

Abstract
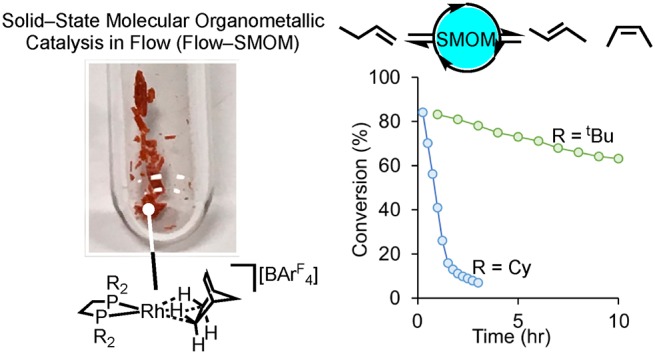
The use of solid–state molecular organometallic chemistry (SMOM–chem) to promote the efficient double bond isomerization of 1-butene to 2-butenes under flow–reactor conditions is reported. Single crystalline catalysts based upon the σ-alkane complexes [Rh(R2PCH2CH2PR2)(η2η2-NBA)][BArF4] (R = Cy, tBu; NBA = norbornane; ArF = 3,5-(CF3)2C6H3) are prepared by hydrogenation of a norbornadiene precursor. For the tBu-substituted system this results in the loss of long-range order, which can be re-established by addition of 1-butene to the material to form a mixture of [Rh(tBu2PCH2CH2PtBu2)(cis-2-butene)][BArF4] and [Rh(tBu2PCH2CH2PtBu2)(1-butene)][BArF4], in an order/disorder/order phase change. Deployment under flow-reactor conditions results in very different on-stream stabilities. With R = Cy rapid deactivation (3 h) to the butadiene complex occurs, [Rh(Cy2PCH2CH2PCy2)(butadiene)][BArF4], which can be reactivated by simple addition of H2. While the equivalent butadiene complex does not form with R = tBu at 298 K and on-stream conversion is retained up to 90 h, deactivation is suggested to occur via loss of crystallinity of the SMOM catalyst. Both systems operate under the industrially relevant conditions of an isobutene co-feed. cis:trans selectivites for 2-butene are biased in favor of cis for the tBu system and are more leveled for Cy.
Keywords: molecular organometallic, single-crystal to single-crystal, isomerization, 1-butene, rhodium
Introduction
The double bond isomerization of simple alkenes is a truly atom efficient process that is used in industry and fine chemical synthesis to add value to chemical feedstock streams.1,2 For example, the Shell higher olefins process relies on an isomerization/metathesis step to maximize the production of desired C12–20 α-olefin fractions,3 while “on-purpose” olefin conversion technologies allow for propene to be generated from ethene/butenes, via isomerization from 1-butene to 2-butenes followed by metathesis with ethene, Scheme 1.4,5 Given the world-wide demand for propene,6 the energy-efficient isomerization of butenes is thus particularly important in an industrial context. The majority of metal-catalysts for butene isomerization are heterogeneous,7,8 and operate at elevated temperatures which can result in selectivity and coking issues. Only a few room temperature catalysts have recently been reported.9,10
Scheme 1. “On Purpose” Olefin Conversion from Butene.

Stereochemical selectivity in butene isomerization is also important, as different isomers can have different relative reactivities in onward processes, such as metathesis, that can affect on-stream conversions.11,12 While the thermodynamic product of isomerization is trans-2-butene,13 kinetically-controlled selectivity for cis-2-butene has been reported, for example by photolytic activation at ambient tempertaure,10 using single-crystal Pt-surfaces,14 or at the early stages of conversion in flow.15 Operating at lower temperatures in the kinetic regime, and potentially under ligand control, homogeneous systems offer more opportunities for selective alkene isomerization. While only a few homogeneous catalysts have been reported for butene isomerization,16−18 systems that show ligand-controlled cis-selectivity for simple alkene-isomerization more generally are known.19 The accepted mechanisms for isomerization in homogeneous systems are via π-allyl/hydride or alkyl intermediates, and normally (although not exclusively20) metal hydride or alkyl precatalysts promote the latter.2,21
We have recently reported the development of solid-state molecular organometallic (SMOM) chemistry in which precisely defined, molecular, organometallic systems undergo reactivity in single-crystal to single-crystal (SC–SC) transformations in the absence of solvent.22 This allows for the synthesis of a range of cationic σ-alkane complexes.23,24 For example, [Rh(Cy2PCH2CH2PCy2)(η2η2–NBA)][BArF4], 2, Scheme 2A, comes from hydrogenation of a diene-precursor, i.e., [Rh(Cy2PCH2CH2PCy2)(η2η2–NBD)][BArF4] 1 (NBA = norbornane, NBD = norbornadiene, ArF= (CF3)2C6H3).23,25−27 This reactivity and stability is facilitated by the [BArF4]− anions that form a well–defined (often) octahedral cage in the solid–state, providing a nanoreactor28 around the reactive cation. Moreover, the alkane ligand in 2 acts as a weakly bound “token” ligand29 that can be displaced in a SC–SC transformation by simple alkenes (e.g., propene and butene) to form, in the solid-state, complexes exemplified by [Rh(Cy2PCH2CH2PCy2)(propene)][BArF4], 3, in which the alkene binds as a π-ligand with supporting agostic Rh···H3C interactions (Scheme 2B).30 In the solid-state at 298 K the propene ligand undergoes a rapid, reversible, degenerate 1,3-hydride shift via an π-allyl intermediate (I), a key step in alkene isomerization.21
Scheme 2. (A) SMOM Technique for Synthesis of σ-Alkane Complexes and (B) 1,3-Hydride Shift in Bound Propene Using the SMOM-catalyst 2.
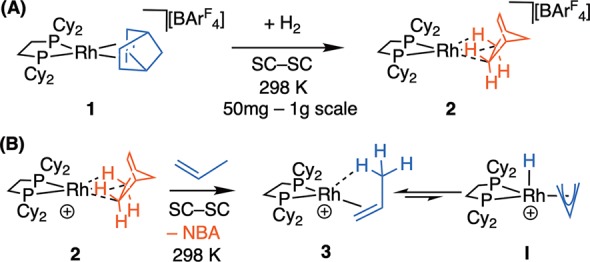
[BArF4]− anions are not shown. SC–SC = single-crystal to single-crystal.
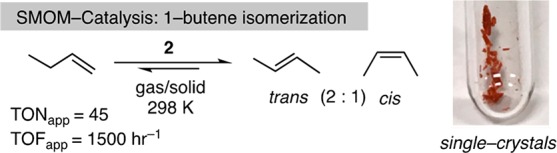
These observations suggest that complex 2 might mediate the solid/gas isomerization of butenes, and under batch conditions single crystalline 2 catalyzes the double bond isomerization of 1-butene to a mixture of 2-butenes (97%) in close to the thermodynamic trans:cis ratio of 2:1, Scheme 3.30,31 It does this quite efficiently, with apparent turnover frequencies (ToFapp32) of ∼1500 h–1, albeit with small TONapp (∼45) due to experimental limitations (NMR tube scale). As microcrystalline finely powdered samples significantly outperformed larger crystalline samples we propose a surface, or near surface, catalytic regime. Recent studies using Xe(g) as a probe have shown that substrates/products likely move in and out of the crystalline lattice via hydrophobic channels created by the [BArF4]− anions.33
Scheme 3. SMOM Butene Solid/Gas Isomerization Catalysis under Batch Conditions.
We now report that complex 2 acts as a SMOM-catalyst under the industrially attractive conditions of flow, at room temperature and pressure, to isomerize 1-butene to 2-butenes. The performance of this molecular heterogeneous catalyst can be tuned by the simple ligand modification of replacing Cy with tBu on the chelating phosphine. This leads to significant improvements in both catalyst stability and cis:trans selectivity. These catalysts also operate in the presence of other C4 alkenes (isobutene) that are often in feed streams from crackers.34 We believe this is the first time that SMOM techniques have been deployed for continuous flow catalysis. This heterogeneous methodology has the advantages of precisely defined and controllable single-site reaction centers, and complements surface organometallic (SOMC) techniques in which molecular catalysts are grafted onto platform support materials such as silica35 or functionalized-MOFs that can act as single-site catalysts,36,37 both of which have been deployed in flow-reactor settings.38,39
Results and Discussion
[Rh(Cy2PCH2CH2PCy2)(η2η2-NBA)][BArF4] Precatalyst: Deactivation to a Butadiene Complex under Flow Conditions
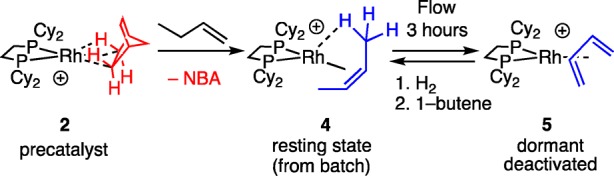
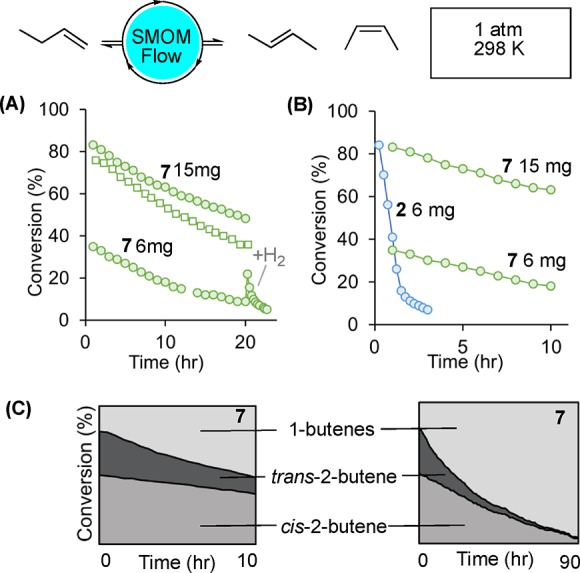
Using a bespoke flow-reactor,40 conditions were optimized in which small amounts of microcrystalline catalyst (6–15 mg of 70–150 μm sieved material that is then ground further) is supported on glass wool and exposed to 2% 1-butene balanced with N2 (atmospheric pressure, 298 K). This methodology was shown to be repeatable over different samples and operators (Figure S73). Using orange complex 2 (6 mg) gave an initial on-stream conversion to 2-butenes of 49%: entry 1, Table 1, Figure 1A, open squares. Deactivation occurred over the period of 3 h (kd6 = 1.04 h–1). Analysis of the, now purple-red, catalyst after this time using 31P{1H} SSNMR (298 K, SSNMR = solid-state NMR) and solution 31P{1H}/1H NMR spectroscopy (CD2Cl2, 183 K) showed that the butadiene complex [Rh(Cy2PCH2CH2PCy2)(η4-C4H6)][BArF4], 5,30 had been formed, Scheme 4. Based on our previous observations during batch isomerization,30 it is likely that the resting state at early times in flow is the cis-2-butene complex, 4.
Table 1. Representative Catalytic Performance for Complexes 2 and 7 in 1-Butene Isomerization under Flow Conditionsa.
| entry | catalyst, mass (mg), moles | flowb (mL/min) | isobutene (%)c | time (h) | conversion (%)d | cis:trans 2-butened | WHSV (h–1)e | apparent specific activity (mol 2-but. mol cat–1 hr–1)f | kdg (h–1) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 (6), 4.0 × 10–6 | 6.4 | 0 | 3 | 49–4 | 1.3:1 – 1:1 | 3.2 | 38.8–3.2 | 1.04 |
| 2 | 2 (6), 4.0 × 10–6 | 7.6 | 15 | 3 | 84–7 | 1.1:1 – 1.3:1 | 3.2 | 66.5–5.5 | 1.42 |
| 3 | 7 (6), 4.3 × 10–6 | 7.6 | 15 | 20 | 35–9 | 2.9:1 – 3.5:1 | 3.2 | 26.0–6.7 | 0.08 |
| 4 | 7 (15), 1.1 × 10–5 | 7.6 | 15 | 20 | 83–48 | 1.6:1 – 3.0:1 | 1.3 | 24.6–14.3 | 0.08 |
| 5 | 7 (15), 1.1 × 10–5 | 7.6 | 15 | 90 | 83–4 | 1.6:1 – 5.2:1 | 1.3 | 24.6–1.3 | 0.05 |
| 6 | 7 (15), 1.1 × 10–5 | 6.4 | 0 | 20 | 76–38 | 1:1 – 3.2:1 | 1.3 | 22.5–11.1 | 0.08 |
298 K, 1 atm, crushed microcrystalline samples. NBD-precatalyst 7 activated in situ with H2 for 10 min, then N2 flush.
1-Butene feed = 6.4 mL/min of a 2% mixture in N2.
15% isobutene = 1.2 mL/min of a 2% mixture in N2.
% Butenes determined by gas chromatography. Range is start and end values.
WHSV = weight hourly space velocity = mass 1-butene/(mass catalyst × time).
Apparent specific activity = (moles 2-butenes)/(total moles cat. × time). Range is start and end activities.
kd = [ln((1 – conv. end)/(conv. end)) – ln((1 – conv. start)/(conv. start))]/time, and assumes a first order deactivation mechanism.6
Figure 1.
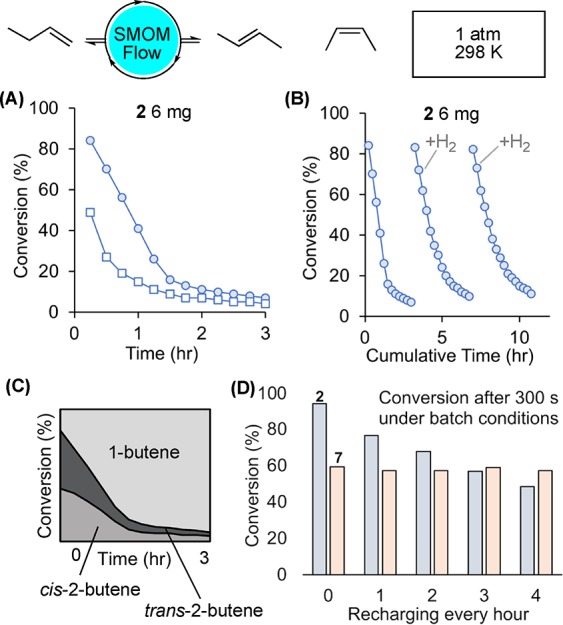
On stream catalytic performance of the 1-butene isomerization catalyst 2 in solid/gas flow, 298 K, 1 atm. Data plotted are total 2-butenes (GC conversion). 1-Butene (2%)/isobutene co-feed (15% relative to 1-butene)/balance N2, unless otherwise stated. (A) 2 6 mg with (○) and without (□) isobutene co-feed. (B) Recycling of pre-catalyst 2 by periodic addition of H2 after 3 h (×2). (C) cis/trans ratios on flow using 2 (6 mg). (D) Comparison of 2 and 7 (H2 activation) under multiple batch-recharges with 1–butene (298 K, 1 atm = 81 μmol, 2 mg of catalyst ∼1.8 μmol).
Scheme 4. Formation of Butadiene Complex and Reactivation.
[BArF4]− anions not shown.
Non-thermodynamic13 cis:trans ratios of close to 1:1 were measured across the 3 h onstream time (Figure 1C, Table 1), similar to those reported for 2 at the very early stages of batch isomerization.31 This suggests that cis-2-butene is the kinetic product in flow. We have previously suggested that loss of the trans-2-butene from the Rh-center in the solid-state represents the highest overall energy span within the system leading to enrichment in the gas phase of 2-cis-butene.31
Conducting the same catalysis in flow but now with 15% co-feed of isobutene, to replicate more closely industrial feed streams34 (entry 2, Figure 1A filled circles), resulted in an increase in the initial on-stream conversion to 84%, but deactivation still occurred over a 3 h period (kd = 1.4 h–1). While we have recently reported that the isobutene complex [Rh(Cy2PCH2CH2PCy2)(η2-C4H8)][BArF4], 6, can be prepared in a SC–SC transformation from 2,41 isobutene clearly does not compete for coordination at the metal site preferentially with either cis- or trans-2-butene, as no significant change in selectivity, or decrease in conversion, is observed with an isobutene co-feed, unlike for some homogeneous systems using closely-related alkenes.19 We tentatively suggest that reversible reaction with isobutene to form 6 may result in increased microcracking of the crystalline-material.42 This would expose more surface, or near surface, active sites resulting in an increase in initial conversion but no change in the rate of deactivation. Consistent with this, use of finely ground material results in higher initial conversions than when larger crystals are used (greater than 150 μm) and there is no significant change in rate of decomposition, Figure S74.
When prepared independently, the butadiene complex 5 is a poor catalyst under flow conditions, presumably as the butadiene ligand is relatively strongly bound to the metal center. Thus, while the formation of complex 5 effectively stops catalysis, this is a dormant state and the catalyst can be reactivated by simply passing H2 through the flow-reactor for 10 min. As we have shown more generally,23 this hydrogenates the diene to an alkane ligand. In this case a butane σ-complex is likely formed similar to the crystallographically characterized pentane analog.43 The catalyst is now primed for reaction with 1-butene, and isomerization catalysis in flow is reinstated to over 80% conversion but is still followed by deactivation over 3 h (kd = 1.1 h–1). This process can be repeated (Figure 1B) showing that the catalyst, in principle, can be regenerated.44
The formation of dormant butadiene complex 5 in flow is similar to that found for single-charge batch conditions, where 5 forms from 4 over 48 h when aged in the solid-state under an atmosphere of 2-butenes.30 The faster deactivation under flow conditions supports our proposal that 1-butene also acts as a transfer dehydrogenation substrate in this process. Consistent with this, when the 1-butene concentration is kept high under batch conditions, by multiple hourly recharges, a drop in performance (as measured by conversion at 300 s) over a similar time scale to flow is observed (Figure 1D).
While these results are encouraging, showing that SMOM-systems can be deployed, and regenerated in situ, using flow conditions, the relatively rapid deactivation is not ideal. We thus turned to explore the effect that the identity of the chelating phosphine ligand has on activity and stability.
[Rh(tBu2PCH2CH2PtBu2)(η2η2-NBD)][BArF4]. Addition of H2, Order/Disorder/Order Phase Changes, and Butene Complexes
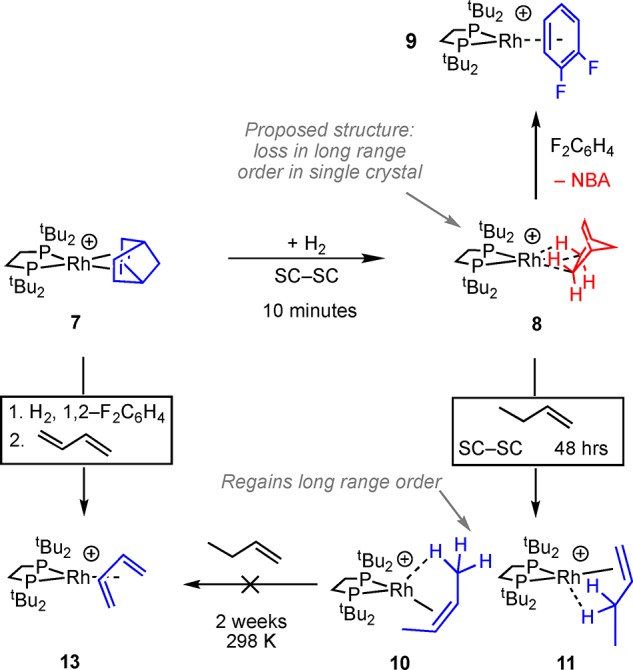
Reasoning that the formation of a butadiene complex, such as 5, would be hindered both kinetically and thermodynamically by increasing the local steric bulk at the metal center, we synthesized the tButyl analog to 1, [Rh(tBu2PCH2CH2PtBu2)(η2η2-NBD)][BArF4], 7, Scheme 5. Complex 7 is conveniently isolated as a red crystalline material in 79% yield and can be prepared from commercially available starting materials on a ∼3–5 g scale. The solid-state structure, as determined by single crystal X-ray diffraction (C121 space group, R = 2.7%), confirms the formulation and shows a pseudo Oh arrangement of [BArF4]− anions, Figure S15. In the 298 K 31P{1H} SSNMR spectrum a closely spaced pair of doublets centered at ∼δ 78 [J(RhP) ≈ 140 Hz] is observed, while in the 13C{1H} SSNMR spectrum signals due to the NBD alkene groups are observed at δ 85.5, δ 72.2. Addition of H2 (10 min for finely crushed samples45) to crystalline 7 resulted in a color change to purple–red, and a new species is now observed in the 31P{1H} SSNMR spectrum as a closely spaced pair of doublets centered at ∼δ 126 [J(RhP) ≈ 190 Hz]. This downfield shift and increase in coupling constant is also noted for when 2 is formed from the corresponding NBD precursor27 and signals the formation of a σ-alkane complex. This is formulated as [Rh(tBu2PCH2CH2PtBu2)(NBA)][BArF4] 8, Scheme 5. The alkene region of 13C{1H} SSNMR spectrum of complex 8 is featureless, signaling hydrogenation of the C=C bonds. Dissolving in 1,2-F2C6H4 resulted in the formation of the arene adduct,46 [Rh(tBu2PCH2CH2PtBu2)(η6–F2C6H4)][BArF4] 9, δ(31P) 117.7, and the liberation of free NBA as measured by 1H NMR spectroscopy.
Scheme 5. Reactivity of Complexes 7 and 8.
Anions not shown.
Attempts to confirm the structure of 8 by single crystal X-ray diffraction were frustrated by the loss of a diffraction pattern (i.e., discrete Bragg peaks, Figure S63) on hydrogenation of 7, and fracturing of larger crystals into smaller ones. This points to a loss of coherence in long-range order and/or crystalline integrity. Remarkably, addition of 1-butene to a sample of thus prepared 8 in a solid-gas reaction (48 h, not optimized, 298 K) restores long-range order in the bulk sample, and the Bragg peaks return. The resulting X-ray structure (Figure 2A,B) shows a well ordered [BArF4]− anion (R = 4.1%), with the cation approximately 60:40 disordered between 1-butene and cis-2-butene being bound at crystallographically indistinguishable {RhP2}+ fragments: [Rh(tBu2PCH2CH2PtBu2)(cis-2-butene)][BArF4], 10, and [Rh(tBu2PCH2CH2PtBu2)(1-butene)][BArF4], 11. There is a space group change to P21/c. Both alkene isomers bind through a Rh···C=C π bond and an agostic Rh···H–C interaction, as previously reported for Cy-analogs 3 and 4.30 While crystallographically characterized examples of 1-butene complexes are known,47 none have a supporting agostic interaction, and no group 9 examples have been reported.
Figure 2.
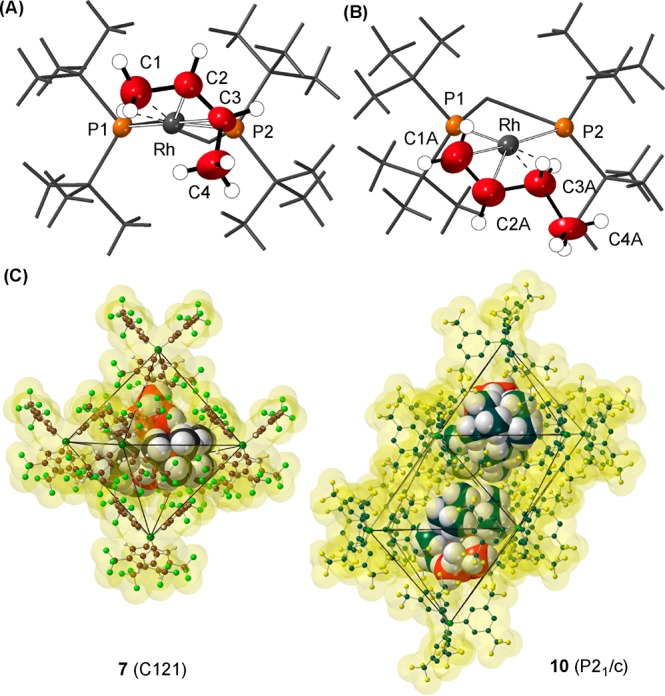
Solid–state structures of the cation 10 (A) and 11 (B). [BArF4]− anions and most hydrogen atoms omitted. Displacement ellipsoids shown at the 50% probability level. Selected bond lengths (Å), 10: Rh–C2, 2.168(6); Rh–C3, 2.356(9); Rh–C1, 2.418(9); C1–C2, 1.474(13); C2–C3, 1.347(13); C3–C4. 1.347(13). 11: Rh–C1A, 2.250(8); Rh–C2A, 2.154(6); Rh–C3A, 2.349(9); C1A–C2A, 1.363(14); C2A–C3A, 1.525(14); C3A–C4A, 1.363(14). (C) Packing diagram of 7 and 10 showing the arrangement of [BArF4]− anions. Cations and isosurface shown at van der Waals radii.
The loss of long-range order on addition of H2 to 7 to form 8, but retention of short-range order that allows for relatively sharp SSNMR spectra to be observed,48 may well be due to the [BArF4]− aryl-groups becoming randomly disordered on formation of the σ-alkane complex. We suggest this is due to internal steric pressure in the anionic cage on formation of the NBA ligand. This forces a loss of coherence in the long-range structure but retains approximately the same local environment around each metal center. Addition of butene replaces the bulky NBA, and the system relaxes to regain long-range order. Figure 2C highlights the difference in extended packing motifs on this order/disorder/order phase change, i.e., 7 to 10/11: moving from an approximate Oh environment (7) to a bifastigium (10/11). The aryl region signals associated with [BArF4]− in the 13C{1H} SSNMR spectrum also changes significantly moving between 7 and 10/11 (e.g., Figures S13 and S29).
Similar order/disorder/order phase changes have been observed in the temperature-promoted dehydration of co-ordinatively flexible copper-based MOFs in which partial loss of water initially results in loss of long-range order, which is restored on complete dehydration.49 We have also commented on a related amorphous to crystalline phase change (non SC–SC) in [Rh(iBu2PCH2CH2PiBu2)(η6-(CF3)2C6H3)BArF3].25 The 298 K 31P{1H} SSNMR spectrum of this mixture of 10/11 shows signals consistent with two sets of overlapping inequivalent 31P environments between δ 117 and 111. While the 298 K 13C{1H} SSNMR spectrum is featureless in the alkene region, cooling to 183 K reveals a set of signals at 90.5, 88.8, and δ 88.3, consistent with two different alkene complexes. These data signal a low-energy alkene isomerization process occurring in the solid-state at 298 K, as we have discussed in detail for complexes 3 and 4.30
Complexes 10 and 11 are stable at 183 K in CD2Cl2 solution for ∼3 h, and low temperature solution NMR data are fully consistent with the solid-state single-crystal X-ray data, being similar to those reported for the Cy-congeners of cis-butene, 4, and propene 3.30 On warming a CH2Cl2 solution of 10/11 or addition of CH2Cl2 to complex 8 at 183 K, decomposition occurs to give a CH2Cl2-activated dimer, Scheme 6, identified by single crystal X-ray diffraction as [Rh2(tBu2PCH2CH2PtBu2)2(μ-Cl)2(μ-CH2)][BArF4]2, 12 (Figure S40). In solution complex 12 is identified by a single environment in the 31P{1H} NMR spectrum [δ 90.8 J(RhP) = 150 Hz], while the bridging methylene is observed at δ 2.18 in the 1H NMR spectrum. Complex 12 comes from overall oxidative addition of one CH2Cl2 molecule to two {Rh(tBu2PCH2CH2PtBu2)}+ fragments. Such reactivity has been noted previously for related systems26,50,51 and is a deactivation pathway for Rh-catalysts in CH2Cl2 solvent.
Scheme 6. Decomposition to form CH2Cl2-Activated Product in Solution.

Anions not shown.
In the solid-state at 298 K there no evidence for the formation of the butadiene complex, [Rh(tBu2PCH2CH2PtBu2)(η4-C4H6)][BArF4] 13, even after 2 weeks. Heating 10/11 to 70 °C for 48 h in the solid-state results in formation of 13 and loss of crystallinity, as measured by X-ray diffraction. Complex 13 can also be prepared independently using solution technqiues, from addition of butadiene to in situ formed 9 (Scheme 5). Complex 13 is then unlikely to form under the conditions used for flow catalysts (298 K).
[Rh(tBu2PCH2CH2PtBu2)(η2η2-NBD)][BArF4]. Use as a Precatalyst under Catalytic Batch and Flow Conditions, Slow Deactivation, and cis/trans Selectivity
With thetBu-substituted complex 7 (and thus 8) in hand, we explored its use for 1-butene isomerization. Preliminary experiments under batch conditions (solid/gas reaction, NMR tube, 1 atm 1-butene, 2 mg 8, TONapp ≈ 45) showed that complex 7 (after pretreatment with H2 to form 8) is a competent isomerization catalyst, so that 2 mg of crushed crystalline material will convert 1-butene to 91% 2-butenes in 2000 s (TOFapp ≈ 80 h–1),53 as measured by gas-phase52 NMR spectroscopy. This is slower than for complex 2 under these same conditions (TOFapp ≈ 800 h–1, Figure S59). However, the tBu functionalized catalyst is significantly more stable than the Cy-analogue, and recharging under butenes results only in a modest reduction in catalytic activity for 8, whereas for 2 significant attenuation is seen due to the formation of the butadiene complex 5 (Figure 1D). At the early stages of catalysis (120 s, 50% conversion) the cis:trans ratio of 2-butene is 3:1. Comparing the relative rates of isomerization of the pure 2-butenes under batch conditions (Figure S60) shows that catalyst 8 isomerizes cis-2-butene more slowly to trans than trans to cis. Both are considerably slower than initial 1-butene isomerization. These factors combine to kinetically favor the production of cis-2-butene at the early stages of catalysis. After 24 h the thermodynamic ratio of 1:3 cis:trans is established.
Having thus established that in situ generated complex 8 was a competent 1-butene isomerization catalyst under batch conditions it was then deployed under flow conditions. The lower intrinsic reactivity of 8 compared with 2 meant that more catalyst was used to achieve equivalent on-stream conversion using our standard conditions (15 mg catalyst supported on glass wool). Under these conditions, an initial on-stream conversion of 83% was measured (Figure 3A, entry 4 in Table 1). This dropped only slightly when the isobutene co-feed was not used (entry 6). It is interesting to note that on forming complex 8 crystal fracturing is observed (vide supra and Figure 4B). This may attenuate the relative influence of isobutene in contrast with the Cy-system. The tBu catalyst, 8, is far more resilient than its Cy-analogue, 2, and significant activity is still retained over 20 h, with conversion only dropping to 48% after this time. Figure 3B compares both catalyst systems and shows that 8 has 1 order of magnitude slower rate of deactivation than 2 (kd = 0.08 h–1). Indeed 8 remains active over nearly 4 days (greater than 90 h) on stream (entry 5, Figure S72). Unlike for catalyst 2, addition of H2 to the deactivated catalyst has a negligible effect in reactivating the system, Figure 3A. We suggest any small resulting increase in conversion is due to a small amount of residual precatayst 7 still present, that is activated on addition of H2, as the equivalent butadiene complex to 5 (i.e., 13) is not formed. The deactivation products of the tBu system are discussed next.
Figure 3.
(A) 7 at 15 mg [(○) and without (□) isobutene co-feed] and 6 mg loadings (+ H2 addition after 20 h). (B) Comparison of 2 (6 mg) and 7 (15, 6 mg, isobutene co-feed) on stream for 3 and 10 h, respectively. (C) cis/trans ratios on flow using 7 (15 mg).
Figure 4.
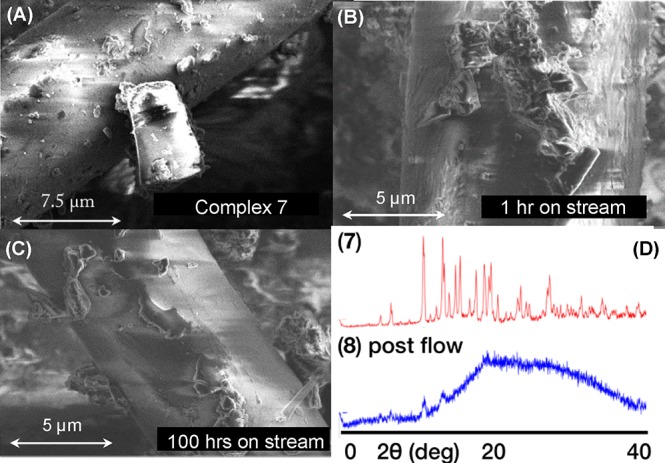
(A) SEM image of catalyst 7 supported on glass wool before activation with H2; (B) SEM image of complex 8 after H2 activation and 1 h on stream (conditions as Table 1); and (C) SEM image of complex 8 after 100 h on stream. (D) Powder X-ray diffraction patterns for precatalyst 7 and 8 post flow (100 h).
Selectivity for cis- over trans-2-butene is observed in the flow experiments but interestingly both the amount of catalyst deployed (6 mg versus 15 mg, Table 1, entries 3 and 4) and the time on stream (Figure 3C) have a marked effect. With less catalyst deployed a greater cis:trans ratio is measured initially, but both loadings show an increase in the ratio of cis-2-butene produced over time. For example, 15 mg of precatalyst 7 initially provides a cis:trans ratio of 1.6:1, which increases to 3:1 after 20 h on stream, whereas for 6 mg loading the initial ratio is 2.9:1 which increases to 3.5:1 after 20 h. Thus, for precatalyst 7 while total conversion is dropping with time, it is the reduction in the trans isomer that dominates this. At the limit of productive catalysis (4% conversion, 90 h, 15 mg catalyst) the cis:trans ratio is 5.2:1. Similar ratios are observed without an isobutene co-feed (entry 6), showing that this additive does not dramatically influence selectivity. Selectivity for alkene isomerization between cis and trans isomers has been demonstrated in homogeneous19,54 and heterogeneous9,10 systems.
Catalyst Speciation and Deactivation for the tBu System in Flow
The speciation and deactivation of complex 8 under flow conditions was interrogated by a combination of solid-state and solution NMR spectroscopy, powder X-ray diffraction and scanning electron microscopy. These show that the catalyst changes with time. After 1 h under flow conditions analysis of the catalyst by 31P{1H} SSNMR spectroscopy showed about 90% unreacted 8 with ∼10% 10/11 (Scheme 7 and Figure S66). This demonstrates that catalysis is likely a surface, or near-surface, process52 as the majority of the catalyst is unchanged, and on-stream conversion at this point is high, ∼80%. This also means that the apparent specific activity measured for this catalyst (∼25 mol 2-butene mol of cat–1 h–1, Table 1) is a lower limit and the real activity may well be 1 order of magnitude higher.
Scheme 7. Complex 8 in Flow.
[BArF4]− anions not shown.
Solution 31P{1H} NMR spectroscopy of this material (183 K, CD2Cl2) showed decomposition to the CH2Cl2 activated product 12, consistent with the behavior of both 8 and 10/11 in solution (i.e., Scheme 6). After 90 h on stream, when conversion has dropped to less than 5%, only a relatively weak and broad spectrum was observed in the 31P{1H} SSNMR spectrum of the recovered catalyst that showed a mixture of products, one of them 10/11 (Figure S67). Such a broad spectrum may also point to the system becoming amorphous, and we speculate that this may be due to coordination of the [BArF4]− anion to a low coordinate Rh-center.27 Solution 31P{1H} NMR spectroscopy (183 K, CD2Cl2) showed multiple products, none of which could be positively identified and may be a result of reaction with CD2Cl2. No butadiene complex 13 was observed. In the more benign solvent 1,2-F2C6H4 the arene adduct, 9, is formed as the only observable organometallic species by 31P{1H} NMR spectroscopy, Scheme 7. Thus, whatever the products formed at the end of flow catalysis, they have weakly enough bound ligands to be displaced by 1,2-F2C6H4 which suggests other factors may be important in deactivation such as loss of crystallinity and the well-defined anion microenvironment.
Further clues to the fate of the catalyst 8 were provided by a combination of powder X-ray diffraction and scanning electron microscopy (SEM). SEM images taken of finely crushed 7 supported on glass wool (before H2 addition) show well-defined crystalline material (Figure 4A). After H2 addition and 1 h on-stream microcrystalline, but slightly fractured, material is still present (Figure 4B), consistent with 31P{1H} SSNMR spectroscopy that shows complex 8 to be dominant. Extended time on stream (100 h) shows significant crystal degradation has occurred (Figure 4C), so that ill-defined, possibility amorphous, material is left – consistent with the very broad 31P{1H} SSNMR spectrum observed after this time. Analysis by powder X-ray diffraction of complex 7 and post-flow material support this, Figure 4D, showing a loss of diffraction in the latter. Combined, these observations might suggest that the crystalline microenvironment of the [BArF4]− anions is an essential prerequisite for catalysis in the solid-state in these systems – protecting and supporting the reactive cationic Rh-center.55 It is interesting to note that after 100 h on stream the SEM analysis of the catalyst shows that the remaining crystalline material has changed in its morphology compared with at the start of catalysis (compare panels B and C in Figure 3). That cis selectivity increases with time may point to different crystalline sites on the surface and/or particle shapes (e.g., terrace, step, and kink) promoting different selectivities for isomerization. Such behavior is commonly invoked in heterogeneous catalysis56 and also specifically in alkene isomerization.14 This behavior is also related to that observed by Blum and co-workers in a crystalline Co-catalysts for ethylene oxide polymerization,57 where single loci on the surface of individual crystals were identified as the active sites for polymerization. At the later stages of catalysis the lower intrinsic activity will also simply further detune any slow 2-butene trans–cis isomerization, and would favor the cis-kinetic product from initial faster isomerization of 1-butene.
Comparison with Solution Catalysis
The arene adducts [Rh(tBu2PCH2CH2PtBu2)(η6-F2C6H4)][BArF4] 9 and [Rh(Cy2PCH2CH2PCy2)(η6-F2C6H4)][BArF4]30 were tested as homogeneous isomerization catalysts in 1,2-F2C6H4 solution. Both isomerize 1-butene to 2-butenes rapidly (1 mol %, TON = 96, 5 min), and recover the fluorobenzene precatalyst at the end as the only organometallic species. These are stable for 48 h in this solvent. No butene, or butadiene, complexes were observed. The thermodymanic ∼1:3 cis:trans ratio of 2-butenes is observed for both. By contrast, in CD2Cl2 solvent rapid decomposition to butadiene (R = Cy, 5)30 and chloride bridged (12, R = tBu) occurs. This shows a role for the fluoroarene as a chaperone ligand in homogeneous catalysis by stabilizing the reactive metal center but being labile enough to reveal reactivity.58 These experiments also demonstrate a significant difference between solution and molecular solid-state reactivity, in terms of speciation, comparative stabilities of the two catalysts and the ratio of 2-butene isomers formed. They also indicate that the deactivation of single-crystalline complex 8 under flow is likely associated with a structural change that quenches activity of the metal sites in the solid-state.
Conclusions
We show here that well-defined single-crystal solid-state molecular organometallic (SMOM) systems can be deployed under flow-reactor conditions for the industrially relevant isomerization of 1-butene under the attractive conditions of room temperature and low pressures. The SMOM approach to catalysis provides precise definition and control of the identity of the reactive lattice points in a molecular solid. This takes advantage of the benefits that homogeneous catalysis offers (ligand control at the steric and electronic level, atomic-level resolution of active sites) and couples these into a heterogeneous environment. This is demonstrated here in that a relatively subtle change in ligand results in a dramatic difference in on-stream stability that can be traced back to how the bulk crystalline material behaves at both a molecular (butadiene formation with PCy2 groups) and macro-level (PtBu2 analog loses crystallinity over time). We have recently shown how such ligand design affects the resulting structures of σ-alkane complexes in the solid-state,26,31 and here we extend this to catalysis. Central to the ability for these systems to operate, both in terms of the synthesis and catalysis, is the microenvironment provided by the [BArF4]− anions that supports and stabilizes the reactive metal center. This offers potential future design strategies that involve both the metal-ligand cation and the extended anion environment. When coupled with the opportunities to exploit the benefits of flow-catalysis conditions (i.e., flow rate and residence time, catalyst support, and multiple feed streams) we suggest that, so-called, flow-SMOM may be a powerful strategy for catalysis.
Acknowledgments
We thank the EPSRC (EP/M024210) and SCG Chemicals Co., Ltd, Thailand for funding. We thank Dr. Dana Crivoi for help in obtaining the SEM images, Dr. Nicholas Rees for SSNMR experiments, Drs. F. Mark Chadwick and Alasdair I. McKay for the initial synthesis of complex 7, and Professors Andrew Goodwin and Dermot O’Hare (Oxford) for insightful comments on aspects of the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.9b03727.
Author Present Address
∥ CIQSO Centre for Research in Sustainable Chemistry, CIQSO, University of Huelva, 21007 Huelva, Spain.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Larsen C. R.; Grotjahn D. B. In Applied Homogeneous Catalysis with Organometallic Compounds; Cornils B., Hermann W. A., Beller M., Paciello R., Eds.; Wiley-VCH: Weinheim, 2018. [Google Scholar]
- Larionov E.; Li H.; Mazet C. Well-Defined Transition Metal Hydrides in Catalytic Isomerizations. Chem. Commun. 2014, 50, 9816–9826. 10.1039/C4CC02399D. [DOI] [PubMed] [Google Scholar]
- Skupinska J. Oligomerization of Alpha-Olefins to Higher Oligomers. Chem. Rev. 1991, 91, 613–648. 10.1021/cr00004a007. [DOI] [Google Scholar]
- Mol J. Industrial Applications of Olefin Metathesis. J. Mol. Catal. A: Chem. 2004, 213, 39–45. 10.1016/j.molcata.2003.10.049. [DOI] [Google Scholar]
- Zhao P.; Ye L.; Sun Z.; Lo B. T. W.; Woodcock H.; Huang C.; Tang C.; Kirkland A. I.; Mei D.; Edman Tsang S. C. Entrapped Single Tungstate Site in Zeolite for Cooperative Catalysis of Olefin Metathesis with Brønsted Acid Site. J. Am. Chem. Soc. 2018, 140, 6661–6667. 10.1021/jacs.8b03012. [DOI] [PubMed] [Google Scholar]
- Sattler J. J. H. B.; Ruiz-Martinez J.; Santillan-Jimenez E.; Weckhuysen B. M. Catalytic Dehydrogenation of Light Alkanes on Metals and Metal Oxides. Chem. Rev. 2014, 114, 10613–10653. 10.1021/cr5002436. [DOI] [PubMed] [Google Scholar]
- Puriwat J.; Chaitree W.; Suriye K.; Dokjampa S.; Praserthdam P.; Panpranot J. Elucidation of the Basicity Dependence of 1-Butene Isomerization on MgO/Mg(OH)2 Catalysts. Catal. Commun. 2010, 12, 80–85. 10.1016/j.catcom.2010.08.015. [DOI] [Google Scholar]
- Thomas J. M.; Thomas W. J.. Principles and Practice of Heterogeneous Catalysis, 2nd ed.; VCH Publishers Inc.: Weinheim, 2014. [Google Scholar]
- Li Y.; Ma C.; Yang H.; Zhang Z.; Zhang X.; Qiao N.; Wang J.; Hao Z. Room-Temperature Isomerization of 1-Butene to 2-Butene over Palladium-Loaded Silica Nanospheres Catalyst. Chem. Eng. J. 2016, 299, 1–7. 10.1016/j.cej.2016.04.040. [DOI] [Google Scholar]
- Hashimoto K.; Masuda Y.; Kominami H. Photocatalytic Isomerization of 1-Butene over Palladium-Loaded Titanium(IV) Oxide Particles: Lewis Acid-Like Features of the Photocatalyst. ACS Catal. 2013, 3, 1349–1355. 10.1021/cs4001514. [DOI] [Google Scholar]
- Bilhou J. L.; Basset J. M.; Mutin R.; Graydon W. F. A Stereochemical Study of Metathesis and Cis-Trans Isomerization of 2-Pentenes. J. Am. Chem. Soc. 1977, 99, 4083–4090. 10.1021/ja00454a029. [DOI] [Google Scholar]
- Poovarawan N.; Suriye K.; Ayudhya S. K. N.; Punpranot J.; Aires F. J. C. S.; Praserthdam P. Effect of 2-Butene Cis/Trans Isomers in the Metathesis of Ethylene and 2-Butene over WO3/SiO2 Catalysts. Catal. Lett. 2014, 144, 920–927. 10.1007/s10562-014-1230-8. [DOI] [Google Scholar]
- Kapteijn F.; van der Steen A. J.; Mol J. C. Thermodynamics of the Geometrical Isomerization of 2-Butene and 2-Pentene. J. Chem. Thermodyn. 1983, 15, 137–146. 10.1016/0021-9614(83)90152-0. [DOI] [Google Scholar]
- Lee I.; Delbecq F.; Morales R.; Albiter M. A.; Zaera F. Tuning Selectivity in Catalysis by Controlling Particle Shape. Nat. Mater. 2009, 8, 132. 10.1038/nmat2371. [DOI] [PubMed] [Google Scholar]
- Salameh A.; Baudouin A.; Soulivong D.; Boehm V.; Roeper M.; Basset J.-M.; Copéret C. CH3-ReO3 on -Al2O3: Activity, Selectivity, Active Site and Deactivation in Olefin Metathesis. J. Catal. 2008, 253, 180–190. 10.1016/j.jcat.2007.10.007. [DOI] [Google Scholar]
- Tolman C. A. Chemistry of Tetrakis(Triethyl Phosphite)Nickel Hydride, HNi[P(OEt)3]4+. IV. Mechanism of Olefin Isomerization. J. Am. Chem. Soc. 1972, 94, 2994–2999. 10.1021/ja00764a016. [DOI] [Google Scholar]
- Kanai H.; Choe S. B.; Klabunde K. J. Isomerization of 1-Butene Catalyzed by ((η6-arene)NiR2. A Very Active Homogeneous Catalyst System. J. Am. Chem. Soc. 1986, 108, 2019–2023. 10.1021/ja00268a048. [DOI] [Google Scholar]
- Sen A.; Lai T. W. Mechanism of Palladium(II)-Catalyzed Carbon-Carbon Double Bond Isomerization in Olefins. Inorg. Chem. 1984, 23, 3257–3258. 10.1021/ic00188a046. [DOI] [Google Scholar]
- Chen C.; Dugan T. R.; Brennessel W. W.; Weix D. J.; Holland P. L. Z-Selective Alkene Isomerization by High-Spin Cobalt(II) Complexes. J. Am. Chem. Soc. 2014, 136, 945–955. 10.1021/ja408238n. [DOI] [PubMed] [Google Scholar]
- Biswas S.; Huang Z.; Choliy Y.; Wang D. Y.; Brookhart M.; Krogh-Jespersen K.; Goldman A. S. Olefin Isomerization by Iridium Pincer Catalysts. Experimental Evidence for an η3-Allyl Pathway and an Unconventional Mechanism Predicted by DFT Calculations. J. Am. Chem. Soc. 2012, 134, 13276–13295. 10.1021/ja301464c. [DOI] [PubMed] [Google Scholar]
- Hartwig J. F.Organotransition Metal Chemistry; University Science Books: Sausalito, CA, 2010. [Google Scholar]
- Pike S. D.; Weller A. S. Organometallic Synthesis, Reactivity and Catalysis in the Solid State Using Well-Defined Single-Site Species. Philos. Trans. R. Soc., A 2015, 373, 20140187. 10.1098/rsta.2014.0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller A. S.; Chadwick F. M.; McKay A. I. Transition Metal Alkane-Sigma Complexes. Adv. Organomet. Chem. 2016, 66, 223–276. 10.1016/bs.adomc.2016.09.001. [DOI] [Google Scholar]
- Hall C.; Perutz R. N. Transition Metal Alkane Complexes. Chem. Rev. 1996, 96, 3125–3146. 10.1021/cr9502615. [DOI] [PubMed] [Google Scholar]
- Pike S. D.; Thompson A. L.; Algarra A. G.; Apperley D. C.; Macgregor S. A.; Weller A. S. Synthesis and Characterization of a Rhodium(I) rrAlkane Complex in the Solid State. Science 2012, 337, 1648–1651. 10.1126/science.1225028. [DOI] [PubMed] [Google Scholar]
- Martínez-Martínez A. J.; Tegner B. E.; McKay A. I.; Bukvic A. J.; Rees N. H.; Tizzard G. J.; Coles S. J.; Warren M. R.; Macgregor S. A.; Weller A. S. Modulation of σ-Alkane Interactions in [Rh(L2)(Alkane)]+ Solid-State Molecular Organometallic (SMOM) Systems by Variation of the Chelating Phosphine and Alkane: Access to η2, η2-σ-Alkane Rh(I), η1-σ-Alkane Rh(III) Complexes, and Alkane Encapsulation. J. Am. Chem. Soc. 2018, 140, 14958–14970. 10.1021/jacs.8b09364. [DOI] [PubMed] [Google Scholar]
- Pike S. D.; Chadwick F. M.; Rees N. H.; Scott M. P.; Weller A. S.; Krämer T.; Macgregor S. A. Solid-State Synthesis and Characterization of σ-Alkane Complexes, [Rh(L2)(η2, η2-C7H12)][BArF4] (L2 = Bidentate Chelating Phosphine). J. Am. Chem. Soc. 2015, 137, 820–833. 10.1021/ja510437p. [DOI] [PubMed] [Google Scholar]
- Petrosko S. H.; Johnson R.; White H.; Mirkin C. A. Nanoreactors: Small Spaces, Big Implications in Chemistry. J. Am. Chem. Soc. 2016, 138, 7443–7445. 10.1021/jacs.6b05393. [DOI] [PubMed] [Google Scholar]
- Dobson G. R.; Hodges P. M.; Healy M. A.; Poliakoff M.; Turner J. J.; Firth S.; Asali K. J. Time-Resolved IR Characterization of Cis-and Trans-[LW(CO)4(Solvent)] [L= PPh3, P(OCHMe2)3, and P(OEt)3] in n-Heptane Solution. The Solvent as a Token Ligand in Short-Lived Reaction Intermediates. J. Am. Chem. Soc. 1987, 109, 4218–4224. 10.1021/ja00248a015. [DOI] [Google Scholar]
- Chadwick F. M.; McKay A. I.; Martinez-Martinez A. J.; Rees N. H.; Krämer T.; Macgregor S. A.; Weller A. S. Solid-State Molecular Organometallic Chemistry. Single-Crystal to Single-Crystal Reactivity and Catalysis with Light Hydrocarbon Substrates. Chem. Sci. 2017, 8, 6014–6029. 10.1039/C7SC01491K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay A. I.; Martínez-Martínez A. J.; Griffiths H. J.; Rees N. H.; Waters J. B.; Weller A. S.; Krämer T.; Macgregor S. A. Controlling Structure and Reactivity in Cationic Solid-State Molecular Organometallic Systems Using Anion Templating. Organometallics 2018, 37, 3524–3532. 10.1021/acs.organomet.8b00215. [DOI] [Google Scholar]
- Crooks A. B.; Yih K.-H.; Li L.; Yang J. C.; Özkar S.; Finke R. G. Unintuitive Inverse Dependence of the Apparent Turnover Frequency on Precatalyst Concentration: A Quantitative Explanation in the Case of Ziegler-Type Nanoparticle Catalysts Made from [(1,5-COD)Ir(μ-O2C8H15)]2 and AlEt3. ACS Catal. 2015, 5, 3342–3353. 10.1021/acscatal.5b00347. [DOI] [Google Scholar]
- Martínez-Martínez A. J.; Rees N. H.; Weller A. S. Reversible Encapsulation of Xenon and CH2Cl2 in a Solid-State Molecular Organometallic Framework (Guest@SMOM). Angew. Chem., Int. Ed. 2019, 58, 16873–16877. 10.1002/anie.201910539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenaus F.; Droste W.; Neumeister J.. Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, 2014. [Google Scholar]
- Copéret C.; Comas-Vives A.; Conley M. P.; Estes D. P.; Fedorov A.; Mougel V.; Nagae H.; Núñez-Zarur F.; Zhizhko P. A. Surface Organometallic and Coordination Chemistry toward Single-Site Heterogeneous Catalysts: Strategies, Methods, Structures, and Activities. Chem. Rev. 2016, 116, 323–421. 10.1021/acs.chemrev.5b00373. [DOI] [PubMed] [Google Scholar]
- Burgun A.; Coghlan C. J.; Huang D. M.; Chen W.; Horike S.; Kitagawa S.; Alvino J. F.; Metha G. F.; Sumby C. J.; Doonan C. J. Mapping-out Catalytic Processes in a Metal-Organic Framework with Single-Crystal X-Ray Crystallography. Angew. Chem. 2017, 129, 8532–8536. 10.1002/ange.201611254. [DOI] [PubMed] [Google Scholar]
- Gonzalez M. I.; Bloch E. D.; Mason J. A.; Teat S. J.; Long J. R. Single-Crystal-to-Single-Crystal Metalation of a Metal-Organic Framework: A Route Toward Structurally Well-Defined Catalysts. Inorg. Chem. 2015, 54, 2995–3005. 10.1021/acs.inorgchem.5b00096. [DOI] [PubMed] [Google Scholar]
- Li Z.; Peters A. W.; Bernales V.; Ortuño M. A.; Schweitzer N. M.; DeStefano M. R.; Gallington L. C.; Platero-Prats A. E.; Chapman K. W.; Cramer C. J.; Gagliardi L.; Hupp J. T.; Farha O. K. Metal-Organic Framework Supported Cobalt Catalysts for the Oxidative Dehydrogenation of Propane at Low Temperature. ACS Cent. Sci. 2017, 3, 31–38. 10.1021/acscentsci.6b00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searles K.; Chan K. W.; Mendes Burak J. A.; Zemlyanov D.; Safonova O.; Copéret C. Highly Productive Propane Dehydrogenation Catalyst Using Silica-Supported Ga-Pt Nanoparticles Generated from Single-Sites. J. Am. Chem. Soc. 2018, 140, 11674–11679. 10.1021/jacs.8b05378. [DOI] [PubMed] [Google Scholar]
- See the Supporting Information.
- McKay A. I.; Bukvic A. J.; Tegner B. E.; Burnage A. L.; Martínez-Martínez A. J.; Rees N. H.; Macgregor S. A.; Weller A. S. Room Temperature Acceptorless Alkane Dehydrogenation from Molecular M-Alkane Complexes. J. Am. Chem. Soc. 2019, 141, 11700–11712. 10.1021/jacs.9b05577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliván M.; Marchenko A. V.; Coalter J. N.; Caulton K. G. Gas/Solid Reactivity of Unsaturated Ruthenium-Containing Molecular Solids. J. Am. Chem. Soc. 1997, 119, 8389–8390. 10.1021/ja971608j. [DOI] [Google Scholar]
- Chadwick F. M.; Rees N. H.; Weller A. S.; Krämer T.; Iannuzzi M.; Macgregor S. A. A Rhodium-Pentane Sigma-Alkane Complex: Characterization in the Solid State by Experimental and Computational Techniques. Angew. Chem., Int. Ed. 2016, 55, 3677–3681. 10.1002/anie.201511269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Continuous addition of diluted H2 (5% with N2) gave a lower initial conversion ∼50% and similar decomposition profile to entry 1 (Table 1), providing no advantage to periodic spiking with H2.
- Longer reaction times under H2 (24 h) resulted in the formation of a different complex, which when dissolved in CH2Cl2 results in the dicationic dimer [(Rh(tBu2PCH2CH2PtBu2)H)2(μ-H)2][BArF4]2 complex 14. See the Supporting Information for the solid-state structure. It is likely that in the solid-state alkane loss by subsitution by H2 occurs to give a monometallic {RhH2}+ fragment that then dimerises on dissolution in CH2Cl2. Interestingly, the solid-state prepared material retains activity for butene isomerization while the recrystallized material does not (Figure S66), suggesting dissoloving in CH2Cl2 quenches vacant sites on the metal centre by dimerization. For an iridium analogue to 14 see:; Chadwick F. M.; Olliff N.; Weller A. S. A convenient route to a norbornadiene adduct of iridium with chelating phosphines, [Ir(R2PCH2CH2PR2)(NBD)][BArF4] and a comparison of reactivity with H2 in solution and the solidestate. J. Organomet. Chem. 2016, 812, 268–271. 10.1016/j.jorganchem.2015.12.001. [DOI] [Google Scholar]
- Pike S. D.; Crimmin M. R.; Chaplin A. B. Organometallic Chemistry Using Partially Fluorinated Benzenes. Chem. Commun. 2017, 53, 3615–3633. 10.1039/C6CC09575E. [DOI] [PubMed] [Google Scholar]
- Hirsekorn K. F.; Veige A. S.; Marshak M. P.; Koldobskaya Y.; Wolczanski P. T.; Cundari T. R.; Lobkovsky E. B. Thermodynamics, Kinetics, and Mechanism of (Silox)3M(Olefin) to (Silox)3M(Alkylidene) Rearrangements (Silox = tBu3SiO; M = Nb, Ta). J. Am. Chem. Soc. 2005, 127, 4809–4830. 10.1021/ja046180k. [DOI] [PubMed] [Google Scholar]
- NMR Crystallography; Harris R. K., Wasylishen R. E., Duer M. J., Eds.; John Wiley & Sons: Chichester, U.K., 2009. [Google Scholar]
- Allan P. K.; Xiao B.; Teat S. J.; Knight J. W.; Morris R. E. In Situ Single-Crystal Diffraction Studies of the Structural Transition of Metal-Organic Framework Copper 5-Sulfoisophthalate, Cu-Sip-3. J. Am. Chem. Soc. 2010, 132, 3605–3611. 10.1021/ja910600b. [DOI] [PubMed] [Google Scholar]
- Mannu A.; Drexler H.-J.; Thede R.; Ferro M.; Baumann W.; Rüger J.; Heller D. Oxidative Addition of CH2Cl2 to Neutral Dimeric Rhodium Diphosphine Complexes. J. Organomet. Chem. 2018, 871, 178–184. 10.1016/j.jorganchem.2018.07.011. [DOI] [Google Scholar]
- Choi J.-C.; Kohno K.; Otsuka M.; Yasuda H.; Sakakura T. Synthesis of a Rhodium(III) Diethyl μ-Carbonato Complex in the Reaction of CO2, H2O, and Ethylene. Organometallics 2011, 30, 6060–6062. 10.1021/om200303w. [DOI] [Google Scholar]
- Pike S. D.; Krämer T.; Rees N. H.; Macgregor S. A.; Weller A. S. Stoichiometric and Catalytic Solid--Gas Reactivity of Rhodium Bis-Phosphine Complexes. Organometallics 2015, 34, 1487–1497. 10.1021/om5013133. [DOI] [Google Scholar]
- By using a larger reaction vessel (see Figure S61), but only a single single-crystal of 8 (∼1 mg), ToNapp of ∼6500 can be achieved in a single batch experiment.
- Fu S.; Chen N.-Y.; Liu X.; Shao Z.; Luo S.-P.; Liu Q. Ligand-Controlled Cobalt-Catalyzed Transfer Hydrogenation of Alkynes: Stereodivergent Synthesis of Z- and E-Alkenes. J. Am. Chem. Soc. 2016, 138, 8588–8594. 10.1021/jacs.6b04271. [DOI] [PubMed] [Google Scholar]
- In this context, recrystallization of bulk complex 7 gives well-defined crystalline material and ∼10% of oil that is chemcially indistiguishable from the crystalline material by NMR spectrocopy. This oily material is significantly less active than single crystalline samples in flow, consistent with the requirement for crystallinity for supporting catalysis.
- Thomas J. M.; Raja R.; Lewis D. W. Single-Site Heterogeneous Catalysts. Angew. Chem., Int. Ed. 2005, 44, 6456–6482. 10.1002/anie.200462473. [DOI] [PubMed] [Google Scholar]
- Fast A.; Esfandiari N. M.; Blum S. A. Small Number of Active Sites and Single-Locus Kinetics Revealed in (Salph)Co-Catalyzed Ethylene Oxide Polymerization. ACS Catal. 2013, 3, 2150–2153. 10.1021/cs400640g. [DOI] [Google Scholar]
- Rifat A.; Kociok-Köhn G.; Steed J. W.; Weller A. S. Cationic Iridium Phosphines Partnered with [Closo-CB11H6Br6]-: (PPh3)2Ir(H)2(closo-CB11H6Br6) and [(PPh3)2Ir(η2-C2H4)3][closo-CB11H6Br6]. Relevance to Counterion Effects in Olefin Hydrogenation. Organometallics 2004, 23, 428–432. 10.1021/om034177o. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.