

Abstract
Refractive errors, in particular myopia, are a leading cause of morbidity and disability world-wide. Genetic investigation can improve understanding of the molecular mechanisms underlying abnormal eye development and impaired vision. We conducted a meta-analysis of genome-wide association studies involving 542,934 European participants and identified 336 novel genetic loci associated with refractive error. Collectively, all associated genetic variants explain 18.4% of heritability and improve the accuracy of myopia prediction (AUC=0.75). Our results suggest that refractive error is genetically heterogeneous, driven by genes participating in the development of every anatomical component of the eye. In addition, our analyses suggest that genetic factors controlling circadian rhythm and pigmentation are also involved in the development of myopia and refractive error. These results may make possible predicting refractive error and the development of personalized myopia prevention strategies in the future.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Introduction
Refractive errors occur when converging light rays from an image do not clearly focus on the retina. They are the seventh most prevalent clinical condition1 and the second leading cause of disability in the world2. The prevalence of refractive error is rapidly increasing, mostly driven by a dramatic rise in the prevalence of one of its forms, myopia (near-sightedness). Although the causes of such a rise over a short time are likely due to environmental and cultural changes from the mid-20th century3, refractive errors are highly heritable4. Several studies5,6 have previously sought to identify genes controlling molecular mechanisms leading to refractive error and myopia. However, the variance and heritability that can be attributed to known genetic factors is modest7 and our knowledge of pathogenic mechanisms remains partial. Here, we conduct a meta-analysis combining data from quantitative spherical equivalent and myopia status from large and previously unpublished genome-wide association studies (GWAS) of more than half a million subjects from the UK Biobank, 23andMe and the Genetic Epidemiology Research on Adult Health and Aging (GERA) cohorts, with subsequent replication and meta-analysis with data previously reported from the Consortium for Refractive Error and Myopia (CREAM).
Results
Association Results.
Analyses were restricted to subjects of European ancestry (Extended Data Figure 1) and combined results from quantitative measures of spherical equivalent and categorical myopia status. Spherical equivalent quantifies refractive error; a negative spherical equivalent, below a certain threshold defines myopia. We used results obtained from GWAS of directly measured spherical equivalent in 102,117 population-based UK Biobank participants8, and 34,998 subjects participating in the GERA Study9 and combined them with results of analyses of self-reported myopia in 106,086 cases and 85,757 controls from the customer base of 23andMe, Inc. (Mountain View, CA), a personal genomics company10. Additionally, we included results from an analysis on the refractive status inferred using demographic and self-reported information on age at first use of prescription glasses among the UK Biobank participants not contributing to the quantitative GWAS (108,956 likely myopes to 70,941 likely non-myopes, see Online Methods). All analyses were adjusted for age, sex and main principal components. To obtain an overall association with refractive error, we meta-analyzed the results from all studies by using the z-scores from the GWAS of the spherical equivalent and the negative values of z-scores from the case-control studies (23andMe and UK Biobank), since myopia is negatively correlated with spherical equivalent. As expected, the large total sample size of the discovery meta-analysis (N=508,855) led to a nominally large genomic inflation factor (λ=1.94). The LD score regression intercept was (1.17), and the (intercept-1)/(mean(chî2)-1) ratio of 0.097 is fully in line with the expectations of polygenicity11.
We found associations for 438 discrete genomic regions (Figure 1, Supplementary Table 1), defined by markers contiguously associated at conventional level of GWAS significance12,13 of p<5×10−08, separated by more than 1 Mbp from other GWAS-associated markers, as recommended elsewhere14. Among them, 308 loci, including 14 on chromosome X, were not described in previous GWAS studies of refractive error7. The observed effect sizes were consistent across all the studies (Supplementary Table 1 and Supplementary Data 1). The association with refractive error was statistically strongest for rs12193446 (p=9.87×10−328), within LAMA2, a gene previously associated with refractive error5,6, mutations of which cause muscular dystrophy15. Consistent with these LAMA2 properties, polymorphisms located within the genes coding for both major LAMA2 receptors, DAG116 (p=1.67×10−08 for rs111327216) and ITGA717 (p=8.57×10−09 for rs17117860) which are also known causes of muscular dystrophy18,19, were significantly associated with refractive error in the discovery meta-analysis.
Figure 1.

All GWAS-associated regions from the main meta-analysis. Each band is a true scale of genomic regions associated with refractive error listed in Supplementary Table 1 (+250kbp on each side to make smaller regions more visible). The different color codes represent the significance (p-value) for the genetic variant within that region that displays the strongest evidence for association.
We compared our discovery meta-analysis findings with GWAS results from 34,079 participants in the CREAM consortium, who were part of a previously reported meta-analysis7. To avoid any potential overlap with the UK Biobank participants, only non-UK European CREAM participants were used for replication. Despite the vast power differential, 55 of the SNPs that showed the strongest association in their respective regions in the discovery meta-analysis were significant after Bonferroni correction in the replication sample. A further 142 had a false discovery rate (FDR) < 0.05 and 192 were nominally significant at P < 0.05 (Supplementary Table 2). The effect sizes observed in the discovery and replication samples were strongly correlated (Pearson’s r=0.91, Extended Data Figure 2). Meta-analysis of all five cohorts (discovery and replication) expanded the number to 449 associated of regions of variable length and number of SNPs (Extended Data Figure 3), of which 336 regions were novel (Supplementary Table 3).
Most of the 449 refractive error-associated regions contained at least one gene linked to severe ocular manifestations in the Online Mendelian Inheritance In Man (OMIM) resource or other genes with interesting link to eye disease (Supplementary Table 4). Although most loci identified through our meta-analyses were novel, several of them hosted genes that harbor mutations leading to myopia or other refractive error phenotypes (Supplementary Data 2). Several genes significantly associated with refractive error were linked to Mendelian disorders affecting corneal structure, some of which code for transcription factors involved in corneal development20 (Supplementary Table 5). Mutations in these genes cause corneal dystrophies (SLC4A11, p=5.81×10−11 for rs41281858, TCF4, p=4.14×10−08, rs41396445; LCAT, p=1.26×10−10, rs5923; and DCN, p=3.67×10−09, rs1280632), megalocornea (LTBP2, p=1.91×10−24, rs73296215) and keratoconus (FNDC3B, p=1.89×10−14, rs199771582, previously described7). Eleven refractive error-associated genes were linked to anomalies of the crystalline lens (Supplementary Table 6), including genes linked to autosomal dominant cataracts (PAX6 previously linked to myopia21, p=8.31×10−11, rs1540320; PITX3, p=1.05×10−10, rs7923183; MAF, p=5.50×10−09, rs16951312; CHMP4B, p=9.95×10 −11 , rs6087538; TDRD7, p=4.79×10−08, rs13301794) and lens ectopia (FBN1, p=3.30×10−24, rs2017765; ADAMTSL4, p=8.19×10−14, rs12131376). Some of the genes affected several eye components. For example, LTBP2 variants are also associated with congenital glaucoma22, and COL4A3 (rs7569375, p=1.14×10−08) causes Alport syndrome, which manifests with abnormal lens shape (lenticonus) and structural changes in the retina.
Association was also observed within or near 13 genes known to harbor mutations causing microphthalmia (Supplementary Table 7), including TENM3 (p=2.48×10−11, rs35446926); OTX2 (p=6.15×10−11, rs928109); VSX2, (p=4.60×10−10, rs35797567); MFRP, (p=2.85×10−16, rs10892353) and the previously identified6 TMEM98, (p=3.49×10−43, rs62067167). Association was also found for VSX1 (p=4.59×10−08 for rs6050351), a gene that is closely regulated by VSX223 and believed to play important roles in eye development24. Many of the genes nearest associated SNPs have been linked to inherited retinal disease (Supplementary Table 8), including 32 genes linked to cone-rod dystrophies, night blindness and retinitis pigmentosa, and age-related macular degeneration (HTRA1/ARMS2). Among genes in novel regions associated with refractive error, ABCA4 (p=3.20×10−10 for rs11165052), and ARMS2/HTRA1 (p=5.72×10−23 for rs2142308) are linked to macular disorders and numerous others to retinitis pigmentosa, retinal dystrophy and other retinal diseases, such as FBN2, (p=8.63×10−11, rs6860901) , TRAF3IP1 (p=5.71×10−16, rs7596847), CWC27 (p=1.84×10−18, rs1309551). Significant association was found near other genes of interest such as DRD1 (p=4.51×10−16, rs13190379), a dopamine receptor. Together, these results are consistent with previous suggestions of light transmission and transduction in refractive error7,25.
Wnt signaling has previously been implicated in experimental myopia26. We found significant association near several Wnt protein-coding genes (WNT7B, a gene previously associated with axial length27, p=1.42×10−26 for rs73175083; WNT10A, previously associated with central corneal thickness28, p=1.65×10−17 for rs121908120 and WNT3B, p=8.52×10−16 for rs70600), suggesting that organogenesis through Wnt signaling is likely to be involved in refractive error. Significant association were found at genes coding for key canonical (e.g. rs13072632 within the CTNNB1 gene, p=7.30×10−27; AXIN2, rs9895291, p=1.40×10−08) and non-canonical Wnt pathway members (NFATC3, rs147561310, p=1.493×10−12) or at genes coding for both (RHOA, rs7623687, p=1.81×10−11 or the previously described7 TCF7L2, rs56299331, p=9.38×10−46; Supplementary Table 9).
Similar to previous published analyses25, we found associations for genes involved in sodium, potassium, calcium magnesium and other cation transporters (Supplementary Table 10). The involvement of genes related to glutamatergic synaptic transmission was also notable (Supplementary Table 11). Glutamate is a first synapse transmitter released by photoreceptors towards bipolar cells and is the main excitatory neurotransmitter of the retina, and expression of genes participating in glutamate signaling pathways is significantly altered in myopia models29. These associations support the involvement in refractive error pathogenesis of neurotransmission and neuronal depolarization and hyperpolarization that was also suggested before7. Associations with POU6F2 gene intronic variants (rs2696187, p=1.11×10−11) also suggests involvement of factors related to development of amacrine and ganglion cells30. Other genes at refractive error-associated loci were annotated to infantile epilepsy, microcephaly, severe learning difficulty, or other inborn diseases affecting the central nervous system (CNS) in OMIM (Supplementary Table 12).
Polymorphisms in genes linked to oculocutaneous albinism (OCA) were significantly associated with refractive error (Supplementary Table 13), although typically association was found for SNPs not strongly associated with other pigmentation traits31. Strong association with refractive error was found near the OCA2 gene causing OCA type 2 (p=1.37×10−15, rs79406658), OCA3 (TYRP1, p=1.18×10−11, rs62538956), OCA5 (SLC39A8, p=4.03×10−17, rs13107325), OCA6 (C10orf11, p=1.73×10−16, rs12256171). In addition, significant association was found near genes linked to ocular albinism (OA) on chromosome X (TBL1X and GPR14332, p=2.20×10−18, rs34437079) and Hermansky-Pudlak Syndrome albinism (BLOC1S1, p=2.4610−22, for rs80340147; note that this gene forms a conjoint read-through transcript the BLOC1S1-RDH5 with RDH5). Other associated markers were located within genes involved in systemic pigmentation also previously associated with refractive error7, such as RALY (p=3.14×10−18, rs2284388), TSPAN10 (p=2.22×−50, rs9747347), as well as melanoma (MCHR2, p=2.37×10−15 for rs4839756).
Functional properties of the associated markers
Among the significantly associated markers, 367 unique markers were frameshift or missense variants (Supplementary Table 14). Several are non-synonymous, such as the Arg141Leu mutation (rs1048661) within LOXL1, a gene that causes pseudoexfoliation syndrome and glaucoma33 and Ala69Ser (rs10490924) in ARMS2, associated with increased susceptibility to age-related macular degeneration34. Other associated variants with predicted deleterious consequences were located in several genes, such as RGR (p=6.89×10–68, rs1042454), a gene previously associated with refractive error7,10 and also retinitis pigmentosa35, and within the FBN1 gene, near clusters of mutations that cause Marfan Syndrome and anterior segment dysgenesis36.
Because the functional link between other associated variants and development of refractive error phenotypes is less obvious, we next performed gene-set enrichment analyses to identify properties that are significantly shared by genes identified by the meta-analysis. An enrichment analysis of Gene Ontology processes (Supplementary Table 15) found enrichment for genes participating in RNA Polymerase II transcription regulation (p=1×10−06) and nucleic acid binding transcription factor activity (p=1.10×10−06), suggesting that many of the genetic associations we identified interfere with gene expression. “Eye development” (p=6.10×10−06) and “Circadian regulation of gene expression” (p=1.10×10−04) were also significantly enriched.
A transcription factor binding site (TFBS) enrichment analysis identified significant (FDR < 0.05) over-representation of sites targeted by GATA4, EP300, RREB1, for which association was observed in the meta-analyses (Supplementary Table 16). Binding sites of transcription factors involved in eye morphogenesis and development such as MAF (whose mutations cause autosomal cataract), FOXC1 and PITX2 (anterior segment dysgenesis) or CRX (cone-rod dystrophy) were also enriched. CRX and PAX4, binding sites were also significantly enriched; these transcription factors are two of the regulators of circadian rhythm and melatonin synthesis37 alongside OTX2, for which SNP significant association was observed in our refractive error meta-analysis (p=6.15×10−11 for rs928109). All of these enriched gene-sets are observed for the first time in a GWAS analysis, although the presence of some of the mechanisms that relate them to refractive error and myopia were hypothesized before38.
Many of the variants associated with refractive error in our analyses were located within or near genes that are expressed in numerous body tissues (Extended Data Figure 4), and in particular from the nervous system, consistent with our evidence of extraocular, central nervous system involvement in refractive error. Within the eye, these genes were particularly strongly expressed in eye tissues such as cornea, ciliary body, trabecular meshwork39 and retina40 (Extended Data Figure 5, Supplementary Table 17). A stratified LD score regression applied to specifically expressed genes (LDSC-SEG)41 revealed the results of the GWAS are most strongly correlated with genes expressed in the retina and basal ganglia in the central nervous system but these correlations are not significant after multiple testing correction (Extended Data Figure 6 and Supplementary Table 18). It is possible that the strength of these correlations was constrained by the fact that in most cases, available expression levels were measured in adult samples, while refractive error and myopia are primarily developed in younger ages.
A Summary data-based Mendelian Randomization (SMR) analysis42 integrating GWAS with eQTL data from peripheral blood43 and brain tissues44 found concomitant association with refractive error and eQTL transcriptional regulation effects for 159 and 97 genes respectively (Supplementary Tables 19 and 20). A similar analysis integrating GWAS summary data with methylation data from brain tissues found association with both refractive error and changes in methylation for 134 genes (Supplementary Table 21).
Genetic effects shared between refractive error and other conditions
Examining the GWAS Catalog45, some of the genetic variants reported here were previously associated with refractive error, and with other traits, in particular intraocular pressure, intelligence and education; the latter two are known myopia risk factors (Supplementary Table 22). We used LD score regression to assess the correlation of genetic effects between refractive error and other phenotypes from GWAS summary statistics (Supplementary Table 23). refractive error genetic risk was significantly correlated with intelligence, both in childhood46 (rg=−0.27, p=4.76×10−09) and adulthood (fluid intelligence score rg=−0.25, p=1.56×10−39), educational attainment (defined as the number of years spent in formal education, rg=−0.24, p=3.36×10−54), self-reported cataract (rg=−0.31, p=4.70×10−10) and intraocular pressure (IOP, rg=−0.14, p=1.04×10−12).
Higher educational attainment appears to cause myopia as demonstrated by Mendelian randomization (MR) studies47. A gene by environment interaction GWAS for spherical equivalent and educational attainment (using age at completion of formal full-time education as a proxy) was conducted in 66,242 UK Biobank participants. Despite the relatively well-powered sample, only one locus yielded evidence of statistically significant interaction (rs536015141 within TRPM1, p=2.35×10−09, Supplementary Table 24), suggesting that the true relationship between refractive error and education is compounded by several factors and may not be linear in nature, as suggested recently48. TRPM1 is localized in rod ON bipolar cell dendrites, and rare mutations cause congenital stationary night blindness49, often associated with high myopia.
To further explore the nature of the relationship between refractive error and IOP, we built MR models using genetic effects previously reported for IOP50. On average, every 1 mmHg increase in IOP predicts a 0.05–0.09 diopters decrease in spherical equivalent (Supplementary Table 25, Extended Data Figure 7). We also built a MR model to assess the relationship between intelligence and spherical equivalent, but statistical evidence in this case points towards genetic pleiotropy rather than causation (Supplementary Table 26). This suggests that both myopia and intelligence are often influenced by the same factors, but without direct causal path linking one to the other. We found no significant genetic correlations between refractive error and the glaucoma endophenotype vertical cup to disc ratio (rg=−0.01, p=0.45), or hair pigmentation (rg=−0.03, p=0.35). Therefore, refractive error and pigmentation may have different allelic profiles with limited sharing of genetic risk.
Conditional analysis and risk prediction
We subsequently carried out a conditional analysis51 on the meta-analysis summary results and found a total of 904 independent SNPs significantly associated with refractive error. 890 of these markers were available in the EPIC-Norfolk Study, an independent cohort that did not participate in the refractive error meta-analysis (Extended Data Figure 8). These markers alone explained 12.1% of the overall spherical equivalent phenotypic variance in a regression model or 18.4% (SE=0.04) of the spherical equivalent heritability. Newly associated markers found in our meta-analysis, but not in the previous large GWAS7, explain 4.6% (SE=0.01) of the spherical equivalent phenotypic variance in EPIC-Norfolk Study, which is an improvement of one third compared to heritability explained by previously associated markers7.
Predictive models, based on the above-mentioned 890 SNPs, along with age and sex, were predictive of myopia (versus all non-myopia controls) with areas under the receiving operating characteristic curve (AUC) of 0.67, 0.74 and 0.75 (Figure 2), depending on the severity cutoff for myopia (≤ −0.75D, ≤ −3.00D and ≤ −5.00D respectively). The performance of the predictions appears not to improve for myopia definitions of −3.00D or worse, suggesting that the information extracted from our meta-analysis is more representative of the genetic risk for common myopia seen in the general population, than for more severe forms of myopia, which may have a distinctive genetic architecture.
Figure 2.

Receiver Operating Characteristic (ROC) curves for myopia predictions, using information from 890 SNP markers identified in the meta-analysis. The three different colors represent three different curves for each of the different definition of myopia: green – all myopia (< −0.75D), magenta – moderate myopia (< −3.00 D) and brown - severe myopia (defined as < −5.00 D).
Further exploration of refractive error genetic architecture
Using information from over half a million population-based participants SNPs identified in these analyses still only explain 18.4% of the spherical equivalent heritability. We next assessed how many common SNPs are likely to explain the entire heritable component of refractive error, and what sample sizes are likely to be needed in the future to identify them, using the likelihood-based approach described elswhere52. We estimate that approximately 13,808 (SE=969) polymorphic variants are likely to be behind the full refractive error heritability. Similar to other quantitative phenotypic traits that are previously published52, our analyses estimate that 10.3% (SE=1.0%) of the phenotypic variance is likely explained by a batch of approximately 543 (SE=81) common genetic variants of relatively large effect size and a further 20.8% (SE=0.9%) of the entire phenotypic variance explained by the remainder. With increased sample sizes, we project that the proportion of variance explained will continue to improve fast but will start plateauing for sample sizes above one million, after which further increases in sample size will likely yield ever diminishing additional phenotypic variance (Extended Data Figure 9).
Discussion
Our results provide evidence for at least two major sets of mechanisms in the pathogenesis of refractive error. The first affect intraocular pressure, eye structure, ocular development and physiology, and the second are CNS-related, including circadian rhythm control. Contributors to refractive error include all anatomical factors that alter refractive power relative to eye size, light transmittance, photoconductance and higher cerebral functions.
The findings implicate almost every single anatomical components of the eye, which along with the central nervous system participate in the development of refractive error. The healthy cornea contributes to 70% of the optical refractive power of the eyes53 and genes involved in corneal structure, topography and function may directly contribute to refractive error through direct changes in the corneal refraction. Our results show that several genes involved in lens development also contribute to refractive error in the general population. It is unclear if their contribution is mediated through alterations in biomechanical properties that affect eyes’ ability to accommodate, changes to the lens refractive index, or alterations in light transmission properties that impair the ability to focus images on the retina.
Many retinal genes are implicated in the development of refractive error, reflecting the role of light in mediating eye growth and the importance of the retina’s role in light transduction and processing7. Associations with refractive error at genes coding for gated ion channels and glutamate receptors point to the photoreceptor-bipolar cell interface as a potentially key factor in refractive error. Rare mutations in several of our associated genes cause night blindness, implicating the rod system in the pathophysiology of refractive error, but many also affect cone pathways. The TRPM1 gene, important for rod ON bipolar cell polarity54, is also implicated in the gene-education interaction analysis. Associations observed for the VSX1 and VSX2, its negative regulator, genes implicate the cone bipolar cells55.
The association with genes involved in pigmentation, including most of the OCA-causing genes, raises questions about the relationship between melanin, pigmentation and eye growth and development. These associations are unlikely to be influenced by any cryptic population structure in our samples, which our analyses were designed to control. None of the major pigmentation-associated SNPs31 was directly associated with refractive error and there was no significant correlation of genetic effects between refractive error and pigmentation.
The mechanisms linking pigmentation with refractive error are unclear. Foveal hypoplasia56 and optic disc57 dysplasias are common in all forms of albinism58. Although melanin synthesis is disrupted in albinism, both melanin and dopamine are synthesized through shared metabolic pathways. Disc and chiasmal lesions in albinism are often attributed to dopamine59, but we found limited evidence supporting an association with refractive error for genetic variants involved in dopamine signaling. The scarcity of association with refractive error for genes involved in dopamine-only pathways contrasts with the abundance of association for genes involved in pigmentation and melanin synthesis. This may suggest that melanin metabolism is connected to refractive error through other mechanisms that are independent from the metabolic pathways it shares with dopamine production. Melanin reaches the highest concentrations in the retinal pigment epithelium at the outmost layer of the retina, and anteriorly, in the iris and variations in pigmentation may affect the intensity of the light reaching the retina. Light exposure is a major protective factor for development of myopia60,61 It is possible that pigmentation plays a role in light signal transmission and transduction.
Animal model experiments suggest that in addition to local ocular mechanisms, emmetropization (the process by which the eye develops to minimize refractive error) is strongly influenced by the CNS62. The strong correlation of genetic risks between refractive error and intelligence and association found for genes linked to severe learning disability support an involvement of the CNS in emmetropization and refractive error pathogenesis.
Results from gene-set enrichment analysis demonstrate an interesting evolution with increasing sample sizes. While smaller previous studies were sufficiently powered to discover enrichment of low, cell-level properties, such as cation channel activity and participation in the synaptic space structures25, significantly more powered recent studies have found additional evidence for enrichment and involvement of more integrated physiological functions, such as light signal processing in retinal cells and others7. Beyond the identification of a much larger number of genes and explaining significantly higher proportions of heritability, our results, based in a considerably more statistically powered sample, uphold the previous findings and support the involvement of the same molecular and physiological mechanisms that were previously described.
In line with expectations from a higher power of association to discover genes and gene sets individually responsible for even smaller proportions of the refractive error variance63, we find evidence for even higher regulatory mechanisms, that act more holistically over the eye development or integrate eye growth and homeostasis with other processes of extraocular nature. For example, we found evidence that binding sites of transcription factors involved in the control of circadian rhythm are significantly enriched among genes associated with refractive error. Circadian rhythm is important in emmetropization and its disruption leads to myopia in animal knock-out models38, potentially through dopamine-mediated mechanisms, or changes in IOP and diurnal variations.
Most of the loci identified through our meta-analysis are not subject to particularly strong and systematic evolutionary pressures (Extended Data Figure 10). The variability in minor allele frequencies observed across loci associated with refractive error may therefore be the result of genetic drift. However, given the variety of the different visual components whose disruptions can result in refractive error, this variability may also be the result of overall balancing forces which encourage high allelic diversity of genes involved in refractive error, providing additional buffering capacity to absorb environmental pressures48 or genetic disruptions on any of the individual components of the visual system.
Our results cast light on potential mechanisms that contribute to refractive error in the general population and have identified the genetic factors that explain a considerable proportion of the heritability and phenotypic variability of refractive error. This allows us to improve significantly our ability to make predictions of myopia risk and generate novel hypotheses on how multiple aspects of visual processing affect emmetropization, which may pave the way to personalized risk management and treatment of refractive error in the population in the future.
Online Methods
Study Participants
The UK Biobank
The UK Biobank is a multisite cohort study of UK residents aged 40 to 69 years who were registered with the National Health Service (NHS) and living up to 25 miles from a study center. Detailed study protocols are available online (http://www.ukbiobank.ac.uk/resources/ and http://biobank.ctsu.ox.ac.uk/crystal/docs.cgi). It was conducted with the approval of the North-West Research Ethics Committee (ref 06/MRE08/65), in accordance with the principles of the Declaration of Helsinki, and all participants gave written informed consent.
Two separate groups of UK Biobank participants were included in these analyses. The first included participants whose refractive error was directly measured (non-cycloplegic autorefraction using the Tomey RC 5000 Auto Refkeratometer, Tomey Corp., Nagoya, Japan). Direct measurements of refractive errors were available for 22.7% of the UK Biobank sample. To ensure reliable and accurate refractive error data, previously published QC criteria were applied64. The spherical equivalent was calculated as spherical refractive error (UK Biobank codes 5084 and 5085) plus half the cylindrical error (UK Biobank 5086 and 5087) for each eye.
The second UK Biobank group included participants without direct measurement of refractive error. These participants refractive error status was inferred using questionnaire and other indirect data. Available demographic and clinical information were used to obtain an estimate about the individual’s likely myopia status. A Support Vector Machine (SVM) model, with age, sex, age of first spectacle wear and year of birth as prediction parameters was used to infer participants’ myopia status. Initial training took place in 80% randomly selected UK Biobank participants of European descent for whom direct spherical equivalent and refractive error status were available. Then the performance was assessed in the remaining 20% of UK Biobank participants of European descent for whom direct spherical equivalent and refractive error status were available. Finally, the SVM predictions in the remaining individuals with no direct spherical error measurements available using the model developed for the training data.
All UK Biobank genotypes were obtained as described elsewhere65. The UK Biobank team then performed imputation from a combined Haplotype Reference Consortium (HRC) and UK10K reference panel. Phasing on the autosomes was carried out using a modified version of the SHAPEIT266 program modified to allow for very large sample sizes. Only HRC-imputed variants were used for the purpose our analyses of the UK Biobank participants. The variant-level quality control exclusion metrics applied to imputed data for GWAS included the following: call rate < 95%, Hardy–Weinberg equilibrium P <1 × 10−6, posterior call probability < 0.9, imputation quality < 0.4, and MAF < 0.005. The Y chromosome and mitochondrial genetic data were excluded from this analysis. In total, 10,263,360 imputed DNA sequence variants were included in our analysis. Non-European ancestry and participants with relatedness corresponding to third-degree relatives or closer, samples with excess of missing genotype calls or heterozygosity were excluded. In total, genotypes were available for 102,117 participants of European ancestry with spherical equivalent data.
Association models in the first UK Biobank subset used the average of spherical equivalent as the outcome and allele dosages at each genetic locus as predictors. Mixed linear regressions, adjusting for age, sex and the first 10 principal components, implemented in the Bolt-LMM software67 were used.
For the second UK Biobank subset, for which no direct spherical equivalent measurement was available, the mixed linear model was built with the predicted myopia status as outcome and using the same covariates as for the previously described linear regression analysis on spherical equivalent. Odds Ratios were obtained from the beta regression coefficient using the equation:
where μ is the fraction of the cases in the sample (μ=0.606). Genotypes with MAF <0.01 and MAC< 400 were removed from analyses in this group.
23andMe
Participating subjects were all volunteers from the 23andMe (Mountain View, CA, USA) personal genomics company customer base. All participants provided informed consent and answered surveys online according to the approved 23andMe human subjects protocol, which was reviewed and approved by Ethical & Independent Review Services, a private institutional review board (http://www.eandireview.com). The participants were identified as myopia cases if they self-reported a diagnosis of myopia or suffering from symptoms of myopia (see Supplementary Notes for more detail).
DNA extraction and genotyping were performed on saliva samples by CLIA-certified and CAP-accredited clinical laboratories of Laboratory Corporation of America. Samples were genotyped on one of four genotyping platforms and batches (Illumina HumanHap550, BeadChip, SNPs, Illumina OmniExpress, plus a variable number of custom SNP assays). Only samples with more than 98.5% genotyping success rate were included. Ethnic categorization was conducted using a support vector machine (SVM) which classified individual haplotypes into one of the 31 reference populations derived from public datasets (the Human Genome Diversity Project, HapMap, and 1000 Genomes), as well as 23andMe customers who have reported having four grandparents from the same country. Genotypes were imputed against the September 2013 release of 1000 Genomes Phase1 reference haplotypes using a Beagle haplotype graph-based phasing algorithm for the autosomal and Minimac268 for X Chromosome loci.
Association test results were computed by linear regression assuming additive allelic effects using imputed allele dosages. Covariates for age, gender, the first ten principal components to account for residual population structure were also included into the model.
The Genetic Epidemiology Research in Adult Health and Aging (GERA) cohort
GERA is part of the Kaiser Permanente Research Program on Genes, Environment, and Health (RPGEH) and has been described in detail elsewhere69. It comprises adult men and women consenting members of Kaiser Permanente Northern California (KPNC), an integrated health care delivery system, with ongoing longitudinal records from vision examinations. For this analysis, 34,998 adults (25 years and older), who self-reported as non-Hispanic white, and who had at least one assessment of spherical equivalent obtained between 2008 and 2014 were included. All study procedures were approved by the Institutional Review Board of the Kaiser Foundation Research Institute. Participants underwent vision examinations, and most subjects had multiple measures for both eyes. Spherical equivalent was assessed as the sphere + cylinder/2. The spherical equivalent was selected from the first documented assessment, and the mean of both eyes was used. Individuals with histories of cataract surgery (in either eye), refractive surgery, keratitis, or corneal diseases were excluded from further analyses.
DNA samples from GERA individuals were extracted from Oragene kits (DNA Genotek Inc., Ottawa, ON, Canada) at KPNC and genotyped at the Genomics Core Facility of the University of California, San Francisco (UCSF). DNA samples were genotyped using the Affymetrix Axiom arrays (Affymetrix, Santa Clara, CA, USA). SNPs with initial genotyping call rate ≥97%, allele frequency difference ≤0.15 between males and females for autosomal markers, and genotype concordance rate >0.75 across duplicate samples were included. In addition, SNPs with genotype call rates <90% were removed, as well as SNPs with a minor allele frequency (MAF) < 1%.
Imputation pre-phasing of genotypes was done using Shape-IT v2.r7271966, variants were imputed from the cosmopolitan 1000 Genomes Project reference panel (phase I integrated release; http://1000genomes.org) using IMPUTE2 v2.3.070. Variants with an imputation IMPUTE r2 < 0.3 were excluded, and analyses were restricted to SNPs that had a minor allele count (MAC) ≥ 20.
For each SNP locus, linear regressions of each individual’s spherical equivalent were performed with the following covariates: age at first documented spherical equivalent assessment, sex, and genetic principal components using PLINK v1.9 (www.cog-genomics.org/plink/1.9/). Data from each SNP were modeled using additive dosages to account for the uncertainty of imputation. The top 10 ancestry PCs were included as covariates, as well as the percentage of Ashkenazi ancestry to adjust for genetic ancestry, as described previously69.
The Consortium for Refractive Error And Myopia (CREAM)
All participants selected for this study were of European descent, 25 years of age or older. refractive error was represented by measurements of refraction and spherical equivalent (Spherical equivalent = spherical refractive error +1/2 cylinder refractive error) was the outcome variable for CREAM. Participants with conditions that could alter refraction, such as cataract surgery, laser refractive procedures, retinal detachment surgery, keratoconus, or ocular or systemic syndromes were excluded from the analyses. Recruitment and ascertainment strategies varied by study and were previously published elsewhere71.
The genotyping process has been described elsewhere71. Samples were genotyped on different platforms, and study-specific QC measures of the genotyped variants were implemented before association analysis. Genotypes were imputed with the appropriate ancestry-matched reference panel for all cohorts from the 1000 Genomes Project (Phase I version 3, March 2012 release). Quality control criteria used for SNP and sample inclusions These metrics were similar to those described in a previous GWAS analyses and detailed information for each cohort is described elsewhere71.
To avert sample overlap, cohorts from the United Kingdom (1985BBC, ALSPAC-Mothers, EPIC-Norfolk, ORCADES and Twins UK) were excluded from the GWAS meta-analysis. Association analyses were performed as described elsewhere71For each individual cohort, a single-marker analysis for the phenotype of SphE (in diopters) was carried out with linear regression with adjustment for age, sex and up to the first five principal components. For all non-family-based cohorts, one of each pair of relatives was removed. In family-based cohorts, mixed model-based tests of association were used to adjust for within-family relatedness.
The European Prospective Investigation into Cancer (EPIC) Study
The EPIC-EPIC is one of the UK arms of a broad pan-European prospective cohort study designed to investigate the etiology of major chronic diseases72. This study was conducted following the principles of the Declaration of Helsinki and the Research Governance Framework for Health and Social Care. The study was approved by the Norfolk Local Research Ethics Committee (05/Q0101/191) and East Norfolk & Waveney NHS Research Governance Committee (2005EC07L). All participants gave written, informed consent. Refractive error was measured in both eyes using a Humphrey Auto-Refractor 500 (Humphrey Instruments, San Leandro, California, USA). Spherical equivalent was calculated as spherical refractive error plus half the cylindrical error for each eye.
The EPIC-Norfolk participants were genotyped using the Affymetrix UK Biobank Axiom Array (the same array as used in UK Biobank); 7,117 contributed to the current study. SNP exclusion criteria included: call rate < 95%, abnormal cluster pattern on visual inspection, plate batch effect evident by significant variation in minor allele frequency, and/or Hardy-Weinberg equilibrium P < 10-7. Sample exclusion criteria included: DishQC < 0.82 (poor fluorescence signal contrast), sex discordance, sample call rate < 97%, heterozygosity outliers (calculated separately for SNPs with minor allele frequency >1% and <1%), rare allele count outlier, and impossible identity-by-descent values. Individuals with relatedness corresponding to third-degree relatives or closer across all genotyped participants were also removed from further analyses. Following these steps all participants were of European descent. Data were pre-phased using SHAPEIT66 version 2 and imputed to the Phase 3 build of the 1000 Genomes project74 (October 2014) using IMPUTE70 version 2.3.2.
The relationship between allele dosage and mean spherical equivalent was analyzed using linear regression adjusted for age, sex and the first 5 principal components. Analyses were carried out using SNPTEST version 2.5.1.
Statistical analyses
We conducted two meta-analyses. For the initial meta-analysis (discovery), we used summary statistic results from the UK Biobank 1st and 2nd subset, the GERA and 23andMe Studies.
For the final meta-analysis, we used all available information (UK Biobank 1 and 2, the GERA, 23andMe and CREAM Consortium).
For all meta-analyses we applied a Z-score method, weighted by the effective population sample size, as implemented in METAL75. No genomic control adjustment was applied during the meta-analysis.
The effective population size was calculated per each locus and as was equal to the total sample size if a linear regression or linear mixed model were used. For case-control studies the effective population was calculated as:
as recommended before76, where N.eff is the effective sample size, N.cases is the number of cases considered to have myopia and N.controls is the number of subjects considered not to have myopia. Following this method, we calculated that for the full-sample analysis of 542,934 subjects, due to the presence of two case-control cohort, our effective sample sizes was 379,227.
Only SNPs with minor allele frequency of at least 1%, which were available from at least 70% of the maximum number of participants across all studies and that were not missing in more than one strata (cohorts), were considered further.
Conditional analyses were conducted using the conditional and joint analysis on summary data (COJO) as implemented in the GCTA program77 to identify independent effects within associated loci as well as the calculation of the phenotypic variance explained78 by all polymorphisms associated with the trait after the conditional analyses. The threshold of significance was set at 5×10−08 and the collinearity threshold was set at r2=0.9.
Genomic inflation was assessed using the package ‘gap’ in R (https://cran.r-project.org/) and to distinguish between the effect of polygenicity and those arising from sample stratification or uncontrolled population admixture, the LD score regression intercepts were calculated using the program LD Score (https://github.com/bulik/ldsc).
Bivariate genetic correlations between refractive error and other complex traits whose summary statistics are publicly available were assessed following previously described methodologies79, using the program LD Score (https://github.com/bulik/ldsc).
To assess the potential value of the loci associated with refractive error to predict myopia, regression-based models were trained and tested separately in two separate groups. The training set comprises the European UK Biobank participants for whom the spherical equivalent measurements were available. The models were tested in the EPIC-Norfolk cohort, which was not part of any of the analyses through which the genetic associations were identified.
The model in included age, sex, and the major genetic variants associated with refractive error after the conditional analysis. Three different definitions of myopia were used based on sliding spherical equivalent thresholds: M1 ≤ −0.75D, M2 ≤ −3.00 D and M3 ≤ −5.00D. These three different definitions of myopia were chosen to correspond to the generally accepted definitions of “any myopia”, “moderate myopia” and “high myopia”. For the latter, we opted for the −5.00D, because definitions based on the more stringent threshold of ≤ −6.00D would have not allowed for a sufficient number of cases in the testing set. For the purpose of these analyses, a “control” was any subject who did not have myopia, defined by a mean spherical equivalent ≥ −0.5D.
A Receiver Operating Characteristic (ROC) curve was drawn for each case and the Area Under the Curve (AUC) was calculated. R programming language and software environment for statistical computing (https://cran.r-project.org/) was used for both the logistic regression models (‘glm’) and to evaluate the performance of the model (‘ROCR’).
Polymorphisms associated at a GWAS level (P<5×10−08) were clustered within an “associated genomic region”, defined as a contiguous genomic region where GWAS-significant markers were within 1 million base pairs from each other. Significant polymorphisms were annotated with the gene inside whose transcript-coding region they are located, or alternatively, if located between two genes, with the gene nearest to it. The associated genomic regions were collectively annotated with the gene overlapping, or nearest the most significantly associated variant within that region.
The known relationships between identified genetic loci and other phenotypic traits were derived from two datasets: the Online Mendelian Inheritance In Man (OMIM, https://omim.org), which is a continuously curated catalog of human genes and phenotypic changes their polymorphic forms cause in humans and the GWAS Catalog80 which is a curated catalog of previous GWAS association of SNPs or genes with other phenotypic traits.
The R (https://cran.r-project.org) package MendelianRandomization v3.4.4 was used for Mendelian randomization analyses.
Disease-relevant tissues and cell types were identified by analyzing gene expression data together with summary statistics from the meta-analysis of refractive error in all five cohorts, as described elsewhere81. Expression data was obtained from the following sources: 1) the GTEx release v7 (https://gtexportal.org/home/datasets) 2) Fetal and adult corneal, trabecular meshwork and ciliary body RNA sequencing data previously described 82 and 3) data from the subset of subjects with presumed healthy adult retinas (AMD=1) from datasets described elsewhere83.
As the transcription data were heterogeneous and in different units, expression levels for all tissues were rank-transformed. Hierarchical clustering was used to help visualize similarities and differences of patterns of transcript expression across different tissues (‘hclust’ package in R).
SMR (Summary data–based Mendelian randomization) uses GWAS variants as instrumental variables and gene expression levels or methylation levels as mediating traits, in order to test whether the causal effect of a specific variant on the phenotype-of-interest acts via a specific gene84. The SMR tests were performed used three different: the summary statistics of eQTL associations in the untransformed peripheral blood samples of 5,311 subjects85, as well as eQTL effects and cis- methylation analysis (cis-mQTL), both in brain tissues86.
The Gene-Set Enrichment Analysis (GSEA) was implemented in the MAGENTA software87. We used the versions from September 2017.
Results of three statistical tests for natural selection were imported from the 1000 Genomes Selection Browser88.
Extended Data
Extended Data Fig. 1:

Principal components plots of the subjects in the main participating cohorts. a) UK Biobank (including the 102,117 subjects with direct refraction measurement and the imputed 108,956 likely myopes to 70,941 likely non-myopes, for a total of 179,897 subjects) , B) Genetic Epidemiology Research on Adult Health and Aging (GERA, N=34,998 ), C) 23andMe (106,086 cases and 85,757 controls, or 191,843 subjects in total).
Extended Data Fig. 2:

Correlation of effect sizes between the discovery cohort meta-analysis. Effect sizes are from two analyses, discovery (UK Biobank analysis on spherical equivalent + GERA, spherical equivalent + 23andMe, self-reported myopia cases and controls + UK Biobank inferred myopia cases and controls, for a total of N=508,855 subjects) and the replication from the non-British CREAM Consortium participants (N=34,079), used as replication. The z-scores for the discovery are on the y-axis and those from the CREAM cohort in the x-axis.
Extended Data Fig. 3:

Distribution of the base-pair length (red) of the 449 regions associated in the meta-analysis of all available cohorts (from Supplementary Table 3), alongside the distribution of number of SNPs (blue) for each region. Numbers in each of the axes in the figure are differentially colored to match the density curve they correspond to: red for the length of the region and blue for the number of SNPs.
Extended Data Fig. 4:
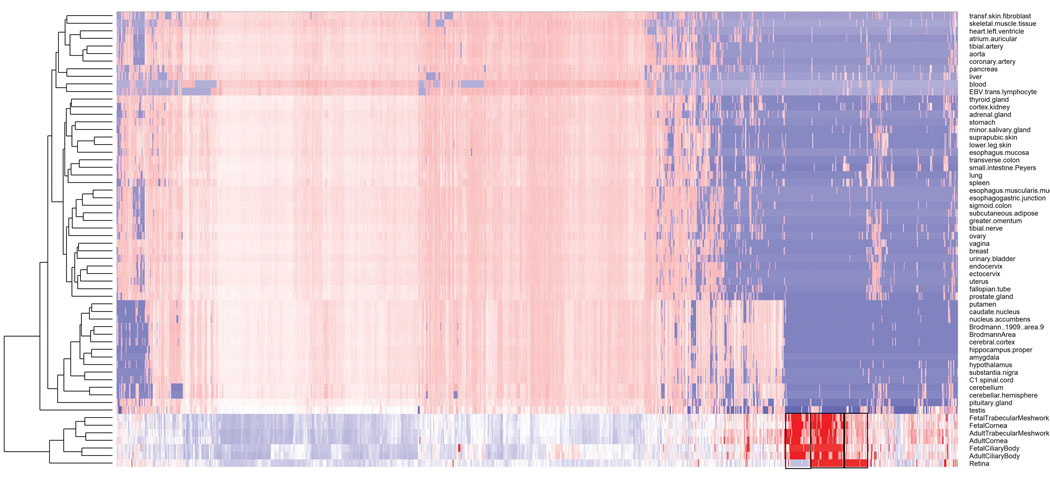
Expression of genes located in the associated loci (from Supplementary Table 3) along the x-axis, across several human body tissues (y-axis). The colors represent the centile ranking of the expression level of the gene in the tissue of interest. The hotter colors represent higher ranking of the gene expression and the colder colors low expression. Both genes and tissues are clustered in accordance with their pattern similarity. The symbol of all the genes could not be visualized and therefore are removed for the sake of clarity. Eye tissues, whether fetal or adult, appear to have similar patterns of gene expressions (clustered together at the bottom of the figure). Genes that are highly expressed in eye tissues fall in three clusters, shown with a black box. These clusters are displayed in more detail in Figure 4A, B and C.
Extended Data Fig. 5:

Genes from the regions associated with RE (from Supplementary Table 3) that are particularly expressed in eye tissues, compared to non-ocular tissues. These clusters are those highlighted in Supplementary Figure 3, but for the sake of clarity they are shown in transposed orientation compared to the previous figure (here genes in the y-axis and eye tissues in the x-axis), but same color codes as before. The dendrograms represent the degree of similarity observed for both tissues and gene expressions. The clusters are given in the order in which they were clustered together, from left to right: A) genes that are expressed more in other ocular tissues (fetal and adult) but much less in the adult retina. B) genes that are highly expressed in the retina and other ocular tissues, and C) genes that are expressed in the retina, but less in the other ocular tissues tested.
Extended Data Fig. 6:

Results of the LD score regression analysis applied to specifically expressed genes (LDSC-SEG) on multiple tissue for the meta-analysis results. Each point represents one tissue or cell line (along the x-axis) and the log10 value of the p-value for the enrichment of the meta-analysis results among genes expressed in these tissues. There were 205 tests carried out, one in each tissue and cell line, therefore only tissues with a correlation p-value< 0.00025 (Log_P> 3.6 in this figure), would have been significant after multiple testing. This condition was not fulfilled for any of the available tissues.
Extended Data Fig. 7:

Mendelian randomization results on causality of IOP over refractive error. Single points in the graph represent coordinates determined by the effect of each specific SNP over IOP (x-axis, mmHg) and spherical equivalent (y-axis, Diopter units). A total of 73 SNPs associated with IOP, but not directly associated with refractive error (i.e. p> 0.05) were selected as instruments. Values of associations with IOP were obtained from a meta-analysis of 139,555 European participants (Reference 50 in the manuscript) and the refractive error associations from 102,117 UK Biobank subjects. The lines represent the regression lines from each model, as specified in the figure legend. In some cases, these lines may not visible because they overlap (please refer to the values underneath the figure).
Extended Data Fig. 8:

Venn’s Diagram of the number of SNPs considered in each of the stages of this study. The different circles represent various stages, inclusion in the meta-analysis (blue), identification of significant loci (green), conditional analysis results identifying independent effects (red) and the total number of SNPs available for inclusion in prediction and heritability estimation in the independent (i.e. not part of the original meta-analysis) EPIC-Norfolk cohort (orange).
Extended Data Fig. 9:

Prediction for the total number of SNPs and phenotypic variance explained as a function of GWAS sample size in future studies, based on the distribution of effects observed in the current meta-analysis. The plot lines show the predicted relationship between the number of loci associated with refractive error (left vertical axis, blue line) and the variance they help explain (red line, right vertical axis), as a function of the sample size (x-axis) used in future GWAS or meta-analyses. These projections are consistent with the observed results, where an effective sample of 379,227 identified 904 independent signals after a conditional analysis, explaining 12–16% of refractive error variability.
Extended Data Fig. 10:

The distribution of various natural selection test scores for SNPs associated with refractive error. The values on the x-axis represent the ranking in terms of natural selection observed and the y-axis the density of that rank. The different tests shown are iHS, XP-EHH (CEU vs YRI), XP-EHH average score, XP-EHH maximum score and Tajima scores (black, green, red, blue and yellow respectively).
Supplementary Material
Acknowledgements
P.T.K. and P.J.F oversaw the UK Biobank eye data acquisition with support from The National Institute for Health Research (NIHR); Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology. The UK Biobank Eye and Vision Consortium was supported by grants from UK NIHR (BRC3_026), Moorfields Eye Charity (ST 15 11 E), Fight for Sight (1507/1508), The Macular Society, The International Glaucoma Association (IGA, Ashford UK) and Alcon Research Institute. V.V. is supported by a core UK Medical Research Council grant MC_UU_00007/10.
23andMe thanks research participants and employees of 23andMe for making this work possible (list of contributing staff in the Supplementary Note).
Genotyping of the GERA cohort was funded by the US National Institute on Aging; National Institute of Mental Health and National Institute of Health Common Fund (RC2 AG036607); data analyses by the National Eye Institute (NEI R01 EY027004, E.J.), National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK116738, E.J.).
The CREAM GWAS meta-analysis was supported by European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Programme (grant 648268 to C.C.W.K), the Netherlands Organisation for Scientific Research (NWO, 91815655 to C.C.W.K) and the National Eye Institute (R01EY020483). V.J.M.V. acknowledges funding from the Netherlands Organisation for Scientific Research (NWO, grant 91617076).
S.M. acknowledges support from the National Health and Medical Research Council (NHMRC) of Australia (grants 1150144, 1116360, 1154543, 1121979).
EPIC-Norfolk infrastructure and core functions are supported by the Medical Research Council (G1000143) and Cancer Research UK (C864/A14136). Genotyping funded by the UK MRC (MC_PC_13048). A.K.P. is supported by a Moorfields Eye Charity grant. P.J.F. received support from the Richard Desmond Charitable Trust, the National Institute for Health Research to Moorfields Eye Hospital and the Biomedical Research Centre for Ophthalmology.
RW and PGH were supported by the National Eye Institute of the National institutes of Health under award number R21EY029309. M.J.S. is a recipient of a Fight for Sight PhD studentship. K.P. is a recipient of a Fight for Sight PhD studentship. P.G.H. the recipient of a FfS ECI fellowship. P.G.H. and C.J.H. acknowledge the TFC Frost Charitable Trust Support for the KCL Department of Ophthalmology. Statistical analyses were run in King’s College London Rosalind HPC LINUX Clusters and cloud servers. The UK Biobank data was accessed as part of the UK Biobank projects 669 and 17615.
J.S.R. is supported in part by the NIHR Biomedical Research Centres at Moorfields Eye Hospital/UCL Institute of Ophthalmology and at the UCL Institute of Child Health/Great Ormond Street Hospital and is an NIHR Senior Investigator. P.M.C. was funded by the Ulverscroft Foundation. O.A.M is supported by Wellcome Trust grant 206619_Z_17_Z and the NIHR Biomedical Research Centre at Moorfields Eye Hospital and the UCL Institute of Ophthalmology.
Footnotes
Competing interests
23andMe is a consumer genomics company.
Data availability
Summary statistics from the cohorts participating in the meta-analysis can be downloaded from ftp://twinr-ftp.kcl.ac.uk/Refractive_Error_MetaAnalysis_2020/ and public repositories such as the GWAS Catalogue (https://www.ebi.ac.uk/gwas/downloads/summary-statistics). These freely downloadable summary statistics are calculated using all cohorts described in this manuscript, except for the 23andMe participants. This is due to a non-negotiable clause in the 23andMe data transfer agreement, intended to protect the privacy of the 23andMe research participants.
To fully recreate our meta-analytic results, all bona fide researchers can obtain the 23andMe summary statistics by emailing 23andMe (dataset-request@23andme.com) and subsequently meta-analyzing them along the freely accessible summary statistics for all the other cohorts.
References:
- 1.Vos T et al. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. The Lancet 390, 1211–1259 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.WHO. The Global Burden of Disease. 2004 Update ISBN-13: 9789241563710. ISBN-10 651629118(2008). [Google Scholar]
- 3.Williams KM et al. Increasing Prevalence of Myopia in Europe and the Impact of Education. Ophthalmology 122, 1489–97 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanfilippo PG, Hewitt AW, Hammond CJ & Mackey DA The heritability of ocular traits. Surv Ophthalmol 55, 561–83 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Kiefer AK et al. Genome-wide analysis points to roles for extracellular matrix remodeling, the visual cycle, and neuronal development in myopia. PLoS Genet 9, e1003299 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verhoeven VJ et al. Genome-wide meta-analyses of multiancestry cohorts identify multiple new susceptibility loci for refractive error and myopia. Nat Genet 45, 314–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tedja MS et al. Genome-wide association meta-analysis highlights light-induced signaling as a driver for refractive error. Nat Genet 50, 834–848 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cumberland PM et al. Frequency and Distribution of Refractive Error in Adult Life: Methodology and Findings of the UK Biobank Study. PLoS One 10, e0139780 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kvale MN et al. Genotyping Informatics and Quality Control for 100,000 Subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) Cohort. Genetics 200, 1051–60 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pickrell JK et al. Detection and interpretation of shared genetic influences on 42 human traits. Nat Genet 48, 709–17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bulik-Sullivan BK et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 47, 291–5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dudbridge F & Gusnanto A Estimation of significance thresholds for genomewide association scans. Genet Epidemiol 32, 227–34 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pe’er I, Yelensky R, Altshuler D & Daly MJ Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet Epidemiol 32, 381–5 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Wood AR et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet 46, 1173–86 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oliveira J et al. LAMA2 gene analysis in a cohort of 26 congenital muscular dystrophy patients. Clin Genet 74, 502–12 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Colognato H et al. Identification of dystroglycan as a second laminin receptor in oligodendrocytes, with a role in myelination. Development 134, 1723–36 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Burkin DJ & Kaufman SJ The alpha7beta1 integrin in muscle development and disease. Cell Tissue Res 296, 183–90 (1999). [DOI] [PubMed] [Google Scholar]
- 18.Ervasti JM & Campbell KP Dystrophin-associated glycoproteins: their possible roles in the pathogenesis of Duchenne muscular dystrophy. Mol Cell Biol Hum Dis Ser 3, 139–66 (1993). [DOI] [PubMed] [Google Scholar]
- 19.Mayer U et al. Absence of integrin alpha 7 causes a novel form of muscular dystrophy. Nat Genet 17, 318–23 (1997). [DOI] [PubMed] [Google Scholar]
- 20.Jean D, Ewan K & Gruss P Molecular regulators involved in vertebrate eye development. Mech Dev 76, 3–18 (1998). [DOI] [PubMed] [Google Scholar]
- 21.Hammond CJ, Andrew T, Mak YT & Spector TD A susceptibility locus for myopia in the normal population is linked to the PAX6 gene region on chromosome 11: a genomewide scan of dizygotic twins. Am J Hum Genet 75, 294–304 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ali M et al. Null mutations in LTBP2 cause primary congenital glaucoma. Am J Hum Genet 84, 664–71 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clark AM et al. Negative regulation of Vsx1 by its paralog Chx10/Vsx2 is conserved in the vertebrate retina. Brain Res 1192, 99–113 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heon E et al. VSX1: a gene for posterior polymorphous dystrophy and keratoconus. Hum Mol Genet 11, 1029–36 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Hysi PG et al. Common mechanisms underlying refractive error identified in functional analysis of gene lists from genome-wide association study results in 2 European British cohorts. JAMA Ophthalmol 132, 50–6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma M et al. Wnt signaling in form deprivation myopia of the mice retina. PLoS One 9, e91086 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miyake M et al. Identification of myopia-associated WNT7B polymorphisms provides insights into the mechanism underlying the development of myopia. Nat Commun 6, 6689 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Cuellar-Partida G et al. WNT10A exonic variant increases the risk of keratoconus by decreasing corneal thickness. Hum Mol Genet 24, 5060–8 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Stone RA et al. Image defocus and altered retinal gene expression in chick: clues to the pathogenesis of ametropia. Invest Ophthalmol Vis Sci 52, 5765–77 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou H, Yoshioka T & Nathans J Retina-derived POU-domain factor-1: a complex POU-domain gene implicated in the development of retinal ganglion and amacrine cells. J Neurosci 16, 2261–74 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hysi PG et al. Genome-wide association meta-analysis of individuals of European ancestry identifies new loci explaining a substantial fraction of hair color variation and heritability. Nat Genet 50, 652–656 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fabian-Jessing BK et al. Ocular albinism with infertility and late-onset sensorineural hearing loss. Am J Med Genet A 176, 1587–1593 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Thorleifsson G et al. Common sequence variants in the LOXL1 gene confer susceptibility to exfoliation glaucoma. Science 317, 1397–400 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Rivera A et al. Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk. Hum Mol Genet 14, 3227–36 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Morimura H, Saindelle-Ribeaudeau F, Berson EL & Dryja TP Mutations in RGR, encoding a light-sensitive opsin homologue, in patients with retinitis pigmentosa. Nat Genet 23, 393–4 (1999). [DOI] [PubMed] [Google Scholar]
- 36.Robinson PN et al. Mutations of FBN1 and genotype-phenotype correlations in Marfan syndrome and related fibrillinopathies. Hum Mutat 20, 153–61 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Rohde K, Moller M & Rath MF Homeobox genes and melatonin synthesis: regulatory roles of the cone-rod homeobox transcription factor in the rodent pineal gland. Biomed Res Int 2014, 946075 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chakraborty R et al. Circadian rhythms, refractive development, and myopia. Ophthalmic Physiol Opt 38, 217–245 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carnes MU, Allingham RR, Ashley-Koch A & Hauser MA Transcriptome analysis of adult and fetal trabecular meshwork, cornea, and ciliary body tissues by RNA sequencing. Exp Eye Res 167, 91–99 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Ratnapriya R et al. Retinal transcriptome and eQTL analyses identify genes associated with age-related macular degeneration. Nat Genet 51, 606–610 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Finucane HK et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat Genet 50, 621–629 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu Z et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet 48, 481–7 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Westra HJ et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet 45, 1238–1243 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qi T et al. Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nat Commun 9, 2282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buniello A et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 47, D1005–D1012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benyamin B et al. Childhood intelligence is heritable, highly polygenic and associated with FNBP1L. Mol Psychiatry 19, 253–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mountjoy E et al. Education and myopia: assessing the direction of causality by mendelian randomisation. BMJ 361, k2022 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pozarickij A et al. Quantile regression analysis reveals widespread evidence for gene-environment or gene-gene interactions in myopia development. Commun Biol 2, 167 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Audo I et al. TRPM1 is mutated in patients with autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet 85, 720–9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Khawaja AP et al. Genome-wide analyses identify 68 new loci associated with intraocular pressure and improve risk prediction for primary open-angle glaucoma. Nat Genet 50, 778–782 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang J et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet 44, 369–75, S1–3 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y, Qi G, Park JH & Chatterjee N Estimation of complex effect-size distributions using summary-level statistics from genome-wide association studies across 32 complex traits. Nat Genet 50, 1318–1326 (2018). [DOI] [PubMed] [Google Scholar]
- 53.Zadnik K et al. Normal eye growth in emmetropic schoolchildren. Optom Vis Sci 81, 819–28 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Li Z et al. Recessive mutations of the gene TRPM1 abrogate ON bipolar cell function and cause complete congenital stationary night blindness in humans. Am J Hum Genet 85, 711–9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chow RL et al. Vsx1, a rapidly evolving paired-like homeobox gene expressed in cone bipolar cells. Mech Dev 109, 315–22 (2001). [DOI] [PubMed] [Google Scholar]
- 56.Struck MC Albinism: Update on ocular features. Current Ophthalmology Reports 3, 232–237 (2015). [Google Scholar]
- 57.Mohammad S et al. Characterization of Abnormal Optic Nerve Head Morphology in Albinism Using Optical Coherence Tomography. Invest Ophthalmol Vis Sci 56, 4611–8 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Yahalom C et al. Refractive profile in oculocutaneous albinism and its correlation with final visual outcome. Br J Ophthalmol 96, 537–9 (2012). [DOI] [PubMed] [Google Scholar]
- 59.Lopez VM, Decatur CL, Stamer WD, Lynch RM & McKay BS L-DOPA is an endogenous ligand for OA1. PLoS Biol 6, e236 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Karouta C & Ashby RS Correlation between light levels and the development of deprivation myopia. Invest Ophthalmol Vis Sci 56, 299–309 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Wu PC, Tsai CL, Wu HL, Yang YH & Kuo HK Outdoor activity during class recess reduces myopia onset and progression in school children. Ophthalmology 120, 1080–5 (2013). [DOI] [PubMed] [Google Scholar]
- 62.Troilo D, Gottlieb MD & Wallman J Visual deprivation causes myopia in chicks with optic nerve section. Curr Eye Res 6, 993–9 (1987). [DOI] [PubMed] [Google Scholar]
- 63.de Leeuw CA, Neale BM, Heskes T & Posthuma D The statistical properties of gene-set analysis. Nat Rev Genet 17, 353–64 (2016). [DOI] [PubMed] [Google Scholar]
References
- 64.Cumberland PM et al. Frequency and Distribution of Refractive Error in Adult Life: Methodology and Findings of the UK Biobank Study. PLoS One 10, e0139780 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bycroft C et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Delaneau O, Marchini J & Zagury JF A linear complexity phasing method for thousands of genomes. Nat Methods 9, 179–81 (2011). [DOI] [PubMed] [Google Scholar]
- 67.Loh P-R, Kichaev G, Gazal S, Schoech AP & Price AL Mixed-model association for biobank-scale datasets. Nature genetics, 1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fuchsberger C, Abecasis GR & Hinds DA minimac2: faster genotype imputation. Bioinformatics 31, 782–4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Banda Y et al. Characterizing Race/Ethnicity and Genetic Ancestry for 100,000 Subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) Cohort. Genetics 200, 1285–95 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Howie BN, Donnelly P & Marchini J A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5, e1000529 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tedja MS et al. Genome-wide association meta-analysis highlights light-induced signaling as a driver for refractive error. Nat Genet 50, 834–848 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Riboli E & Kaaks R The EPIC Project: rationale and study design. European Prospective Investigation into Cancer and Nutrition. Int J Epidemiol 26 Suppl 1, S6–14 (1997). [DOI] [PubMed] [Google Scholar]
- 73.Hayat SA et al. Cohort profile: A prospective cohort study of objective physical and cognitive capability and visual health in an ageing population of men and women in Norfolk (EPIC-Norfolk 3). Int J Epidemiol 43, 1063–72 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Delaneau O, Marchini J, Genomes Project, C. & Genomes Project, C. Integrating sequence and array data to create an improved 1000 Genomes Project haplotype reference panel. Nat Commun 5, 3934 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Willer CJ, Li Y & Abecasis GR METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–1 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Winkler TW et al. Quality control and conduct of genome-wide association meta-analyses. Nat Protoc 9, 1192–212 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yang J, Lee SH, Goddard ME & Visscher PM GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88, 76–82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang J et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet 42, 565–9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bulik-Sullivan B et al. An atlas of genetic correlations across human diseases and traits. Nat Genet 47, 1236–41 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Buniello A et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 47, D1005–D1012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Finucane HK et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat Genet 50, 621–629 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Carnes MU, Allingham RR, Ashley-Koch A & Hauser MA Transcriptome analysis of adult and fetal trabecular meshwork, cornea, and ciliary body tissues by RNA sequencing. Exp Eye Res 167, 91–99 (2018). [DOI] [PubMed] [Google Scholar]
- 83.Ratnapriya R et al. Retinal transcriptome and eQTL analyses identify genes associated with age-related macular degeneration. Nat Genet 51, 606–610 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhu Z et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet 48, 481–7 (2016). [DOI] [PubMed] [Google Scholar]
- 85.Westra HJ et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet 45, 1238–1243 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Qi T et al. Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nat Commun 9, 2282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Segre AV et al. Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet 6(2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pybus M et al. 1000 Genomes Selection Browser 1.0: a genome browser dedicated to signatures of natural selection in modern humans. Nucleic Acids Res 42, D903–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.