

Abstract
Gene expression is precisely controlled in a stage and cell-type-specific manner, largely through the interaction between cis-regulatory elements and their associated trans-acting factors. Where these components aggregate in promoters and enhancers, they are able to cooperate to modulate chromatin structure and support the engagement in long-range 3D superstructures that shape the dynamics of a cell's genomic architecture. Recently, the term ‘super-enhancer’ has been introduced to describe a hyper-active regulatory domain comprising a complex array of sequence elements that work together to control the key gene networks involved in cell identity. Here, we survey the unique characteristics of super-enhancers compared to other enhancer types and summarize the recent advances in our understanding of their biological role in gene regulation. In particular, we discuss their capacity to attract the formation of phase-separated condensates, and capacity to generate three-dimensional genome structures that precisely activate their target genes. We also propose a multi-stage transition model to explain the evolutionary pressure driving the development of super-enhancers in complex organisms, and highlight the potential for involvement in tumorigenesis. Finally, we discuss more broadly the role of super-enhancers in human health disorders and related potential in therapeutic interventions.
INTRODUCTION
Enhancers are well known for their genomic location and orientation independent activity in the regulation of gene expression. The first eukaryotic enhancer was identified in a primate virus SV40 in the early 1980s. Banerji et al. observed that the remote viral element containing a 72-bp repeat sequence could enhance recombinant β-globin expression 200-fold when cohabitating in a plasmid construct transfected into mammalian cells (1). Two years later, several independent groups reported that enhancers located in the mouse immunoglobin (Ig) heavy chain gene loci could activate nearby Ig promoter in cis specifically in lymphocytes, which demonstrated for the first time that enhancers function in a tissue specific manner (2–4).
The term ‘super-enhancer’ was first used in 2004 by Chen and colleagues to describe a 651 bp segment of baculoviral genomic DNA designated hr3. They observed that this regulatory domain could stimulate activity of the ie-1 reporter gene promoter up to 7000-fold in transfected cells (5). Almost a decade later, Young and colleagues used the term ‘super-enhancer’ (SE) to characterize large genomic domains, conferring a key role in control of cell identity and disease (6–8). Using ChIP-seq data from the multiple tissue types available from the ENCODE and Roadmap Epigenome Projects (9,10), they were able to demonstrate that SEs span tens of kilobases (kb) of DNA sequence and are densely occupied by master transcription factors (TFs) and mediators. Collectively, these observations suggested that SEs play a key role in organizing the gene expression patterns that regulate cell identity (6–8). The Young definition of SE, in relation to developmentally important genomic segments, extends well beyond the early usage, which related to their performance in expression assays in vitro, and has become the established use of the terminology.
While there is now very substantial support for a paradigm in which SEs are a major regulatory component of the gene expression that shapes cell identity, there is an alternative view held by some researchers, that SEs are no more than clusters of enhancers (11,12) that contribute with additive effect on their target genes in a manner more similar to previously described locus control regions (LCRs) (13). Therefore, in view of this controversy, we consider it is timely to review our current knowledge of SEs and discuss the evidence in support of the range of opinions. In this work, we also recapitulate genome-wide identification and characterization of SEs and provide an online repository of a high-quality collection of SEs with meta-analysis. Furthermore, we explore the biological support for a role of SEs in gene regulation in light of the phase separation and three-dimensional (3D) genome organization models for SE action. We also propose an evolutionary framework to explain the emergence of SEs in complex organisms. Finally, we will discuss the involvement of SEs in human health disorders and their potential as targets for therapeutic interventions.
GENOME-WIDE IDENTIFICATION OF SUPER-ENHANCERS
Young and colleagues originally identified SEs at the genome-wide scale, based on ChIP-seq signal enrichment of Mediator subunit MED1 or master TFs, such as MyoD, T-bet and C/EBPα, in mouse embryonic stem cells (mESCs) and other tissues including myotube, T helper and macrophages (6,7). A similar strategy was also applied to other cell types using ChIP-seq data of acetylated histone H3 lysine 27 (H3K27ac), which is a surrogate epigenomic marker of active enhancers (8). The Young group also developed the ROSE software tool to facilitate SE identification in silico (7,8). This algorithm stitches closely-distributed enhancers identified from H3K27ac (or MED1/master TF) ChIP-seq data, ranks the stitched enhancers by their input-subtracted ChIP-seq signal, and finally separates SEs from typical enhancers by a graphic elbow point identified on the ranked ChIP-seq signal plot (Figure 1A). The output is slightly different for the different kinds of data input, such that the elbow points are usually sharper with MED1 than H3K27ac, and the final SE collections identified by the two marks are not in 100% agreement. To exclude the possibility of transcription start sites (TSS) overlapping with regions of SE calling, constituent enhancers are usually excluded from stitching if they are located within a ±2000 bp window flanking an annotated TSS (8).
Figure 1.

Identification and characteristics of super-enhancers. (A) Call of SEs with MED1 or H3K27ac ChIP-seq data, using ROSE algorithm which takes into account enhancer ranks and ChIP signals (6). Y-axis gives input-subtracted MED1 or H3K27ac ChIP-seq coverage, and x-axis shows the rank of ‘superness’ based on the value given on y-axis. Dashed lines in both directions indicate the cutoffs for separating SEs from typical enhancer. (B) Box-plots comparing the median size (upper) and the number (bottom) between SEs and typical enhancers (TEs) in 30 cell lines, 11 primary cells and 24 tissues available from the ENCODE project. (C) Box-plots comparing the median size (left) and the number (right) of SEs, stretch enhancers (StrEs), and typical enhancers (TEs) in eight selected cell lines. (D) Characteristic summary of SEs, in comparison to stretch enhancers (StrEs) and typical enhancers (TEs).
To the best of our knowledge there are currently three SE databases which gather published SEs and implement the ROSE algorithm to mine available ChIP-seq data, including dbSUPER (14), SEA (15), and SEdb (16). The most recent of these, SEdb, contains a collection of more than 331,000 SEs derived from 541 human cell lines/tissues. We also provide an online data repository of SE data, including a core collection of human SEs with comparative and exploratory analyses (discussed below in this Survey and Summary) to further support the biological investigation of these structures. This resource is available at https://sunlightwang.github.io/Super-Enhancers/ and will be continuously updated and expanded going forward.
UNIQUE CHARACTERISTICS OF SUPER-ENHANCERS
SEs are comprised of a small number of genomic loci of extremely large size
In a comparison of SEs and typical enhancers (TEs) in 30 cell lines, 24 tissues and 11 primary cell types available from the ENCODE project (10), it was noted that the median size of SEs in general spreads from 10 kb to over 60 kb, whereas the median size of TEs ranges from 1 kb to 4 kb, smaller by approximately one order of magnitude (6,7) (Figure 1B, upper). By contrast, when looking at the number of SEs and TEs in each cell type, the trend is exactly the opposite: SEs are fewer than TEs by one to two orders of magnitude (Figure 1B, bottom).
Around the time that these SE were described, another group independently reported enhancers of size >3 kb, and used the alternative nomenclature ‘stretch enhancer’ (StrE) to characterize their extraordinary length (17). Similar to SEs, StrEs are also found cell type specific and important in programming cell identity gene expression (17). Although SEs and StrEs share some properties, they are conceptually and functionally different in at least two respects. Firstly, while StrEs are determined by an arbitrary cut-off in genomic size (3 kb), SEs are discriminated from other enhancers in a parameter-free manner after clustered enhancers stitching (Figure 1A), which gives more weight to the biological essence of SEs. Secondly, extraordinarily strong TF binding and associated Mediator complex signals endow SEs with special biochemical properties, for example, facilitating them to form liquid-liquid phase-separated condensates (discussed later in this article). By contrast, any large enhancer can be designated a StrE, regardless of their biological activity.
In a comparison of SEs with TEs, StrEs in eight cell lines where collections of all the three enhancer types are available, both the small-number and extremely-large-size of SEs become more apparent (Figure 1C). For example, the median size of StrEs is in the order of a thousand base pairs, 2- to 3-fold larger than that of TEs, but still much smaller than their SE counterpart. While SEs are usually stitched up by constituent enhancers, between which there can be gaps of up to 12.5 kb (a cut-off used in the original SE paper), StrEs are defined as large enhancers or enhancer-like chromatin states based on hidden Markov model inference. Therefore, some caution should be applied where large gaps may prevent an account of synergistic influences from nearby domains in a StrE. When looking at the number of the three types of enhancers in each cell line, the StrEs are numerous, much more comparable to TEs than SEs (Figure 1C, D).
SEs specify cell identity
Within the high-quality SE collections derived from the ENCODE project, including the data from 65 samples described above, we charted the genome-wide SE landscape as shown in Figure 2A. The vast majority of SEs only appear in few or individual cell types (Figure 2B), supporting the assertion that SEs are highly cell type specific. To further explore this specificity, we performed hierarchical clustering based on the Jaccard distance that measures the SE location dissimilarity between every pair of all the 65 samples. A larger Jaccard distance is indicative of fewer overlapped SEs relative to the total number of all SEs occurring in the two sample comparison, and vice versa. As shown in Figure 2C, the hierarchical clustering indicates a clear separation between tissue samples (except for only three outliers) and cell lines / primary cells, indicating that SEs are also sensitive to the cellular growth environment. Indeed, environment-specific SEs were observed in resident macrophages (18). Principal component analysis (PCA) based on SE occurrence matrix of the 65 samples also confirmed the same phenomenon, even with a much clearer segregation of tissue samples from the others (Figure 2D). Primary cells scatter between tissue samples and cell lines when looking at the first component (i.e. the x-axis of Figure 2D), which accounts for nearly one third of the variance, confirming that both the growth potential and immortality of primary culture cells lie in between the tissue samples and cell lines. Intriguingly, the PCA visualization of the 65 samples based on SE occurrence is analogous to the analysis based on global gene expression, where cell lines show distinct gene expression profiles compared to normal tissues (19). It is also interesting to note that sub-clusters comprised of only primary cells or cell lines are observed, despite the fact that they are, in general, more intermingled (Figure 2C, D). When looking at a finer resolution in Figure 2C, cell lines derived from similar cell origins (e.g. prostate or blood), tend to be grouped together immediately, which is consistent with literature reports (8). Such phenomenon is also observed in tissues developed from a common pathway (e.g. thoracic aorta, ascending aorta and coronary artery).
Figure 2.

Super-enhancers and cell identity. (A) Genome-wide SE landscape of in total 65 samples (30 cell lines, 11 primary cells and 24 tissues) available from the ENCODE project. Bars on the chromosomes indicate the genomic loci of SEs, and colors indicate each SE appearing in how many different cell lines, primary cells, and tissues, respectively. (B) The number of SEs (y-axis) co-occurring in different number (x-axis) of cell lines, primary cells and tissues, respectively. The vast majority of SEs are not shared by different samples. (C) Hierarchical clustering of the 65 samples based on co-occurrence of SEs measured by the pairwise Jaccard distance. The colors in the dendrogram indicate the three major clusters, and the text colors of the sample names indicate the sample types (i.e. cell lines, primary cells or tissues). (D) Principal component analysis (PCA) of the 65 samples based on the SE co-occurrence matrix. The scatter plot shows the first two PCs, and the dot colors indicate the sample types (i.e. cell lines, primary cells, or tissues).
These observations suggest that SE profile can be used as a biomarker or proxy to categorize cell types and their cell growth conditions or status. Moreover, the fact that SEs differ among the same cell type in different niches also suggests that they are sensitive to external environmental signals (18). To date, SEs are becoming commonly used as molecular markers to sub-classify complex diseases in precision medicine, and this will be discussed later in this article.
SEs are densely bound by master TFs
It has been shown that enhancers can interact with promoters via long-range chromatin loops to activate gene transcription (20). These structures are mediated and stabilized by the complex cooperation between an array of cis-acting regulatory elements and trans-acting proteins that co-localize in each chromatin domain. In both human and eukaryotic model organisms, TFs are found densely bound to enhancers in a combinatorial manner and synergistically responsible for regulatory activity (21–26). SEs are densely co-occupied by master TFs and Mediator complex, and these master TFs establish auto-regulatory networks (6,27,28). In mouse ESCs, the three well-known pluripotent TFs, Oct4, Sox2 and Nanog are identified at SEs at extraordinarily elevated levels (6). In comparison to StrEs, SEs have a significantly higher level of chromatin accessibility surveyed by DNase I hypersensitivity assay and also denser master TF binding signals (29).
Interestingly, TF binding sites inside SEs are not evenly distributed but form a series of dense clusters, termed ‘constituents’ (6), ‘epicenters’ (30) or ‘hotspots’ (27). In a more dynamic system, Adam and colleagues took advantage of the hair follicle stem cell differentiation model, and observed the remodeling of SEs and their epicenters during lineage commitment (30). The cohort of TF binding sites was also altered following the change of SE locations. The location alteration was reversible and remarkably sensitive to microenvironment, allowing high plasticity for cells to adapt the environment for proliferation and lineage commitment. In this system, the pioneer factor SOX9 took the major responsibility to govern the landscape of SEs by protecting against H3K27me3 (histone H3 lysine 27 tri-methylation, a marker for inactive chromatin) and initiating enrichment of H3K27ac. In the aforementioned example, macrophages residing in five different tissue environments share only ∼40–50% of SEs (18). The pioneer factor PU.1 is a fate-determining TF for transmitting the environmental signal to commitment of environment-specific SEs in macrophages.
Several lines of evidence suggest that master TFs are capable of directing active SE formation (18,30,31) and this is supported by extensive co-localization of TFs within SE ‘hotspot’ regions either sequentially or simultaneously in response to developmental stimuli (27). Hnisz et al. demonstrated that terminal TFs of multiple signaling pathways frequently co-occurred in SEs but not their smaller TEs cousins, suggesting that SEs may provide a ‘hub’ for cells to be hyper-sensitive to divergent environmental signals (32).
High occupancy of mediator at SEs
Defined by the enhancer clusters with high Mediator occupancy and/or strong enhancer activity, SEs are expected to largely overlap with known functional chromatin domains, such as differential methylation regions (DMRs), locus control regions (LCRs) and transcription initiation platforms (TIPs) (8). Compared to stretch enhancers and typical enhancers, SEs exhibit significantly higher enrichment of active chromatin marks and binding of chromatin remodelers and organizers (29). The Mediator complex, composed of 26 core subunits, is known to be associated with enhancers and mediate enhancer functions by transmitting regulatory signal to the associated transcription machinery (33–36). Mediator contributes to stable assembly of transcription pre-initiation complex, regulation of RNA Polymerase II (PolII) pausing and elongation, and the formation of enhancer-promoter looping and three-dimensional (3D) genome organization (reviewed in (34)). SEs are heavily loaded with the Mediator complex at least one order of magnitude greater than any typical enhancers (6), giving rise to extraordinary activity and specific biochemical characteristics (7).
Altogether, SEs demonstrate their ‘superness’ not only by their enormous size, achieved though clustering closely localized enhancers, but also their super-strong transcriptional activity due to dense interaction with transcription factors, chromatin remodelers, transcription co-activators, and Pol II holoenzyme (Figure 1D). These properties are further supported by their capacity to drive short and long range interaction through phase separation and 3D genomic association, highlighting the qualitative difference to typical enhancers (see below), and suggest a mechanism to explain why transcriptional activation by SEs is greater than the sum of their constituent enhancer components (12).
THE ROLE OF SUPER-ENHANCERS IN TRANSCRIPTIONAL REGULATION
Given the unique characteristics of SEs, it is interesting to speculate why complex cells need them? The most likely answer to this question is that SEs specify and maintain cell identity, which is a vital biological attribute of complex multicellular organisms that need to be able to developmentally regulate the formation and maintenance of cell type specific tissue compartment. While mechanisms for transcriptional regulation through promoter activation were first identified in unicellular prokaryotes (37), the level of sophistication in vertebrate systems probably required more mechanistic complexity including long-range chromatin interactions from enhancers to target genes (20). Cell type specificity, in particular, has been shown to be associated with both master TFs and epigenetic chromatin marks. However, very little is known about how tissue-specific enhancers and, in particular, SEs emerge to support lineage commitment during development (38).
Phase separated condensates formed at SEs
SEs are extraordinarily densely bound by master TFs and highly occupied by the Mediator complex and other transcriptional coactivators, which raises the very interesting possibility that the genome can generate unusual physicochemical properties at their site of action. For example the Young lab has recently shown that the transcriptional coactivators BRD4 and MED1 are components of the liquid-liquid phase separated transcriptional condensates (39,40). They provided additional evidence that BRD4 and MED1 condensates co-localized with SEs (39) (Figure 3A). In a parallel study, Cho et al. showed further evidence that mediator and Pol II were co-existing in stable subcellular compartments, forming condensates and associated with SE elements (41). In both studies it was demonstrated that the formation of phase-separated liquid condensates were impaired in cells treated with 1,6-hexanediol, a compound known to disrupt these complexes (39,41). In addition, ChIP-seq with antibodies against BRD4 and MED1 revealed that the treatment of 1,6-hexanediol resulted in reduced BRD4 and MED1 binding at enhancers, and the effect was more profound at SEs (39). Mechanistically, SE-bound proteins such as BRD4 and MED1 typically contain large intrinsically disordered regions (IDRs), which multivalently but weakly interact with a large number of TFs and cofactors that also contain IDRs (40,42,43). The multi-valent interaction between IDRs facilitates condensation and liquid–liquid phase separation (LLPS) (39,44,45), where the highly concentrated transcriptional machinery guarantees the robust expression of cell identity genes. In support of this observation, Gibson et al. experimentally observed that adding BRD4 promoted formation of a new liquid phase of acetylated chromatin (46).
Figure 3.

Phase-separated transcriptional condensates at super-enhancers. (A) Immunofluorescence and DNA-FISH showing the co-localization between BRD4 or MED1 condensates (magenta) and the Nanog gene locus (green) in fixed mouse ESCs. The blue lines indicate the nuclear boundaries, and the overlapping signal in the merged channels is shown in white (figure was adapted from (39) with permission from AAAS). (B) Theoretical curves showing the dependence of transcriptional activity on the increase of valency for SEs (parameterized as N = 50 molecules) and typical enhancers (N = 10) based on a phase-separation model proposed by Sharp et al. (50). While SEs reach a high transcriptional activity at relatively low valency, typical enhancers need higher valency to achieve the same transcriptional activity (figure was adapted from (50) with permission from Elsevier). (C) A schematic chart showing the energy homeostasis in formation of LLPS at SEs. Structured Interaction and multivalent interaction occur, providing energy to compensate the loss of entropy when LLPS forms and the system gets ordered.
This is in concordance with related work in the field suggesting that the phase separation process plays a prominent role in 3D genome organization and is involved in organizing cell identity (47). The involvement of SEs in formation of LLPS also supports the assertion that SEs contribute more to transcriptional regulation than the additive effect of its multiple component TEs. More broadly it has been suggested that membraneless cellular organelles formed via phase separation play critical roles in a variety of cellular processes (reviewed in (48)). The localized protein concentration in these compartments is thought to be vital for the formation of phase-separated liquid droplets (49). The hub-like characteristics of SEs make the ideal biomolecular substrates to form phase-separated condensates comprising highly cooperative TFs, chromatin remodelers, transcription co-activators and RNA Pol II that elevate the local density ∼10-fold the molecular density of the components at typical enhancers (6–8). As a result, Sharp and colleagues hypothesized that formation of a phase separated assembly more likely occurred at SEs than at TEs (50). Based on this hypothesis, they proposed a kinetic model of transcriptional control, and explored the dynamic property of transcriptional activity by varying the number (N) of interacting molecules (i.e. the enhancer element and its associated factors) in a fixed-volume system, and the valency of each molecule. In this example, the transcriptional activity was quantitatively approximated by the relative size of the largest molecule cluster connected via cross-linking interactions (i.e. maximum size of clusters/N). When the transcriptional activity was reaching the value of 1, all molecules in the system formed a single interacting cluster, very likely resulting in the phase-separated assembly (50). By modeling SEs as a system containing 50 molecules while typical enhancers as a system consisting of 10 molecules, they revealed a divergent relationship between transcriptional activity and the change of valency between the two systems (Figure 3B). Where SEs reached a high transcriptional activity at low valency, typical enhancers needed higher valency to achieve the same transcriptional activity. This result suggests that SEs may undergo phase separation at a lower level of valency than typical enhancers. Moreover, a steeper increase of transcriptional activity of SEs was observed (Figure 3B), indicating that SEs likely behaved as binary switch in regulating key gene expression and could rapidly facilitate the establishment and maintenance of cell identity. In addition, the model also successfully explained a number of important observations in enhancer-mediated transcriptional control, including the transitional bursting patterns of enhancers and the ability of SEs to simultaneously activate multiple genes (50).
In terms of energy homeostasis, Chakraborty et al. claimed that formation of LLPS drives the chromatin in order and therefore leads to loss of entropy. This biophysical change requires compensation from energetic gain to maintain stable condensates. Where these form at enhancers, there are two ways to achieve this: (i) strong structured covalent master TF–DNA interaction and (ii) weak multivalent protein–protein (mostly TF-coactivators) interactions via IDRs (51). Therefore, SEs, exceeding other regulatory elements in both master TF binding sites and co-activator concentration, are more prone to the formation of transcriptional condensates (Figure 3C).
3D genome organization concerning SEs and their targets
The 3D genome organization has been shown to play critical roles in gene regulation and cell functions, also exhibiting cell-type specificity (47,52,53). In particular, insulated neighborhoods within chromosomal loop structures formed by CTCF-mediated interaction of two chromatin domains, provide a powerful paradigm for precise gene expression control (54). In other words, the superior transcriptional activity of SEs has to be strictly restricted within insulated neighborhoods such that they can be precisely and specifically tethered to their target genes. Even compared to stretch enhancers, relatively higher occupancy of cohesin and CTCF, the two factors mediating long-range DNA interaction and looping, has been found in constituents of SEs (29), supporting the notion that the chromosomal loops connecting SEs and promoters are more strictly controlled and maintained. An important question related to this is how the cell identity genes and their associated SEs are organized throughout the entire genome at the nucleotide sequence level?
Human genome contains many gene poor regions, called gene deserts that range in size from 5% to 40% of the entire chromosome (55). These segments often referred to as ‘junk DNA’ comprising 716 Mb, can be classified into two different categories based on their sequence conservation: stable gene deserts (>2% conserved) and variable gene deserts (<2% conserved). Intriguingly, gene ontology (GO) analysis shows that the tissue specific or developmentally regulated genes are moderately expressed and most of them are located in the stable gene deserts (55,56). In addition to the key cell identity gene bodies, their cis-regulatory elements are also embedded throughout the gene deserts. Comparative analysis has revealed that the density of transcriptional regulatory elements is three times higher in stable gene deserts than variable gene deserts and other intergenic regions (55). These cis-regulatory elements are generally linked to the neighboring genes, supported by a substantial depletion of synteny breakpoints in between. For example, the murine ESC pluripotent gene Sox2 locus at chromosome 3qA3 is flanked by 1.5 Mb stable gene deserts and regulated by its SE 130 kb downstream of the gene body in the same gene desert (57,58,59) (Figure 4A). Another case is the human proto-oncogene MYC locus, which is located within a 1.2 Mb stable gene deserts at chromosome 8q24, regulated by a few distinct SEs in different tissues. Genome-wide association studies (GWAS) of multiple cancer and metabolic disease cohorts have identified single nucleotide polymorphisms (SNPs) in the gene deserts tightly linked to high risks of breast cancer, colorectal cancer, prostate cancer, ovarian cancer and so on (summarized in (60)). Some of these SNPs are located inside TF binding clusters (21), which are recognized as constituents of MYC gene SEs (8,32,61). A few more gene loci with similar genome arrangement have been reported, including loci of KLF4, OTX2 and DACH1 (62–64).
Figure 4.

Chromatin 3D Organization and Insulated Neighborhoods. (A) Comparison of topologically associating domain (TAD) structure observed by Hi-C and insulated neighborhoods organized by two CTCF binding at borders. Upper panel shows the classical Hi-C interaction heatmap. Darker color indicates stronger normalized interaction frequency, represented by color key on the left side. Lower panel shows ChIP-seq of CTCF and H3K27ac within one TAD structure that encompasses Sox2 and its SE. Note that CTCF binds to both sides of TAD boundaries (asterisk) which is also borders of insulated neighborhoods. The Hi-C data used for generating this plot were taken from Yan et al. (59). (B) A model cartoon showing the organization of insulated neighborhood. An SE (blue) and its target gene (red) are demarcated in a segment bordered by CTCF and associated cohesin complex. Due to the insulated environment, genes outside the neighborhood keep silent while inside genes are activated by the SEs.
These observations suggest that the key genes tend to be independently organized from other chromosomal territories, which is likely to be evolutionarily favored for two non-mutually exclusive reasons, if not more: (i) enclosed genes are essential for cell identity and function, and thus require precise expression without interference by other signals other than its own SEs; (ii) SEs are so powerful that they need to be prevented from driving unrelated neighboring genes unexpectedly. Interestingly, in ESCs, Dowen et al. found that these cell identity genes not only occurred in gene deserts but also were restricted within insulated neighborhoods enclosed by CTCF and its associated cohesin, termed as SE domains (SDs) (65). The same phenomenon was described in a study on T-lymphoblastic leukemia (TLL) cells where an SE was restricted within the same CTCF-organized neighborhood of its target locus IL7R, a key gene for normal T cell development and pathogenesis of TLL (66). Deleting a CTCF binding site at one border of the SD caused dysregulation of internal cell identity genes and activation of external genes nearby the SDs (64,65). Disruption of insulated chromatin neighborhoods set for repressing proto-oncogene expression could activate oncogene and lead to severe health problems, further supporting a role of SD for precise control of essential gene expression (67).
The transcriptionally insulated neighborhoods, at a median size of ca. 200 kb, are formed by flanked CTCF binding in convergent orientations (64,65) (Figure 4B). Similar genome organization is observed as ‘contact domains’ (median size: 180 kb) identified from deeply sequenced in situ Hi-C data (68). The ‘contact domains’ are associated with distinct patterns of histone modifications across the domain borders. Taken together, this strongly deduces that ‘contact domains’ (by in situ Hi-C) and ‘insulated neighborhoods’ (by ChIA-PET) may refer to the same 3D chromatin structure units or at least with partial overlapping (64). When comparing the Hi-C data and ChIA-PET pairs carried out in the same primed human ESC (H1) line, Ji et al. found that the insulated neighborhoods were the fundamental units (subdomains) that constituted the megabase-sized topologically associating domains (TADs), which was also mediated by cohesin-associated CTCF–CTCF loops. Thus, they propose a model that genome is partitioned into many large and physically close TADs, which are constituted by multiple transcriptionally insulated neighborhoods. These subdomains delimit the effective range of enhancers, SEs and repressors (64).
However, forming the 3D genome configuration is a complex and dynamic process, which has yet to be completely understood (69). With combined efforts from multiple disciplines, a few models of transcriptional regulation and genome organization have been proposed. For example, a recent model based on physical principles suggests that forming clusters of active RNA polymerase and associated TFs is the elementary feature of 3D chromatin structures. These clusters are surrounded by DNA loops, and thus the large domains, TADs and chromatin A/B compartments are simply a single or multiple cluster with loops. This model also explains the extraordinary activity of SEs in transcriptional regulation, because Mediator and master TFs bound to SEs would increase the time of an associated promoter staying closely to the active polymerase clusters (70).
A multi-stage transition model of the SE formation
Experimental evidence supports that binding of master TFs not only attracts more TFs, but also facilitates the recruitment of chromatin remodelers and transcriptional co-activators that possess enzymatic activity for histone modifications (71–76). Early biochemical studies uncovered that a single TF, C/EBP β, could compete against histones for DNA binding and mono-nucleosome formation (77), which was also confirmed at the chromatin level. A single master TF, HNF3 or GATA4, could compete against assembled nucleosome arrays (chromatin fibers) with assistance from its C-terminal DNA binding domain, and consequently makes the DNA accessible to other proteins including chromatin remodelers and modification enzymes (78). In a recent study, it has been found that immediately after DNA replication in each cell cycle, nucleosomes are repositioned at promoters and enhancers, followed by re-establishment of DNA accessibility led by TFs and chromatin remodelers in a fast mode (accomplished in hours) (79). As a result, phase-separated ‘transcription factories’ are formed around the protein-crowded chromatin foci, which can also be stably inherited along cellular division (80–83). Such ‘factory’ organization greatly increases the efficiency for multiple genes being coordinately regulated, and is supported by the recent observation that promoter-promoter interaction takes up 42% of total long-range interactions (84).
In some cases, one TF may be insufficient for nucleosome exclusion and chromatin opening, requiring instead cooperativity of multiple master TFs (85–87). For example, temporally persistent hierarchical binding of SOX2/OCT4/KLF4 prior to c-MYC was found in mammalian ESCs (87). Despite the importance of a cluster of TF binding for enhancer activity, the TF–DNA binding itself remains short-lived and highly dynamic. Chen et al. took advantage of super-resolution microscopical single-molecule tracking technology (SMT) and observed that TFs employed a 3D diffusion-dominated searching mode assisted by 1D sliding to locate their targeting sites, which took over 6 min (88). By contrast, the residential time for SOX2 binding to its stable recognition site lasted for only ∼12–14 s. These results suggest that TF binding alone could not fully explain the establishment of enhancers, whereas more stable and inheritable events are required, although TF binding per se is an initial step.
Collectively, we introduce a ‘multi-stage transition’ model to describe the consecutive drift of evolution of the regulatory landscape from TF–DNA binding to SE formation (Figure 5). The initiation of the regulatory element takes place when a master TF searches for its target site, and competes against local nucleosomes for stable binding (Stage I). To achieve a more stable stage, such an event must be followed by recruitment of chromatin remodelers and histone modification enzymes to re-organize and maintain the more accessible and inheritable chromatin environment (Stage II). Along the evolution of cellular function, a few key genes demand synergistic regulation from multiple convergent regulatory elements and thus drive the formation of SEs (Stage III), the final destination for active transformation of cis-regulatory components (Figure 5).
Figure 5.
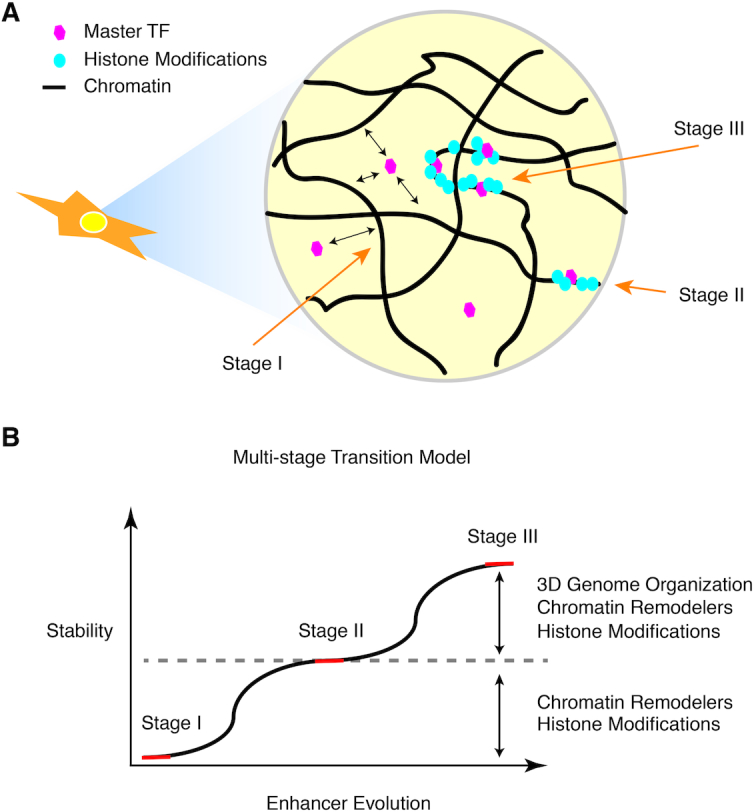
Multi-stage transition model of super-enhancer formation. (A) A schematic cartoon showing different stages of SE formation. Stage I: dynamic binding of the master TF; Stage II: accumulation of histone modifications and other TFs and stabilization of typical enhancers; Stage III: convergent evolution of synergistic typical enhancers to form SEs. (B) A diagram showing the process of enhancer evolution, represented in (A). Red line indicates the steady states while the black curve indicates the transition stages. Note that earlier stages are less stable than later stages where the enhancer is becoming larger in size and stronger in various protein binding signals and histone biochemical modifications.
A variety of evolutionary pressures separate the formation of SEs from TEs in different stages of the model. During Stages I and II, stable binding of a master TF is required initially to compete with nucleosomes, this is followed by the subsequent binding of other transcriptional activators and chromatin remodelers. As shown before in other examples, master TF binding at TEs is far less prevalent than at SEs. The lack of abundant master TF binding leads to a reduced enrichment of chromatin remodelers and co-activators (Stage II). The binding of BRD4 can facilitate the formation of LLPS between acetylated and un-acetylated chromatin (46). Upon establishment of LLPS, the nascent SEs become more stable than other foci. In the last stage, selection pressure eventually ‘selects’ the most functional SEs: (i) if a gene driven by a new SE is toxic to the cells, the corresponding SE hence becomes deleterious and will experience negatively selection and will tend to be removed from the cell population; (ii) when a gene driven by an SE is non-essential for cell function, the neutral genetic drift will dilute its presence in a cell population. Therefore, only the key genes that equip cells with proper functions will be positively preserved and retain their corresponding SEs during evolution. Analogously, a selective process could help oncogenes quickly develop SEs that predisposes them to uncontrolled growth advantage, and suggests a mechanism that could drive tumor cells to acquire SEs in tumor pathogenesis (8).
SUPER-ENHANCERS AND DISEASES
SEs and their defects have been linked to multiple genetic diseases (8), including cancer (7,89), metabolic (8,90,91) and immune diseases (92) (reviewed in (93)). In this section, we will discuss current knowledge of SE-associated human health conditions, and the potential applications for disease diagnosis, prognosis, and treatment.
The vast majority of risk SNPs that confer genetic diseases rarely alter protein coding but mostly reside in non-coding regulatory loci (94,95). Significantly larger disease-associated mutation repertoire has been found in SEs compared to other open chromatin regions, such as promoters and typical enhancers (8). In some cases, small mutations and indels have been found to unexpectedly generate new SEs or rewire SEs to oncogenes that drive tumor pathogenesis (8,67,89,96).
Cancers
SEs possess the capability of not only specifying cell identity but also maintaining cancer cell identity and discriminating carcinoma subtypes (97). The application of SE analysis to medulloblastoma is one of the first cases showing efficient way of using epigenetic data to trace the cellular origin of poorly characterized cancer malignancies (98,99). By querying their respective SEs together with their associated master TFs, Lin et al. managed to (i) locate the novel targets of the aberrant transcriptional system in different cancer subtypes and (ii) more importantly, identify cell-of-origin for cancer subgroups.
Multiple cancer types show either a prominent mutation rate or distorted regulatory landscape at the SEs of driver oncogenes in disease-relevant cell types. One particularly interesting example was discovered in T-cell acute lymphoblastic leukemia (T-ALL). For a long time, a helix-loop-helix TF TAL1 has been associated with bi-allelic activation in some T-ALL patients, which could be caused by loss of function of an upstream repressive regulatory element (100). In addition to these individuals with bi-allelic ectopic expression, it was found that there were a substantial number of patients and cell lines carrying mono-allelic overexpression of TAL1 (100). When the same team revisited the T-ALL subgroup with mono-allelic activation of TAL1, they discovered that the aberrant allele acquired a 20-kb long SE accommodating binding sites for a few major leukemogenic TFs, including RUNX1, GATA-3 and TAL1 itself (96). Strikingly, this oncogenic alteration was simply caused simply by a 12 bp insertion which generated a new MYB binding site that did not exist in cells without gaining the SE (Figure 6A).
Figure 6.

Three models of cancer-acquired super-enhancers. (A) Model 1: Due to mutation occurrence, a MYB binding motif is generated next to a silent oncogene, and the motif recruits MYB protein binding. Following the multi-stage transition model, the master TF binding accumulates more stable signals of other TFs, chromatin remodelers and histone modifications, resulting in formation of a stable and strong SE. Hence, the adjacent oncogene is activated (black arrow) by the newly acquired SE and causes oncogenesis. (B) Model 2: Unlike Model 1, the mutation does not create a novel binding site for master TFs but erases a binding site of CTCF, which is also an anchor site for an insulated neighborhood (brown circle). The formerly silent oncogene is activated by a juxtaposed SE and consequently results in tumorigenesis. Note that CTCF/Cohesin binding site mutation is significantly enriched in cancer genome. (C) Model 3: The activation-induced cytidine deaminase (AID) is accumulated to the largely permissive chromatin surrounding an SE by convergent transcripts from enhancer RNA and mRNA transcribed from the SE and gene body, respectively. Binding of AID triggers instability of genome and concomitantly translocation, which brings an oncogene next to the SE and promotes lymphomagenesis.
Mutation or insertions that inappropriately activate enhancers to reboot a repressed disease driver gene, is not a novel finding in the pathogenesis of malignancy. In fact it is particularly common at the MYC locus (101,102). Whole-genome sequencing analysis of the widely-studied human cervical cancer cell line HeLa uncovered an active genomic fragment from Human Papilloma Virus type 18 (HPV-18) insertion into an originally silent constituent of a MYC super-enhancer, and thereafter drove upregulation of the proto-oncogene MYC promoting oncogenesis (103). However, in the TAL1 example, the short insertion for the de novo MYB binding site that creates a broad SE is rather unexpected. MYB binding itself is dynamic and thus may not be able to boost the activation of TAL1 expression. However, MYB, as a pioneer factor, draws chromatin remodelers, coactivators and other TFs in to establish a relatively stable regulatory niche. Accumulation of more and more executive activation factors to the mutated locus promotes formation of an SE that is powerful enough to drive oncogenic TAL1 expression and subsequent carcinogenesis. This model provides a novel mechanism for tumorigenesis through gaining binding sites of master TFs, which results in greater-than-expected gene expression and upregulation of oncogenes.
Cancers associated mutations are also frequently detected at CTCF and cohesin bound sequences, which are also enriched in SEs (104). With the whole genome sequencing of 231 colorectal cancer (CRC) patient tumors and paired normal intestinal tissues, Katainen et al. reported overall elevated mutation frequency in CTCF binding sites (CBSs) by an order of magnitude compared to average genome. Through further scrutiny of public cancer genome databases, they found that the CBSs were the somatic mutational hotspots in the non-coding cancer genome of multiple tumor types. These mutated CBSs were highly overlapping with CTCF-CTCF anchor sites that formed insulated neighborhoods in human ESCs. This is also consistent with another study by Hnisz et al. in which microdeletions caused loss of the boundary CTCF binding and consequently defeated the insulated neighborhood for repressed oncogenes expression in T-ALL (67) (Figure 6B).
During the process of somatic hypermutation and class-switch recombination, B cells have to undergo a series of DNA double strand breakage and highly frequent mutations triggered by activation-induced deaminase (AID) (105). Such a process, can aberrantly cause genome instability and malignancies. AID, which does not have any DNA sequencing binding motifs, has been shown with a high binding rate at extensively permissive SEs and transcriptionally active gene bodies (106,107). Comprehensive analyses dissecting the molecular mechanism involved show that Pol II stalled by convergent transcription collision recruits off-targeted AID to the largely permissive environment of SEs, which subsequently initiates mutations and translocations. Translocation can bring proto-oncogenes under the control of SEs resulting in over-expression and B cell lymphomagenesis (108) (Figure 6C).
Developmental defects
Besides cancer, the proto-oncogene MYC is also linked to various developmental defects including common congenital malfunction, cleft lip with or without cleft palate (CL/P) (109–111). The MYC locus synteny is conserved between human and mouse. More interestingly, most of the mutation hotspots are enriched in cell type specific H3K27ac peaks, representing tissue-specific MYC enhancers and SEs (102,112–114). Using genome editing tools to manipulate the mouse Myc locus, Uslu et al. confirmed that a 300-kb medionasal super-enhancer (MNE) was responsible for 30% of Myc expression in face and limb during development, but not in liver or heart (102).
In comparison, manipulation of a typical enhancer may only have a marginal effect, considering its low regulatory activity (58,59,113,115). Thus, deletion of larger regulatory elements like SEs could help in the detection of such effects and better understanding relevant mechanisms. Systematically genome-wide validation of SE function for development and diseases is intriguing. The fast development of genome editing tool CRISPR/Cas9 (116,117) makes in vivo validation studies more feasible and should be more broadly applied in the near future (57,58,115).
A recent work published by the Mundlos lab showed in vivo evidence from animal model that CTCF organized topology of 3D chromatin structure was responsible for SE functions (118). Firstly, they identified three heterozygous deletions on neighboring chromosome regions in patients with three different limb genetic abnormalities – brachydactyly, F-syndrome and polydactyly. Subsequently, they used the CRISPR/Cas9 editing tool to reconstruct the chromosome deletions and successfully reproduced the disease phenotypes in mouse models. A 150-kb SE fragment was found to be the target as disruption of chromatin structure by deletion, inversion or duplication rewired this regulatory element to genes in neighboring TAD structure, which was originally insulated in wild-type cells. This mechanism is very similar to what was observed for oncogenes TAL1 and LMO2 in T-ALL as TAD structure itself is an insulated neighborhood (100).
Potential therapeutic targets
Since SEs are heavily modified biochemically by various enzymes such as acetyltransferases or methyltransferases, they exhibit the potential ‘drug targetable’ characteristics. Two unique characteristics of SEs that we discussed above further make them particularly relevant targets for cancer therapy: (i) SEs are more sensitive to external signals than any other genomic regions; (ii) SEs are controlling cell identity genes in both healthy and disease cells. Both features could potentially guard the specificity of drugs, one of the most challenging issues in cancer therapy.
The epigenetic modification enzymes are found mutated at high frequency in cancer patients (∼20–40%), such as DNMT3A (DNA cytosine de novo methyltransferase 3A) in AML and MDS, and CBP/EP300 (lysine acetyltransferases) in bladder cancer and lymphomas (reviewed in (119)). The DNMT inhibitors, 5-azacytidine and decitabine, have been clinically used to effectively treat high risk myelodysplastic syndrome and AML (120). Among others, inhibitors of histone lysine deacetylases (HDACs), acetyltransferases (CBP) and chromatin remodelers are all available in the pharmaceutical market for cancer treatment (119). In addition to histone modification enzymes, targeting transcription coactivators is another feasible therapeutic strategy. Drugs such as JQ1 and THZ1, inhibiting BRD4 (7) and CDK7 (121), respectively, have been shown to specifically target tumor-specific SEs, providing an efficient way for targeting only cancer cells.
BRD4 and JQ1
The first compound reported to target SEs is ‘JQ1’, designed to specifically inhibit Bromodomain and Extraterminal (BET) superfamily member BRD4 that highly occupies SEs in cancer cells (7,122,123). BRD4 is known as a Mediator-interacting partner since the 1990s (124) and it is also a drug target in leukemia (125). Treating multiple myeloma tumor cells with JQ1 causes disproportional loss of BRD4 binding to the genome, more pronounced at SEs than TEs and other regions (7). Therefore, the SE controlled genes that include mostly oncogenes in cancer such as MYC, get more affected than other genes in tumor cells. Following independent studies, using JQ1 to target BRD4 and SEs, have demonstrated similar effects in a broad spectrum of cancer types, such as colorectal cancer (126), ovarian cancer (127), Merkel cell carcinoma (128), B cell lymphoma (129), and alveolar rhabdomyosarcoma (123). Of note, JQ1 is currently still being evaluated in phase I and II clinical trials, and resistance to JQ1 has been reported in a few cancer cases (130–132).
CDK7 and THZ1
Mediator super-enrichment in SEs also contributes to accumulation of Pol II holoenzyme and other subunits of transcription machinery. Cyclin Dependent Kinase 7 (CDK7) is a kinase subunit of a general transcription factor TFIIH, required for transcription machinery assembly. It can phosphorylate C-terminal domain (CTD) of Pol II and facilitate transcription initiation. Targeting CDK7 by its small molecule covalent inhibitor THZ1, can effectively inhibit master TF RUNX1 expression in T-ALL by disrupting its SE-associated transcription regulation circuitry (121). THZ1 shows a strong inhibitory effect on tumor growth in a human xenograft mouse model in a dose dependent manner. Similarly, THZ1 downregulates amplified MYCN expression in a high-risk neuroblastoma mouse model, notably, without any systemic toxicity (133). Strikingly, when the same strategy is applied to small-cell lung cancer (SCLC), on which there has been no significant therapeutic progress since chemotherapy was introduced in the 1970s, SE-associated genes, including MYC family genes, are highly vulnerable to the THZ1 treatment (134).
These two examples show that SE-associated protein factors are ideal options for drug targets. Besides the recent advances in genome editing technologies, CRISPR/Cas9, in particular, offer a possibility of novel gene therapies that directly correct pathogenic SEs (93). Of note, not only SEs and their directly interacting factors, but the pathways and master genes identified associated with disease-specific SEs also provide potential therapeutic targets, like transcription factors LMX1A, EOMES and LHX2 for group 4 medulloblastoma (98,99).
CONCLUSIONS
In this Survey and Summary, we have highlighted the unique characteristics of SEs and their biological role in transcriptional regulation in health and disease. In particular, we addressed the ‘elephant in the room’: are SEs clusters of additive enhancers or a novel type of synergistic regulatory element? To address this question, we provide multiple lines of additional evidence suggesting that they are highly likely to be a unique biological entity in transcriptional regulation. Firstly, this is supported by a number of distinct characteristics of SEs compared to other enhancers, including their sequence composition, genomic size, regulatory activity, proteins bound, and genes under their regulation (also reviewed previously (135)). One of the most striking features of SE loci to emerge recently is that they are likely to be isolated from other chromatin domains by forming an insulated nuclear compartment through liquid-liquid phase-separated, membraneless condensates. In these focused compartments, they are more likely to have the necessary autonomy to precisely drive the regulation of genes controlling cell identity (40,136). Secondly, knockout experiments targeting SE components, furthermore demonstrate that functional hierarchy and synergistic interactions exist among different constituents within an SE (32,137). For instance, in a study targeting the constituents of the α-globin gene-associated SE (11), a linear-logistic model, which allowed for interactions between constituent enhancers, explained the knockout results better than a simple linear model (12), suggesting that the regulatory relationship between individual constituents are ‘synergistic’. Finally, SE constituents display the tissue specificity as an entire group. For example, SEs with multiple constituents in MYC-locus are located in a non-overlapping manner among different cell types (8,115): CRC tumors generally harbor an SE of MYC ∼300 kb upstream of its promoter, while in acute leukemia an SE 1.7 Mb downstream of the TSS plays a primary role of activating c-MYC expression (138,139). These observations further suggest the synergistic role of SE constituents in regulating gene expression. We therefore conclude that SEs form a distinct regulatory entity beyond additive clustering of independent enhancers, although more in vivo evidence is still required to further support this concept. Given the involvement of SEs in human pathology, future studies that generate greater understanding of their function as holistic entities, will be important for the development of new biomarkers and treatments that target these powerful genomic regulatory structures.
DATA AVAILABILITY
A core collection of SEs used for the meta-analyses in this Survey and Summary and the computational scripts are available in the GitHub repository (https://sunlightwang.github.io/Super-Enhancers/).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Prof. Bing Ren and Dr Miao Yu from Ludwig Institute for Cancer Research for helpful discussions, and also Dr Inderpreet Kaur Sur from Karolinska Institutet for constructive comments during the revision of the manuscript. We apologize to our colleagues for citing reviews instead of original research articles because of space limitations.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [81873642 to J.Y.]; City University of Hong Kong [7200595, 7005314, 9667188, 9610424]; Opening Foundation of Key Laboratory of Resource Biology and Biotechnology in Western China (Northwest University), Ministry of Education; NHMRC Senior Research Fellowship [1121474 to M.J.C.]; The Hong Kong Epigenomics Project (Lo Ka Chung Charitable Foundation) (J.Y.). Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Banerji J., Rusconi S., Schaffner W.. Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell. 1981; 27:299–308. [DOI] [PubMed] [Google Scholar]
- 2. Mercola M., Wang X.F., Olsen J., Calame K.. Transcriptional enhancer elements in the mouse immunoglobulin heavy chain locus. Science. 1983; 221:663–665. [DOI] [PubMed] [Google Scholar]
- 3. Banerji J., Olson L., Schaffner W.. A lymphocyte-specific cellular enhancer is located downstream of the joining region in immunoglobulin heavy chain genes. Cell. 1983; 33:729–740. [DOI] [PubMed] [Google Scholar]
- 4. Gillies S.D., Morrison S.L., Oi V.T., Tonegawa S.. A tissue-specific transcription enhancer element is located in the major intron of a rearranged immunoglobulin heavy chain gene. Cell. 1983; 33:717–728. [DOI] [PubMed] [Google Scholar]
- 5. Chen Y., Yao B., Zhu Z., Yi Y., Lin X., Zhang Z., Shen G.. A constitutive super-enhancer: homologous region 3 of Bombyx mori nucleopolyhedrovirus. Biochem. Biophys. Res. Commun. 2004; 318:1039–1044. [DOI] [PubMed] [Google Scholar]
- 6. Whyte W.A., Orlando D.A., Hnisz D., Abraham B.J., Lin C.Y., Kagey M.H., Rahl P.B., Lee T.I., Young R.A.. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013; 153:307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Loven J., Hoke H.A., Lin C.Y., Lau A., Orlando D.A., Vakoc C.R., Bradner J.E., Lee T.I., Young R.A.. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013; 153:320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-Andre V., Sigova A.A., Hoke H.A., Young R.A.. Super-enhancers in the control of cell identity and disease. Cell. 2013; 155:934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Roadmap Epigenomics C., Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J. et al.. Integrative analysis of 111 reference human epigenomes. Nature. 2015; 518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Consortium E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hay D., Hughes J.R., Babbs C., Davies J.O.J., Graham B.J., Hanssen L., Kassouf M.T., Marieke Oudelaar A.M., Sharpe J.A., Suciu M.C. et al.. Genetic dissection of the alpha-globin super-enhancer in vivo. Nat. Genet. 2016; 48:895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dukler N., Gulko B., Huang Y.F., Siepel A.. Is a super-enhancer greater than the sum of its parts. Nat. Genet. 2016; 49:2–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moorthy S.D., Davidson S., Shchuka V.M., Singh G., Malek-Gilani N., Langroudi L., Martchenko A., So V., Macpherson N.N., Mitchell J.A.. Enhancers and super-enhancers have an equivalent regulatory role in embryonic stem cells through regulation of single or multiple genes. Genome Res. 2017; 27:246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Khan A., Zhang X.. dbSUPER: a database of super-enhancers in mouse and human genome. Nucleic Acids Res. 2016; 44:D164–D171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wei Y., Zhang S., Shang S., Zhang B., Li S., Wang X., Wang F., Su J., Wu Q., Liu H. et al.. SEA: a super-enhancer archive. Nucleic Acids Res. 2016; 44:D172–D179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jiang Y., Qian F., Bai X., Liu Y., Wang Q., Ai B., Han X., Shi S., Zhang J., Li X. et al.. SEdb: a comprehensive human super-enhancer database. Nucleic Acids Res. 2019; 47:D235–D243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Parker S.C., Stitzel M.L., Taylor D.L., Orozco J.M., Erdos M.R., Akiyama J.A., van Bueren K.L., Chines P.S., Narisu N., Program N.C.S. et al.. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:17921–17926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gosselin D., Link V.M., Romanoski C.E., Fonseca G.J., Eichenfield D.Z., Spann N.J., Stender J.D., Chun H.B., Garner H., Geissmann F. et al.. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell. 2014; 159:1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lukk M., Kapushesky M., Nikkila J., Parkinson H., Goncalves A., Huber W., Ukkonen E., Brazma A.. A global map of human gene expression. Nat. Biotechnol. 2010; 28:322–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Levine M. Transcriptional enhancers in animal development and evolution. Curr. Biol. 2010; 20:R754–R763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yan J., Enge M., Whitington T., Dave K., Liu J., Sur I., Schmierer B., Jolma A., Kivioja T., Taipale M. et al.. Transcription factor binding in human cells occurs in dense clusters formed around cohesin anchor sites. Cell. 2013; 154:801–813. [DOI] [PubMed] [Google Scholar]
- 22. Boyer L.A., Lee T.I., Cole M.F., Johnstone S.E., Levine S.S., Zucker J.P., Guenther M.G., Kumar R.M., Murray H.L., Jenner R.G. et al.. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005; 122:947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moorman C., Sun L.V., Wang J., de Wit E., Talhout W., Ward L.D., Greil F., Lu X.J., White K.P., Bussemaker H.J. et al.. Hotspots of transcription factor colocalization in the genome of Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:12027–12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roy S., Ernst J., Kharchenko P.V., Kheradpour P., Negre N., Eaton M.L., Landolin J.M., Bristow C.A., Ma L., Lin M.F. et al.. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science. 2010; 330:1787–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stanojevic D., Small S., Levine M.. Regulation of a segmentation stripe by overlapping activators and repressors in the Drosophila embryo. Science. 1991; 254:1385–1387. [DOI] [PubMed] [Google Scholar]
- 26. Liu Z., Merkurjev D., Yang F., Li W., Oh S., Friedman M.J., Song X., Zhang F., Ma Q., Ohgi K.A. et al.. Enhancer activation requires trans-recruitment of a mega transcription factor complex. Cell. 2014; 159:358–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Siersbaek R., Rabiee A., Nielsen R., Sidoli S., Traynor S., Loft A., La Cour Poulsen L., Rogowska-Wrzesinska A., Jensen O.N., Mandrup S.. Transcription factor cooperativity in early adipogenic hotspots and super-enhancers. Cell Rep. 2014; 7:1443–1455. [DOI] [PubMed] [Google Scholar]
- 28. Saint-Andre V., Federation A.J., Lin C.Y., Abraham B.J., Reddy J., Lee T.I., Bradner J.E., Young R.A.. Models of human core transcriptional regulatory circuitries. Genome Res. 2016; 26:385–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Khan A., Mathelier A., Zhang X.. Super-enhancers are transcriptionally more active and cell type-specific than stretch enhancers. Epigenetics. 2018; 13:910–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adam R.C., Yang H., Rockowitz S., Larsen S.B., Nikolova M., Oristian D.S., Polak L., Kadaja M., Asare A., Zheng D. et al.. Pioneer factors govern super-enhancer dynamics in stem cell plasticity and lineage choice. Nature. 2015; 521:366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brown J.D., Lin C.Y., Duan Q., Griffin G., Federation A.J., Paranal R.M., Bair S., Newton G., Lichtman A.H., Kung A.L. et al.. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol. Cell. 2014; 56:219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hnisz D., Schuijers J., Lin C.Y., Weintraub A.S., Abraham B.J., Lee T.I., Bradner J.E., Young R.A.. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol. Cell. 2015; 58:362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kagey M.H., Newman J.J., Bilodeau S., Zhan Y., Orlando D.A., van Berkum N.L., Ebmeier C.C., Goossens J., Rahl P.B., Levine S.S. et al.. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010; 467:430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Allen B.L., Taatjes D.J.. The Mediator complex: a central integrator of transcription. Nat. Rev. Mol. Cell Biol. 2015; 16:155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Davis J.A., Takagi Y., Kornberg R.D., Asturias F.A.. Structure of the yeast RNA polymerase II holoenzyme: Mediator conformation and polymerase interaction. Mol. Cell. 2002; 10:409–415. [DOI] [PubMed] [Google Scholar]
- 36. Holstege F.C., Jennings E.G., Wyrick J.J., Lee T.I., Hengartner C.J., Green M.R., Golub T.R., Lander E.S., Young R.A.. Dissecting the regulatory circuitry of a eukaryotic genome. Cell. 1998; 95:717–728. [DOI] [PubMed] [Google Scholar]
- 37. Browning D.F., Busby S.J.. The regulation of bacterial transcription initiation. Nat. Rev. Microbiol. 2004; 2:57–65. [DOI] [PubMed] [Google Scholar]
- 38. Peng X.L., So K.K., He L., Zhao Y., Zhou J., Li Y., Yao M., Xu B., Zhang S., Yao H. et al.. MyoD- and FoxO3-mediated hotspot interaction orchestrates super-enhancer activity during myogenic differentiation. Nucleic Acids Res. 2017; 45:8785–8805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sabari B.R., Dall’Agnese A., Boija A., Klein I.A., Coffey E.L., Shrinivas K., Abraham B.J., Hannett N.M., Zamudio A.V., Manteiga J.C. et al.. Coactivator condensation at super-enhancers links phase separation and gene control. Science. 2018; 361:eaar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Boija A., Klein I.A., Sabari B.R., Dall’Agnese A., Coffey E.L., Zamudio A.V., Li C.H., Shrinivas K., Manteiga J.C., Hannett N.M. et al.. Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell. 2018; 175:1842–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cho W.K., Spille J.H., Hecht M., Lee C., Li C., Grube V., Cisse I.I.. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science. 2018; 361:412–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu J., Perumal N.B., Oldfield C.J., Su E.W., Uversky V.N., Dunker A.K.. Intrinsic disorder in transcription factors. Biochemistry. 2006; 45:6873–6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Staby L., O'Shea C., Willemoes M., Theisen F., Kragelund B.B., Skriver K.. Eukaryotic transcription factors: paradigms of protein intrinsic disorder. Biochem. J. 2017; 474:2509–2532. [DOI] [PubMed] [Google Scholar]
- 44. Chong S., Dugast-Darzacq C., Liu Z., Dong P., Dailey G.M., Cattoglio C., Heckert A., Banala S., Lavis L., Darzacq X. et al.. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science. 2018; 361:eaar2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cramer P. Organization and regulation of gene transcription. Nature. 2019; 573:45–54. [DOI] [PubMed] [Google Scholar]
- 46. Gibson B.A., Doolittle L.K., Schneider M.W.G., Jensen L.E., Gamarra N., Henry L., Gerlich D.W., Redding S., Rosen M.K.. Organization of chromatin by intrinsic and regulated phase separation. Cell. 2019; 179:470–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stadhouders R., Filion G.J., Graf T.. Transcription factors and 3D genome conformation in cell-fate decisions. Nature. 2019; 569:345–354. [DOI] [PubMed] [Google Scholar]
- 48. Boeynaems S., Alberti S., Fawzi N.L., Mittag T., Polymenidou M., Rousseau F., Schymkowitz J., Shorter J., Wolozin B., Van Den Bosch L. et al.. Protein phase separation: a new phase in cell biology. Trends Cell Biol. 2018; 28:420–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Alberti S. Phase separation in biology. Curr. Biol. 2017; 27:R1097–R1102. [DOI] [PubMed] [Google Scholar]
- 50. Hnisz D., Shrinivas K., Young R.A., Chakraborty A.K., Sharp P.A.. A phase separation model for transcriptional control. Cell. 2017; 169:13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shrinivas K., Sabari B.R., Coffey E.L., Klein I.A., Boija A., Zamudio A.V., Schuijers J., Hannett N.M., Sharp P.A., Young R.A. et al.. Enhancer features that drive formation of transcriptional condensates. Mol. Cell. 2019; 75:549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tan L., Xing D., Daley N., Xie X.S.. Three-dimensional genome structures of single sensory neurons in mouse visual and olfactory systems. Nat. Struct. Mol. Biol. 2019; 26:297–307. [DOI] [PubMed] [Google Scholar]
- 53. Tan L., Xing D., Chang C.H., Li H., Xie X.S.. Three-dimensional genome structures of single diploid human cells. Science. 2018; 361:924–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hnisz D., Day D.S., Young R.A.. Insulated neighborhoods: structural and functional units of mammalian gene control. Cell. 2016; 167:1188–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ovcharenko I., Loots G.G., Nobrega M.A., Hardison R.C., Miller W., Stubbs L.. Evolution and functional classification of vertebrate gene deserts. Genome Res. 2005; 15:137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Taylor J. Clues to function in gene deserts. Trends Biotechnol. 2005; 23:269–271. [DOI] [PubMed] [Google Scholar]
- 57. Li Y., Rivera C.M., Ishii H., Jin F., Selvaraj S., Lee A.Y., Dixon J.R., Ren B.. CRISPR reveals a distal super-enhancer required for Sox2 expression in mouse embryonic stem cells. PLoS One. 2014; 9:e114485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhou H.Y., Katsman Y., Dhaliwal N.K., Davidson S., Macpherson N.N., Sakthidevi M., Collura F., Mitchell J.A.. A Sox2 distal enhancer cluster regulates embryonic stem cell differentiation potential. Genes Dev. 2014; 28:2699–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yan J., Chen S.A., Local A., Liu T., Qiu Y., Dorighi K.M., Preissl S., Rivera C.M., Wang C., Ye Z. et al.. Histone H3 lysine 4 monomethylation modulates long-range chromatin interactions at enhancers. Cell Res. 2018; 28:204–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ghoussaini M., Song H., Koessler T., Al Olama A.A., Kote-Jarai Z., Driver K.E., Pooley K.A., Ramus S.J., Kjaer S.K., Hogdall E. et al.. Multiple loci with different cancer specificities within the 8q24 gene desert. J. Natl. Cancer Inst. 2008; 100:962–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Heyn H., Vidal E., Ferreira H.J., Vizoso M., Sayols S., Gomez A., Moran S., Boque-Sastre R., Guil S., Martinez-Cardus A. et al.. Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer. Genome Biol. 2016; 17:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nobrega M.A., Ovcharenko I., Afzal V., Rubin E.M.. Scanning human gene deserts for long-range enhancers. Science. 2003; 302:413. [DOI] [PubMed] [Google Scholar]
- 63. Kimura-Yoshida C., Kitajima K., Oda-Ishii I., Tian E., Suzuki M., Yamamoto M., Suzuki T., Kobayashi M., Aizawa S., Matsuo I.. Characterization of the pufferfish Otx2 cis-regulators reveals evolutionarily conserved genetic mechanisms for vertebrate head specification. Development. 2004; 131:57–71. [DOI] [PubMed] [Google Scholar]
- 64. Ji X., Dadon D.B., Powell B.E., Fan Z.P., Borges-Rivera D., Shachar S., Weintraub A.S., Hnisz D., Pegoraro G., Lee T.I. et al.. 3D chromosome regulatory landscape of human pluripotent cells. Cell Stem Cell. 2016; 18:262–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dowen J.M., Fan Z.P., Hnisz D., Ren G., Abraham B.J., Zhang L.N., Weintraub A.S., Schuijers J., Lee T.I., Zhao K. et al.. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell. 2014; 159:374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang H., Zang C., Taing L., Arnett K.L., Wong Y.J., Pear W.S., Blacklow S.C., Liu X.S., Aster J.C.. NOTCH1-RBPJ complexes drive target gene expression through dynamic interactions with superenhancers. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:705–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hnisz D., Weintraub A.S., Day D.S., Valton A.L., Bak R.O., Li C.H., Goldmann J., Lajoie B.R., Fan Z.P., Sigova A.A. et al.. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science. 2016; 351:1454–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rao S.S., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S. et al.. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014; 159:1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zheng H., Xie W.. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 2019; 20:535–550. [DOI] [PubMed] [Google Scholar]
- 70. Cook P.R., Marenduzzo D.. Transcription-driven genome organization: a model for chromosome structure and the regulation of gene expression tested through simulations. Nucleic Acids Res. 2018; 46:9895–9906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Puri P.L., Sartorelli V., Yang X.J., Hamamori Y., Ogryzko V.V., Howard B.H., Kedes L., Wang J.Y., Graessmann A., Nakatani Y. et al.. Differential roles of p300 and PCAF acetyltransferases in muscle differentiation. Mol. Cell. 1997; 1:35–45. [DOI] [PubMed] [Google Scholar]
- 72. Rampalli S., Li L., Mak E., Ge K., Brand M., Tapscott S.J., Dilworth F.J.. p38 MAPK signaling regulates recruitment of Ash2L-containing methyltransferase complexes to specific genes during differentiation. Nat. Struct. Mol. Biol. 2007; 14:1150–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Simone C., Forcales S.V., Hill D.A., Imbalzano A.N., Latella L., Puri P.L.. p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nat. Genet. 2004; 36:738–743. [DOI] [PubMed] [Google Scholar]
- 74. Cao Y., Kumar R.M., Penn B.H., Berkes C.A., Kooperberg C., Boyer L.A., Young R.A., Tapscott S.J.. Global and gene-specific analyses show distinct roles for Myod and Myog at a common set of promoters. EMBO J. 2006; 25:502–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yuan W., Condorelli G., Caruso M., Felsani A., Giordano A.. Human p300 protein is a coactivator for the transcription factor MyoD. J. Biol. Chem. 1996; 271:9009–9013. [DOI] [PubMed] [Google Scholar]
- 76. Blum R., Vethantham V., Bowman C., Rudnicki M., Dynlacht B.D.. Genome-wide identification of enhancers in skeletal muscle: the role of MyoD1. Genes Dev. 2012; 26:2763–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Svaren J., Klebanow E., Sealy L., Chalkley R.. Analysis of the competition between nucleosome formation and transcription factor binding. J. Biol. Chem. 1994; 269:9335–9344. [PubMed] [Google Scholar]
- 78. Cirillo L.A., Lin F.R., Cuesta I., Friedman D., Jarnik M., Zaret K.S.. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell. 2002; 9:279–289. [DOI] [PubMed] [Google Scholar]
- 79. Ramachandran S., Henikoff S.. Transcriptional regulators compete with nucleosomes post-replication. Cell. 2016; 165:580–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Xu M., Wang W., Chen S., Zhu B.. A model for mitotic inheritance of histone lysine methylation. EMBO Rep. 2012; 13:60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Audergon P.N., Catania S., Kagansky A., Tong P., Shukla M., Pidoux A.L., Allshire R.C.. Epigenetics. Restricted epigenetic inheritance of H3K9 methylation. Science. 2015; 348:132–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ragunathan K., Jih G., Moazed D.. Epigenetics. Epigenetic inheritance uncoupled from sequence-specific recruitment. Science. 2015; 348:1258699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hattori N., Niwa T., Kimura K., Helin K., Ushijima T.. Visualization of multivalent histone modification in a single cell reveals highly concerted epigenetic changes on differentiation of embryonic stem cells. Nucleic Acids Res. 2013; 41:7231–7239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Li G., Ruan X., Auerbach R.K., Sandhu K.S., Zheng M., Wang P., Poh H.M., Goh Y., Lim J., Zhang J. et al.. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012; 148:84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Moyle-Heyrman G., Tims H.S., Widom J.. Structural constraints in collaborative competition of transcription factors against the nucleosome. J. Mol. Biol. 2011; 412:634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Adams C.C., Workman J.L.. Binding of disparate transcriptional activators to nucleosomal DNA is inherently cooperative. Mol. Cell Biol. 1995; 15:1405–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Soufi A., Garcia M.F., Jaroszewicz A., Osman N., Pellegrini M., Zaret K.S.. Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell. 2015; 161:555–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Chen J., Zhang Z., Li L., Chen B.C., Revyakin A., Hajj B., Legant W., Dahan M., Lionnet T., Betzig E. et al.. Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell. 2014; 156:1274–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Vaharautio A., Taipale J.. Cancer. Cancer by super-enhancer. Science. 2014; 346:1291–1292. [DOI] [PubMed] [Google Scholar]
- 90. Ounzain S., Pedrazzini T.. Super-enhancer lncs to cardiovascular development and disease. Biochim. Biophys. Acta. 2015; 1863:1953–1960. [DOI] [PubMed] [Google Scholar]
- 91. Pacheco M.P., John E., Kaoma T., Heinaniemi M., Nicot N., Vallar L., Bueb J.L., Sinkkonen L., Sauter T.. Integrated metabolic modelling reveals cell-type specific epigenetic control points of the macrophage metabolic network. BMC Genomics. 2015; 16:809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Herranz D., Ambesi-Impiombato A., Palomero T., Schnell S.A., Belver L., Wendorff A.A., Xu L., Castillo-Martin M., Llobet-Navas D., Cordon-Cardo C. et al.. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat. Med. 2014; 20:1130–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Shin H.Y. Targeting Super-Enhancers for disease treatment and diagnosis. Mol. Cells. 2018; 41:506–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J. et al.. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012; 337:1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Tuupanen S., Yan J., Turunen M., Gylfe A.E., Kaasinen E., Li L., Eng C., Culver D.A., Kalady M.F., Pennison M.J. et al.. Characterization of the colorectal cancer-associated enhancer MYC-335 at 8q24: the role of rs67491583. Cancer Genet. 2012; 205:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Mansour M.R., Abraham B.J., Anders L., Berezovskaya A., Gutierrez A., Durbin A.D., Etchin J., Lawton L., Sallan S.E., Silverman L.B. et al.. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014; 346:1373–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Sengupta S., George R.E.. Super-enhancer-driven transcriptional dependencies in cancer. Trends Cancer. 2017; 3:269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Lin C.Y., Erkek S., Tong Y., Yin L., Federation A.J., Zapatka M., Haldipur P., Kawauchi D., Risch T., Warnatz H.J. et al.. Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature. 2016; 530:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Northcott P.A., Korshunov A., Pfister S.M., Taylor M.D.. The clinical implications of medulloblastoma subgroups. Nat. Rev. Neurol. 2012; 8:340–351. [DOI] [PubMed] [Google Scholar]
- 100. Ferrando A.A., Herblot S., Palomero T., Hansen M., Hoang T., Fox E.A., Look A.T.. Biallelic transcriptional activation of oncogenic transcription factors in T-cell acute lymphoblastic leukemia. Blood. 2004; 103:1909–1911. [DOI] [PubMed] [Google Scholar]
- 101. Edelmann J., Holzmann K., Miller F., Winkler D., Buhler A., Zenz T., Bullinger L., Kuhn M.W., Gerhardinger A., Bloehdorn J. et al.. High-resolution genomic profiling of chronic lymphocytic leukemia reveals new recurrent genomic alterations. Blood. 2012; 120:4783–4794. [DOI] [PubMed] [Google Scholar]
- 102. Uslu V.V., Petretich M., Ruf S., Langenfeld K., Fonseca N.A., Marioni J.C., Spitz F.. Long-range enhancers regulating Myc expression are required for normal facial morphogenesis. Nat. Genet. 2014; 46:753–758. [DOI] [PubMed] [Google Scholar]
- 103. Adey A., Burton J.N., Kitzman J.O., Hiatt J.B., Lewis A.P., Martin B.K., Qiu R., Lee C., Shendure J.. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature. 2013; 500:207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Katainen R., Dave K., Pitkanen E., Palin K., Kivioja T., Valimaki N., Gylfe A.E., Ristolainen H., Hanninen U.A., Cajuso T. et al.. CTCF/cohesin-binding sites are frequently mutated in cancer. Nat. Genet. 2015; 47:818–821. [DOI] [PubMed] [Google Scholar]
- 105. Nussenzweig A., Nussenzweig M.C.. Origin of chromosomal translocations in lymphoid cancer. Cell. 2010; 141:27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Meng F.L., Du Z., Federation A., Hu J., Wang Q., Kieffer-Kwon K.R., Meyers R.M., Amor C., Wasserman C.R., Neuberg D. et al.. Convergent transcription at intragenic super-enhancers targets AID-initiated genomic instability. Cell. 2014; 159:1538–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Qian J., Wang Q., Dose M., Pruett N., Kieffer-Kwon K.R., Resch W., Liang G., Tang Z., Mathe E., Benner C. et al.. B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell. 2014; 159:1524–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Alt F.W., Zhang Y., Meng F.L., Guo C., Schwer B.. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell. 2013; 152:417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Nesbit C.E., Tersak J.M., Prochownik E.V.. MYC oncogenes and human neoplastic disease. Oncogene. 1999; 18:3004–3016. [DOI] [PubMed] [Google Scholar]
- 110. Jackson T., Allard M.F., Sreenan C.M., Doss L.K., Bishop S.P., Swain J.L.. The c-myc proto-oncogene regulates cardiac development in transgenic mice. Mol. Cell Biol. 1990; 10:3709–3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ludwig K.U., Mangold E., Herms S., Nowak S., Reutter H., Paul A., Becker J., Herberz R., AlChawa T., Nasser E. et al.. Genome-wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci. Nat. Genet. 2012; 44:968–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Sotelo J., Esposito D., Duhagon M.A., Banfield K., Mehalko J., Liao H., Stephens R.M., Harris T.J., Munroe D.J., Wu X.. Long-range enhancers on 8q24 regulate c-Myc. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:3001–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Sur I.K., Hallikas O., Vaharautio A., Yan J., Turunen M., Enge M., Taipale M., Karhu A., Aaltonen L.A., Taipale J.. Mice lacking a Myc enhancer that includes human SNP rs6983267 are resistant to intestinal tumors. Science. 2012; 338:1360–1363. [DOI] [PubMed] [Google Scholar]
- 114. Tuupanen S., Turunen M., Lehtonen R., Hallikas O., Vanharanta S., Kivioja T., Bjorklund M., Wei G., Yan J., Niittymaki I. et al.. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat. Genet. 2009; 41:885–890. [DOI] [PubMed] [Google Scholar]
- 115. Dave K., Sur I., Yan J., Zhang J., Kaasinen E., Zhong F., Blaas L., Li X., Kharazi S., Gustafsson C. et al.. Mice deficient of Myc super-enhancer region reveal differential control mechanism between normal and pathological growth. Elife. 2017; 6:e23382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A. et al.. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013; 339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M.. RNA-guided human genome engineering via Cas9. Science. 2013; 339:823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Lupianez D.G., Kraft K., Heinrich V., Krawitz P., Brancati F., Klopocki E., Horn D., Kayserili H., Opitz J.M., Laxova R. et al.. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015; 161:1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Geutjes E.J., Bajpe P.K., Bernards R.. Targeting the epigenome for treatment of cancer. Oncogene. 2012; 31:3827–3844. [DOI] [PubMed] [Google Scholar]
- 120. Kelly T.K., De Carvalho D.D., Jones P.A.. Epigenetic modifications as therapeutic targets. Nat. Biotechnol. 2010; 28:1069–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Kwiatkowski N., Zhang T., Rahl P.B., Abraham B.J., Reddy J., Ficarro S.B., Dastur A., Amzallag A., Ramaswamy S., Tesar B. et al.. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014; 511:616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Anders L., Guenther M.G., Qi J., Fan Z.P., Marineau J.J., Rahl P.B., Loven J., Sigova A.A., Smith W.B., Lee T.I. et al.. Genome-wide localization of small molecules. Nat. Biotechnol. 2014; 32:92–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Gryder B.E., Yohe M.E., Chou H.C., Zhang X., Marques J., Wachtel M., Schaefer B., Sen N., Song Y., Gualtieri A. et al.. PAX3-FOXO1 establishes myogenic super enhancers and confers BET bromodomain vulnerability. Cancer Discov. 2017; 7:884–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Jiang Y.W., Veschambre P., Erdjument-Bromage H., Tempst P., Conaway J.W., Conaway R.C., Kornberg R.D.. Mammalian mediator of transcriptional regulation and its possible role as an end-point of signal transduction pathways. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:8538–8543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Dawson M.A., Prinjha R.K., Dittmann A., Giotopoulos G., Bantscheff M., Chan W.I., Robson S.C., Chung C.W., Hopf C., Savitski M.M. et al.. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011; 478:529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Togel L., Nightingale R., Chueh A.C., Jayachandran A., Tran H., Phesse T., Wu R., Sieber O.M., Arango D., Dhillon A.S. et al.. Dual targeting of bromodomain and extra-terminal domain proteins, and WNT or MAPK signaling, inhibits c-MYC expression and proliferation of colorectal cancer cells. Mol. Cancer Ther. 2016; 15:1217–1226. [DOI] [PubMed] [Google Scholar]
- 127. Zhang Z., Ma P., Jing Y., Yan Y., Cai M.C., Zhang M., Zhang S., Peng H., Ji Z.L., Di W. et al.. BET bromodomain inhibition as a therapeutic strategy in ovarian cancer by downregulating FoxM1. Theranostics. 2016; 6:219–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Sengupta D., Kannan A., Kern M., Moreno M.A., Vural E., Stack B. Jr, Suen J.Y., Tackett A.J., Gao L.. Disruption of BRD4 at H3K27Ac-enriched enhancer region correlates with decreased c-Myc expression in Merkel cell carcinoma. Epigenetics. 2015; 10:460–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Chapuy B., McKeown M.R., Lin C.Y., Monti S., Roemer M.G., Qi J., Rahl P.B., Sun H.H., Yeda K.T., Doench J.G. et al.. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013; 24:777–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Pawar A., Gollavilli P.N., Wang S., Asangani I.A.. Resistance to BET inhibitor leads to alternative therapeutic vulnerabilities in castration-resistant prostate cancer. Cell Rep. 2018; 22:2236–2245. [DOI] [PubMed] [Google Scholar]
- 131. Jin X., Yan Y., Wang D., Ding D., Ma T., Ye Z., Jimenez R., Wang L., Wu H., Huang H.. DUB3 promotes bet inhibitor resistance and cancer progression by deubiquitinating BRD4. Mol. Cell. 2018; 71:592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Yin Y., Sun M., Zhan X., Wu C., Geng P., Sun X., Wu Y., Zhang S., Qin J., Zhuang Z. et al.. EGFR signaling confers resistance to BET inhibition in hepatocellular carcinoma through stabilizing oncogenic MYC. J. Exp. Clin. Cancer Res. 2019; 38:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Chipumuro E., Marco E., Christensen C.L., Kwiatkowski N., Zhang T., Hatheway C.M., Abraham B.J., Sharma B., Yeung C., Altabef A. et al.. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell. 2014; 159:1126–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Christensen C.L., Kwiatkowski N., Abraham B.J., Carretero J., Al-Shahrour F., Zhang T., Chipumuro E., Herter-Sprie G.S., Akbay E.A., Altabef A. et al.. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell. 2014; 26:909–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Pott S., Lieb J.D.. What are super-enhancers. Nat. Genet. 2015; 47:8–12. [DOI] [PubMed] [Google Scholar]
- 136. Zamudio A.V., Dall’Agnese A., Henninger J.E., Manteiga J.C., Afeyan L.K., Hannett N.M., Coffey E.L., Li C.H., Oksuz O., Sabari B.R. et al.. Mediator condensates localize signaling factors to key cell identity genes. Mol. Cell. 2019; doi:10.1016/j.molcel.2019.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Shin H.Y., Willi M., HyunYoo K., Zeng X., Wang C., Metser G., Hennighausen L.. Hierarchy within the mammary STAT5-driven Wap super-enhancer. Nat. Genet. 2016; 48:904–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Bahr C., von Paleske L., Uslu V.V., Remeseiro S., Takayama N., Ng S.W., Murison A., Langenfeld K., Petretich M., Scognamiglio R. et al.. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature. 2018; 553:515–520. [DOI] [PubMed] [Google Scholar]
- 139. Shi J., Whyte W.A., Zepeda-Mendoza C.J., Milazzo J.P., Shen C., Roe J.S., Minder J.L., Mercan F., Wang E., Eckersley-Maslin M.A. et al.. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013; 27:2648–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
A core collection of SEs used for the meta-analyses in this Survey and Summary and the computational scripts are available in the GitHub repository (https://sunlightwang.github.io/Super-Enhancers/).