

Abstract
Identifying the root of a phylogenetic tree is important because incorrectly rooted phylogenetic trees may mislead evolutionary and taxonomic inferences. Many techniques for inferring the root have been proposed, but each has shortcomings that may make it inappropriate for any particular dataset. Here we outline the various ways to root phylogenetic trees, which include: outgroup, midpoint rooting, molecular clock rooting, and Bayesian molecular clock rooting. In addition, we discuss the pros and cons and also list software availability for each of the rooting methods.
Keywords: Bayesian molecular clock rooting, Midpoint rooting, Molecular clock, Outgroup rooting, Phylogeny, Rooted tree, Rooted versus unrooted, Unrooted tree
Rooted versus Unrooted
Phylogenetic trees are either rooted or unrooted, depending on the research questions being addressed. The root of the phylogenetic tree is inferred to be the oldest point in the tree and corresponds to the theoretical last common ancestor of all taxonomic units included in the tree. The root gives directionality to evolution within the tree (Baldauf, 2003). Accurate rooting of a phylogenetic tree is important for directionality of evolution and increases the power of interpreting genetic changes between sequences (Pearson et al., 2013).
Many techniques such as molecular clock, Bayesian molecular clock, outgroup rooting, or midpoint rooting methods tend to estimate the root of a tree using data and assumptions (Boykin et al., 2010). However, Steel (2012) discusses root location in random trees and points out that information in the prior distribution of the topology alone can convey the location of the root of the tree. These results show that the tree models that treat all taxa equally and are sampling consistently convey information about the location of the ancestral root in unrooted trees (Steel, 2012).
Why Do We Need a Rooted Tree?
We are interested in rooting a phylogenetic tree in order to show the path of evolution of biological species. Therefore most users of phylogenetic trees want rooted trees because they give an indication of the directionality of evolutionary change. The root of phylogenetic tree is crucial in evolutionary interpretation of the tree (Williams, 2014), because an unrooted tree species shows only the relationships among the taxa and does not define the evolutionary path (Figure 1(a)).
Figure 1.


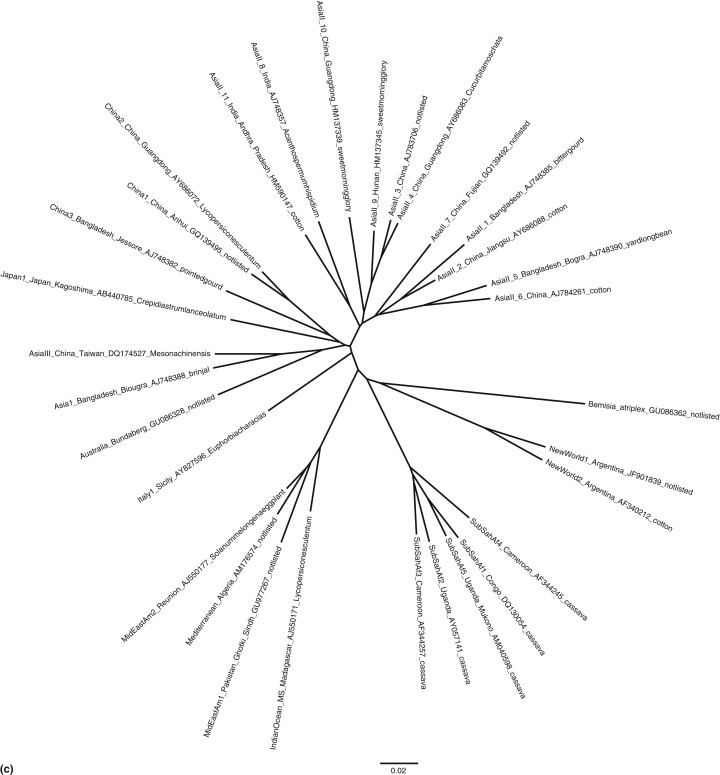
(a) Outgroup rooted phylogenetic tree of the Bemisia tabaci species complex (whiteflies) from a modified dataset (Boykin et al., 2013). Tip labels correspond to geographic location_sublocation_GenBank accession number_host. (b) Unrooted phylogenetic tree of the Bemisia tabaci species complex (whiteflies) from a modified dataset (Boykin et al., 2013). Tip labels correspond to geographic location_sublocation_GenBank accession number_host. (c) Unrooted star phylogenetic tree of the Bemisia tabaci species complex (whiteflies) from a modified dataset (Boykin et al., 2013). Tip labels correspond to geographic location_sublocation_GenBank accession number_host.
When Do We Need an Unrooted Tree?
An unrooted tree is desired when we do not have a distantly related group (sequence) for comparison or when primary interest is focused only on relationships among the taxa rather than on the directionality of evolutionary change. Unrooted trees are beneficial in depicting clusters of related sequences. Unrooted gene trees have also become more prevalent within the multispecies coalescent phylogenetic framework, leading to systematic approaches for inferring unrooted species trees from unrooted gene tree topologies (Liu and Yu, 2011). Gene trees and species trees can have similar topologies but often there is considerable discordance between gene tree and species trees (Degnan, 2013). For this reason, understanding how to root gene trees will have implications for accurate species tree inference (Figures 1(b) and 1(c)).
How Do You Root a Phylogenetic Tree?
Outgroup Rooting
There are several rooting methods (Table 1 ), however the most popular and widely used is the outgroup method (Wheeler, 1990, Tarrío et al., 2000, Hess and De Moraes Russo, 2007, Boykin et al., 2010). Outgroup method assumes that one or more of the taxa are divergent from the rest of the taxa (ingroup). The branch linking the ingroup and outgroup becomes the starting point, and defines all subsequent evolutionary events within the tree (Brady et al., 2011, Williams, 2014). In addition to providing evolutionary information of the ingroup, the outgroup has other additional functions. It allows the identification of distinct features within ingroup sequences (Wheeler, 1990). An important aspect of outgroup method is the need for a priori knowledge on the appropriate outgroup to use for the set of sequences (Wheeler, 1990, Hess and De Moraes Russo, 2007). However, this is also the main bottleneck for this method, especially within higher taxonomic groups such as angiosperms, birds, and mammals where a consensus outgroup is lacking (Qiu et al., 2001). As a consequence, many authors are forced to choose between different sorts of outgroups that are either phylogenetically close or phylogenetically distant (Rota-Stabelli and Telford, 2008).
Table 1.
Four methods for rooting phylogentic trees
| Rooting method | Pros | Cons | Software |
|---|---|---|---|
| Outgroup | Accurate | Must have an outgroup | PAUP |
| Long branch attraction | Figtree | ||
| R package: ape (http://www.inside-r.org/packages/cran/ape/docs/unroot) | |||
| Midpoint | Fast | Dependent on clocklike data | PAUP |
| No outgroup needed | Not good with unbalanced trees | R package: phangorn (https://cran.r-project.org/web/packages/phangorn/phangorn.pdf) | |
| Molecular clock | No outgroup needed | Computationally intense | PAUP |
| Robust to violations of the clock | PAML | ||
| Bayesian molecular clock | Alternative rootings uncovered | Must customize the prior | Post Root (http://www.stat.osu.edu/~lkubatko/software/phyl_util.html) |
| No outgroup needed | Root annotator (http://sourceforge.net/projects/rootannotator/) |
Lack of an appropriate outgroup results in drawbacks such as the long branch attraction (LBA). LBA occurs mainly when the outgroup taxa are distantly related to the ingroup due to either large divergence time and/or increased rate of evolution (Tarrío et al., 2000). This results in homoplastic changes occurring at rapidly evolving sites thus resulting in artifactual rooting (random rooting) (Wheeler, 1990, Hendy and Penny, 2011, Maddison et al., 1984). Several criteria have been proposed to prevent LBA within phylogenetic trees, through a multistep process as proposed by Rota-Stabelli and Telford (2008) to assist in outgroup selection especially in the case of arthropod classes. They include: (1) low substitution rate; (2) ingroup like G+C composition; (3) new strand bias estimators ‘skew index’; (4) the tendency of the outgroup to avoid ‘random branding effect’; and (5) phylogenetic proximity to the arthropod.
An alternative approach to assess the importance of an outgroup in rooting the tree is explored by Graham et al. (2002); this is by establishing whether the outgroup provides sufficient signal in response to root location, indicative of historic linkage or due to LBA. Using Pontederiaceae, an aquatic monocot, as the case study they assessed how the nearest outgroup provides for rooting Pontederiaceae compared to those less closely related relatives and further investigate the role of LBA when determining the optimal rooting of Pontederiaceae. However, they concluded that LBA may influence rooting, and may be supporting the wrong outgroup. To further reduce LBA and to ensure robustness of the outgroup rooting method they recommend multiple sampling of outgroups within the sister group rather than sampling within less closely related taxa.
Midpoint Rooting
Midpoint rooting calculates tip to tip distances and then places the root halfway between the two longest tips (Swofford et al., 1996). The ancestral point will be identified if the tree has constant rates of evolution. The method is exclusively dependent on the branch length of the phylogenetic tree and the assumption of the molecular clock (Holland et al., 2003). Homogeneity is assumed across the branch and that the two most divergent taxa evolved at equal rates (Holland et al., 2003; Swofford et al., 1996). If the tree is balanced, midpoint rooting works well. However, a major limitation of midpoint rooting is the dependence on having clocklike data and a balanced topology.
The midpoint rooting method is often applied to viral genetic datasets because in many cases outgroups are unknown. For example, Stavrinides and Guttman (2004) utilize midpoint rooting to establish the evolutionary relationship of severe acute respiratory syndrome (SARS) coronaviruses. They carried out phylogenetic analysis of the viral genes encoding for viral structural proteins specifically, envelope matrix (M) and nucleocapsid (N) proteins. The midpoint rooting of the trees generated using the M protein data shows two groups, one that consisted of porcine, feline, and canine and one that contained bovine and murine coronaviruses (Coronaviridae). On the other hand, the N protein tree is midpoint rooted on the branch leading to the group 1 coronaviruses. Moreover, the appropriateness of midpoint rooting is supported by the results of Tajima’s relative rate test (Tajima, 1993), indicating no rate heterogeneity among the coronavirus groups (Stavrinides and Guttman, 2004).
This method should be used as an alternative to the outgroup rooting method and could be adopted as the default method when the outgroup method is difficult to apply either due to problems with available outgroups, such as LBA or lack of a priori knowledge of the outgroup (Hess and De Moraes Russo, 2007).
Molecular Clock Rooting
The molecular clock rooting method has one assumption: the rate of evolution is constant for the sequences of interest (Yang and Rannala, 2012). The rate is typically expressed in substitutions per site per year or substitutions per site per million years (Brown and Yang, 2011). The strict clock is often used in analyses of sequences sampled at the intraspecific level, for which usually there is an exceptionally low rate of variation (Brown and Yang, 2011, Ho and Duchêne, 2014). The molecular clock assumption becomes problematic for distantly related species because there is a linear relationship between the genetic distances and approximate divergence. The slope of the line directly corresponds to the evolutionary rate variation among species especially among divergent taxa (Welch and Bromham, 2005). Before utilizing the molecular clock method for rooting a phylogenetic tree users should test if a molecular clock is appropriate to describe the data. Testing for the molecular clock entails generating two maximum likelihood trees, one computed with the molecular clock enforced and one without the molecular clock enforced and then utilizing the likelihood ratio test (Felsenstein, 1983, Holder and Lewis, 2003).
Bayesian Molecular Clock Rooting
Huelsenbeck et al. (2002) proposed the use of Bayesian inference under the molecular clock assumption to infer the root of a phylogenetic tree. After obtaining the posterior distribution of trees under Bayesian inference, the root of the tree is inferred to be the root position with the highest posterior probability. This method also provides the posterior probability that the root lies on any branch of the ingroup topology. Another advantage of the Bayesian method is that it allows the user to evaluate alternative rootings. Other rooting methods only return one rooting for a particular dataset, without any numerical assessment of confidence in that rooting. A Bayesian molecular clock analysis successfully identified the root of Orcuttieae (Poaceae) (Boykin et al., 2010) when all other methods failed. Post_root was developed to analyze the output from MrBayes (Ronquist et al., 2012) or ExaBayes (Aberer et al., 2014) runs. The output from Post_Root will give the number of unique roots and also the most probable root position.
Most recently, Calvignac-Spencer et al. (2014) have further developed Post_Root to a web-based interface in their quest to identify the branch root posterior probability (RPP) of the most recent Ebola outbreak in West Africa. They were forced to rely on Bayesian molecular clock rooting because there is no known outgroup for Ebola. It is often the case when analyzing viral sequences that no outgroup is known; therefore the Bayesian molecular clock rooting is a very useful alternative, especially when rooting is crucial for viral outbreaks.
Acknowledgment
Research in the Boykin lab is provided by the Bill & Melinda Gates Foundation via a subcontract (B04265) from the National Research Institute, University of Greenwich, UK. An Australian PhD Award to J Wainaina. The Plant Biosecurity Cooperative Research Centre, Australia (61056) supported S Maina.
See also
Bayesian Phylogenetic Methods. Phylogenetic Tree
Biographies

Tonny Kinene is a PhD student in Bioinformatics at The University of Western Australia, funded by Bill and Melinda Gates Foundation to protect cassava from whiteflies in East Africa. His research focuses on systematics and rates of evolution amongst the African cassava whitefly species. He has a strong background in applied mathematics and while at Lappeenranta University of Technology (Finland), he did courses in Technomathematics. He holds an MSc degree in Mathematical modeling (Makerere University, Uganda) and his research at masters level focused on formulating a mathematical model to investigate the transmission dynamics of cassava brown streak disease from Bemisia tabaci to the plant and vice versa and also quantify the cost effectiveness of the proposed control measures. He also holds a PGD in Information Technology from Amity University Uttar Pradesh (India) and a BSc with Education in Mathematics and Physics (Makerere University, Uganda).

James Wainaina is Australian Award PhD receipt from Kenya. James research focus is on genomics and computational biology of whitefly and whitefly transmitted viruses within smallholder cropping systems in Eastern and Central Africa. James completed his MSc at Jomo Kenyatta University of Agriculture and Technology (JKUAT) and a Combating Disease of Poverty Consortium (CPDC) Fellowship at the National University of Ireland (Maynooth). More recently, James has been involved in the development and implementation of an aflatoxin diagnosis platform at the Biosciences eastern and central Africa hub (BecA-ILRI) hub, Nairobi Kenya a joint initiative with the Agency for International Development (AusAID) and the Commonwealth Scientific and Industrial Research Organisation (CSIRO).

Solomon is currently a PhD student at The University of Western Australia (UWA), working in the Plant Biosecurity CRC Early Warning research program with the support of a UWA SIRF scholarship. He holds a BSc Biotechnology from Periyar University (India) and MSc Biotechnology from the Kenyatta University (Kenya). He has also been awarded a BecA training in Next generation sequencing training in Dubai–UAE. Asia-Africa Science platform program (AASPP) supported by the Japan Society of the Promotion of Science (JSPS) Japan. Before commencing his studies at UWA, Solomon worked at the Biosciences Eastern and Central Africa-International Livestock Research Institute Hub (BecA-ILRI Hub), Nairobi (a CGIAR Research Center in Kenya) as Research Assistant in Capacity Building and Genomics Platforms. He was responsible for capacity building, which involved training and providing technical support to visiting scientists and students, technology transfer, and conducting training and organizing workshops.

Laura M. Boykin is an American computational biologist who uses genomics and supercomputing to help smallholder farmers in sub-Saharan Africa control whiteflies, which have caused devastation of local cassava crops. Using genetic data to understand the whitefly’s evolution, Boykin’s research has proven important genetic differences in various whitefly species. Boykin also works to equip African scientists with a greater knowledge of genomics and high-performance computing skills to tackle future insect outbreaks. Boykin completed her PhD in biology at the University of New Mexico while working at Los Alamos National Laboratory in the Theoretical Biology and Biophysics group, and is currently an Assistant Professor at the University of Western Australia and TED Fellow. For more information see the lab webpage: www.lauraboykinresearch.com.
References
- Aberer A.J., Kobert K., Stamatakis A. ExaBayes: Massively parallel bayesian tree inference for the whole-genome era. Molecular Biology and Evolution. 2014;31(10):1–8. doi: 10.1093/molbev/msu236. Available at: http://www.ncbi.nlm.nih.gov/pubmed/25135941 (accessed 13.10.15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldauf S.L. The deep roots of eukaryotes. Science. 2003;300(5626):1703–1706. doi: 10.1126/science.1085544. [DOI] [PubMed] [Google Scholar]
- Boykin L.M., Bell C.D., Evans G., Small I., DeBarro P. Is agriculture driving the diversification of the Bemisia tabaci species complex (Hemiptera: Sternorrhyncha: Aleyrodidae)?: Dating, diversification and biogeographic evidence revealed. BMC Evolutionary Biology. 2013;13:228. doi: 10.1186/1471-2148-13-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boykin L.M., Kubatko L.S., Lowrey T.K. Comparison of methods for rooting phylogenetic trees: A case study using Orcuttieae (Poaceae: Chloridoideae) Molecular Phylogenetics and Evolution. 2010;54(3):687–700. doi: 10.1016/j.ympev.2009.11.016. [DOI] [PubMed] [Google Scholar]
- Brady S.G., Litman J.R., Danforth B.N. Rooting phylogenies using gene duplications: An empirical example from the bees (Apoidea) Molecular Phylogenetics and Evolution. 2011;60(3):295–304. doi: 10.1016/j.ympev.2011.05.002. [DOI] [PubMed] [Google Scholar]
- Brown R.P., Yang Z. Rate variation and estimation of divergence times using strict and relaxed clocks. BMC Evolutionary Biology. 2011;11(1):271. doi: 10.1186/1471-2148-11-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvignac-Spencer S., Schulze J.M., Zickmann F., Renard B.Y. Clock rooting further demonstrates that Guinea 2014 EBOV is a member of the Zaïre lineage. PLoS Currents. 2014 doi: 10.1371/currents.outbreaks.c0e035c86d721668a6ad7353f7f6fe86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan J.H. Anomalous unrooted gene trees. Systematic Biology. 2013;62(4):574–590. doi: 10.1093/sysbio/syt023. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Statistical inference of phylogenies. Journal of the Royal Statistical Society. 1983;A 146:246–272. [Google Scholar]
- Graham S.W., Olmstead R.G., Barrett S.C.H. Rooting phylogenetic trees with distant outgroups: a case study from the commelinoid monocots. Molecular Biology and Evolution. 2002;19(10):1769–1781. doi: 10.1093/oxfordjournals.molbev.a003999. [DOI] [PubMed] [Google Scholar]
- Hendy M.D., Penny D. A framework for the quantitative study of evolutionary trees. Systematic Zoology. 2011;38(4):297–309. Society of systematic biologists a framework for the quantitative study of evolutionary trees. [Google Scholar]
- Hess P.N., De Moraes Russo C.A. An empirical test of the midpoint rooting method. Biological Journal of the Linnean Society. 2007;92(4):669–674. doi: 10.1111/j.1095-8312.2007.00864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho S.Y.W., Duchêne S. Molecular-clock methods for estimating evolutionary rates and timescales. Molecular Ecology. 2014;23(24):5947–5965. doi: 10.1111/mec.12953. [DOI] [PubMed] [Google Scholar]
- Holder M., Lewis P.O. Phylogeny estimation: Traditional and Bayesian approaches. Nature Reviews. Genetics. 2003;4(4):275–284. doi: 10.1038/nrg1044. [DOI] [PubMed] [Google Scholar]
- Holland B.R., Penny D., Hendy M.D. Outgroup misplacement and phylogenetic inaccuracy under a molecular clock – a simulation study. Systematic Biology. 2003;52(2):229–238. doi: 10.1080/10635150390192771. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck J.P., Larget B., Miller R.E., Ronquist F. Potential applications and pitfalls of Bayesian inference of phylogeny. Systematic Biology. 2002;51(5):673–688. doi: 10.1080/10635150290102366. [DOI] [PubMed] [Google Scholar]
- Liu L., Yu L. Estimating species trees from unrooted gene trees. Systematic Biology. 2011;60(5):661–667. doi: 10.1093/sysbio/syr027. [DOI] [PubMed] [Google Scholar]
- Maddison W.P., Donoghue M.J., Maddison D.R. Outgroup analysis and parsimony. Systematic Zoology. 1984;33(1):83–103. Society of systematic biologists outgroup analysis and parsimony. [Google Scholar]
- Pearson T., Hornstra H.M., Sahl J.W. When outgroups fail; Phylogenomics of rooting the emerging pathogen, Coxiella burnetii. Systematic Biology. 2013;62(5):752–762. doi: 10.1093/sysbio/syt038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y.L., Lee J., Whitlock B.A., Bernasconi-Quadroni F., Dombrovska O. Was the ANITA rooting of the angiosperm phylogeny affected by long-branch attraction? Amborella, Nymphaeales, Illiciales, Trimeniaceae, and Austrobaileya. Molecular Biology and Evolution. 2001;18(9):1745–1753. doi: 10.1093/oxfordjournals.molbev.a003962. [DOI] [PubMed] [Google Scholar]
- Ronquist F., Teslenko M., van der Mark P. Mrbayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Systematic Biology. 2012;61(3):539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota-Stabelli O., Telford M.J. A multi criterion approach for the selection of optimal outgroups in phylogeny: Recovering some support for Mandibulata over Myriochelata using mitogenomics. Molecular Phylogenetics and Evolution. 2008;48(1):103–111. doi: 10.1016/j.ympev.2008.03.033. [DOI] [PubMed] [Google Scholar]
- Stavrinides J., Guttman D.S. Mosaic evolution of the severe acute respiratory syndrome coronavirus mosaic evolution of the severe acute respiratory syndrome coronavirus. Journal of Virology. 2004;78(1):76–82. doi: 10.1128/JVI.78.1.76-82.2004. Available at: http://jvi.asm.org/content/78/1/76.full.pdf+html (accessed 13.10.15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel M. Root location in random trees: A polarity property of all sampling consistent phylogenetic models except one. Molecular Phylogenetics and Evolution. 2012;65(1):345–348. doi: 10.1016/j.ympev.2012.06.022. [DOI] [PubMed] [Google Scholar]
- Swofford D.L., Olsen G.J., Waddell P.J., Hillis D.M. Phylogenetic inference. In: Hiillis D.M., Moritz D., Mable B.K., editors. Molecular Systematics. Sinauer Associates; Sunderland, MA: 1996. pp. 407–514. [Google Scholar]
- Tajima F. Simple methods for testing the molecular evolutionary clock hypothesis. Genetics. 1993;135(2):599–607. doi: 10.1093/genetics/135.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarrío R., Rodríguez-Trelles F., Ayala F.J. Tree rooting with outgroups when they differ in their nucleotide composition from the ingroup: The Drosophila saltans and willistoni groups, a case study. Molecular Phylogenetics and Evolution. 2000;16(3):344–349. doi: 10.1006/mpev.2000.0813. [DOI] [PubMed] [Google Scholar]
- Welch J.J., Bromham L. Molecular dating when rates vary. Trends in Ecology and Evolution. 2005;20(6):320–327. doi: 10.1016/j.tree.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Wheeler W.C. Nucleic acid sequence phylogeny and random outgroups. Cladistics. 1990;6(4):363–367. doi: 10.1111/j.1096-0031.1990.tb00550.x. Available at: http://doi.wiley.com/10.1111/j.1096–0031.19900.tb00550.x (accessed 13.10.15). [DOI] [PubMed] [Google Scholar]
- Williams T.A. Evolution: Rooting the eukaryotic tree of life. Current Biology. 2014;24(4):R151–R152. doi: 10.1016/j.cub.2014.01.026. [DOI] [PubMed] [Google Scholar]
- Yang Z., Rannala B. Molecular phylogenetics: principles and practice. Nature Reviews Genetics. 2012;13(5):303–314. doi: 10.1038/nrg3186. [DOI] [PubMed] [Google Scholar]