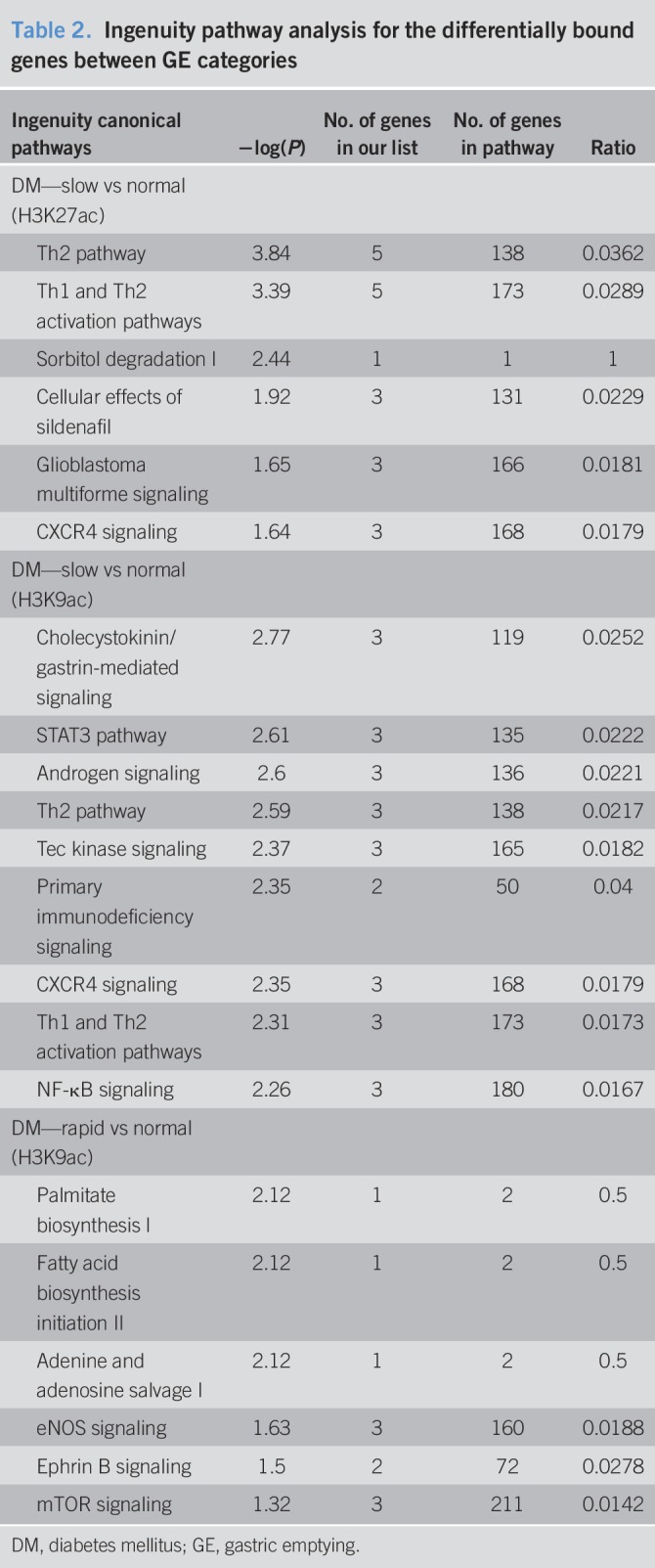
Figure 2.

A comparison of H3K27ac binding at selected genes between patients with DM with delayed (red) and normal (blue) gastric emptying. The histograms, marked with an *, show the magnitude of binding. The horizontal bars, marked with #, denote significant peaks detected by the MACS2 algorithm. For several inflammatory genes, H3K27ac peaks were greater in patients with DM with delayed GE than normal GE—CXCR4 (data range: 0–82), CCR3 (0–50), IL34 (0–10), IL1RL1 (0–10), PDE4A (0–20), PTGDR2 (0–20), PRDM1 (0–50), CRLF2 (0–30), FBP1 (0–40), and CMKLR1 (0–40). DM, diabetes mellitus; GE, gastric emptying.
Table 3.
Functionally important genes with differential binding with specific histone marks between GE categories

Alteration of H3K9ac marks
Compared with patients with normal GE, delayed GE was associated with differential H3K9ac binding at 286 sites. Of these, 54 were located at the promoters of known genes; 32 and 22 genes, respectively, had higher and lower H3K9ac binding in patients with delayed GE (Figure 3, Supplementary Digital Content 8, http://links.lww.com/CTG/A208). The pathway analysis implicated intracellular signaling by cholecystokinin/gastrin, STAT3, androgens, Tec kinase, and inflammatory processes (Th2 activation, CXCR4 signaling, and NF-κB pathway, Table 2). Compared to patients with normal GE, patients with delayed GE had increased H3K9ac binding at several genes, including cytokine and chemokine receptors (IL5RA [2.5], IL7R [1.5], and CCR3 [2.4]), RELB [2.7], the NF-κB subunit, genes involved in T-cell signaling (ICOS [2.1] and PRKCQ [1.6]), and other inflammatory genes (RGS1 [1.6], CLEC2D [2.0], CD300LB [1.6], and JAML [2.5]) (Figure 3, Table 3). There were 639 differentially bound peaks with H3K9ac between patients with rapid and normal GE. Of these, 100 peaks were located at the promoters of known genes; 55 had higher and 45 had lower binding with H3K9ac in patients with rapid GE (Figure 4, Supplementary Digital Content 9, http://links.lww.com/CTG/A209). The differentially occupied genes are implicated in fatty acid and nucleotide metabolism and signaling by eNOS and mTOR (Table 2). Potentially relevant genes include immune-related genes (CXCR4 [1.7], RGS1 [1.9], TNFRSF25 [−1.3], MNDA [2.0], ALOX5AP [1.1], PELI3 [−1.6], and CD55 [1.4]), PEIZO1 (−1.5), the mechanosensitive ion channel, and FASN (−1.3), which catalyzes the synthesis of long-chain fatty acids (Figure 4, Table 3).
Figure 3.

Differential binding of H3K9ac between DM patients with delayed and normal GE. The affinity heat map (a) shows 286 individual sites (rows) that are differentially bound with H3K9ac between patients with normal and rapid GE. The inset shows the color scale for the binding affinity. Observe that the samples from patients with DM with delayed and normal GE are clustered separately. In the volcano plot (b), only a small fraction of all peaks are differentially bound with H3K27ac between patients with DM with delayed and normal GE. Red, green, and gray symbols are as used in Figure 1. (c) A comparison of H3K9ac binding at inflammatory genes between patients with DM with delayed (red) and normal (blue) gastric emptying—IL7R (data range: 0–20), RGS1 (0–20), CD300LB (0–30), CLEC2D (0–10), and JAML (0–30). DM, diabetes mellitus; FDR, false discovery rate; GE, gastric emptying.
Figure 4.

Differential binding of H3K9ac between DM patients with rapid and normal GE. The affinity heat map (a) shows 639 individual sites (rows) that are differentially bound with H3K9ac between patients with normal and rapid GE. The inset shows the color scale for the binding affinity. Observe that the samples from patients with DM with rapid and normal GE are clustered separately. In the volcano plot (b), only a small fraction of all peaks are differentially bound with H3K9ac between patients with DM with rapid and normal GE. Red, green, and gray symbols are as used in Figure 1. (c) A comparison of H3K9ac binding at selected inflammatory genes between patients with DM with rapid (green) and normal (blue) gastric emptying: RGS1 (data range: 0–10), TNFRSF25 (0–10), PELI3 (0–50), PIEZO1 (0–50), and FASN (0–40). DM, diabetes mellitus; GE, gastric emptying.
Alteration of H3K4me3 marks
Compared with patients with normal GE, patients with rapid GE had higher and lower binding with H3K4me3, at the promoter site, in 6 and 4 genes, respectively (log2FC ≥ |1.5|) (Supplementary Digital Content 10, http://links.lww.com/CTG/A210). There were no differentially bound sites for H3K4me3 between patients with delayed and normal GE.
Motif analysis
Supplementary Digital Content 11, http://links.lww.com/CTG/A211, enlists the significantly enriched motifs in the differentially bound sites for all the 3 comparisons. This includes the binding sites for TFs belonging to the myocyte enhancer factor-2 (MEF2) family (MEF2A, 2B, 2C, and 2D), the forkhead box (FOX) family (FOXC2 and D3), interferon signaling (STAT1–STAT2 and IRF1), Kruppel-like factor (KLF9), specificity proteins (SP1 and SP2), zinc finger proteins (ZNF384 and ZNF263), and other TFs RARG and RREB1, which were enriched in the differential H3K27ac and H3K9ac peaks in delayed GE compared with normal GE. Additional TFs enriched in the differentially bound sites for H3K9ac between patients with rapid vs normal GE include other FOX family members, KLFs, E26 transformation-specific family factors (ETV6, SPIC, and SPIB), and USF1.
DISCUSSION
This study suggests the new paradigm that histone modifications, which are associated with active transcription, discriminate between patients with DM with normal and delayed GE and between normal and rapid GE. These epigenetic marks may offer insights into the pathogenesis of abnormal GE in DM. By comparison, the diabetic phenotype and the specific upper GI symptoms were less useful for discriminating between patients with DM with rapid and separately delayed GE from normal GE in this and previous studies (37).
The differentially enriched genes are broadly associated with immune cell activation and hematopoiesis, cytokine signaling, glucose homeostasis, and neuronal function. For 95 of 108 differentially bound genes with H3K27ac, there was higher binding in patients with delayed GE than normal GE. H3K27ac is a robust mark of active enhancers and promoters and is strongly correlated with binding of transcription factors and gene expression (32). These genes include CXCR4, which is a key molecule in chemotaxis of lymphocytes, T-cell proliferation, and B-cell differentiation, IL-34, which promotes differentiation and viability of monocytes and macrophages, IL-5R and CCR3, which are found on eosinophils and basophils and activate eosinophils, IL1RL1, the receptor for IL33, which activates the MyD88/NF-κB signaling pathway to stimulate mast cells, Th2 and Treg cells, and type 2 innate lymphoid cells (38), PTGDR2 (aka CRTH2), the receptor for prostaglandin D2, which mediates chemotaxis of Th2 cells, eosinophils, and basophils (39), CRLF2, the receptor for thymic stromal lymphopoietin (TSLP), which regulates differentiation of hematopoietic cells (40) and drives the development of inflammatory Th2 cells (41), PRDM1 (aka BLIMP1), which plays a role in the development, retention, and long-term establishment of tissue-resident T-lymphocyte cells in nonlymphoid organs, such as the skin and gut (42), and TLR5, a pattern recognition receptor that activates innate immune response through the NF-κB pathway. Several of these genes (i.e., CCR3, CRLF2, CXCR4, IL5RA, IL1RL1, and PTGDR2) are related to the type 2 immune response, specifically Th2 cell activation and function.
The binding of H3K9ac at the promoter sites of several inflammation-related genes was also higher in patients with delayed GE than normal GE. In addition to IL5RA and CCR3, for which H3K27ac binding was higher, the list includes IL7R, RELB, ICOS, PRKCQ, CLEC2D, CD300LB, and JAML. IL7R mediates the effects of TSLP by forming heterodimers with CRLF2, which also had higher H3K27ac binding in delayed GE. IL7, the other cytokine that binds to IL7R, promotes T- and B-cell development and maintains intraepithelial lymphocytes in the gut (43). Some of these genes are related to T-cell activation, i.e., ICOS, which is upregulated by T-cell activation and facilitates differentiation of multiple T-cell lineages (44), PRKCQ, which links the TCR signaling complex to TFs that mediate T-cell activation (45), and JAML, which is a vital costimulatory receptor for epithelial γδ T cells (46). Other functionally relevant genes include RELB, a member of the Rel/NF-κB family of TFs, RGS1, which regulates T- and B-cell chemokine signaling and is implicated in the pathogenesis of autoimmunity (47), CLEC2D, which induces IFN-gamma secretion from NK cells (48), and CD300LB, which promotes myeloid cell efferocytosis (49).
These findings are generally consistent with the concept that immune-mediated damage to the neuromuscular apparatus may at least partly mediate delayed GE in animal models and patients with diabetic gastroparesis (5,6,50,51). IL-34 is of particular interest because infiltration of monocytes/macrophages in the gastric myenteric plexus has been implicated to damage the ICC and delay GE in a model (i.e., nonobese diabetic [NOD] mice) of type 1 DM (6).
Additional genes that are enriched with H3K27ac in patients with delayed vs normal GE include those that are linked to glucose homeostasis and the complications of DM, i.e., PDE4A (52,53), FBP1 (54,55), SORD (56), CMKLR1 (57), and BACE2 (58,59). In particular, CXCR4 mediates pain signaling and central sensitization in 4 animal models, including streptozotocin-induced painful diabetic neuropathy (60–62). CXCR4 has also been implicated in cardiac fibrosis in rats with DM (63). It is also increased and may serve as a renoprotective mediator in rats and humans with diabetic nephropathy (64). By contrast, MIR874 and TIMP3, which were differentially bound with H3K27ac in patients with DM with delayed (vs normal) GE, were protective against nephropathy in diabetic animals (65,66). Many of the genes with differential H3K9ac binding are also related to the pathogenesis, i.e., RGS1 (47), PRKCQ (67), ICOS (68), and IL7R (69), and complications of DM, i.e., RELB (70,71), ICOS (72), PRKCQ (73), MGAT4A (74), and SCN9A (75).
The pathogenesis of rapid GE in patients with DM is essentially unknown. Despite the small sample size, epigenetic alterations distinguished between patients with DM with rapid and normal GE. Compared with normal GE, 100 genes were differentially bound with H3K9ac at the promoter site in patients with rapid GE. This includes several inflammatory genes, such as TNFRSF25, the receptor for TL1A, which mediates costimulation of T cells to produce cytokines and promote proliferation of activated and regulatory T cells (76), CXCR4 and RGS1, which also had higher binding with H3K27ac and H3K9ac, respectively, in delayed GE, PELI3, which mediates c-JUN and ELK1 activation in innate immune signaling pathways (77), CD55 the complement decay-accelerating factor, ALOX5AP, which is required for leukotriene synthesis (78), MNDA, the myeloid cell nuclear differentiation antigen, which mediates granulocyte and monocyte cell–specific responses to interferons, and CARMIL2 (aka RLTPR), which functions as a scaffold to promote CD28 costimulation in T cells (79). In addition, genes related to insulin sensitivity, such as AHNAK (80), PELI3 (81), and FASN (82), pancreatic β-cell function, such as KLF11 (83), and glucose metabolism, such as AQP9 (84) and PGP (85), were also among the genes differentially bound with H3K9ac in patients with DM with rapid GE. H3K9ac binding at the promoter site of the mechanosensitive ion channel PIEZO1, which regulates vascular development, blood pressure, and red cell volume, is also present in the gastrointestinal tract (86). Its close homologue PIEZO2 mediates serotonin release from enterochromaffin cells in the gut in response to mechanical stimuli (87). Deletion of PIEZO1 is associated with increased adipose tissue expression of proinflammatory genes and insulin resistance (88).
Contrary to earlier observations, which suggested that rapid GE is associated with early type 2 DM, whereas delayed GE is associated with long-lasting type 1 DM, a larger study observed that the DM phenotype (i.e., type and duration of DM and presence of complications) did not distinguish between rapid and normal GE (37,89). However, neuropathy and insulin use were associated with a higher and lower risk, respectively, of rapid GE in DM. Rapid GE has been described in 3 animal models of DM, i.e., streptozotocin-treated rodents and genetically diabetic BB/Wor rats (90), mice with mutation of the leptin receptor (Leprdb/db) (91), and NOD LtJ mice (92). Two weeks after developing DM, NOD mice transiently develop rapid gastric GE, followed by normal and then delayed GE (92). Because the NOD LtJ mice and BB/Wor rats have immune-mediated DM, it is not inconceivable that the immune-mediated mechanisms contribute to rapid GE. However, the immune mechanisms responsible for rapid GE in these mice and diabetic BB/Wor rats are unknown.
Motif analysis revealed several potentially relevant TFs that were enriched in the differential peaks for H3K27ac and H3K9ac. Of these motifs, the MEF2 family of TFs, MEF2A and 2C have been implicated in diabetic cardiomyopathy (93–95), the ras-responsive TF RREB1 variants were associated with end-stage renal disease in type 2 DM, the Fox family of TFs is related to insulin signaling and resistance (i.e., FoxO1 and O4, FoxC2, and FoxK1 and K2) (96–98), and glucose sensing and homeostasis (i.e., FoxA2 and FoxP1) (99,100), and USF1 is a risk gene for type 2 DM (101).
This is the first study to compare the epigenome in patients with DM with normal, rapid, and delayed GE. Although the findings are exciting, there are several limitations. Although these patients had typical features of DGE, the sample size was relatively small; selection bias cannot be excluded. The ChIP-seq data for 1 or 2 but not all 3 marks were available in 5 patients. The actual sample size was lower than the ideal sample size based on a post hoc analysis of the observed differences in epigenetic marks between groups. For H3K27ac, the ideal (actual) sample size was 16 (13) patients with either delayed or normal GE. Corresponding values were 19 (11) patients with rapid or normal GE for H3K9ac and 54 (12) patients with rapid or normal GE for H3K4me3. Because full-thickness gastric biopsies were not available, the blood epigenome was evaluated. The peripheral blood is arguably a biosensor for systemic metabolic, endocrine, and inflammatory changes, which contribute to the gastrointestinal manifestations of DM. Although epigenetic changes related to H3K27 acetylation (102), H3K4 trimethylation (103), and H3K9ac acetylation (104) are correlated with the predicted changes in transcription, gene expression was not confirmed in this study. The extent to which the observed differences are related to abnormal GE vs other complications and severity of DM is unclear. It is conceivable that not all differentially expressed genes are relevant to the pathogenesis of GE disturbances, and the mechanism(s) by which these genes affect GE is unclear. Besides the stomach, the epigenetic alterations may also affect extragastric factors (e.g., autonomic nervous system), which contribute to GE disturbances in DM (105).
In summary, there are several differentially bound histone modifications in whole blood between patients with DM with normal and delayed GE and between normal and rapid GE. These modifications, which pertain to dysimmunity, glucose metabolism, and other complications of DM, may provide clues to the pathogenesis and the impetus for further studies that investigate the contribution of epigenetic and inflammatory mechanisms to gastrointestinal sensorimotor dysfunctions in DM.
CONFLICTS OF INTEREST
Guarantor of the article: Adil E. Bharucha, MBBS, MD.
Specific author contributions: A.E.B. and T.O designed the study. J.-H.L. performed the experiments. H.Y., A.B., S.P.N., and S.K. analyzed the data. S.P.N., S.K., and A.E.B. wrote the manuscript. All authors approved the final version of the manuscript.
Financial support: This study was supported by the USPHS NIH Grant R01 DK68055 (A.E.B., T.O., and J.H.L.).
Potential competing interests: None to report.
Writing assistance: None to report.
Study Highlights.
WHAT IS KNOWN
✓ Patients with DM and GI symptoms generally have normal or delayed and less frequently rapid GE.
✓ GI symptoms and the diabetic phenotype are of limited utility for predicting GE in such patients.
✓ Epigenetic mechanisms explain the deleterious consequences of hyperglycemia on non-GI complications of DM (metabolic memory).
WHAT IS NEW HERE
✓ Differential binding with H3K27ac and H3K9ac in whole blood discriminated between patients with DM with normal vs delayed GE and H3K9ac discriminated between normal vs rapid GE.
✓ The differentially bound genes regulate hematopoiesis, inflammation, and immunity. They are also associated with glucose metabolism, neuronal functions, and other complications of DM.
TRANSLATIONAL IMPACT
✓ These epigenetic alterations might provide clues to the pathogenesis of GI manifestations of DM and be targeted to predict the presence of or treat GE disturbances in DM.
Supplementary Material
Footnotes
SUPPLEMENTARY MATERIAL accompanies this paper at http://links.lww.com/CTG/A201, http://links.lww.com/CTG/A202, http://links.lww.com/CTG/A203, http://links.lww.com/CTG/A204, http://links.lww.com/CTG/A205, http://links.lww.com/CTG/A206, http://links.lww.com/CTG/A207, http://links.lww.com/CTG/A208, http://links.lww.com/CTG/A209, http://links.lww.com/CTG/A210, http://links.lww.com/CTG/A211
References
- 1.Bharucha AE, Kudva YC, Basu A, et al. Relationship between glycemic control and gastric emptying in poorly controlled type 2 diabetes. Clin Gastroenterol Hepatol 2014;13(3):466–76.e461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bharucha AE, Batey-Schaefer B, Cleary PA, et al. Delayed gastric emptying is associated with early and long-term hyperglycemia in type 1 diabetes mellitus. Gastroenterology 2015;149:330–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chakraborty S, Halland M, Burton D, et al. GI dysfunctions in diabetic gastroenteropathy, their relationships with symptoms, and effects of a GLP-1 antagonist. J Clin Endocrinol Metab 2019;104(6):1967–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bharucha AE, Kudva YC, Prichard DO. Diabetic gastroparesis. Endocr Rev 2019;40(5):1318–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grover M, Farrugia G, Lurken MS, et al. Cellular changes in diabetic and idiopathic gastroparesis. Gastroenterology 2011;140(5):1575–85.e1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cipriani G, Gibbons SJ, Miller KE, et al. Change in populations of macrophages promotes development of delayed gastric emptying in mice. Gastroenterology 2018;154(8):2122–36.e2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gibbons SJ, Grover M, Choi KM, et al. Repeat polymorphisms in the Homo sapiens heme oxygenase-1 gene in diabetic and idiopathic gastroparesis. PLoS One 2017;12(11):e0187772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kilpatrick ES, Rigby AS, Atkin SL. The diabetes control and complications trial: The gift that keeps giving. Nat Rev Endocrinol 2009;5(10):537–45. [DOI] [PubMed] [Google Scholar]
- 9.Writing Team for the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Effect of intensive therapy on the microvascular complications of type 1 diabetes mellitus. JAMA 2002;287(19):2563–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Writing Team for the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Sustained effect of intensive treatment of type 1 diabetes mellitus on development and progression of diabetic nephropathy: The epidemiology of diabetes interventions and complications (EDIC) study. JAMA 2003;290(16):2159–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin CL, Albers JW, Pop-Busui R; DCCT/EDIC Research Group. Neuropathy and related findings in the diabetes control and complications trial/epidemiology of diabetes interventions and complications study. Diabetes Care 2014;37(1):31–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosen ED, Kaestner KH, Natarajan R, et al. Epigenetics and epigenomics: Implications for diabetes and obesity. Diabetes 2018;67(10):1923–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kato M, Natarajan R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat Rev Nephrol 2019;20:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miao F, Chen Z, Genuth S, et al. Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes 2014;63(5):1748–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Z, Miao F, Paterson AD, et al. Epigenomic profiling reveals an association between persistence of DNA methylation and metabolic memory in the DCCT/EDIC type 1 diabetes cohort. Proc Natl Acad Sci U S A 2016;113(21):E3002–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharma U, Rando OJ. Metabolic inputs into the epigenome. Cell Metab 2017;25(3):544–58. [DOI] [PubMed] [Google Scholar]
- 17.Bhandare R, Schug J, Le Lay J, et al. Genome-wide analysis of histone modifications in human pancreatic islets. Genome Res 2010;20(4):428–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miao F, Chen Z, Zhang L, et al. Profiles of epigenetic histone post-translational modifications at type 1 diabetes susceptible genes. J Biol Chem 2012;287(20):16335–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mishra M, Zhong Q, Kowluru RA. Epigenetic modifications of Nrf2-mediated glutamate-cysteine ligase: Implications for the development of diabetic retinopathy and the metabolic memory phenomenon associated with its continued progression. Free Radic Biol Med 2014;75:129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Marinis Y, Cai M, Bompada P, et al. Epigenetic regulation of the thioredoxin-interacting protein (TXNIP) gene by hyperglycemia in kidney. Kidney Int 2016;89(2):342–53. [DOI] [PubMed] [Google Scholar]
- 21.Kimball AS, Joshi A, Carson WFt, et al. The histone methyltransferase MLL1 directs macrophage-mediated inflammation in wound healing and is altered in a murine model of obesity and type 2 diabetes. Diabetes 2017;66(9):2459–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marks DL, Olson RL, Urrutia R, et al. Epigenetics of gastrointestinal diseases: Notes from a workshop. Epigenetics 2018;13(4):449–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Desai A, O'Connor M, Neja B, et al. Reproducibility of gastric emptying assessed with scintigraphy in patients with upper GI symptoms. Neurogastroenterol Motil 2018;30(10):e13365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tack J, Talley NJ, Camilleri M, et al. Functional gastroduodenal disorders [erratum appears in Gastroenterology. 2006 Jul;131(1):336]. Gastroenterology 2006;130(5):1466–79. [DOI] [PubMed] [Google Scholar]
- 25.Rentz AM, Kahrilas P, Stanghellini V, et al. Development and psychometric evaluation of the patient assessment of upper gastrointestinal symptom severity index (PAGI-SYM) in patients with upper gastrointestinal disorders. Qual Life Res 2004;13(10):1737–49. [DOI] [PubMed] [Google Scholar]
- 26.Camilleri M, Iturrino J, Bharucha AE, et al. Performance characteristics of scintigraphic measurement of gastric emptying of solids in healthy participants. Neurogastroenterol Motil 2012;24(12):1076–e1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhong J, Ye Z, Lenz SW, et al. Purification of nanogram-range immunoprecipitated DNA in ChIP-seq application. BMC Genomics 2017;18(1):985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yan H, Evans J, Kalmbach M, et al. HiChIP: A high-throughput pipeline for integrative analysis of ChIP-seq data. BMC Bioinformatics 2014;15:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25(14):1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-seq (MACS). Genome Biol 2008;9(9):R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ross-Innes CS, Stark R, Teschendorff AE, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012;481(7381):389–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Creyghton MP, Cheng AW, Welstead GG, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 2010;107(50):21931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McLeay RC, Bailey TL. Motif enrichment analysis: A unified framework and an evaluation on ChIP data. BMC Bioinformatics 2010;11:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pham TH, Benner C, Lichtinger M, et al. Dynamic epigenetic enhancer signatures reveal key transcription factors associated with monocytic differentiation states. Blood 2012;119(24):e161–171. [DOI] [PubMed] [Google Scholar]
- 35.Karmodiya K, Krebs AR, Oulad-Abdelghani M, et al. H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genomics 2012;13:424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dincer A, Gavin DP, Xu K, et al. Deciphering H3K4me3 broad domains associated with gene-regulatory networks and conserved epigenomic landscapes in the human brain. Translational Psychiatry 2015;5:e679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bharucha AE, Camilleri M, Forstrom LA, et al. Relationship between clinical features and gastric emptying disturbances in diabetes mellitus. Clin Endocrinol (Oxf) 2009;70(3):415–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Griesenauer B, Paczesny S. The ST2/IL-33 Axis in immune cells during inflammatory diseases. Front Immunol 2017;8:475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hirai H, Tanaka K, Yoshie O, et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med 2001;193(2):255–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhong J, Sharma J, Raju R, et al. TSLP signaling pathway map: A platform for analysis of TSLP-mediated signaling. Database (Oxford) 2014;2014:bau007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ochiai S, Jagot F, Kyle RL, et al. Thymic stromal lymphopoietin drives the development of IL-13(+) Th2 cells. Proc Natl Acad Sci U S A 2018;115(5):1033–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zundler S, Becker E, Spocinska M, et al. Hobit- and Blimp-1-driven CD4(+) tissue-resident memory T cells control chronic intestinal inflammation. Nat Immunol 2019;20(3):288–300. [DOI] [PubMed] [Google Scholar]
- 43.Yang H, Madison B, Gumucio DL, et al. Specific overexpression of IL-7 in the intestinal mucosa: The role in intestinal intraepithelial lymphocyte development. Am J Physiol Gastrointest Liver Physiol 2008;294(6):G1421–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simpson TR, Quezada SA, Allison JP. Regulation of CD4 T cell activation and effector function by inducible costimulator (ICOS). Curr Opin Immunol 2010;22(3):326–32. [DOI] [PubMed] [Google Scholar]
- 45.Zhang EY, Kong KF, Altman A. The yin and yang of protein kinase C-theta (PKCtheta): A novel drug target for selective immunosuppression. Adv Pharmacol 2013;66:267–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Witherden DA, Verdino P, Rieder SE, et al. The junctional adhesion molecule JAML is a costimulatory receptor for epithelial gammadelta T cell activation. Science 2010;329(5996):1205–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caballero-Franco C, Kissler S. The autoimmunity-associated gene RGS1 affects the frequency of T follicular helper cells. Genes Immun 2016;17:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mathew PA, Chuang SS, Vaidya SV, et al. The LLT1 receptor induces IFN-gamma production by human natural killer cells. Mol Immunol 2004;40(16):1157–63. [DOI] [PubMed] [Google Scholar]
- 49.Voss OH, Tian L, Murakami Y, et al. Emerging role of CD300 receptors in regulating myeloid cell efferocytosis. Mol Cell Oncol 2015;2(4):e964625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bernard CE, Gibbons SJ, Mann IS, et al. Association of low numbers of CD206-positive cells with loss of ICC in the gastric body of patients with diabetic gastroparesis. Neurogastroenterol Motil 2014;26(9):1275–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grover M, Bernard CE, Pasricha PJ, et al. Diabetic and idiopathic gastroparesis is associated with loss of CD206-positive macrophages in the gastric antrum. Neurogastroenterol Motil 2017;29(6):e13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wouters EF, Bredenbroker D, Teichmann P, et al. Effect of the phosphodiesterase 4 inhibitor roflumilast on glucose metabolism in patients with treatment-naive, newly diagnosed type 2 diabetes mellitus. J Clin Endocrinol Metab 2012;97(9):E1720–1725. [DOI] [PubMed] [Google Scholar]
- 53.Vollert S, Kaessner N, Heuser A, et al. The glucose-lowering effects of the PDE4 inhibitors roflumilast and roflumilast-N-oxide in db/db mice. Diabetologia 2012;55(10):2779–88. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Y, Xie Z, Zhou G, et al. Fructose-1,6-bisphosphatase regulates glucose-stimulated insulin secretion of mouse pancreatic beta-cells. Endocrinology 2010;151(10):4688–95. [DOI] [PubMed] [Google Scholar]
- 55.van Poelje PD, Potter SC, Erion MD. Fructose-1, 6-bisphosphatase inhibitors for reducing excessive endogenous glucose production in type 2 diabetes. Handb Exp Pharmacol 2011(203):279–301. [DOI] [PubMed] [Google Scholar]
- 56.Carr IM, Markham AF. Molecular genetic analysis of the human sorbitol dehydrogenase gene. Mamm Genome 1995;6(9):645–52. [DOI] [PubMed] [Google Scholar]
- 57.Ernst MC, Haidl ID, Zuniga LA, et al. Disruption of the chemokine-like receptor-1 (CMKLR1) gene is associated with reduced adiposity and glucose intolerance. Endocrinology 2012;153(2):672–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alcarraz-Vizan G, Castano C, Visa M, et al. BACE2 suppression promotes beta-cell survival and function in a model of type 2 diabetes induced by human islet amyloid polypeptide overexpression. Cell Mol Life Sci 2017;74(15):2827–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Southan C. BACE2 as a new diabetes target: A patent review (2010 - 2012). Expert Opin Ther patents 2013;23(5):649–63. [DOI] [PubMed] [Google Scholar]
- 60.Menichella DM, Abdelhak B, Ren D, et al. CXCR4 chemokine receptor signaling mediates pain in diabetic neuropathy. Mol Pain 2014;10:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jayaraj ND, Bhattacharyya BJ, Belmadani AA, et al. Reducing CXCR4-mediated nociceptor hyperexcitability reverses painful diabetic neuropathy. J Clin Invest 2018;128(6):2205–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu D, Fan T, Huo X, et al. Progressive increase of inflammatory CXCR4 and TNF-alpha in the dorsal root ganglia and spinal cord maintains peripheral and central sensitization to diabetic neuropathic pain in rats. Mediators Inflamm 2019;2019:4856156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chu PY, Walder K, Horlock D, et al. CXCR4 antagonism attenuates the development of diabetic cardiac fibrosis. PLoS One 2015;10(7):e0133616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Siddiqi FS, Chen LH, Advani SL, et al. CXCR4 promotes renal tubular cell survival in male diabetic rats: Implications for ligand inactivation in the human kidney. Endocrinology 2015;156(3):1121–32. [DOI] [PubMed] [Google Scholar]
- 65.Yao T, Zha D, Gao P, et al. MiR-874 alleviates renal injury and inflammatory response in diabetic nephropathy through targeting toll-like receptor-4. J Cell Physiol 2018;234(1):871–9. [DOI] [PubMed] [Google Scholar]
- 66.Basu R, Lee J, Wang Z, et al. Loss of TIMP3 selectively exacerbates diabetic nephropathy. Am J Physiol Ren Physiol 2012;303(9):F1341–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li Y, Soos TJ, Li X, et al. Protein kinase C Theta inhibits insulin signaling by phosphorylating IRS1 at Ser(1101). J Biol Chem 2004;279(44):45304–7. [DOI] [PubMed] [Google Scholar]
- 68.Hawiger D, Tran E, Du W, et al. ICOS mediates the development of insulin-dependent diabetes mellitus in nonobese diabetic mice. J Immunol 2008;180(5):3140–7. [DOI] [PubMed] [Google Scholar]
- 69.Monti P, Bonifacio E. Interleukin-7 and type 1 diabetes. Curr Diab Rep 2014;14(9):518. [DOI] [PubMed] [Google Scholar]
- 70.Starkey JM, Haidacher SJ, LeJeune WS, et al. Diabetes-induced activation of canonical and noncanonical nuclear factor-κB pathways in renal cortex. Diabetes 2006;55(5):1252–9. [DOI] [PubMed] [Google Scholar]
- 71.Mollah ZU, Pai S, Moore C, et al. Abnormal NF-kappa B function characterizes human type 1 diabetes dendritic cells and monocytes. J Immunol 2008;180(5):3166–75. [DOI] [PubMed] [Google Scholar]
- 72.Zhang HY, Ruan LB, Li Y, et al. ICOS/ICOSL upregulation mediates inflammatory response and endothelial dysfunction in type 2 diabetes mellitus. Eur Rev Med Pharmacol Sci 2018;22(24):8898–908. [DOI] [PubMed] [Google Scholar]
- 73.Li Z, Abdullah CS, Jin ZQ. Inhibition of PKC-theta preserves cardiac function and reduces fibrosis in streptozotocin-induced diabetic cardiomyopathy. Br J Pharmacol 2014;171(11):2913–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.López-Orduña E, Cruz M, García-Mena J. The transcription of MGAT4A glycosyl transferase is increased in white cells of peripheral blood of Type 2 Diabetes patients. BMC Genet 2007;8(1):73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li QS, Cheng P, Favis R, et al. SCN9A variants may be implicated in neuropathic pain associated with diabetic peripheral neuropathy and pain severity. Clin J Pain 2015;31(11):976–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Meylan F, Richard AC, Siegel RM. TL1A and DR3, a TNF family ligand-receptor pair that promotes lymphocyte costimulation, mucosal hyperplasia, and autoimmune inflammation. Immunol Rev 2011;244(1):188–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jensen LE, Whitehead AS. Pellino3, a novel member of the Pellino protein family, promotes activation of c-Jun and Elk-1 and may act as a scaffolding protein. J Immunol 2003;171(3):1500–6. [DOI] [PubMed] [Google Scholar]
- 78.Evans JF, Ferguson AD, Mosley RT, et al. What's all the FLAP about?: 5-lipoxygenase-activating protein inhibitors for inflammatory diseases. Trends Pharmacol Sci 2008;29(2):72–8. [DOI] [PubMed] [Google Scholar]
- 79.Roncagalli R, Cucchetti M, Jarmuzynski N, et al. The scaffolding function of the RLTPR protein explains its essential role for CD28 co-stimulation in mouse and human T cells. J Exp Med 2016;213(11):2437–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shin JH, Kim IY, Kim YN, et al. Obesity resistance and enhanced insulin sensitivity in ahnak-/- mice fed a high fat diet are related to impaired adipogenesis and increased energy expenditure. PLoS One 2015;10(10):e0139720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang S, Wang B, Humphries F, et al. The E3 ubiquitin ligase Pellino3 protects against obesity-induced inflammation and insulin resistance. Immunity 2014;41(6):973–87. [DOI] [PubMed] [Google Scholar]
- 82.Fernandez-Real JM, Menendez JA, Moreno-Navarrete JM, et al. Extracellular fatty acid synthase: A possible surrogate biomarker of insulin resistance. Diabetes 2010;59(6):1506–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Neve B, Fernandez-Zapico ME, Ashkenazi-Katalan V, et al. Role of transcription factor KLF11 and its diabetes-associated gene variants in pancreatic beta cell function. Proc Natl Acad Sci U S A 2005;102(13):4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Spegel P, Chawade A, Nielsen S, et al. Deletion of glycerol channel aquaporin-9 (Aqp9) impairs long-term blood glucose control in C57BL/6 leptin receptor-deficient (db/db) obese mice. Physiol Rep 2015;3(9):e12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mugabo Y, Zhao S, Seifried A, et al. Identification of a mammalian glycerol-3-phosphate phosphatase: Role in metabolism and signaling in pancreatic beta-cells and hepatocytes. Proc Natl Acad Sci U S A 2016;113(4):E430–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Alcaino C, Farrugia G, Beyder A. Mechanosensitive piezo channels in the gastrointestinal tract. Curr Top Membr 2017;79:219–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Alcaino C, Knutson KR, Treichel AJ, et al. A population of gut epithelial enterochromaffin cells is mechanosensitive and requires Piezo2 to convert force into serotonin release. Proc Natl Acad Sci U S A 2018;115(32):E7632–e7641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhao C, Sun Q, Tang L, et al. Mechanosensitive ion channel Piezo1 regulates diet-induced adipose inflammation and systemic insulin resistance. Front Endocrinol (Lausanne) 2019;10:373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kuwelker S, Muthyala A, O'Connor M, et al. Clinical features and disturbances of gastrointestinal transit in patients with rapid gastric emptying. Neurogastroenterol Motil 2020:e13779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nowak TV, Roza AM, Weisbruch JP, et al. Accelerated gastric emptying in diabetic rodents: Effect of insulin treatment and pancreas transplantation. J Lab Clin Med 1994;123(1):110–6. [PubMed] [Google Scholar]
- 91.Hayashi Y, Toyomasu Y, Saravanaperumal SA, et al. Hyperglycemia increases interstitial cells of cajal via MAPK1 and MAPK3 signaling to ETV1 and KIT, leading to rapid gastric emptying. Gastroenterology 2017;153(2):521–35.e520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Choi KM, Zhu J, Stoltz GJ, et al. Determination of gastric emptying in nonobese diabetic mice. Am J Physiol Gastrointest Liver Physiol 2007;293(5):G1039–1045. [DOI] [PubMed] [Google Scholar]
- 93.Razeghi P, Young ME, Cockrill TC, et al. Downregulation of myocardial myocyte enhancer factor 2C and myocyte enhancer factor 2C-regulated gene expression in diabetic patients with nonischemic heart failure. Circulation 2002;106(4):407–11. [DOI] [PubMed] [Google Scholar]
- 94.Chen X, Liu G, Zhang W, et al. Inhibition of MEF2A prevents hyperglycemia-induced extracellular matrix accumulation by blocking Akt and TGF-beta1/Smad activation in cardiac fibroblasts. Int J Biochem Cell Biol 2015;69:52–61. [DOI] [PubMed] [Google Scholar]
- 95.Chen XY, Lv RJ, Zhang W, et al. Inhibition of myocyte-specific enhancer factor 2A improved diabetic cardiac fibrosis partially by regulating endothelial-to-mesenchymal transition. Oncotarget 2016;7(21):31053–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 2004;117(4):421–6. [DOI] [PubMed] [Google Scholar]
- 97.Ridderstrale M, Carlsson E, Klannemark M, et al. FOXC2 mRNA Expression and a 5' untranslated region polymorphism of the gene are associated with insulin resistance. Diabetes 2002;51(12):3554–60. [DOI] [PubMed] [Google Scholar]
- 98.Sakaguchi M, Cai W, Wang CH, et al. FoxK1 and FoxK2 in insulin regulation of cellular and mitochondrial metabolism. Nat Commun 2019;10(1):1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang H, Gauthier BR, Hagenfeldt-Johansson KA, et al. Foxa2 (HNF3beta) controls multiple genes implicated in metabolism-secretion coupling of glucose-induced insulin release. J Biol Chem 2002;277(20):17564–70. [DOI] [PubMed] [Google Scholar]
- 100.Zou Y, Gong N, Cui Y, et al. Forkhead box P1 (FOXP1) transcription factor regulates hepatic glucose homeostasis. J Biol Chem 2015;290(51):30607–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Meex SJ, van Vliet-Ostaptchouk JV, van der Kallen CJ, et al. Upstream transcription factor 1 (USF1) in risk of type 2 diabetes: Association study in 2000 Dutch caucasians. Mol Genet Metab 2008;94(3):352–5. [DOI] [PubMed] [Google Scholar]
- 102.Hamdane N, Juhling F, Crouchet E, et al. HCV-induced epigenetic changes associated with liver cancer risk persist after sustained virologic response. Gastroenterology 2019;156(8):2313–29.e2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pu M, Wang M, Wang W, et al. Unique patterns of trimethylation of histone H3 lysine 4 are prone to changes during aging in Caenorhabditis elegans somatic cells. PLoS Genet 2018;14(6):e1007466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kelley DZ, Flam EL, Izumchenko E, et al. Integrated analysis of whole-genome ChIP-seq and RNA-seq data of primary head and neck tumor samples associates HPV integration sites with open chromatin marks. Cancer Res 2017;77(23):6538–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bharucha AE, Camilleri M, Low PA, et al. Autonomic dysfunction in gastrointestinal motility disorders. Gut 1993;34(3):397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.