

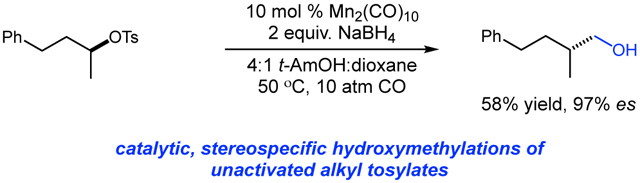
Abstract
The development of a stereospecific hydroxymethylation of alkyl tosylates using an inexpensive, first-row catalyst is described. The transformation proceeds under mild conditions with low pressure to deliver homologated alcohols as products. Chiral, non-racemic β-branched primary alcohols are obtained with high enantiospecificity from easily accessed secondary alkyl substrates. Simple modification of the reaction system also permits access to α-d2 alcohols. These studies use anionic metal carbonyl catalysis to access a synthetic equivalent of the challenging hydroxymethyl anion from carbon monoxide.
Graphical Abstract
The homologation of carbon chains by a single unit is featured in a number of synthetic organic reactions. Classic transformations of carbonyl compounds such as the Killiani-Fisher and Arndt-Eistert syntheses are valued for their simplicity and convenience and find widespread use in synthesis.1-7 Conversely, few methods are available for the formal homologation of alcohols, either directly or via their conversion to alkyl halides or pseudohalides.8,9 Recent work has demonstrated the potential of radical-mediated methods to achieve this goal (Figure 1). For example, Ryu and co-workers have developed a hydromethylation using formaldehyde and cyanoborohydride in a radical chain process.10 More recently, Mankad and co-workers reported a copper-catalyzed, radical-mediated transformation of alkyl iodides to silyl ethers, which upon deprotection with tetrabutylammonium fluoride yields homologated alcohols.11 In each of these processes, the intermediacy of carbon-centered radicals dictates that stereocontrol in reactions of secondary substrates is a significant challenge.
Figure 1.

Hydroxymethylations of alkyl electrophiles.
We targeted the development of an alternative, stereospecific approach to the homologation of alkyl electrophiles using anionic metal carbonyl catalysis.12 Chiral, non-racemic branched primary alcohols are important building blocks in asymmetric synthesis. This strategy would facilitate their synthesis from easily accessed, chiral, non-racemic secondary alkyl tosylates. Herein, we report the successful development of a stereospecific hydroxymethylation using a commercially available manganese carbonyl dimer. This mild, catalytic transformation represents a unique and concise approach to the one carbon homologation of alkyl tosylates with excellent stereocontrol.
Our investigation commenced with the hydroxymethylation of primary tosylate 1 (Table 1). We determined that a catalytic system comprised of 10 mol % Mn2(CO)10 and two equivalents of NaBH4 provided the homologated alcohol 2 in good yield (67%, entry 1). Substitution of Mn2(CO)10 with the putative catalytic nucleophile Na[Mn(CO)5] was similarly effective (entry 2). Interestingly, the use of Co2(CO)8–the precatalyst used in previous studies of stereospecific anionic metal carbonyls catalysis–significantly reduced reaction efficiency, likely owing to the lower nucleophilicity of Na[Co(CO)4] (entry 3).12, 13 Decreasing the catalyst loading to 5 mol % (entry 4) or the CO pressure to 1 atm (balloon, entry 5) slightly lowered efficiency. Increasing the CO pressure to 20 atm provided little improvement (entry 6). Performing the reaction at room temperature decreased conversion (entry 7), while excluding ambient light had little effect (entry 8). Notably, omitting the dioxane co-solvent did not impact the yield in this case, but was important with other tosylates (entry 9).14 No product was formed in the absence of the catalyst (entry 10).
Table 1.
Manganese-catalyzed hydroxymethylation of an unactivated alkyl tosylate.
 | ||
|---|---|---|
| entry | variation from standard conditions above | yield (%)a |
| 1 | none | 67 |
| 2 | 20 mol % Na[Mn(CO)5] | 68 |
| 3 | 10 mol % Co2(CO)8 | 50 |
| 4 | 5 mol % Mn2(CO)10 | 61 |
| 5 | 1 atm CO | 60 |
| 6 | 20 atm CO | 70 |
| 7 | rt | 48 |
| 8 | dark | 64 |
| 9 | no dioxane | 66 |
| 10 | no Mn2(CO)10 | 0 |
Reactions were performed with [1]0 = 0.5 M. aYields determined by 1H NMR spectroscopy of crude reaction mixture using an internal standard.
Having identified a viable catalytic system, we turned our attention to the scope of the hydroxymethylation, starting with primary alkyl tosylates (Table 2). The hydroxymethylation of the tosylate derived from the monoterpenoid citronellol provided homologated alcohol 4, demonstrating compatibility with alkenyl substrates (entry 2). Common polar functionality such as esters and Boc-protected amines are also tolerated in the hydroxymethylation (entries 3 and 4). The homologation of indolyl tosylate 9 yielded alcohol 10, demonstrating the efficiency of the reaction in the presence of electron-rich heterocycles (entry 5). Notably, the hydroxymethylation of a lithocholic acid derivative is successful in the presence of a silyl ether, which would undergo deprotection using a previously reported copper-catalyzed hydroxymethylation protocol (entry 6).11
Table 2.
Manganese-catalyzed hydroxymethylation of primary alkyl tosylates.
| entry | substrate | product | yield (%)a |
|---|---|---|---|
| 1 | 66 | ||
| 2 |  |
52 | |
| 3 | 57 | ||
| 4 | 53 | ||
| 5 |  |
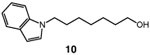 |
58 |
| 6 | 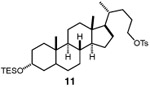 |
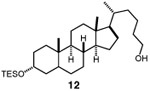 |
56 |
See Table 1 for conditions.
Isolated yields.
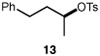
We continued with the hydroxymethylations of chiral, non-racemic secondary tosylates (Table 3). We view the capability of our polar catalytic manifold to enable stereospecific hydroxymethylations as a powerful, unique aspect of our approach. The hydroxymethylation of chiral, non-racemic tosylate 13 delivered alcohol 14 in 58% yield and with excellent enantiospecificity (97%, entry 1). Importantly, the reaction is not limited to methyl-branched substrates as demonstrated by the reaction of tosylate 15, which although less efficient (40% yield) proceeds in >99% es (entry 2). The homologations of tosylates derived from chiral, non-racemic 1,3-diols proceeded efficiently with high enantiospecificities (entries 3 and 4) and demonstrated reaction tolerance of electron-poor arenes. Alkyl tosylate 21 containing thiophene underwent hydroxymethylations in 54% yield. Finally, a simple aliphatic tosylate (23) was also a viable substrate, and provided the hydroxymethylation product with good enantiospecificity (entry 7). Generally, the remaining mass balance contained a mixture of unreacted starting material, alkene, and alkane byproducts. While the results of Tables 2 and 3 demonstrate that the reaction yields of the hydroxymethylation are moderate, the uniformly high stereoselectivities are an attractive feature of this catalytic process.
Table 3.
Stereospecific, manganese-catalyzed hydroxymethylation of chiral, non-racemic secondary alkyl tosylates.
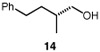
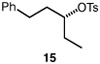
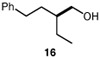
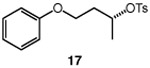

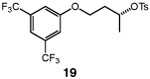
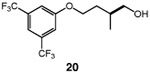
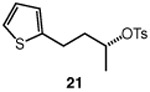
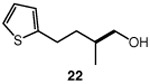
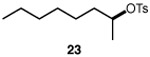
See Table 1 for conditions.
Isolated yields unless otherwise noted. Enantiospecificity (es) = (eeproduct/eesubstrate) x 100%, determined by chiral HPLC.
Reaction yield determined by 1H NMR spectroscopy of crude reaction mixtures using an internal standard.
31% isolated yield.
Enantiospecificity determined from the tosylated product (see Supporting Information).
α-Deuterated alcohols are important compounds due to their use as drug analogs and internal standards in proteomic, metabolomic, and LADMET studies.15 Common routes to these α-deuterated products proceed via reduction of carboxylic acid derivatives using LiAlD4 or highly reactive single-electron reductants.16 An alternative approach via direct α-deuteration of an alcohol requires precious ruthenium catalysts and can provide product regioisomers.17-19 Given its commercial availability, we sought to apply NaBD4 in the hydroxymethylation to achieve α-deuterium incorporation under our mild catalytic conditions. As an initial demonstration of our approach to α-deuterated alcohols, we performed the hydroxymethylation of primary tosylate 1 with 2 equiv NaBD4. The hydroxymethylation proceeded in 50% isolated yield and 94% deuterium incorporation (eq 1). This modification of our catalytic system involves a nucleophilic substitution with a formal deuterated hydroxymethyl anion equivalent and offers a new concise approach to α-deuterated alcohols under mild conditions.
 |
(1) |
We sought to uncover details regarding the reaction mechanism by studying the reactivity of a putative acylmanganese intermediate. The reaction of substrate 13 with 1 equiv of Na[Mn(CO)5]-the active manganate formed in situ–in the absence of NaBH4 provided the acylmanganese 25 in 48% yield. This intermediate was subsequently reduced with NaBH4 to deliver the homologated alcohol 14 in 56% 1H NMR yield (eq 3), consistent with the viability of the acylmanganese as a precursor to the hydroxymethylation product. Furthermore, comparison of hydroxymethylation product 14 (Table 3, entry 1) to an independently prepared sample indicated that the reaction proceeded with inversion of configuration at the stereogenic center.
Synthesis of Acyl Manganese Intermediate
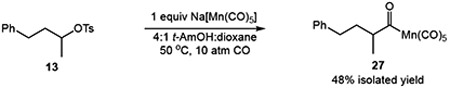 |
(2) |
Reduction of Acyl Manganese Intermediate
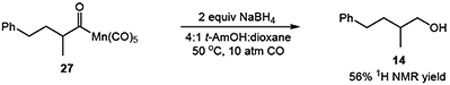 |
(3) |
A mechanistic proposal for the catalytic hydroxymethylation is illustrated in Scheme 1. The dimanganese decacarbonyl precatalyst is reduced in situ by NaBH4 to provide the active sodium pentacarbonylmanganate species. Subsequent nucleophilic attack on the substrate forms an alkylmanganese intermediate, which undergoes migratory insertion of CO with retention of configuration. The resulting acyl manganese is reduced by NaBH4 to regenerate the active catalyst. The aldehyde initially formed in this step is further reduced to give the hydroxymethylation product.
Scheme 1.
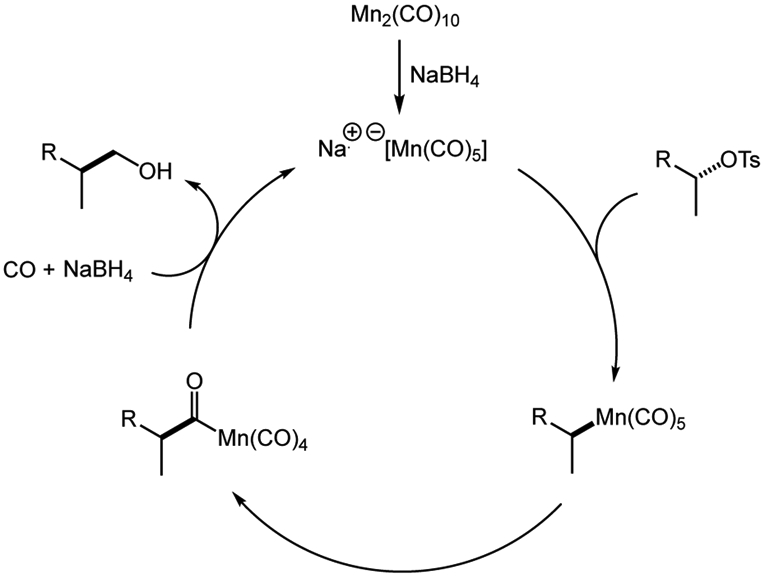
Plausible catalytic cycle for the stereospecific hydroxymethylation.
In conclusion, we have developed a stereospecific hydroxymethylation of alkyl tosylates using manganese catalysis. This approach leverages the reactivity of anionic metal carbonyl catalysis to access a formal hydroxymethyl anion equivalent from CO and hydride. A mild, stereospecific homologation of alkyl electrophiles is achieved, providing direct access to chiral, non-racemic β-branched primary alcohols—and α-deuterated derivatives—from simple starting materials. Future studies will target the further development of valuable synthetic methods using this unique mode of metal catalysis.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by Award No. R35 GM131708 from the National Institute of General Medical Sciences. We thank the UNC Department of Chemistry Mass Spectrometry Core Laboratory for assistance with MS analysis, supported by National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM118055 and the National Science Foundation under Grant No. CHE1726291. We thank Justin Marcum (University of North Carolina-Chapel Hill) for assistance with SFC and helpful discussions.
Footnotes
Supporting Information. Experimental procedures and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Kowalski CJ, Haque MS, Fields KW Ester Homologation Via Alpha-Bromo Alpha-Keto Dianion Rearrangement, J. Am. Chem. Soc 1985, 107, 1429–1430. [Google Scholar]
- 2.Ford A, Miel H, Ring A, Slattery CN, Maguire AR, McKervey MA Modern Organic Synthesis with α-Diazocarbonyl Compounds. Chem. Rev 2015, 115, 9981–10080. [DOI] [PubMed] [Google Scholar]
- 3.Levine SG A New Aldehyde Synthesis, J. Am. Chem. Soc 1958, 80, 6150–6151. [Google Scholar]
- 4.Dinizo SE, Freerksen RW, Pabst WE, Watt DS A One-Carbon Homologation of Carbonyl Compounds to Carboxylic Acids, Esters, And Amides, J. Am. Chem. Soc 1977, 99, 182–186. [Google Scholar]
- 5.Kluge AF, Cloudsdale IS Phosphonate Reagents for the Symthesis of Enol Ethers and One-Carbon Homologation to Aldehydes, J. Org. Chem 1979, 44, 4847–4852. [Google Scholar]
- 6.McNulty J, Das P Development of A One-Pot Method for the Homologation of Aldehydes to Carboxylic Acids. Tetrahedron 2009, 65, 7794–7800. [Google Scholar]
- 7.Cukalovic A, Monbaliu J-CM, Heynderickx GJ, Stevens CV User Friendly and Flexible Kiliani Reaction on Ketoses Using Microreaction Technology, J. Flow Chem 2012, 2, 43–46. [Google Scholar]
- 8.Kagawa N, Nibbs AE, Rawal VH One-Carbon Homologation of Primary Alcohols to Carboxylic Acids, Esters, and Amides via Mitsunobu Reactions with MAC Reagents. Org. Lett 2016, 18, 2363–2366. [DOI] [PubMed] [Google Scholar]
- 9.Soundararajan R, Li G, Brown HC Homologation of representative boronic esters using in situ generated (halomethyl)lithiums: A comparative study. Tetrahedron Lett. 1994, 35, 8957–8960. [Google Scholar]
- 10. a).Kawamoto T, Fukuyama T, Ryu I Radical Addition of Alkyl Halides to Formaldehyde in the Presence of Cyanoborohydride as a Radical Mediator. A New Protocol for Hydroxymethylation Reaction, J. Am. Chem. Soc 2012, 134, 875–877. [DOI] [PubMed] [Google Scholar]; b) For an earlier example of radical-mediated hydroxymethylation, see: Kobayashi S; Kawamoto T; Uehara S; Fukuyama T; Ryu I Black-Light-Induced Radical/Ionic Hydroxymethylation of Alkyl Iodides with Atmospheric CO in the Presence of Tetrabutylammonium Borohydride. Org. Lett 2013, 12, 1548–1551. [DOI] [PubMed] [Google Scholar]
- 11.Zhao S, Mankad N Cu-Catalyzed Hydroxymethylation of Unactivated Alkyl Iodides with CO to Provide One-Carbon-Extended Alcohols. Angew. Chem. Int. Ed 2018, 57, 5867–5870. [DOI] [PubMed] [Google Scholar]
- 12.a) For prior work in stereospecific carbonylation of alkyl tosylates, see: Sargent BT; Alexanian EJ; Cobalt-Catalyzed Carbonylative Cross-Coupling of Alkyl Tosylates and Dienes: Stereospecific Synthesis of Dienones at Low Pressure. J. Am. Chem. Soc 2017, 139, 12438–12440. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sargent BT; Alexanian EJ; Cobalt-Catalyzed Aminocarbonylation of Alkyl Tosylates: Stereospecific Carbonylation of Amides. Angew. Chem. Int. Ed 2019, 58, 9533–9536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.King RB Applications of Metal Carbonyl Anions In The Synthesis of Unusual Organometallic Compounds. Acc. Chem. Res 1970, 3, 417–427. [Google Scholar]
- 14.See the Supporting Information for a representative example of the effect of dioxane.
- 15.Michelotti A; Roche M; 40 Years of Hydrogen-Deuterium Exchange Adjacent to Heteroatoms: A Survey. Synthesis 2019, 51, 1319–1328.. [Google Scholar]; Electron Transfer Reduction of Unactivated Esters Using SmI2–H2O. Chem. Commun. 2011, 47, 10254–10256. [DOI] [PubMed] [Google Scholar]
- 16.Szostak M, Spain M, Procter DJ Electron Transfer Reduction of Unactivated Esters Using SmI2-H2O. Chem. Commun 2011, 47, 10254–10256. [DOI] [PubMed] [Google Scholar]
- 17.Chatterjee B, Gunanathan C Ruthenium Catalyzed Selective α- and α,β-Deuteration of Alcohols Using D2O. Org. Lett 2015, 17, 4794–4797. [DOI] [PubMed] [Google Scholar]
- 18.Zhang L, Nguyen DH, Raffa G, Desset S, Paul S, Dumeignil F, Gauvin RM Efficient Deuterium Labelling of Alcohols in Deuterated Water Catalyzed by Ruthenium Pincer Complexes. Catal. Commun 2016, 84, 67–70. [Google Scholar]
- 19.Kar S, Goeppert A, Sen R, Kothandaraman J, Prakash GKS Regioselective Deuteration of Alcohols in D2O Catalysed by Homogeneous Manganese and Iron Pincer Complexes. Green Chem. 2018, 20, 2706–2710. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.