

Abstract

Experimental, spectroscopic, and computational studies are reported that provide an evidence-based mechanistic description of an intermolecular reductive C–N coupling of nitroarenes and arylboronic acids catalyzed by a redox-active main-group catalyst (1,2,2,3,4,4-hexamethylphosphetane P-oxide, i.e., 1·[O]). The central observations include the following: (1) catalytic reduction of 1·[O] to PIII phosphetane 1 is kinetically fast under conditions of catalysis; (2) phosphetane 1 represents the catalytic resting state as observed by 31P NMR spectroscopy; (3) there are no long-lived nitroarene partial-reduction intermediates observable by 15N NMR spectroscopy; (4) the reaction is sensitive to solvent dielectric, performing best in moderately polar solvents (viz. cyclopentylmethyl ether); and (5) the reaction is largely insensitive with respect to common hydrosilane reductants. On the basis of the foregoing studies, new modified catalytic conditions are described that expand the reaction scope and provide for mild temperatures (T ≥ 60 °C), low catalyst loadings (≥2 mol%), and innocuous terminal reductants (polymethylhydrosiloxane). DFT calculations define a two-stage deoxygenation sequence for the reductive C–N coupling. The initial deoxygenation involves a rate-determining step that consists of a (3+1) cheletropic addition between the nitroarene substrate and phosphetane 1; energy decomposition techniques highlight the biphilic character of the phosphetane in this step. Although kinetically invisible, the second deoxygenation stage is implicated as the critical C–N product-forming event, in which a postulated oxazaphosphirane intermediate is diverted from arylnitrene dissociation toward heterolytic ring opening with the arylboronic acid; the resulting dipolar intermediate evolves by antiperiplanar 1,2-migration of the organoboron residue to nitrogen, resulting in displacement of 1·[O] and formation of the target C–N coupling product upon in situ hydrolysis. The method thus described constitutes a mechanistically well-defined and operationally robust main-group complement to the current workhorse transition-metal-based methods for catalytic intermolecular C–N coupling.
1. Introduction
Aryl- and heteroarylamines are common in pharmaceuticals, natural products, agrochemicals, and functional materials.1 Consequently, the efficient construction of C–N bonds has been the target of considerable innovation. In particular, developments in transition-metal-catalyzed C–N coupling chemistry have shaped the dominant approach to arylamine synthesis.2 Chief among these methods is the Buchwald–Hartwig reaction (Figure 1A),3 which enables the net redox-neutral nucleophilic substitution of aryl (pseudo)halide with N-nucleophiles via Pd(0)/Pd(II) activation of the electrophilic partner through oxidative addition.4,5 A growing mastery over this important reaction has been enabled by increasingly detailed mechanistic understanding,6 with progressive optimizations of reaction conditions,7 ligands,8 and catalyst precursors9 resulting in ever-improving scope and efficiency.10
Figure 1.

(A) Redox-neutral C–N cross coupling (Buchwald–Hartwig). (B) Oxidative C–N cross coupling (Chan–Lam). (C) Reductive C–N cross coupling (PIII/PV=O redox catalysis).
In an alternative approach, intermolecular C–N cross coupling can be achieved in an oxidative manner by the reaction of N-nucleophiles with arylboron reagents under aerobic copper catalysis (i.e., Chan–Lam reaction, Figure 1B). In addition to the synthetic complementarity, this approach is supported in a practical sense by the impressive catalog of arylboron derivatives now available both commercially and by synthesis.11 And as with the Buchwald–Hartwig reaction, considerable experimental effort has helped to decrypt significant aspects of the Chan–Lam mechanism,12 providing the basis for an increasingly reliable and predictive model of reactivity with this method.13
As part of an ongoing program aimed at developing designer main-group compounds as biphilic14 organocatalysts in organic synthesis,15 we reported recently a reductive method for intermolecular C–N cross coupling. This method relies on an all-main-group system composed of an organophosphorus P(III)/P(V)=O redox catalyst and hydrosilane terminal reductant to transform nitroarenes and boronic acids into N-arylamines through intermolecular C–N bond formation (Figure 1C).16 The chief attributes of this method include (1) the use of precursors (i.e., nitroarenes) that are distinct from—but no less accessible than—those used in established C–N cross coupling methods, and (2) unique chemoselectivities and functional group tolerance inherent to the all-main-group conditions of the PIII/PV=O catalytic manifold.
To better understand the reductive P(III)/P(V)=O-catalyzed C–N bond-forming process and facilitate its further synthetic development, we were animated by several unresolved questions, including the following: (1) What is the nature of the turnover-limiting step in the catalytic C–N coupling reaction, and what is the role of the organophosphorus catalyst in this step? (2) What is the relationship of the catalytic C–N coupling reaction to related methods involving P(III)/P(V)=O-catalyzed nitroarene deoxygenation, and to what extent do the reactive intermediates coincide? (3) Can further improvements in reaction scope be attained, especially as informed through hypothesis-based experimentation within a mechanistic rationale?
In this Article, we provide an integrated experimental, spectroscopic, and computational description of the biphilic organophosphorus-catalyzed reductive C–N coupling strategy that systematically delineates the nature of deoxygenative events of nitroaromatics, especially in the context of the C–N bond formation. Among the key findings, we present herein: (1) a qualitative description of reaction parameters, culminating in a generally improved set of reaction conditions that enable heretofore challenging coupling reactions of azaheterocyclic nitroarene and boronic acids partners; (2) competition experiments that differentiate the intermolecular C–N cross coupling reaction from previous P(III)/P(V)=O-catalyzed C–N bond-forming methods, and weigh against the intermediacy of veritable arylnitrene intermediates along the C–N coupling pathway, (3) experimental spectroscopic and kinetic evidence that establish a P(III) resting state of the phosphetane catalyst and imply a rapid P(V)=O→P(III) turnover step for this small-ring phosphacycle; (4) a computational description of the overall energy landscape for the C–N coupling reaction pathway with an explicit description of the importance of organophosphorus biphilicity through energy decomposition analysis of the turnover-limiting transition state. Through these results, we establish the P(III)/P(V)=O-catalyzed intermolecular reductive C–N cross coupling of nitroarenes and arylboronic acids as an operationally robust and mechanistically well-defined main-group complement to the established transition-metal-based methods for catalytic intermolecular C–N coupling.
2. Results
2.1. Impact of Reaction Condition Variables
An evaluation of experimental variables for the organophosphorus-catalyzed reductive C–N coupling of nitroarenes and boronic acids was undertaken in order to provide a qualitative description of the parameter space that controls reaction yield and efficiency.
2.1.1. Solvent Dielectric Influences Yield
Prior optimization efforts had identified the high-boiling hydrocarbon m-xylene (ε = 2.6) as a suitable solvent for reductive intermolecular C–N coupling. Specifically, coupling of nitrobenzene (2) and phenylboronic acid (3) in m-xylene proceeds with full conversion of starting material and an 86% yield of product diphenylamine 4 over the course of 4 h at 120 °C. The ethereal solvent di-n-butyl ether (ε = 3.1) performed similarly (Figure 2). However, with increasing solvent polarity a significant and non-monotonic effect of solvent on the reaction outcome was observed. Solvents of moderate polarity, such as cyclopentyl methyl ether (CPME, ε = 4.8) and 1,2-dichlorobenzene (ε = 9.9) lead to improved yields (Table 1, entries 3 and 4), but further increases in solvent polarity (i.e., benzonitrile (PhCN, ε = 26.0), N-methyl-2-pyrrolidone (NMP, ε = 32.0), and dimethyl sulfoxide (DMSO, ε = 46.7) were shown to erode both the conversion and the yield. On the basis of the foregoing experiments, CPME—which exhibits favorable process characteristics17—was selected as the solvent of choice for the further study.
Figure 2.
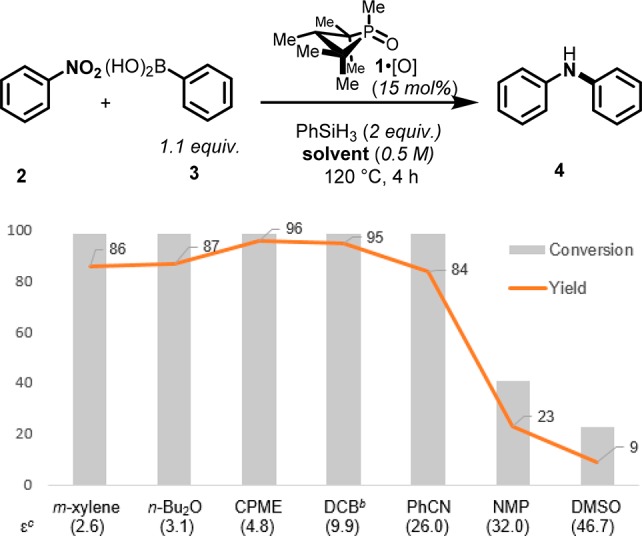
Solvent effect evaluation on the organophosphorus-catalyzed reductive C–N coupling reaction. aYields were determined through analysis by gas chromatography with the use of dodecane as an internal standard. bSolvents: CPME, cyclopentyl methyl ether; DCB, 1,2-dichlorobenzene; PhCN, benzonitrile; NMP, N-methyl-2-pyrrolidone; DMSO, dimethyl sulfoxide. cε is the dielectric constant.
Table 1. Effect of Hydrosilane Loading and Identity on the Organophosphorus-Catalyzed Reductive C–N Coupling Reaction.

| entry | change from “standard conditions” | conv (yield) (%)a |
|---|---|---|
| 1 | none | 99 (96) |
| 2 | 5 mol% of 1·[O], 10 h | 99 (95) |
| 3 | 2 mol% of 1·[O], 36 h | 99 (93) |
| 4 | 80 °C, 20 h | 99 (95) |
| 5 | 60 °C, 96 h | 99 (93) |
| 6 | 0.77 equiv of PhSiH3 | 98 (94) |
| 7 | 0.66 equiv of PhSiH3 | 85 (79) |
| 8 | 0.33 equiv of PhSiH3 | 49 (46) |
| 9 | 3.0 equiv of Ph2SiH2 | 96 (88) |
| 10 | 3.0 equiv of TMDSc | 93 (85) |
| 11 | 1.5 equiv of TMCTSb | 99 (83) |
| 12 | 4.0 equiv of PMHS | 99 (96) |
| 13 | Ph-Bpin instead of PhB(OH)2 | 49 (trace) |
Yields were determined through analysis by gas chromatography with the use of dodecane as an internal standard.
TMCTS = 2,4,6,8-tetramethylcyclotetrasiloxane.
TMDS = 1,1,3,3-tetramethyldisiloxane.
2.1.2. Performance Is Maintained at Low Catalyst Loading and Temperature
The robustness of the phosphetane catalyst 1·[O] under conditions of catalysis allow for significant decreases in its loading. For instance, decrease in loading of 1·[O] to 5 mol% (Table 1, entry 2) or 2 mol% (Table 1, entry 3) permits high conversion and yield, with the provision of a compensatory elongation of the reaction time to 10 and 36 h, respectively. Relatedly, the catalytic transformation is retained with high yield even at temperatures down to 60 °C (Table 1, entries 4 and 5), emphasizing the high reactivity of the phosphetane catalyst.
2.1.3. Numerous Common Hydrosilane Reductants Are Viable
Our “first-generation” conditions for PIII/PV=O-catalyzed reductive C–N coupling called for the use of 2.0 equiv of phenylsilane (PhSiH3) as the terminal reductant with respect to limiting nitrobenzene (2) (Table 1, entry 1), but experiments show that fewer equivalents may be employed. Indeed, an excess of phenylsilane is not inherently required, and loadings as low as 0.77 equiv lead to qualitatively similar reaction outcomes (entry 6); lower loadings do, however, lead to diminished conversion and yield (entries 7 and 8). Taking into consideration that the reductive conversion of nitrobenzene (2) to diphenylamine (4) is a two-fold reduction at N, the inference from these experiments is that all three Si–H reducing equivalents from phenylsilane can be leveraged for productive C–N coupling. With its low molecular weight and low effective mass per Si–H equivalent, phenylsilane could therefore be considered a rather efficient terminal reductant for the PIII/PV=O-catalyzed C–N coupling reaction. We note, moreover, that hydrosilane equivalency shows no influence on reaction time (Table S2, entries 12–26), which has implications for its mechanistic role in mediating PIII/PV=O catalysis (vide infra).
The reaction does not strictly require PhSiH3 as the hydrosilane terminal reductant, but instead a wide range of common silicon-based reducing reagents are able to be interfaced with the PIII/PV=O-catalyzed reductive C–N coupling. Along with Ph2SiH2 (Table 1, entry 9), a variety of siloxane-based reductants including 1,1,3,3-tetramethyldisiloxane (TMDS, Table 1, entry 10), 2,4,6,8-tetramethylcyclotetrasiloxane (TMCTS, Table 1, entry 11), and poly(methylhydro)siloxane (PMHS, Table 1, entry 12) are viable.18 Of these, PMHS is particularly attractive due to its ease of handling and low cost, recommending it for further method development.
As previously observed, the aryl C–N coupling reaction is most effective when arylboronic acid coupling partners are employed. Even under optimal reaction conditions, the use of phenylboronic acid pinacol ester (Ph–Bpin) in place of phenylboronic acid (3) results in only trace formation of coupling product 4 (Table 1, entry 13). The lower overall observed conversion (49%) is connected to substantial catalyst decomposition when the less-efficient boronate partner is employed.
2.1.4. Modified Conditions Enable Coupling of Previously Challenging Partners
With an eye toward an expanded scope for the PIII/PV=O-catalyzed reductive C–N coupling method, we sought to determine if the versatility of the reaction conditions observed in the foregoing sections would provide an opportunity to approach previously problematic classes of coupling partners. The reaction of 1-methyl-5-nitroindole (5) with 4-fluorophenylboronic acid (6) is an illustrative example (Table 2). When applying typical first-generation reaction conditions (Table 2, entry 1), only 13% yield was obtained of the desired reductive coupling product 7. However, consistent with the solvent effect reported in section 2.1.1, a solvent change to CPME resulted in a somewhat improved yield (27%, entry 2). Even more significantly, though, use of the hydrosilane reductant PMHS in m-xylene resulted in significantly improved yields (47%, Table 2, entry 3). The beneficial solvent and hydrosilane effects are synergistic in this case, such that the reaction of 5 and 6 conducted with PMHS in CPME provides coupling product 7 in a preparatively useful yield (68%, Table 2, entry 4).
Table 2. Impact of Reaction Variables on Reductive C–N Coupling of Heterocyclic Nitroarenes.

| entry | SiH (equiv) | solvent | yield (%)a |
|---|---|---|---|
| 1 | PhSiH3 (2) | m-xylene (0.25 M) | 13 |
| 2 | PhSiH3 (2) | CPME (0.25 M) | 27 |
| 3 | PMHS (6) | m-xylene (0.25 M) | 47 |
| 4 | PMHS (6) | CPME (0.25 M) | 68 (66)b |
Yields were determined through analysis by 19F NMR with the aid of 4-fluorotoluene as an internal standard.
Isolated yield in parentheses.
These “second-generation” conditions (i.e., catalyst, 15 mol% 1,2,2,3,4,4-hexamethylphosphetane oxide (1·[O]); reductant, poly(methylhydro)siloxane; solvent, CPME) have been found to provide a general improvement in yield for all C–N coupling reactions we have assayed to date, and especially so for a variety of five- and six-membered heterocyclic nitroarenes that had previously been challenging to the intermolecular reductive PIII/PV=O-catalyzed C–N coupling method (Table 3). In addition to indole 7, a range of heteroarylnitro substrates are converted with reductive C–N coupling into the corresponding heteroarylamines as exemplified by pyrazole 8, 2H-indazole 9, pyrimidine 10, and aminobenzothioazole 11. Furthermore, reactions involving the incorporation of heteroaryl boronic acid coupling partners are similarly advantaged by the modified “second-generation” conditions; for instance, 1H-indazolyl (12), pyrazolyl (13), pyrimidinyl (14), and pyridinyl (15) boronic acids are successfully coupled with (hetero)aryl nitro partners. In all cases, though, the modified “second-generation conditions” afford marked improvements over the previously reported “first-generation conditions” and allow preparatively useful yields of functionally dense heteroarylamines. In instances where the heteroaryl boronic acid is found to be thermally unstable with respect to protodeboronation, a further modification to decrease the reaction temperature (80–100 °C) is found to be permissible (12–14).
Table 3. Examples of Reductive C–N Coupling of Heterocyclic Nitroarenes and/or Boronic Acidsa.


Yields reported for isolated products.
Reaction was conducted at 100 °C.
Reaction was conducted at 80 °C. See Supporting Information for full experimental details.
2.2. Competition Studies: Intermolecular C–N Coupling vs Arylnitrene Reactivity
In an effort to delineate the relationship between the reductive C–N coupling reaction from previously reported P(III)/P(V)=O-catalyzed reactions of nitroarenes, we designed a set of competition experiments as described in Tables 4 and 5. As a point of reference, subjection of 2-nitrobiphenyl (17) to first-generation catalytic conditions with omission of the phenylboronic acid coupling partner resulted in formation of carbazole (19) by intramolecular cyclization (Table 4, entry 1).15c As previously reported, this Csp2–H amination reaction proceeds by two-fold sequential deoxygenation to give an arylnitrene that undergoes insertion to the proximal C–H position.15c,19,58 We postulated that if similar arylnitrene intermediates were involved in the C–N cross coupling reaction with boronic acids, then a competition between intramolecular carbazole cyclization and intermolecular aryl amination with 2-nitrobiphenyl as a probe substrate would favor the former on kinetic grounds. In the event, reaction of 2-nitrobiphenyl (17) in the presence of phenylboronic acid 3 under otherwise identical reaction conditions led preferentially to the intermolecular reductive C–N cross coupling as the dominant reaction product (Table 4, entry 2). Notably, the use of CPME as the solvent (Table 4, entry 3) accentuates the bias in favor of the C–N cross coupling.
Table 4. Competition Studies between Reductive C–N Coupling and Arylnitrene Reactivity Starting from 2-Nitrobiphenyl.

| entry | equiv of boronic acid (3) | solvent | yield of 18 (%)a | yield of 19 (%)a |
|---|---|---|---|---|
| 1 | 0 | m-xylene | — | 82 |
| 2 | 1.1 | m-xylene | 76 | 22 |
| 3 | 1.1 | CPME | 88 | 11 |
Yields were determined through analysis by gas chromatography with the use of 1,3,5-trimethoxybenzene as an internal standard.
Table 5. Competition Studies between Reductive C–N Coupling vs Arylnitrene Reactivity Starting from 4-Nitrobenzonitrile.

| entry | equiv of boronic acid (3) | solvent | yield of 21 (%)a | yield of 22 (%)a |
|---|---|---|---|---|
| 1 | 0 | m-xylene | — | 78 |
| 2 | 1.1 | m-xylene | 87 | <2 |
| 3 | 1.1 | CPME | 93 | <2 |
Yields were determined through analysis by 1H NMR spectroscopy with the aid of dibromomethane as an internal standard.
In a related fashion, intermolecular competition experiments are similarly inconsistent with formation of arylnitrenes on the pathway to C–N cross coupling. Deoxygenation of 4-nitrobenzonitrile (20) under conditions of P(III)/P(V)=O catalysis proves competent for arylnitrene generation, as inferred from in situ trapping with diethylamine to give azepine 22 as the major product (Table 5, entry 1).20 However, when phenylboronic acid is admitted under otherwise identical reaction conditions, the reaction is shunted away from formation of azepine 22, instead providing the diarylamine 21 by C–N coupling in good yield (Table 5, entry 2). As before, CPME as the solvent (Table 5, entry 3) further favors formation of the C–N cross coupling product 21 relative to azepine 22.
The implications of these results are two-fold. First, the C–N cross coupling reaction evidently does not result from amination of the arylboronic acids by a free arylnitrene, but rather the mechanistic branching point along the pathway leading to cyclization or coupling must precede arylnitrene formation. Second, the impact of CPME on the product ratio suggests that the qualitative solvent effect observed in section 2.1.1 may arise through the relative suppression of the nitrene-forming pathway, which is nonproductive with respect to intermolecular C–N bond formation.
2.3. In Situ Spectroscopic Studies
2.3.1. Catalyst Speciation in Reductive C–N Coupling
In order to evaluate the catalyst speciation, in situ31P NMR spectra (161.9 MHz, 100 °C) were recorded under conditions of catalysis for the coupling reaction of nitrobenzene and phenylboronic acid (1.0 equiv of 2, 1.1 equiv of 3, 15 mol% of 1·[O], 2 equiv of phenylsilane, 0.2 M in toluene-d8). These spectra showed that phosphetane oxide 1·[O] (δ 55.9 ppm) is rapidly converted (t1/2 ≈ 5 min) to the corresponding tricoordinate phosphetane epimers anti-1 and syn-1 (δ 32.9 and δ 19.2 ppm, respectively)21 (Figure 3). Over the ensuing reaction time during which 2 is converted to 4, the tricoordinate epimers of 1 remain the only observable phosphorus-containing compounds in solution. Evidently, reduction of the phosphetane oxide 1·[O] is quite swift and the reduced tricoordinate phosphetane 1 represents the resting state with respect to the catalytic phosphorus component. These observations run counter to prevailing notions about the kinetic inertness of phosphine oxides and provide evidence for the exceptional reactivity of phosphetane oxide 1·[O] as a biphilic O-atom transfer catalyst.
Figure 3.

Time-stacked in situ31P NMR spectra (T = 100 °C, toluene-d8) at t = 0, 15, 60, and 360 min. Chemical shifts: 1·[O], δ 55.9 ppm; 1, δ 32.9 (anti) and 19.2 (syn) ppm. Units of chemical shift (δ) are ppm relative to 85% H3PO4 as an external standard.
2.3.2. Reactant Speciation in Reductive C–N Coupling
1H NMR spectra (400 MHz, 100 °C) of a catalytic reaction show consumption of nitrobenzene over ca. 3 h with concomitant appearance of diphenylamine 4 as the major product (Figure 4). 15N NMR spectra (40.5 MHz, 100 °C) collected under identical conditions indicate that isotopically enriched 15N-nitrobenzene (δ 369.4 ppm) is cleanly converted into the product diphenylamine (δ 86.0 ppm), and no long-lived intermediates are observed in the range 950 ppm > δ > −50 ppm (Figure S1).
Figure 4.

Time-stacked in situ15N NMR spectra (T = 100 °C, toluene-d8) at t = 0, 15, 60, and 360 min. Chemical shifts: 2, δ 369.4 ppm; 4, δ 86.0 ppm. Units of chemical shift (δ) are ppm relative to NH3(l) as an external standard.
2.4. Catalytic Kinetics Experiments
The kinetic progress of the catalytic coupling of nitrobenzene 2 and phenylboronic acid 3 was monitored via ex situ HPLC analysis of reaction aliquots drawn at intervals over the course of 7 h. Nitrobenzene 2 is converted to diphenylamine 4 in >95% efficiency with no discernible intermediates (chromatograms in Figure S4), consistent with the observations from NMR spectroscopy. The decrease in concentration of starting material 2 as a function of time fits a first-order kinetic model (Figure 5A), where the initial rates vary linearly with precatalyst 1·[O] concentration in the range 0.02 M ≤ [1·[O]] ≤ 0.08 M (Figure 5B), indicating that the reaction in Figure 3 is first-order with respect to both substrate 2 and precatalyst 1·[O]. Rate constants obtained by the complementary monitoring of increasing product 4 concentration with time at varying precatalyst 1·[O] concentrations (Figure S6) agree within ±10%. Initial reaction rates measured for this catalytic reaction vary neither as a function of phenylsilane concentration (0.2 M < [PhSiH3] < 0.8 M], Figure 5C) nor phenylboronic acid (3) concentration (0.1 M < [3] < 0.7 M], Figure 5D). The empirical rate law for the catalytic C–N coupling therefore is described by the equation:
Figure 5.
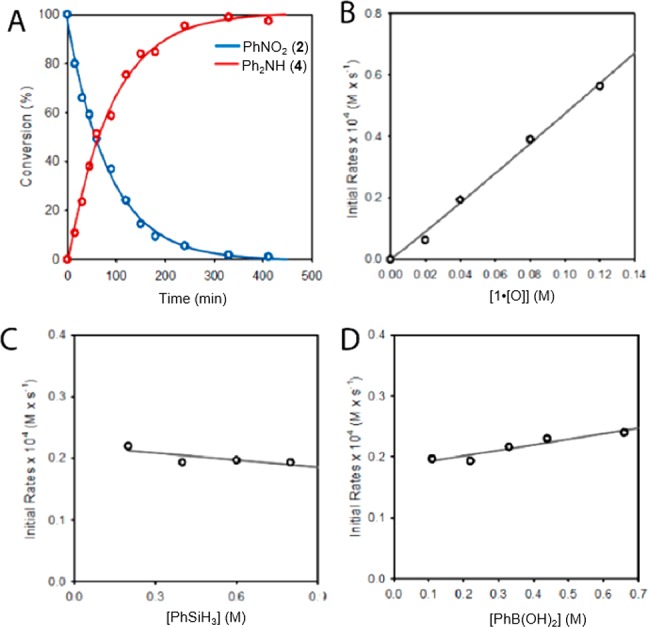
Spectroscopic and experimental mechanistic investigations. (A) Plot of conversion of substrate 2 (blue) to product 4 (red) vs time. (B) Plot of initial rates for substrate 2 consumption vs precatalyst 1·[O] concentration. (C) Plot of initial rates for substrate 2 consumption vs phenylsilane concentration. (D) Plot of initial rates for substrate 2 consumption vs phenylboronic acid 3 concentration.
2.5. Computational Studies
2.5.1. Initial Deoxygenation and Rate-Determining Step
Density functional theory calculations, conducted at the M06-2X/6-311++G(d,p) level with a polarizable continuum model (PCM) for solvation in m-xylene (ε = 2.3478), provide an atomistic-level proposal of mechanism that agrees with spectroscopic and kinetic studies. In accordance with our previous calculations on nitroarene–phosphine reactivity,15c DFT predicts a stepwise pathway for reductive C–N coupling initiated by a (3+1) cheletropic addition of nitrobenzene 2 with phosphetane 1 to form pentacoordinate spiro-bicyclic dioxazaphosphetane Int-1 (Figure 6A). The transition state for the concerted (3+1) addition step can be viewed as a Woodward–Hoffmann allowed [4πs+2ωs] cycloaddition (TS-1, Figure 6B) with a computed barrier of ΔG⧧rel = +31.0 kcal/mol). By virtue of this relatively high barrier, passage through TS-1 represents the slowest step in the computed pathway, kinetically gating all downstream events and providing a rationale for the failure to spectroscopically detect any reaction intermediates. Dioxazaphosphetane Int-1 evolves by a retro-(2+2) fragmentation with a low kinetic barrier via TS-2 (Figure 6B, ΔG⧧rel = +10.8 kcal/mol) to give phosphine oxide 1·[O] and nitrosobenzene (Int-2) (ΔGrel = −31.9 kcal/mol). The lower activation barrier calculated for the collapse of the spirobicyclo Int-1 (via TS-2) relative to its formation (via TS-1) stems from the incipient dissociation of P-oxide 1·[O] and release of ring strain during the fragmentation.
Figure 6.

Mechanistic proposal for catalytic reductive C–N coupling supported by density functional theory (DFT) calculations at the M06-2X/6-311++G(d,p)/PCM(m-xylene) level of theory. Relative free energies (italics) are given in kcal/mol. (A) Proposed mechanism of initial nitrobenzene deoxygenation and rate-determining step. (B) Computed model of TS-1 and TS-2. (C) Proposed mechanism of second deoxygenation and product-forming step. (D) Computed model of TS-3a, TS-4, and TS-5. Phosphorus (orange), oxygen (red), nitrogen (blue), carbon (gray), boron (pink), hydrogen (white). Bond distances in Å.
EDA-NOCV calculations22,23 of the charge flow and pairwise orbital interactions of TS-1 validate the biphilic character of phosphetane 1. Electrostatic (ΔEelstat = −81.1 kcal/mol) and orbital interactions (ΔEorb = −68.2 kcal/mol) between the phosphetane 1 and nitrobenzene 2 fragments are attractive and comparable in magnitude, accounting for 54.3% and 45.7% of the bonding interactions, respectively. Together, ΔEelstat and ΔEorb offset the Pauli electron pair repulsion term (ΔEPauli = 137.8 kcal/mol) to afford a total bonding energy of −11.5 kcal/mol. Analysis of the deformation densities displays both the electron donation from the HOMO of phosphetane 1 to the LUMO of nitrobenzene 2 and the backward electron donation from the HOMO of nitrobenzene 2 to the LUMO of phosphetane 1. The main deformation density (Δqσd = −1.0592) corresponds to a strong σ-donation from the phosphorus lone pair to the nitroarene and contributes to a stabilization of −56.8 kcal/mol (Figure 7A). An additional deformation densities with a smaller contribution (Δqπ = −0.2823) is consistent with π-backdonation from the nitroarene to the P–C σ* antibonding orbitals of the phosphetane and provide a considerable stabilization of −9.0 kcal/mol (Figure 7B).
Figure 7.
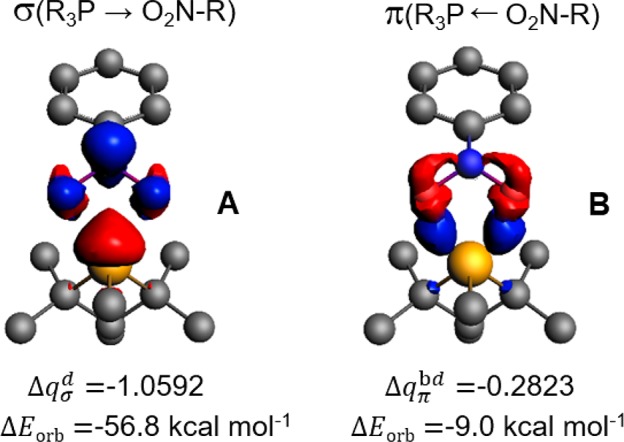
EDA-NOCV results of the orbital interactions for the (3+1) cheletropic addition of nitrobenzene 2 with phosphetane 1 to form pentacoordinate spiro-bicyclic dioxazaphosphetane Int-1. Red = depletion, blue = accumulation. (A) Forward electron donation. (B) Backward electron donation.
A second-order perturbation natural bond orbital (NBO)24,25 analysis of TS-1 affords additional insight into donor–acceptor interactions. Phosphorus lone pair σ-donation is represented by incipient σ P–O bonds polarized toward the oxygen that display an approximate composition of 38.52% P(sp3.64) + 61.48% O(sp23.53). Interestingly, endocyclic σ P–C bonds of the phosphetane, which are polarized toward the carbon and present an approximate composition of 37.41% P(sp2.44) + 62.59% C(sp4.40), also act as donors delocalized into the geminal acceptor σ* P–O bonds. In contrast, π-symmetry back-donation from the nitroarene moiety entails delocalization of both the σ P–O bonds and the O lone pairs into the geminal σ* P–C antibonding orbitals with relative second-order perturbation energies consistent with a 4:1 donor prevalence of the σ P–O bonds over the O lone pairs.
2.5.2. Second Deoxygenation and Product-Forming Step
Once formed, nitrosobenzene (Int-2) itself is subject to reaction with phosphetane 1 (Figure 7C) to give an oxazaphosphirane intermediate Int-3 (ΔGrel = −1.0 kcal/mol). Isomeric transition structures TS-3a and TS-3b, differing in the trajectory for the phosphetane attack on Int-2, were located. Both structures describe an asynchronous (2+1) addition with a P-centered spiro geometry that facilitates the interaction of the phosphorus lone pair with the π* orbital of the N=O group. TS-3a, which corresponds to the attack of the phosphorus on the nitrogen of the N=O group,26 is favored by 8.1 kcal/mol relative to TS-3b, which represents the attack of the phosphorus on the oxygen27 in agreement with a prevalence of the LUMO coefficient of the N=O group at the nitrogen atom.28 Electrophilic ring opening of oxazaphosphirane Int-3 with phenylboronic acid via TS-4 (ΔG⧧rel = +6.2 kcal/mol) coincides with the favorable formation of phosphonium oxyaminoborate betaine Int-4 (ΔGrel = −2.1 kcal/mol), featuring a typical aminoboronate B–N bond length and an intramolecular charge-dipole contact between the phosphorus and the OH group of the aminoborate moiety. As a suitable zwitterionic retron for 1,2-metalate rearrangement,24,29Int-4 represents the immediate precursor to C–N bond formation, evolving via TS-5 (ΔG⧧rel = +11.7 kcal/mol) with departure of phosphine oxide 1·[O] by antiperiplanar migration of the phenyl group from boron to nitrogen to give phenylboramidic acid (Figure 7D).
A DFT analysis of the competition between the Cadogan cyclization and the reductive C–N coupling pathways for 2-nitrobiphenyl (17) qualitatively supports the experimental preference for C–N coupling discussed in section 2.2 (Tables 4 and 5).30 Following a rate-limiting first deoxygenation of the nitro group by phosphetane 1 (TS-6, ΔG⧧rel = +29.7 kcal/mol) to afford 2-nitrosobiphenyl (23) (Figure 8A), reaction of the nitroso group with 1 takes place via a significantly lower barrier (TS-8, ΔG⧧rel = +17.2 kcal/mol) to give the “branching” intermediate oxazaphosphirane Int-6. In the Cadogan cyclization pathway, Int-6 evolves through loss of phosphetane P-oxide 1·[O] (TS-9, ΔG⧧rel = +15.0 kcal/mol) to form the carbazole product (19) via C–H insertion of the biphenylnitrene Int-7 (TS-10, ΔG⧧rel = +8.9 kcal/mol).15c Alternatively, in the reductive C–N coupling pathway, Int-6 reacts with phenylboronic acid (TS-11, ΔG⧧rel = +9.9 kcal/mol) to generate phosphonium oxyaminoborate betaine Int-8, which undergoes 1,2-metalate rearrangement and dissociation of phosphetane P-oxide 1·[O] (TS-12, ΔG⧧rel = +12.2 kcal/mol). Inspection of the nonlimiting steps that intervene in the branching of oxazaphosphirane Int-6 suggests that the experimental preference for reductive C–N coupling can be attributed to the circumvention of the biphenylnitrene pathways that mediates the Cadogan cyclization via a higher energy barrier TS-9 (Figure 8B).
Figure 8.

DFT studies (M06-2X/6-311++G(d,p)/PCM(m-xylene)) for the competition between intramolecular Cadogan cyclization and intermolecular reductive C–N coupling. (A) Initial deoxygenation with 2-nitrobiphenyl. (B) Proposed mechanism of second deoxygenation and product-forming step. (C) Computed models of TS-8, TS-9, TS-11, and TS-12. Phosphorus (orange), oxygen (red), nitrogen (blue), carbon (gray), boron (pink), hydrogen (white). Bond distances in Å.
3. Discussion
As a complement to established net redox neutral (Buchwald–Hartwig and related) and net oxidative (Chan–Lam) transition-metal-catalyzed C–N coupling methods, the current method brings together nitroarene and arylboronic acid coupling partners through net reductive catalysis enabled by the P(III)/P(V)=O redox couple. Nitroarenes are attractive coupling partners because they are readily accessible and easily transformed in synthesis; the nitro functional group is both easily installed and strategically useful due to its powerful inductive effect.31 And while nitroarenes are common precursors to aryl amine and aryl halide substrates for known transition-metal-catalyzed couplings, they are less commonly used as substrates themselves for direct catalytic C–N bond-forming reactions. Precedent within this vein includes the work of Nicholas, who established iron-catalyzed reductive C–N bond construction by reaction of nitroarenes with alkynes;32 Baran, who has discovered an iron-catalyzed synthesis of N-alkylamines by reductive C–N bond formation between nitroarenes with alkenes;33 and Shaver and Thomas, who have described related transformations catalyzed by an iron bis(phenolato)amine catalyst.34 Hu has reported iron- and nickel-catalyzed reductive C–N bond formation by reaction of nitroarenes with alkyl and acyl electrophiles, respectively.35 Apart from these catalytic methods, there exist several reagent-based approaches to direct conversion of nitroarenes to the corresponding N-functionalized anilines. Knochel36 and Kürti37 have demonstrated the use of excess Grignard reagents to convert nitroarenes to N-arylanilines directly. Niggemann has found that the combination of nitroarenes with organozinc reagents in the presence of stoichiometric B2pin2 results in reductive conversion to N-functionalized anilines.38 Recent works from our group39 and Csákÿ40 have validated a stoichiometric, phosphine-mediated reductive coupling of nitroarenes and arylboronic acids. Relatedly, Suárez-Pantiga and Sanz reported that phosphine-mediated reductive coupling of nitroarenes and boronic acids is catalyzed by an oxomolybdenum compound.41 Among these varied approaches, the P(III)/P(V)=O-catalyzed method—with its relatively mild conditions, commercial catalyst, and inexpensive reductant—compares rather favorably.
With regard to the mechanism of the P(III)/P(V)=O-catalyzed reductive C–N coupling reaction, the combined experimental and computational data point toward a catalytic reaction sequence that evolves in two stages—an initial deoxygenation of the nitroarene substrate to the corresponding nitrosoarene (Figure 9, top hemisphere), and a subsequent second deoxygenation that converts the intermediate nitrosoarene into the observed N-arylated product (Figure 9, bottom hemisphere). The common thread uniting these two sequential reduction events is the action of the small-ring phosphacycle 1·[O] to catalyze O-atom transfer by redox cycling in the P(III)/P(V) couple. Since O’Brien’s initial report of an organophosphorus-catalyzed Wittig reaction,42,43 P(III)/P(V) redox catalysis has emerged as an productive area of organophosphorus catalysis,44−46 with work from Woerpel,47 Rutjes and van Delft,48 Werner,49 Mecinović,48g Kwon,50 and Voituriez,51 among others.52−55 In the context of the current C–N coupling method, the observation that the resting state of the catalyst resides at the P(III) oxidation state (i.e., phosphetane 1) confirms the swift deoxygenation kinetics of small-ring phosphine oxides noted by Marsi56 and Keglevich57 and makes clear that P(V)=O→P(III) turnover is not a significant impediment to method development in the P(III)/P(V) couple with these catalytic structures.
Figure 9.

Proposed mechanism for organophosphorus-catalyzed reductive C–N coupling.
The initial nitroarene-to-nitrosoarene deoxygenation event is gated by a (3+1) cheletropic addition of nitrobenzene 2 with phosphetane 1. Consistent with experimental spectroscopy and kinetics, DFT modeling confirms that this step is turnover limiting and highest in energy of any transition state in the entire reductive C–N coupling sequence. Analysis of the transition structure within both the EDA-NOCV and NBO theoretical frameworks validates the notion of pairwise orbital interactions allowing for electron flow both to and from the phosphorus site, in accord with the concept of “biphilic” (i.e., synergistic single-site donor/acceptor) reactivity of the phosphetane. The relative magnitudes of the donor and acceptor interactions suggest that the former predominates, which is consistent with Hammett studies (see SI) indicating a net transfer of electron density to the nitroarene in the transition state.58,59 Once formed, Int-1 evolves via retro-(2+2) fragmentation to liberate phosphetane oxide 1·[O] and nitrosobenzene (Int-2), an obligate albeit unobserved intermediate under catalytic conditions. The phosphetane oxide 1·[O] is itself subject to rapid deoxygenation by hydrosilane to return to the P(III) resting state (1) and close the first catalytic deoxygenation cycle.
The second deoxygenation stage commences with capture of nitrosobenzene (Int-2) by P(III) phosphetane 1 through an asynchronous (2+1) addition to provide an oxazaphosphirane Int-3. On the basis of product distributions obtained from competition studies between intermolecular C–N coupling vs arylnitrene reactivity, we posit that this oxazaphosphirane Int-3 serves as the pivotal “branching” intermediate whose fate is a key determinant of product distribution. Whereas unimolecular loss of phosphetane oxide 1·[O] from Int-3 liberates an arylnitrene reactive intermediate that results in azepine ring expansion or Cadogan cyclization (cf. TS-9), DFT predicts a low-energy bimolecular reaction of oxazaphosphirane Int 3 with arylboronic acid leads to heterolytic ring-opening (cf. TS-4) and formation of betaine Int-4. We surmise that the apparent solvent influence in the competition experiments (section 2.1) operates by stabilization of partial charge build-up in the transition states leading to and from dipolar structure Int-4 (i.e., TS-4 and TS-5), relative to dissociative loss of phosphetane oxide 1·[O]. In analogy to numerous related electrophilic amination reactions of organoboron reagents,24,38a,60−63 an ensuing 1,2-metalate rearrangement of betaine Int-4 results in the formation of the desired C–N bond, which either upon hydrolysis with adventitious water or upon workup gives the target amine. A final hydrosilane-mediated reduction of phosphetane oxide 1·[O] returns the catalyst to the P(III) resting state (1) and closes the second catalytic deoxygenation cycle.
4. Conclusion
P(III)/P(V)=O-catalyzed intermolecular reductive C–N cross coupling of nitroarenes and arylboronic acids is emerging as an operationally robust and mechanistically well-defined main-group complement to the established transition-metal-based methods for catalytic intermolecular C–N coupling. Combined experimental, spectroscopic, and computational experiments provide a description of the biphilic organophosphorus-catalyzed method by systematically differentiating the nature of deoxygenative events of nitroaromatics especially in the context of the C–N bond formation. Namely, the rate-determining step is a (3+1) addition. The product-determining step involves the ring-opening of an oxazaphosphirane. Combined, these findings enrich the fundamental understanding of the biphilic reactivity of phosphetanes as generalized platforms for catalytic reductive O-atom transfer operating in the PIII/PV=O redox manifold and provide an experimentally based mechanistic framework to guide iterative catalyst design and method development.
5. Experimental Section
A full description of the general experimental methods can be found in the Supporting Information.
5.1. Representative Synthetic Procedure for the Reductive C–N Coupling
The appropriate nitro substrate (if solid) and phosphetane oxide precatalyst 1·[O] (15 mol% unless otherwise noted) were added to an oven-dried glass culture tubes with threaded end (20 × 125 mm; Fisher Scientific part no. 14-959-35A), outfitted with a phenolic screw-thread open top cap (Kimble-Chase part no. 73804-15425) and PTFE-lined silicone septum (Thermo Fisher part no. B7995-15) sequentially. Following evacuation and the introduction of nitrogen on a Schlenk line, dry CPME was added via syringe. Lastly, hydrosilane and nitro substrate (if liquid) were added and the reaction mixture was stirred at 120 °C. When complete, the reaction vessel screw cap was unscrewed (note that in some cases pressure release was observed) and 10 mL of distilled water was added. With the aid of ethyl acetate, the reaction mixture was transferred to a separatory funnel. After mixing and separation of the aqueous layer, the organic layer was washed with 10 mL of a 1 M NaOH aqueous solution and 10 mL of brine. Each aqueous phase was back-extracted with 10 mL portions of ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated with the aid of a rotary evaporator. The crude residues were purified via column chromatography to yield pure coupling products. Columns were primarily slurry packed with hexanes, and mobile-phase polarity was increased gradually to the mixture indicated.
5.2. Spectroscopic Investigations
To an oven-dried purged septum-sealed NMR tube were added 15N-labeled nitrobenzene (12 mg, 0.10 mmol, 1.0 equiv), phenylboronic acid 3 (11 mg, 0.11 mmol, 1.1 equiv), and 1·[O] (2.6 mg, 0.015 mmol, 15 mol%) in toluene-d8 (0.5 mL). The tube was inserted into the NMR probe thermostated at 100 °C, and a t = 0 spectrum was obtained. The tube was ejected from the probe, phenylsilane (25 μL, 0.20 mmol, 2.0 equiv) was added via syringe, and the NMR tube was reinjected into the probe. 15N (ppm is relative to NH3(l) external standard) and 31P NMR spectra (ppm is relative to 85% H3PO4 external standard) were collected at 15, 60, 180, and 360 min.
5.3. Kinetics Experiments
For a kinetic run corresponding to a single rate constant, a solution of nitrobenzene (2) and phosphetane P-oxide 1·[O] in m-xylene was prepared under nitrogen in an oven-dried, three-neck round-bottom flask fitted with a silicon-tipped IR probe and a magnetic stir bar. The solution temperature was stabilized at 108 ± 2 °C, and the reaction was initiated by adding PhSiH3. Reaction monitoring started 15 min after the addition of PhSiH3 to ensure full reduction of 1·[O], as determined by the disappearance of the P-oxide IR absorbance at 1199 cm–1. Sample aliquots (20 μL ± 10%) were periodically taken using a calibrated automated sampler,64 diluted at room temperature into acetonitrile (80×), and analyzed using an HPLC system equipped with a C18 column (4.6 × 50 mm) and an SPD-20A/20AV UV–vis detector. Good pseudo-first-order plots were obtained by monitoring the decay of nitrobenzene (2) and growth of diphenylamine (4) relative to a standard calibration curve, and the initial rates (Δ[2]/Δt) were calculated by multiplying the pseudo-first-order reaction rate constants (exponential slopes) by the corresponding concentrations of nitrobenzene (2). Rates were shown to be reproducible within experimental error (±10%).
5.4. Computational Methods
Geometries were optimized in Gaussian 0965 using the M06-2X66 density functional with the 6-311++G(d,p) basis set. The calculated energies (ΔG, 298.15 K, 1.0 atm) result from the sum of electronic and thermal free energies as obtained from the frequency analysis at the same level of theory. Open-shell singlet energies were spin-projected.67 Frequency calculations for all stationary points were carried out to describe them either as minima (i = 0) or as first-order transition states (i = 1). For all transition structures, visualization of the imaginary frequencies corresponded to the expected normal mode for the elementary step under investigation. Intrinsic reaction coordinate calculations were performed from the transition states in forward and reverse directions to confirm the lowest energy reaction pathways that connect the corresponding minima. See Supporting Information for further details.
Acknowledgments
Funding was provided by NIH NIGMS (GM114547), MIT, and Bristol-Myers Squibb.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c01666.
General methods, additional optimization, synthetic procedures; 1H, 13C, 15N, and 31P NMR spectra; kinetics data; spectroscopy data; computational details; and Cartesian coordinates, including Figures S1–S12 and Tables S1–S8 (PDF)
Author Present Address
§ Kallyope Inc., 430 East 29th Street, Suite 1050, New York, NY 10016, United States
The authors declare no competing financial interest.
Supplementary Material
References
- a Vitaku E.; Smith D. T.; Njardarson J. T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 2014, 57, 10257. 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]; b Knölker H.-J., Ed. The Alkaloids: Chemistry and Biology; Elsevier: San Diego, 2011; Vol. 70. [Google Scholar]; c Ćiric-Marjanovic G. Recent advances in polyaniline research: polymerization mechanisms, structural aspects, properties and applications. Synth. Met. 2013, 177, 1. 10.1016/j.synthmet.2013.06.004. [DOI] [Google Scholar]
- a Hartwig J. F. Carbon-Heteroatom Bond Formation Catalysed by Organometallic Complexes. Nature 2008, 455, 314. 10.1038/nature07369. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bariwal J.; Van der Eycken E. C–N Bond Forming Cross-coupling Reactions: an overview. Chem. Soc. Rev. 2013, 42, 9283. 10.1039/c3cs60228a. [DOI] [PubMed] [Google Scholar]
- a Jiang L.; Buchwald S. L.. Palladium-Catalyzed Aromatic Carbon-Nitrogen Bond Formation. In Metal-Catalyzed Cross-Coupling Reactions, 2nd ed.; De Meijere A., Diderich F., Eds.; Wiley-Blackwell: Hoboken, NJ, 2008; pp 699–760. [Google Scholar]; b Dorel R.; Grugel C. P.; Haydl A. M. The Buchwald–Hartwig Amination After 25 Years. Angew. Chem., Int. Ed. 2019, 58, 17118. 10.1002/anie.201904795. [DOI] [PubMed] [Google Scholar]
- For Ni-catalyzed reactions, see:; a Marín M.; Rama R. J.; Nicasio M. C. Ni-Catalyzed Amination Reactions: An Overview. Chem. Rec. 2016, 16, 1819. 10.1002/tcr.201500305. [DOI] [PubMed] [Google Scholar]; b Corcoran E. B.; Pirnot M. T.; Lin S.; Dreher S. D.; DiRocco D. A.; Davies I. W.; Buchwald S. L.; MacMillan D. W. C. Aryl Amination using Ligand-free Ni(II) Salts and Photoredox Catalysis. Science 2016, 353, 279. 10.1126/science.aag0209. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Oderinde M. S.; Jones N. H.; Juneau A.; Frenette M.; Aquila B.; Tentarelli S.; Robbins D. W.; Johannes J. W. Highly Chemoselective Iridium Photoredox and Nickel Catalysis for the Cross-Coupling of Primary Aryl Amines with Aryl Halides. Angew. Chem., Int. Ed. 2016, 55, 13219. 10.1002/anie.201604429. [DOI] [PubMed] [Google Scholar]; d Lim C. H.; Kudisch M.; Liu B.; Miyake G. M. C-N Cross-Coupling via Photoexcitation of Nickel-Amine Complexes. J. Am. Chem. Soc. 2018, 140, 7667. 10.1021/jacs.8b03744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For Cu-catalyzed reactions, see:; a Beletskaya I. P.; Cheprakov A. V. The Complementary Competitors: Palladium and Copper in C–N Cross-Coupling Reactions. Organometallics 2012, 31, 7753. 10.1021/om300683c. [DOI] [Google Scholar]; b Sambiagio C.; Marsden S. P.; Blacker A. J.; McGowan P. C. Copper Catalysed Ullmann Type Chemistry: From Mechanistic Aspects to Modern Development. Chem. Soc. Rev. 2014, 43, 3525. 10.1039/C3CS60289C. [DOI] [PubMed] [Google Scholar]; c Bhunia S.; Pawar G. G.; Kumar S. V.; Jiang Y.; Ma D. Selected Copper-Based Reactions for C-N, C-O, C-S, and C-C Bond Formation. Angew. Chem., Int. Ed. 2017, 56, 16136. 10.1002/anie.201701690. [DOI] [PubMed] [Google Scholar]
- a Hartwig J. F. Transition Metal Catalyzed Synthesis of Arylamines and Aryl Ethers from Aryl Halides and Triflates: Scope and Mechanism. Angew. Chem., Int. Ed. 1998, 37, 2046.. [DOI] [PubMed] [Google Scholar]; b Brusoe A. T.; Hartwig J. F. Palladium-Catalyzed Arylation of Fluoroalkylamines. J. Am. Chem. Soc. 2015, 137, 8460. 10.1021/jacs.5b02512. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Peacock D. M.; Roos C. B.; Hartwig J. F. Palladium-Catalyzed Cross Coupling of Secondary and Tertiary Alkyl Bromides with a Nitrogen Nucleophile. ACS Cent. Sci. 2016, 2, 647. 10.1021/acscentsci.6b00187. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Peacock D. M.; Jiang Q.; Hanley P. S.; Cundari T. R.; Hartwig J. F. Reductive Elimination from Phosphine-Ligated Alkylpalladium(II) Amido Complexes to Form Sp3 Carbon-Nitrogen Bonds. J. Am. Chem. Soc. 2018, 140, 4893. 10.1021/jacs.8b00928. [DOI] [PubMed] [Google Scholar]
- a Dennis J. M.; White N. A.; Liu R. Y.; Buchwald S. L. Breaking the Base Barrier: An Electron-Deficient Palladium Catalyst Enables the Use of a Common Soluble Base in C-N Coupling. J. Am. Chem. Soc. 2018, 140, 4721. 10.1021/jacs.8b01696. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dennis J. M.; White N. A.; Liu R. Y.; Buchwald S. L. Pd-Catalyzed C-N Coupling Reactions Facilitated by Organic Bases: Mechanistic Investigation Leads to Enhanced Reactivity in the Arylation of Weakly Binding Amines. ACS Catal. 2019, 9, 3822. 10.1021/acscatal.9b00981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Park N. H.; Vinogradova E. V.; Surry D. S.; Buchwald S. L. Design of New Ligands for the Palladium-Catalyzed Arylation of α-Branched Secondary Amines. Angew. Chem., Int. Ed. 2015, 54, 8259. 10.1002/anie.201502626. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ruiz-Castillo P.; Blackmond D. G.; Buchwald S. L. Rational Ligand Design for the Arylation of Hindered Primary Amines Guided by Reaction Progress Kinetic Analysis. J. Am. Chem. Soc. 2015, 137, 3085. 10.1021/ja512903g. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Olsen E. P. K.; Arrechea P. L.; Buchwald S. L. Mechanistic Insight Leads to a Ligand Which Facilitates the Palladium-Catalyzed Formation of 2-(Hetero)Arylaminooxazoles and 4-(Hetero)Arylaminothiazoles. Angew. Chem., Int. Ed. 2017, 56, 10569. 10.1002/anie.201705525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ingoglia B. T.; Buchwald S. L. Oxidative Addition Complexes as Precatalysts for Cross-Coupling Reactions Requiring Extremely Bulky Biarylphosphine Ligands. Org. Lett. 2017, 19, 2853. 10.1021/acs.orglett.7b01082. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Uehling M. R.; King R. P.; Krska S. W.; Cernak T.; Buchwald S. L. Pharmaceutical diversification via palladium oxidative addition complexes. Science 2019, 363, 405. 10.1126/science.aac6153. [DOI] [PubMed] [Google Scholar]
- Ruiz-Castillo P.; Buchwald S. L. Applications of Palladium-Catalyzed C-N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564. 10.1021/acs.chemrev.6b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hall D. G.; Wiley-VCH: Weinheim, 2005. [Google Scholar]; b Fyfe J. W. B.; Watson A. J. B. Recent Developments in Organoboron Chemistry: Old Dogs, New Tricks. Chem. 2017, 3, 31. 10.1016/j.chempr.2017.05.008. [DOI] [Google Scholar]
- a King A. E.; Brunold T. C.; Stahl S. S. Mechanistic Study of Copper-Catalyzed Aerobic Oxidative Coupling of Arylboronic Esters and Methanol: Insights into an Organometallic Oxidase Reaction. J. Am. Chem. Soc. 2009, 131, 5044. 10.1021/ja9006657. [DOI] [PubMed] [Google Scholar]; b King A. E.; Ryland B. L.; Brunold T. C.; Stahl S. S. Kinetic and Spectroscopic Studies of Aerobic Copper(II)-Catalyzed Methoxylation of Arylboronic Esters and Insights into Aryl Transmetalation to Copper(II). Organometallics 2012, 31, 7948. 10.1021/om300586p. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Vantourout J. C.; Law R. P.; Isidro-Llobet A.; Atkinson S. J.; Watson A. J. B. Chan-Evans-Lam Amination of Boronic Acid Pinacol (BPin) Esters: Overcoming the Aryl Amine Problem. J. Org. Chem. 2016, 81, 3942. 10.1021/acs.joc.6b00466. [DOI] [PubMed] [Google Scholar]; d Vantourout J. C.; Miras H. N.; Isidro-Llobet A.; Sproules S.; Watson A. J. B. Spectroscopic Studies of the Chan–Lam Amination: A Mechanism-Inspired Solution to Boronic Ester Reactivity. J. Am. Chem. Soc. 2017, 139, 4769. 10.1021/jacs.6b12800. [DOI] [PubMed] [Google Scholar]
- West M. J.; Fyfe J. W. B.; Vantourout J. C.; Watson A. J. B. Mechanistic Development and Recent Applications of the Chan–Lam Amination. Chem. Rev. 2019, 119, 12491. 10.1021/acs.chemrev.9b00491. [DOI] [PubMed] [Google Scholar]
- Kirby A. J.; Warren S. G.. The Organic Chemistry of Phosphorus; Elsevier: Amsterdam, 1967; p 20. [Google Scholar]
- a Zhao W.; Yan P. K.; Radosevich A. T. A Phosphetane Catalyzes Deoxygenative Condensation of α-Keto Esters and Carboxylic Acids via PIII/PV=O Redox Cycling. J. Am. Chem. Soc. 2015, 137, 616. 10.1021/ja511889y. [DOI] [PubMed] [Google Scholar]; b Nykaza T. V.; Harrison T. S.; Ghosh A.; Putnik R. A.; Radosevich A. T. A Biphilic Phosphetane Catalyzes N–N Bond-Forming Cadogan Heterocyclization via PIII/PV=O Redox Cycling. J. Am. Chem. Soc. 2017, 139, 6839. 10.1021/jacs.7b03260. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Nykaza T. V.; Ramirez A.; Harrison T. S.; Luzung M. R.; Radosevich A. T. Biphilic Organophosphorus-Catalyzed Intramolecular Csp2–H Amination: Evidence for a Nitrenoid in Catalytic Cadogan Cyclizations. J. Am. Chem. Soc. 2018, 140, 3103. 10.1021/jacs.7b13803. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Nykaza A. T. V; Li G.; Yang J.; Luzung M. R.; Radosevich A. T. Angew. Chem., Int. Ed. 2020, 59, 4505. 10.1002/anie.201914851. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Ghosh A.; Lecomte M.; Kim-Lee S.-H.; Radosevich A. T. Organophosphorus-Catalyzed Deoxygenation of Sulfonyl Chlorides: Electrophilic (Fluoroalkyl)Sulfenylation by PIII/PV=O Redox Cycling. Angew. Chem., Int. Ed. 2019, 58, 2864. 10.1002/anie.201813919. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Lecomte M.; Lipshultz J. M.; Kim-Lee S.-H.; Li G.; Radosevich A. T. Driving Recursive Dehydration by PIII/PV Catalysis: Annulation of Amines and Carboxylic Acids by Sequential C–N and C–C Bond Formation. J. Am. Chem. Soc. 2019, 141, 12507. 10.1021/jacs.9b06277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nykaza T. V.; Cooper J. C.; Li G.; Mahieu N.; Ramirez A.; Luzung M. R.; Radosevich A. T. Intermolecular Reductive C-N Cross Coupling of Nitroarenes and Boronic Acids by PIII/PV=O Catalysis. J. Am. Chem. Soc. 2018, 140, 15200. 10.1021/jacs.8b10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K.; Yamagiwa N.; Torisawa Y. Cyclopentyl Methyl Ether as a New and Alternative Process Solvent. Org. Process Res. Dev. 2007, 11, 251. 10.1021/op0680136. [DOI] [Google Scholar]
- The number of total hydride equivalents was maintained when comparing different silane reagents.
- Tsao M. L.; Gritsan N.; James T. R.; Platz M. S.; Hrovat D. A.; Borden W. T. Study of the Chemistry of Ortho- and Para-Biphenylnitrenes by Laser Flash Photolysis and Time-Resolved IR Experiments and by B3LYP and CASPT2 Calculations. J. Am. Chem. Soc. 2003, 125, 9343. 10.1021/ja0351591. [DOI] [PubMed] [Google Scholar]
- a Poe R.; Schnapp K.; Young M. J. T.; Grayzar J.; Platz M. S. Chemistry and Kinetics of Singlet (Pentafluorophenyl)Nitrene. J. Am. Chem. Soc. 1992, 114, 5054. 10.1021/ja00039a016. [DOI] [Google Scholar]; b Karney W. L.; Borden W. T. Ab Initio Study of the Ring Expansion of Phenylnitrene and Comparison with the Ring Expansion of Phenylcarbene. J. Am. Chem. Soc. 1997, 119, 1378. 10.1021/ja9635241. [DOI] [Google Scholar]; c Gritsan N. P.; Likhotvorik I.; Tsao M. L.; Çelebi N.; Platz M. S.; Karney W. L.; Kemnitz C. R.; Borden W. T. Ring-Expansion Reaction of Cyano-Substituted Singlet Phenyl Nitrenes: Theoretical Predictions and Kinetic Results from Laser Flash Photolysis and Chemical Trapping Experiments. J. Am. Chem. Soc. 2001, 123, 1425. 10.1021/ja002594b. [DOI] [Google Scholar]
- The 31P NMR chemical shifts of 1 and 1·[O] exhibit temperature-dependent behavior. See the Supporting Information of ref (15b) for plots of 31P NMR chemical shift temperature dependence for syn-1 and anti-1. The temperature dependence of 31P NMR chemical shifts has been noted previously:; Gordon M. D.; Quin L. D. Temperature Dependence of 31P NMR Chemical Shifts of Some Trivalent Phosphorus Compounds. J. Magn. Reson. 1976, 22, 149. 10.1016/0022-2364(76)90171-2. [DOI] [Google Scholar]
- a The EDA-NOCV method, which combines energy decomposition analysis (EDA) with the natural orbital for chemical valence approach (NOCV), was executed as part of the Amsterdam Density Functional software package.; b ADF2014, SCM; Theoretical Chemistry, Vrije Universiteit: Amsterdam, The Netherlands, http://www.scm.com; (See Supporting Information for complete reference).
- a Mitoraj M.; Michalak A.; Ziegler T. A Combined Charge and Energy Decomposition Scheme for Bond Analysis. J. Chem. Theory Comput. 2009, 5, 962. 10.1021/ct800503d. [DOI] [PubMed] [Google Scholar]; b Mitoraj M.; Michalak A.; Ziegler T. On the Nature of the Agostic Bond between Metal Centers and β-Hydrogen Atoms in Alkyl Complexes. An Analysis Based on the Extended Transition State Method and the Natural Orbitals for Chemical Valence Scheme (ETS-NOCV). Organometallics 2009, 28, 3727. 10.1021/om900203m. [DOI] [Google Scholar]
- a Brown H. C.; Kramer G W.; Levy A. B.; Midland M. M.. Organic Syntheses via Boranes; Wiley-Interscience, New York, 1975. [Google Scholar]; b Brown H. C.; Salunkhe A. M.; Argade A. B. Organoboranes. 55. Improved Procedure for the Conversion of Representative Achiral and Chiral Alkyl-, (E)-1-Alkenyl and (Z)-1-Alkenyl-, and Arylboronates into the Corresponding Organyldichloroboranes. Organometallics 1992, 11, 3094. 10.1021/om00045a025. [DOI] [Google Scholar]; c Phanstiel O. IV; Wang W. X.; Powell D. H.; Ospina M. P.; Leeson J B. A. Synthesis of Secondary Amines via N-(Benzoyloxy)amines and Organoboranes. J. Org. Chem. 1999, 64, 803. 10.1021/jo981613l. [DOI] [PubMed] [Google Scholar]; d Matteson D. S.; Kim G. Y. Asymmetric Alkyldifluoroboranes and Their Use in Secondary Amine Synthesis. Org. Lett. 2002, 4, 2153. 10.1021/ol025973d. [DOI] [PubMed] [Google Scholar]; e Bagutski V.; Elford T. G.; Aggarwal V. K. Synthesis of Highly Enantioenriched C-Tertiary Amines from Boronic Esters: Application to the Synthesis of Igmesine. Angew. Chem., Int. Ed. 2011, 50, 1080. 10.1002/anie.201006037. [DOI] [PubMed] [Google Scholar]
- Weinhold F. In Encyclopedia of Computational Chemistry; Schleyer P. v. R., Allinger N. L., Clark T., Gasteiger J., Kollman P. A., Schaefer H. F. III, Schreiner P. R., Eds.; John Wiley & Sons: Chichester, UK, 1998; Vol. 3, pp 1792–1811. [Google Scholar]
- Zhu J. S.; Li C. J.; Tsui K. Y.; Kraemer N.; Son J.-H.; Haddadin M. J.; Tantillo D. J.; Kurth M. J. Accessing Multiple Classes of 2H-Indazoles: Mechanistic Implications for the Cadogan and Davis-Beirut Reactions. J. Am. Chem. Soc. 2019, 141, 6247. 10.1021/jacs.8b13481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khursan V. S.; Shamukaev V. A.; Chainikova E. M.; Khursan S. L.; Safiullin R. L. Kinetics and Mechanism of the Nitrosobenzene Deoxygenation by Trivalent Phosphorous Compounds. Russ. Chem. Bull. 2013, 62, 2477. 10.1007/s11172-013-0359-8. [DOI] [Google Scholar]
- Leach A. G.; Houk K. N. Transition States and Mechanisms of the Hetero-Diels-Alder Reactions of Hyponitrous Acid, Nitrosoalkanes, Nitrosoarenes, and Nitrosocarbonyl Compounds. J. Org. Chem. 2001, 66, 5192. 10.1021/jo0104126. [DOI] [PubMed] [Google Scholar]
- Thomas S. P.; French R. M.; Jheengut V.; Aggarwal V. K. Homologation and Alkylation of Boronic Esters and Boranes by 1,2-Metallate Rearrangement of Boron Ate Complexes. Chem. Rec. 2009, 9, 24. 10.1002/tcr.20168. [DOI] [PubMed] [Google Scholar]
- For analogous calculations using a PCM model for solvation in n-butyl acetate (ε = 4.9941), see ref (15c).
- Ono N.The Nitro Group in Organic Synthesis; Wiley: New York, 2001. [Google Scholar]
- Srivastava R. S.; Nicholas K. M. Kinetics of the Allylic Amination of Olefins by Nitroarenes Catalyzed by [CpFe(CO)2]2. Organometallics 2005, 24, 1563. 10.1021/om049336y. [DOI] [Google Scholar]
- Gui J.; Pan C.-M.; Jin Y.; Qin T.; Lo J. C.; Lee B. J.; Spergel S. H.; Mertzman M. E.; Pitts W. J.; La Cruz T. E.; Schmidt M. A.; Darvatkar N.; Natarajan S. R.; Baran P. S. Practical olefin hydroamination with nitroarenes. Science 2015, 348, 886. 10.1126/science.aab0245. [DOI] [PubMed] [Google Scholar]
- Zhu K.; Shaver M. P.; Thomas S. P. Chemoselective Nitro Reduction and Hydroamination Using a Single Iron Catalyst. Chem. Sci. 2016, 7, 3031. 10.1039/C5SC04471E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cheung C. W.; Hu X. Amine Synthesis via Iron-Catalysed Reductive Coupling of Nitroarenes with Alkyl Halides. Nat. Commun. 2016, 7, 12494. 10.1038/ncomms12494. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cheung C. W.; Ploeger M. L.; Hu X. Direct Amidation of Esters with Nitroarenes. Nat. Commun. 2017, 8, 14878. 10.1038/ncomms14878. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cheung C. W.; Ploeger M. L.; Hu X. Nickel-Catalyzed Reductive Transamidation of Secondary Amides with Nitroarenes. ACS Catal. 2017, 7, 7092. 10.1021/acscatal.7b02859. [DOI] [Google Scholar]
- a Sapountzis I.; Knochel P. A New General Preparation of Polyfunctional Diarylamines by the Addition of Functionalized Arylmagnesium Compounds to Nitroarenes. J. Am. Chem. Soc. 2002, 124, 9390. 10.1021/ja026718r. [DOI] [PubMed] [Google Scholar]; b Dohle W.; Staubitz A.; Knochel P. Mild Synthesis of Polyfunctional Benzimidazoles and Indoles by the Reduction of Functionalized Nitroarenes with Phenylmagnesium Chloride. Chem. - Eur. J. 2003, 9, 5323. 10.1002/chem.200305090. [DOI] [PubMed] [Google Scholar]; c Kopp F.; Sapountzis I.; Knochel P. Preparation of Polyfunctionalized Amines by the Addition of Functionalized Organomagnesium Reagents to Nitrosoarenes. Synlett 2003, 885. 10.1055/s-2003-38738. [DOI] [Google Scholar]; d Sapountzis I.; Knochel P. A New Method for the Selective Amination of 1,3- and 1,4-Dinitrobenzenes and Protected Nitroanilines Leading to Polyfunctional 1,3- and 1,4- Disubstituted Anilines. Synlett 2004, 955. 10.1055/s-2004-820043. [DOI] [Google Scholar]; e Dhayalan V.; Saemann C.; Knochel P. Synthesis of polyfunctional secondary amines by the addition of functionalized zinc reagents to nitrosoarenes. Chem. Commun. 2015, 51, 3239. 10.1039/C4CC08846H. [DOI] [PubMed] [Google Scholar]
- Gao H.; Xu Q.-L.; Yousufuddin M.; Ess D. H.; Kurti L. Transition-Metal-Free, Low-Temperature Intramolecular Amination of Aromatic C-H Bonds: Rapid Synthesis of Fused Heterocycles.. Angew. Chem., Int. Ed. 2014, 53, 2701. 10.1002/anie.201309973. [DOI] [PubMed] [Google Scholar]
- a Rauser M.; Ascheberg C.; Niggemann M. Electrophilic Amination with Nitroarenes. Angew. Chem., Int. Ed. 2017, 56, 11570. 10.1002/anie.201705356. [DOI] [PubMed] [Google Scholar]; b Rauser M.; Ascheberg C.; Niggemann M. Direct Reductive N-Functionalization of Aliphatic Nitro Compounds. Chem. - Eur. J. 2018, 24, 3970. 10.1002/chem.201705986. [DOI] [PubMed] [Google Scholar]; c Rauser M.; Warzecha D. P.; Niggemann M. O2-Mediated Oxidation of Aminoboranes through 1,2-N Migration. Angew. Chem., Int. Ed. 2018, 57, 5903. 10.1002/anie.201803168. [DOI] [PubMed] [Google Scholar]; d Rauser M.; Eckert R.; Gerbershagen M.; Niggemann M. Catalyst-Free Reductive Coupling of Aromatic and Aliphatic Nitro Compounds with Organohalides. Angew. Chem., Int. Ed. 2019, 58, 6713. 10.1002/anie.201814197. [DOI] [PubMed] [Google Scholar]
- Nykaza T. V.; Yang J.; Radosevich A. T. PEt3-Mediated Deoxygenative C–N Coupling of Nitroarenes and Boronic Acids. Tetrahedron 2019, 75, 3248. 10.1016/j.tet.2019.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscales S.; Csáky A. G. Transition-Metal-Free Three-Component Synthesis of Tertiary Aryl Amines from Nitro Compounds, Boronic Acids, and Trialkyl Phosphites. Adv. Synth. Catal. 2020, 362, 111. 10.1002/adsc.201901009. [DOI] [Google Scholar]
- Suárez-Pantiga S.; Hernández-Ruiz R.; Virumbrales C.; Pedrosa M. R.; Sanz R. Reductive Molybdenum-Catalyzed Direct Amination of Boronic Acids with Nitro Compounds. Angew. Chem., Int. Ed. 2019, 58, 2129. 10.1002/anie.201812806. [DOI] [PubMed] [Google Scholar]
- O’Brien C. J.; Tellez J. L.; Nixon Z. S.; Kang L. J.; Carter A. L.; Kunkel S. R.; Przeworski K. C.; Chass G. A. Recycling the Waste: The Development of a Catalytic Wittig Reaction. Angew. Chem., Int. Ed. 2009, 48, 6836. 10.1002/anie.200902525. [DOI] [PubMed] [Google Scholar]
- a O’Brien C. J.; Lavigne F.; Coyle E. E.; Holohan A. J.; Doonan B. J. Breaking the ring through a room temperature catalytic Wittig reaction. Chem. - Eur. J. 2013, 19, 5854. 10.1002/chem.201300546. [DOI] [PubMed] [Google Scholar]; b O’Brien C. J.; Nixon Z. S.; Holohan A. J.; Kunkel S. R.; Tellez J. L.; Doonan B. J.; Coyle E. E.; Lavigne F.; Kang L. J.; Przeworski K. C. The development of the catalytic Wittig reaction. Chem. - Eur. J. 2013, 19, 15281. 10.1002/chem.201301444. [DOI] [PubMed] [Google Scholar]; c Coyle E. E.; Doonan B. J.; Holohan A. J.; Walsh K. A.; Lavigne F.; Krenske E. H.; O’Brien C. J. Catalytic Wittig reactions of semi-and nonstabilized ylides enabled by ylide tuning. Angew. Chem., Int. Ed. 2014, 53, 12907. 10.1002/anie.201406103. [DOI] [PubMed] [Google Scholar]; d Kirk A. M.; O’Brien C. J.; Krenske E. H. Why Do Silanes Reduce Electron-Rich Phosphine Oxides Faster than Electron-Poor Phosphine Oxides?. Chem. Commun. 2020, 56, 1227. 10.1039/C9CC08718D. [DOI] [PubMed] [Google Scholar]
- a Guo H.; Fan Y. C.; Sun Z.; Wu Y.; Kwon O. Phosphine Organocatalysis. Chem. Rev. 2018, 118, 10049. 10.1021/acs.chemrev.8b00081. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lao Z.; Toy P. H. Catalytic Wittig and aza-Wittig Reactions. Beilstein J. Org. Chem. 2016, 12, 2577. 10.3762/bjoc.12.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a For reviews discussing PV=O catalysis, see: Marsden S. P.Catalytic Variants of Phosphine Oxide-Mediated Organic Transformations. In Sustainable Catalysis; Dunn P. J., Hii K. K., Krische M. J., Williams M. T., Eds.; John Wiley & Sons, Inc.: New York, 2013; pp 339–361. [Google Scholar]; b Denmark S. E.; Stavenger R. A. Asymmetric Catalysis of Aldol Reactions with Chiral Lewis Bases. Acc. Chem. Res. 2000, 33, 432. 10.1021/ar960027g. [DOI] [PubMed] [Google Scholar]; c Denmark S. E.; Beutner G. L. Lewis Base Catalysis in Organic Synthesis. Angew. Chem., Int. Ed. 2008, 47, 1560. 10.1002/anie.200604943. [DOI] [PubMed] [Google Scholar]; d Benaglia M.; Rossi S. Chiral Phosphine Oxides in Present-Day Organocatalysis. Org. Biomol. Chem. 2010, 8, 3824. 10.1039/c004681g. [DOI] [PubMed] [Google Scholar]
- a Denton R. M.; An J.; Adeniran B. Phosphine Oxide-Catalysed Chlorination Reactions of Alcohols Under Appel Conditions. Chem. Commun. 2010, 46, 3025. 10.1039/c002825h. [DOI] [PubMed] [Google Scholar]; b Denton R. M.; Tang X.; Przeslak A. Catalysis of Phosphorus(V)-Mediated Transformations: Dichlorination Reactions of Epoxides Under Appel Conditions. Org. Lett. 2010, 12, 4678. 10.1021/ol102010h. [DOI] [PubMed] [Google Scholar]; c Denton R. M.; An J.; Adeniran B.; Blake A. J.; Lewis W.; Poulton A. M. Catalytic Phosphorus(V)-Mediated Nucleophilic Substitution Reactions: Development of a Catalytic Appel Reaction. J. Org. Chem. 2011, 76, 6749. 10.1021/jo201085r. [DOI] [PubMed] [Google Scholar]; d An J.; Tang X.; Moore J.; Lewis W.; Denton R. M. Phosphorus(V)-Catalyzed Deoxydichlorination Reactions of Aldehydes. Tetrahedron 2013, 69, 8769. 10.1016/j.tet.2013.07.100. [DOI] [Google Scholar]; e Yu T.-Y.; Wang Y.; Xu P.-F. An Unusual Triphenylphosphine Oxide Catalyzed Stereoselective 1,3-Dichlorination of Unsaturated Ketoesters. Chem. - Eur. J. 2014, 20, 98. 10.1002/chem.201303688. [DOI] [PubMed] [Google Scholar]; f Tang X.; An J.; Denton R. M. A Procedure for Appel Halogenations and Dehydrations Using a Polystyrene Supported Phosphine Oxide. Tetrahedron Lett. 2014, 55, 799. 10.1016/j.tetlet.2013.11.098. [DOI] [Google Scholar]; g Buonomo J. A.; Aldrich C. C. Mitsunobu Reactions Catalytic in Phosphine and a Fully Catalytic System. Angew. Chem., Int. Ed. 2015, 54, 13041. 10.1002/anie.201506263. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Hirose D.; Gazvoda M.; Košmrlj J.; Taniguchi T. The “Fully Catalytic System” in Mitsunobu Reaction Has Not Been Realized Yet. Org. Lett. 2016, 18, 4036. 10.1021/acs.orglett.6b01894. [DOI] [PubMed] [Google Scholar]; i Jiang L.; Yu J.; Niu F.; Zhang D.; Sun X. A High-Efficient Method for the Amidation of Carboxylic Acids Promoted by Triphenylphosphine Oxide and Oxalyl Chloride. Heteroat. Chem. 2017, 28, e21364 10.1002/hc.21364. [DOI] [Google Scholar]; j Beddoe R. H.; Sneddon H. F.; Denton R. M. The Catalytic Mitsunobu Reaction: A Critical Analysis of the Current State-of-the-Art. Org. Biomol. Chem. 2018, 16, 7774. 10.1039/C8OB01929K. [DOI] [PubMed] [Google Scholar]; k Beddoe R. H.; Andrews K. G.; Magné V.; Cuthbertson J. D.; Saska J.; Shannon-Little A. L.; Shanahan S. E.; Sneddon H. F.; Denton R. M. Redox-Neutral Organocatalytic Mitsunobu Reactions. Science 2019, 365, 910. 10.1126/science.aax3353. [DOI] [PubMed] [Google Scholar]
- Harris J. R.; Haynes M. T. II; Thomas A. M.; Woerpel K. A. Phosphine-catalyzed reductions of alkyl silyl peroxides by titanium hydride reducing agents: Development of the method and mechanistic investigations. J. Org. Chem. 2010, 75, 5083. 10.1021/jo1008367. [DOI] [PubMed] [Google Scholar]
- a van Kalkeren H. A.; van Delft F. L.; Rutjes F. P. J. T. Organophosphorus catalysis to bypass phosphine oxide waste. ChemSusChem 2013, 6, 1615. 10.1002/cssc.201300368. [DOI] [PubMed] [Google Scholar]; b van Kalkeren H. A.; Blom A. L.; Rutjes F. P. J. T.; Huijbregts M. A. J. On the usefulness of life cycle assessment in early chemical methodology development: The case of organophosphorus-catalyzed Appel and Wittig reactions. Green Chem. 2013, 15, 1255. 10.1039/c3gc00053b. [DOI] [Google Scholar]; c van Kalkeren H. A.; Leenders S. H.; Hommersom C. R.; Rutjes F. P.; van Delft F. L. In situ phosphine oxide reduction: A catalytic Appel reaction. Chem. - Eur. J. 2011, 17, 11290. 10.1002/chem.201101563. [DOI] [PubMed] [Google Scholar]; d van Kalkeren H. A.; van Delft F. L.; Rutjes F. P. J. T. Catalytic Appel reactions. Pure Appl. Chem. 2012, 85, 817. 10.1351/PAC-CON-12-06-13. [DOI] [Google Scholar]; e van Kalkeren H. A.; Bruins J. J.; Rutjes F. P. J. T.; van Delft F. L. Organophosphorus-catalysed Staudinger reduction. Adv. Synth. Catal. 2012, 354, 1417. 10.1002/adsc.201100967. [DOI] [Google Scholar]; f van Kalkeren H. A.; te Grotenhuis C.; Haasjes F. S.; Hommersom C. R. A.; Rutjes F. P. J. T.; van Delft F. L. Catalytic Staudinger/aza-Wittig sequence by in situ phosphane oxide. Eur. J. Org. Chem. 2013, 2013, 7059. 10.1002/ejoc.201300585. [DOI] [Google Scholar]; g Lenstra D. C.; Rutjes F. P.; Mecinović J. Triphenylphosphine catalysed amide bond formation between carboxylic acids and amines. Chem. Commun. 2014, 50, 5763. 10.1039/c4cc01861c. [DOI] [PubMed] [Google Scholar]
- a Werner T. Phosphonium salt organocatalysis. Adv. Synth. Catal. 2009, 351, 1469. 10.1002/adsc.200900211. [DOI] [Google Scholar]; b Werner T.; Hoffmann M.; Deshmukh S. First enantioselective catalytic Wittig reaction. Eur. J. Org. Chem. 2014, 2014, 6630. 10.1002/ejoc.201402941. [DOI] [Google Scholar]; c Werner T.; Hoffmann M.; Deshmukh S. First microwaveassisted catalytic Wittig reaction. Eur. J. Org. Chem. 2014, 2014, 6873. 10.1002/ejoc.201403113. [DOI] [Google Scholar]; d Hoffmann M.; Deshmukh S.; Werner T. Scope and limitation of the microwave-assisted catalytic Wittig reaction. Eur. J. Org. Chem. 2015, 2015, 4532. 10.1002/ejoc.201500310. [DOI] [Google Scholar]; e Werner T.; Hoffmann M.; Deshmukh S. Phospholane catalyzed Wittig reaction. Eur. J. Org. Chem. 2015, 2015, 3286. 10.1002/ejoc.201500243. [DOI] [Google Scholar]; f Schirmer M. L.; Adomeit S.; Werner T. First base-free catalytic Wittig reaction. Org. Lett. 2015, 17, 3078. 10.1021/acs.orglett.5b01352. [DOI] [PubMed] [Google Scholar]; g Schirmer M. L.; Adomeit S.; Spannenberg A.; Werner T. Novel base-free catalytic Wittig reaction for the synthesis of highly functionalized alkenes. Chem. - Eur. J. 2016, 22, 2458. 10.1002/chem.201503744. [DOI] [PubMed] [Google Scholar]; h Longwitz L.; Spannenberg A.; Werner T. Phosphetane Oxides as Redox Cycling Catalysts in the Catalytic Wittig Reaction at Room Temperature. ACS Catal. 2019, 9, 9237. 10.1021/acscatal.9b02456. [DOI] [Google Scholar]; i Longwitz L.; Werner T. Reduction of Activated Alkenes by P(III)/P(V) Redox Cycling Catalysis. Angew. Chem., Int. Ed. 2020, 59, 2760. 10.1002/anie.201912991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhang K.; Cai L.; Yang Z.; Houk K. N.; Kwon O. Bridged [2.2.1] bicyclic phosphine oxide facilitates catalytic γ-umpolung addition-Wittig olefination. Chem. Sci. 2018, 9, 1867. 10.1039/C7SC04381C. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cai L.; Zhang K.; Chen S.; Lepage R. J.; Houk K. N.; Krenske E. H.; Kwon O. Catalytic Asymmetric Staudinger-Aza-Wittig Reaction for the Synthesis of Heterocyclic Amines. J. Am. Chem. Soc. 2019, 141, 9537. 10.1021/jacs.9b04803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fourmy K.; Voituriez A. Catalytic cyclization reactions of Huisgen zwitterion with α-ketoesters by in situ chemoselective phosphine oxide reduction. Org. Lett. 2015, 17, 1537. 10.1021/acs.orglett.5b00426. [DOI] [PubMed] [Google Scholar]; b Saleh N.; Voituriez A. Synthesis of 9H-pyrrolo[1,2-a]indole and 3H-pyrrolizine derivatives via a phosphine-catalyzed umpolung addition/intramolecular Wittig reaction. J. Org. Chem. 2016, 81, 4371. 10.1021/acs.joc.6b00473. [DOI] [PubMed] [Google Scholar]; c Saleh N.; Blanchard F.; Voituriez A. Synthesis of nitrogencontaining heterocycles and cyclopentenone derivatives via phosphinecatalyzed Michael addition/intramolecular Wittig reaction. Adv. Synth. Catal. 2017, 359, 2304. 10.1002/adsc.201700313. [DOI] [Google Scholar]
- a Tsai Y.-L.; Lin W. Synthesis of multifunctional alkenes from substituted acrylates and aldehydes via phosphine-catalyzed Wittig reaction. Asian J. Org. Chem. 2015, 4, 1040. 10.1002/ajoc.201500251. [DOI] [Google Scholar]; b Lee C.; Chang T.; Yu J.; Reddy G. M.; Hsiao M.; Lin W. Synthesis of Functionalized Furans via Chemoselective Reduction/Wittig Reaction Using Catalytic Triethylamine and Phosphine. Org. Lett. 2016, 18, 3758. 10.1021/acs.orglett.6b01781. [DOI] [PubMed] [Google Scholar]
- a Wang L.; Sun M.; Ding M.-W. Catalytic intramolecular Wittig reaction based on a phosphine/phosphine oxide catalytic cycle for the synthesis of heterocycles. Eur. J. Org. Chem. 2017, 2017, 2568. 10.1002/ejoc.201601628. [DOI] [Google Scholar]; b Wang L.; Wang Y.; Chen M.; Ding M.-W. Reversible P(III)/P(V) redox: Catalytic aza-Wittig reaction for the synthesis of 4(3H)- quinazolinones and the natural product vasicinone. Adv. Synth. Catal. 2014, 356, 1098. 10.1002/adsc.201300950. [DOI] [Google Scholar]; c Wang L.; Xie Y.-B.; Huang N.-Y.; Yan J.-Y.; Hu W.-M.; Liu M.-G.; Ding M.-W. Catalytic aza-Wittig reaction of acid anhydride for the synthesis of 4H-benzo[d][1,3]oxazin-4-ones and 4-benzylidene-2- aryloxazol-5(4H)-ones. ACS Catal. 2016, 6, 4010. 10.1021/acscatal.6b00165. [DOI] [Google Scholar]
- Rommel S.; Belger C.; Begouin J.; Plietker B. Dual [Fe + Phosphine] catalysis: Application in catalytic Wittig olefination. ChemCatChem 2015, 7, 1292. 10.1002/cctc.201500053. [DOI] [Google Scholar]
- Bel Abed H.; Mammoliti O.; Bande O.; Van Lommen G.; Herdewijn P. Organophosphorus-catalyzed diaza-Wittig reaction: Application to the synthesis of pyridazines. Org. Biomol. Chem. 2014, 12, 7159. 10.1039/C4OB01201A. [DOI] [PubMed] [Google Scholar]
- a Marsi K. L. Stereochemistry of Some Reactions of Phospholane Derivatives. J. Am. Chem. Soc. 1969, 91, 4724. 10.1021/ja01045a025. [DOI] [Google Scholar]; b Marsi K. L. Phenylsilane Reduction of Phosphine Oxides with Complete Stereospecificity. J. Org. Chem. 1974, 39, 265. 10.1021/jo00916a041. [DOI] [Google Scholar]
- Keglevich G.; Fekete M.; Chuluunbaatar T.; Dobo A.; Harmat V.; Toke L. One-pot Transformation of Cyclic Phosphine Oxides to Phosphine–Boranes by Dimethyl Sulfide–borane. J. Chem. Soc., Perkin Trans. 2000, 1, 4451. 10.1039/b005380p. [DOI] [Google Scholar]
- a Cadogan J. I. G.; Cameron-Wood M.; Mackie R. K.; Searle R. J. G. The Reactivity of Organophosphorus Compounds. Part XIX. Reduction of Nitro-Compounds by Triethyl Phosphite: A Convenient New Route to Carbazoles, Indoles, Indazoles, Triazoles, and Related Compounds. J. Chem. Soc. 1965, 0, 4831. 10.1039/jr9650004831. [DOI] [Google Scholar]; b Cadogan J. I. G. Reduction of Nitro- and Nitroso-Compounds by Tervalent Phosphorus Reagents. Q. Rev., Chem. Soc. 1968, 22, 222. 10.1039/qr9682200222. [DOI] [Google Scholar]; c Cadogan J. I. G.; Todd M. J. Reduction of Nitro- and Nitroso-Compounds by Tervalent Phosphorus Reagents. Part IV. Mechanistic Aspects of the Reduction of 2,4,6-Trimethyl-2’-Nitrobiphenyl, 2-Nitrobiphenyl, and Nitrobenzene. J. Chem. Soc. C 1969, 0, 2808. 10.1039/J39690002808. [DOI] [Google Scholar]; d Armour M. A.; Cadogan J. I. G.; Grace D. S. B. Reduction of Nitro- and Nitroso-Compounds by Tervalent Phosphorus Reagents. Part XI. A Kinetic Study of the Effects of Varying the Reagent and the Nitro-Compound in the Conversion of o-Nitrobenzylideneamines to 2-Substituted Indazoles. J. Chem. Soc., Perkin Trans. 2 1975, 2 (11), 1185. 10.1039/p29750001185. [DOI] [Google Scholar]
- Sundberg R. J.; Lang C.-C. Structure-reactivity Studies of Deoxygenation Reactions. J. Org. Chem. 1971, 36, 300. 10.1021/jo00801a012. [DOI] [Google Scholar]
- For amination reactions with more Lewis acidic organoboron reagents, see; a Brown H. C.; Salunkhe A. M.; Singaram B. Chiral Synthesis via Organoboranes. 28. Reaction of α-Chiral Organyldichloroboranes with Organyl Azides Providing a Synthesis of Secondary Amines with Exceptionally High Enantiomeric Purities. J. Org. Chem. 1991, 56, 1170. 10.1021/jo00003a046. [DOI] [Google Scholar]; b Phanstiel O. IV; Wang Q. X.; Powell D. H.; Ospina M. P.; Leeson B. A. Synthesis of Secondary Amines via N-(Benzoyloxy)Amines and Organoboranes. J. Org. Chem. 1999, 64, 803. 10.1021/jo981613l. [DOI] [PubMed] [Google Scholar]; c Kim B. J.; Matteson D. S. Conversion of Alkyltrifluoroborates into Alkyldichloroboranes with Tetrachlorosilane in Coordinating Solvents. Angew. Chem., Int. Ed. 2004, 43, 3056. 10.1002/anie.200453690. [DOI] [PubMed] [Google Scholar]; d Ou L.; Shao J.; Zhang G.; Yu Y. Metal-Free Carbon-Nitrogen Bond-Forming Coupling Reaction between Arylboronic Acids and Organic Azides. Tetrahedron Lett. 2011, 52, 1430. 10.1016/j.tetlet.2010.11.103. [DOI] [Google Scholar]
- For amination reactions with boronic acids, see:; a Zhu C.; Li G.; Ess D. H.; Falck J. R.; Kürti L. Elusive Metal-Free Primary Amination of Arylboronic Acids: Synthetic Studies and Mechanism by Density Functional Theory. J. Am. Chem. Soc. 2012, 134, 18253. 10.1021/ja309637r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Coeffard V.; Moreau X.; Thomassigny C.; Greck C. Transition-Metal-Free Amination of Aryl Boronic Acids and Their Derivatives. Angew. Chem., Int. Ed. 2013, 52, 5684. 10.1002/anie.201300382. [DOI] [PubMed] [Google Scholar]; c Voth S.; Hollett J. W.; Mccubbin J. A. Transition-Metal-Free Access to Primary Anilines from Boronic Acids and a Common +NH2 Equivalent. J. Org. Chem. 2015, 80, 2545. 10.1021/jo5025078. [DOI] [PubMed] [Google Scholar]; d Sun H.; Gong L.; Tian Y.; Wu J.; Zhang X.; Liu J.; Fu Z.; Niu D. Metal- and Base-Free Room-Temperature Amination of Organoboronic Acids with N-Alkyl Hydroxylamines. Angew. Chem., Int. Ed. 2018, 57, 9456. 10.1002/anie.201802782. [DOI] [PubMed] [Google Scholar]
- Starkov P.; Jamison T. F.; Marek I. Electrophilic Amination: The case of Nitrenoids. Chem. - Eur. J. 2015, 21, 5278. 10.1002/chem.201405779. [DOI] [PubMed] [Google Scholar]
- Lower oxygenates of nitrogen or alternative nitrogen reagents are more common in C–N bond formation with boronic acids. Nitrosoarenes:; a Yu Y.; Srogl J.; Liebeskind L. S. Cu(I)-Mediated Reductive Amination of Boronic Acids with Nitroso Aromatics. Org. Lett. 2004, 6, 2631. 10.1021/ol048982q. [DOI] [PubMed] [Google Scholar]; b Roscales S.; Csákÿ A. G. Synthesis of Di(hetero)arylamines from Nitrosoarenes and Boronic Acids: A General, Mild, and Transition-Metal-Free Coupling. Org. Lett. 2018, 20, 1667. 10.1021/acs.orglett.8b00473. [DOI] [PubMed] [Google Scholar]; N-Alkyl hydroxylamines:; c Sarma M. J.; Phukan P. Metal-Free Synthesis of Secondary Amines by the Reaction of Tosyl Triazene and Aryl Boronic Acid. Synth. Commun. 2018, 48, 656. 10.1080/00397911.2017.1410175. [DOI] [Google Scholar]
- Accurate reaction sampling and dilution was performed using a probe-based Mettler-Toledo EasySampler 1210 system, see:; Zawatzky K.; Grosser S.; Welch C. J. Facile Kinetic Profiling of Chemical Reactions Using MISER Chromatographic Analysis. Tetrahedron 2017, 73, 5048. 10.1016/j.tet.2017.05.048. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam N. J.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, revision B.01; Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
- a Zhao Y.; Truhlar D. G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: two New Functionals and Systematic Testing of four M06-class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]; b Zhao Y.; Truhlar D. G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157. 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K.; Takahara Y.; Fueno T.; Houk K. N. Extended Hartree-Fock (EHF) Theory of Chemical Reactions - III. Projected Møller-Plesset (PMP) Perturbation Wavefunctions for Transition Structures of Organic Reactions. Theor. Chim. Acta 1988, 73, 337. 10.1007/BF00527740. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.