

Abstract
The design of alpha-helical tectons for self-assembly is maturing as a science. We have now reached the point where many different coiled-coil topologies can be reliably produced and validated in synthetic systems and the field is now moving on towards more complex, discrete structures and applications. Similarly the design of infinite or fiber assemblies has also matured, with the creation fibers that have been modified or functionalized in a variety of ways. This chapter discusses the progress made in both of these areas as well as outlining the challenges still to come.
Keywords: Self-assembly, designed coiled-coil peptides, designed peptide fibers, bioconjugation, scaffolds, fibrils, building blocks, tectons, synthetic biology, biomolecular design
I. Introduction
The design of self-assembling complex systems offers an excellent opportunity to reflect and improve on our ability to understand, mimic, and redesign natural systems. Knowledge of nature can be advanced through a cycle of observing, deducing the rules by which complex structure is formed, attempting to create synthetic analogs, and comparing the results to the original template. Where this approach has been successful in duplicating natural systems, a platform is created from which new versions can be made, with improved or varied functionality, and with technological applications across society. This area of research falls under the broad umbrella of synthetic biology.
A. Synthetic Biology
The research field of synthetic biology was proposed by Wacław Szybalski in 19741 as the next phase of what had previously been a more descriptive study of molecular biology. The rise of recombinant DNA technology at this time opened the door to the possibility of creating synthetic genomes and organisms, and as such the field has its origins in metabolic and genetic engineering. However, the strength of synthetic biology has been the incorporation of a wide range of ideas from various disciplines including biological, chemical, and conventional engineering, systems biology, and protein design.
The field is united by an interdisciplinary, goal-driven approach that aims to both design and fabricate biological components and systems that do not already exist in the natural world, and to redesign and fabricate existing biological systems.
There are many diverse approaches being used under the umbrella of synthetic biology but they all have a few key points in common. First, these approaches are attempts to produce a system that is nonnatural but exhibits some aspect of natural behavior. Second, they derive from the engineering perspective that biology is modular. Modularity is reflected at many of the length scales at which natural systems are present, including individuals in a population, cells in a tissue, and proteins assembled into transcription machinery all the way down to the individual amino acids in a protein chain. It therefore makes sense to map out attempts to create synthetic biology with reference to the point in the natural hierarchy of structure at which alterations cause divergence from nature.2
The y-axis of the map in Fig. 1 represents the biomolecular and systems hierarchies in natural biology. Starting with basic building blocks—such as the nucleotides, amino acids, carbohydrates, and lipids; moving through oligonucleotides and polypeptides, which we term tectons; onto folded, assembled, and functional biomolecules—including nucleic acids, proteins and assemblies thereof—and lipid vesicles; and up to cells, in which these various components are brought together, encapsulated, organized, and orchestrated. The x-axis represents increasing diversity from nature: the idea being that if nonnatural amino acids are incorporated into a protein, the divergence from the natural system would increase as the mutation was passed up the hierarchy.
Fig. 1.
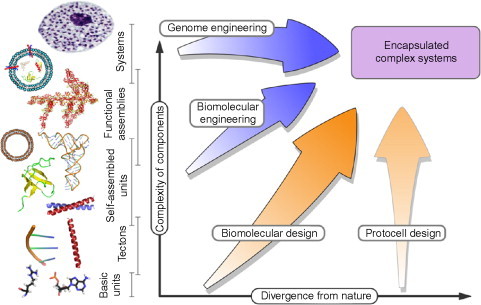
Diagram of synthetic biology space. The divergence of the design components from their natural equivalent is represented on the x-axis and the complexity of constituent components is represented on the y-axis. Several pathways are shown with labels that show the approximate areas of study that they encompass.
Reproduced with permission from Channon et al.3 copyright Elsevier (2008).
Several pathways are indicated in the diagram, which correspond to broad areas of synthetic biology. At the highest level of the hierarchy is the genome engineering approach, in which constructed genomes are inserted into host cells.4 This approach was successfully developed by Venter and colleagues at the J Craig Venter Institute using chemically synthesized fragments of DNA followed by in vivo recombination to produce full-length synthetic chromosomes. The synthetic chromosomes were injected into a host cell which had previously had all genetic material removed. The new DNA took control of the host cell's molecular machinery, causing them to produce progeny which contain only the synthetic chromosomes.5
The next pathway down, biomolecular engineering, uses the same concept of inserting new function into a host cell at the level of individual biomolecular components or pathways.6 A key example of work in this area is that of Keasling and colleagues at Berkley on the antimalarial drug artemisinin.7 In this study, a pathway that creates the drug precursor in plants has been transferred and adapted to function in yeast. The fast growing properties of yeast can then be exploited to produce the drug in far higher quantities at a much lower cost. The critical feature of this approach is that biological pathways are modular and can be cut, transposed, and added to new organisms with a minimal amount of interference with existing pathways. The BioBricks project aims to put this on a sound engineering footing by producing a catalog of genetic building blocks whose properties are well defined and can be plugged into new contexts.8
The next pathway is that of Biomolecular design, which will be discussed in more detail in the rest of this chapter. Essentially, it applies the same principle as above but at the length scale of individual biomolecules rather than whole pathways.
Finally, at the right of the diagram is the protocell engineering approach.9 The idea behind this work is to capture the defining features of natural cells in biomimetic systems: that is, it is (1) an encapsulated system, (2) blueprinted by some molecular-based store of information, and (3) harnesses energy from its environment and performs some form of metabolism. Ultimately, protocells might also have the ability to pass on their blueprint for the construction of successive generations.10., 11. Ideally, though bioinspired, none of these aspects would use natural biomolecules: that is, no DNA/RNA-based information stores or transfers; no carbohydrate-based or similar metabolism; no protein structures, binders, or catalysts; and though this appears to be a less stringent stipulation, no natural lipids as membrane components.
B. Biomolecular Design
Biomolecular designers take the view that stripped-down or de novo biomolecules provide useful modular units for building novel structure and/or function. There are two main types of biomolecule to work with, namely, DNA or proteins, and hence two building blocks that can be considered, namely, DNA bases or amino acids. In this chapter, only the use of proteins and amino acids is discussed; however, it should be noted at this point that there is a great deal of exciting work being done creating new structures and machines out of RNA, DNA, and DNA analogs.12., 13., 14., 15., 16., 17.
The process of protein design involves a cycle of observing natural proteins, deducing the rules that cause them to fold and function, and then using these rules to develop analogs of natural proteins that are amenable to manipulation.
It is possible to look for patterns of amino-acid sequences that give rise to structural motifs at all levels of the protein structural hierarchy; however, this chapter focuses on the α-helical secondary structural element and its onward association to quaternary structures known as “coiled coils.”
C. The α-Helix as a Naturally Occurring Tecton for Self-Assembly
Focusing on α-helices might seem restrictive but, for several reasons, α-helices make excellent tectons. First, they are easier to work with than β-strands as they are stabilized by local interactions. An isolated β-strand is generally not a stable chain configuration, as there are a great many “dangling” hydrogen bonds to be satisfied (see Fig. 2 ). To stabilize the strand, these bonds must be satisfied by another strand and/or other nearby secondary structural element. This property makes the β-sheet a nonlocal secondary structure as many nonadjacent areas of the peptide chain may be involved. These considerations mean that de novo design of all but the simplest β-sheet structures can require a predictive knowledge of the behavior of the entire peptide, something that is not currently possible. However, a single helix can be stable individually, with the hydrogen bond network formed between amino acids that are near to each other in the primary sequence. Specifically, the hydrogen bonds are formed between the carbonyl oxygen of residue i and the amide proton of residue i + 4.
Fig. 2.
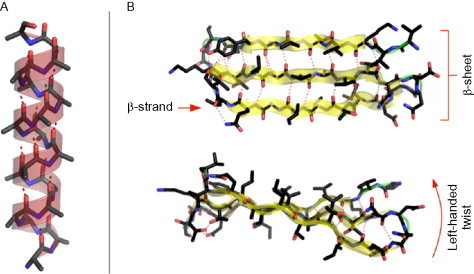
Cartoons of (A) an α-helix and (B) a β-sheet formed from three β-strands. Hydrogen bonds are shown in each structure by dashed lines. The α-helix contains only local backbone hydrogen bonds, between i and i + 4 positions. The β-sheet contains many nonlocal hydrogen bonds.
The α-helix is also relatively rigid, meaning that interactions at one end of the helix are fairly independent of those at the other end or, indeed, anywhere along the length.
Finally, the width of the helix and the length of each amino acid conspire to make a repeating pattern of side chains running up the outside of the helix. Specifically, the α-helix has 3.6 amino acids per turn, which leads to amino acid “i + 7” being almost directly above amino acid “i.” This is a very useful property, as it means that repeated sequences are arranged regularly in space. It provides the opportunity to use a repeated pattern of hydrophobic (h) and polar (p) residues to produce a helix that is amphipathic (that has one polar face and one hydrophobic face). To achieve this in the α-helix, a pattern of (hpphppp)n is used along the chain (see Fig. 3A).
Fig. 3.
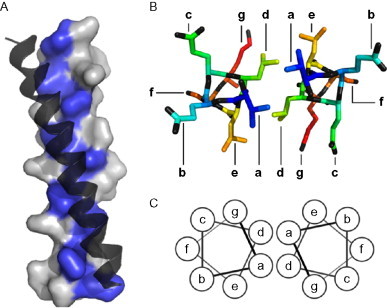
Hydrophobic core interactions in coiled coils. (A) Solvent-accessible surface of an α-helix in a coiled coil illustrating how hydrophobic a and d core residues (highlighted) are aligned into a “hydrophobic stripe” on one side of the helix. The second helix of the coiled coil is shown docked along this stripe, through contact with its own hydrophobic stripe. (B) A view of a single helical repeat (seven residues) of the coiled coil from above, showing the organization of the side chains. The a and d residues point into the core, interacting with their opposite number. (C) The helical wheel diagram used to diagrammatically represent the structure of a coiled coil.
There are many examples of such amphipathic helices being used as tectons for self-assembly in nature. However, perhaps the most directly applicable use is in the coiled coil. In this structure, the hydrophobic stripes of two (or more) helices come together and wrap around each other in order to bury the hydrophobic side chains. Specifically, the side chains in the hydrophobic core pack tightly in a regime known as “knobs into holes” (KIH) packing.18 The details of this packing determine the structure of the resulting coiled coil in terms of the orientation of the helices and the number of helices involved. Many different topologies of coiled coil are found in nature and these have been categorized in two searchable databases.19., 20. Some of the simpler coiled-coil structures are shown in Fig. 4 , although this shows only a small subset of the possible geometries.
Fig. 4.
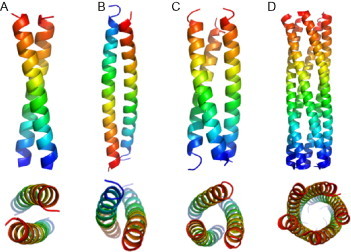
Some examples of coiled-coil structures shown from orthogonal views. (A) A parallel dimer from 2ZTA. (B) An antiparallel dimer from 1HF9. (C) A parallel trimer from 1BB1. (D) A parallel pentamer from 2GUV. Chains are colored from blue at the N-terminus to red at the C-terminus.
As one would expect for such an obvious natural tecton, coiled coils are ubiquitous as they are found in every compartment of plant cells and in all prokaryotic and eukaryotic cells.21 Within these various contexts, coiled coils provide a wide range of structures and functions. Shorter coiled coils offer molecular recognition, bringing together other proteins and hence functions in specifically defined combinations. Examples of biological function within the cell include their use as DNA transcription factors, binding to DNA to either repress or promote gene transcription, and the self-assembly of signaling complexes, including ion channels.22., 23. Molecular recognition by coiled coils is also used to fuse transported vesicles to their target membranes using, for example, proteins of the SNARE family.24
The rigidity of the coiled-coil structure allows the use of longer coiled coils as structural components. Often, these coiled coils have defined lengths set by the length of the sequences, as is the case with bacterial cell wall spacers.25 However, coiled coils also form components of fibrillar assemblies such as the intermediate filaments26 and spectrin which form two- and three-dimensional scaffolds that support the cell.27
Finally, coiled coils within the cell are dynamic, both in terms of their ability to responsively mediate other protein–protein interactions but also more directly in the form of motor proteins. The three main classes of cytoskeletal motor proteins, namely, kinesins, myosins, and dyneins, all contain long coiled-coil domains.23 The function of these coiled coils can be both structural—for example, controlling aspects of the stalks attaching cargo to the motor domains28—or functional—for example, in using rearrangements in the coiled-coil structure to achieve motive force.29
It is clear from the ways in which coiled coils are used in nature that they could provide an extremely useful tool for engineering biology. The following sections explore in more detail the knowledge that has been extracted from natural systems and how this has been used to begin designing new functional components.
II. Designing Discrete Helical Assemblies
In this section, the rules that link sequence to structure in coiled coils, which are the most ubiquitous helical self-assemblies in nature, are explored in more detail.
A. Lessons from Nature
The KIH packing found in coiled coils is a motif that can be searched for computationally in protein structures, using software such as socket. 30 The basis of the packing is that a hydrophobic “knob” residue on one helix slots into a hole formed in the center of a diamond of four “hole” residues displayed on an opposing helix (see Fig. 5 ). By locating structures matching specific criteria, one is able to extract detailed sequence to structure information.31 The repeating pattern of hydrophobic and polar amino acids (hpphppp)n can be examined in more detail and assigned the positional nomenclature abcdefg, with the hydrophobic residues occupying positions a and d. Further, it is often useful to visualize the heptad repeat by displaying it on a helical wheel as shown in Fig. 3C.
Fig. 5.
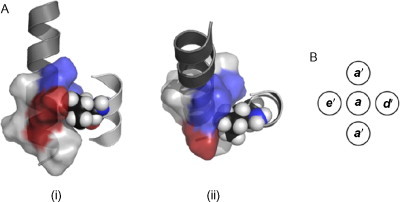
Knobs-into-holes (KIH) interactions. (A) A KIH interaction between two helices, seen from (i) the side and (ii) the top. The hole is shown as a solvent-accessible surface; blue indicates the surface of hydrophobic core residues, and red is a charged lysine in an e' position. The knob is donated by the right-hand helix. A Van der Waals representation of the knob is shown, and it can be seen that there is a very close steric interaction between the knob residue and the hole. (B) The hole comprises residues a', d', e', and a' (from the next helical repeat), corresponding to residues in positions i, i + 3, i + 4, and i + 7, and the knob is formed by residue a.
By examining the frequency with which various hydrophobic residues are used in these positions, it is found that, as well as being the main driving force for helix association, they control the oligomer state of the coiled coil. For example, a combination of isoleucine and asparagine at a and leucine at d favors dimer formation; using isoleucine at both a and d positions preferentially creates trimers; and using leucine at a positions and isoleucine at d positions favors the tetramer.32 Higher order oligomers are also promoted by the inclusion of extra hydrophobic amino acids flanking the core region. The two residues (positions e and g) on either side of the hydrophobic interface tend to be occupied by charged amino acids, allowing a range of ionic interactions both between the helices of the coiled coil and within each helix.33
Recently,34 it has been shown that these rules may not be generally applicable outside the context of the specific sequence originally examined (GCN4). One significant variation on these rules is caused by the existence of trigger sequences that are capable of specifying the oligomer state of the coiled coil.35., 36. These are small pieces of sequence that can fold independently into helical fragments before the oligomerization of the coiled coil occurs. It has recently been shown that, in the case of GCN4, the insertion of trigger sequences specifying oligomeric state may be more important than the use of individual specifying residues in determining the final structure.37 Overall, the current situation is that caution must still be applied when designed peptides are required to exhibit a specific oligomerization state, and that experimental verification of structure is prudent.
As well as influencing oligomer state, the details of the hydrophobic core packing can determine the helix orientation creating both parallel and antiparallel coiled coils. Recently, progress has been made in analyzing how sequences are related to the helix orientation in coiled coils, and prediction algorithms are improving.38 The thermodynamic preferences for various combinations of side chains packing into the core have been explored39 and the design of antiparallel coiled coils is an expanding field.40
The most frequently occurring coiled coils are dimeric, and many of these find function in the cell as transcription factors. In particular, the basic leucine zipper domain proteins (bZIP) are a large collection of parallel dimeric coiled coils. These proteins are of interest to the design process because they exhibit various levels of specificity: that is to say, many of the sequences bind preferentially to only a few partner sequences in the collection. This specificity is a key property necessary in making designed self-assembly that is modular and not promiscuous. An interactome for bZIP proteins has been mapped out by Keating and colleagues using a microscale protein array technique, in which the interactions of 49 human bZIP proteins and 10 from Saccharomyces cerevisiae were measured.41 This technique involves printing plates with each of the proteins under conditions in which they are expected to be monomeric, and then exposing the plates to fluorescently labeled analogs of each of the proteins. The resulting level of fluorescence retained on the plate is used to calculate the interaction strength (Fig. 6 ).
Fig. 6.
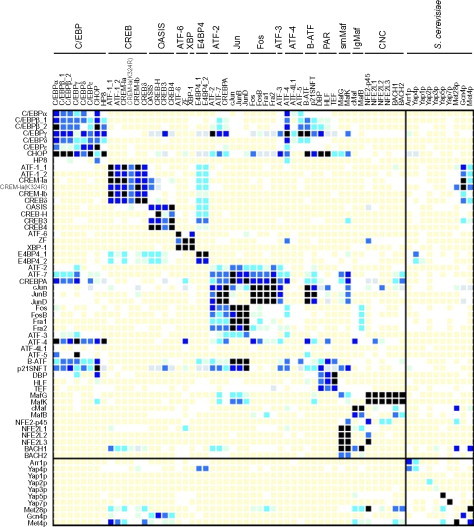
The experimentally determined interactome for human bZIP proteins taken from Newman and Keating41. The darker the squares are the more statistically significant the interaction. Dark squares on the diagonal indicate strong homodimeric interactions, and off-diagonal dark squares heterodimeric interactions (surface-bound proteins are shown on the left and fluorescently labeled probes are shown across the top).
Reproduced with permission from Newman and Keating41, copyright The American Association for the Advancement of Science (2003).
This study provides a wealth of information on how specificity of interaction is achieved, and the Keating group has gone on to produce a number of computer algorithms aimed at using this data to predict binding between bZIP proteins. These algorithms variously use combinations of electrostatic information, empirically determined weighting by sequence, and calculations of structural stability made from atomic resolution models.42., 43.
From these studies and others, it has further been deduced that specificity is achieved through three main methods. First, the use of asparagine at a positions (which also specifies for dimers) is used. The introduction of this polar side chain into the hydrophobic core of the coiled coil is destabilizing, however, this effect can be mitigated by partnering with another asparagine on an opposing helix. This provides a design rule: helices with asparagine will preferentially assemble with other helices possessing asparagine at the same point in the helix.
Second, the positions on either side of the hydrophobic core (e and g) tend to be occupied by charged amino acids. Complimentary pairs of charges can be used to favor specific helix pairing and noncomplementary charge pairs can be used to disfavor unwanted helix pairing.
Third, there is the possibility of using the size of the side chains to produce complementary fitting in the hydrophobic core. Large hydrophobic amino acids forming “knobs” can be accommodated by smaller “hole” residues on the opposing helix.
Many aspects of this statistically derived data have been confirmed experimentally by the group of Vinson using point mutations to a heterodimeric system derived from the PAR family member VBP B-ZIP domain.44 In a second more comprehensive study,45 10 pairs of coiled coils were made, with each pair having a different amino acid at the single mutated a position (I, V, L, N, A, K S, T, E, and R). The thermodynamic stability of all 100 combinations of peptides was then measured. The first conclusion to be drawn from this work is that the most stable homotypic interactions were for isoleucine, followed by valine, leucine, and asparagine. By looking at the coupling energies using double-mutant cycles, it was also deduced that the heterotypic interaction in which asparagine paired with isoleucine was the most repulsive, and that lysine and arginine paired with isoleucine, valine, or leucine, formed the most attractive set of heterotypic interactions.
Vinson's group also investigated the use of charged e and g positions within the same system, again using double-mutant cycles.46., 47. The amino acids alanine, lysine, arginine, glutamine, and glutamic acid were placed in e and g positions, and the stability of all the combinations of heterodimers was considered. The conclusions were that the interaction in which arginine at e paired with glutamic acid at g was the most stabilizing (with respect to alanine–alanine) and also had the highest coupling energy. This interaction was also found to be the least reduced by increased salt concentration, indicating that the charges may be partially buried. Other stabilizing interactions were lysine at e with glutamic acid at g, and the switched e for g versions of these interactions. As is expected, the like charged pairs are destabilizing, with glutamic acid paired with itself being the least stable, followed by arginine–arginine, lysine–arginine, and lysine–lysine pairs.
B. Designed Coiled Coils: Methods for Maximizing Specificity
The lessons learned in the previous section have been put to use. Many examples exist of coiled coils designed to be hetero, homo, dimer, and other oligomer states.
1. Exploiting Natural Specificity
Our first example of exploiting natural specificity is the work of Arndt's group in redesigning the transcription activator protein-1, which includes the coiled-coil heterodimer peptides Fos and Jun.48 This is an interesting target, as it is implicated in various cancers where it can become upregulated or overexpressed. The idea is that a synthetic peptide that could outcompete either half of this interaction would be useful as a drug. In this work, the technique of protein-fragment complementation assays combined with growth competition was used to produce optimized binding partners for both the wild-type Fos and Jun (see Fig. 7 ). The library used in the assay was constructed semirationally using information from the families of Fos and Jun and knowledge of coiled-coil interactions. The winning peptides from the library had higher melting temperatures when mixed with their wild-type partners than the natural interaction, as well as an even tighter heterodimeric interaction with each other.
Fig. 7.
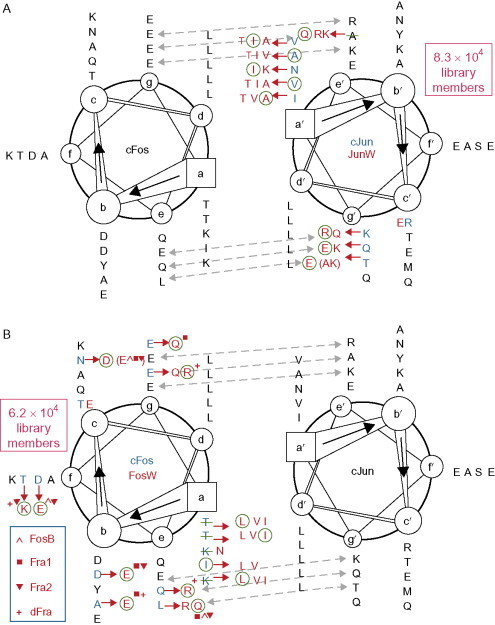
Sequences for wild-type Fos and Jun are shown in black only. Amino acids in red indicate those used in the libraries for the mutant Fos and Jun. (Where the wild-type residue is not present in the library it is struck through in green.) The residues used in the winning peptides are circled in green. Dotted blue lines indicate potentials salt bridges.
Reproduced with permission from Mason et al.48 copyright National Academy of Sciences, USA (2006).
The two winning peptides along with the wild-type peptides and five other related peptides were screened for the melting temperatures of each of the homo or hetero dimeric combinations. The data from this were used to create an algorithm that predicts the melting temperature of coiled-coil dimers (see www.molbiotech.uni-freiburg.de/bCIPA). This algorithm uses empirically derived weights based on helical propensity, electrostatics, and packing of the hydrophobic core.
The designing of synthetic partners for bZIP proteins has also been explored by the Keating group. They have developed an algorithm called CLASSY, which uses a cluster expansion method to convert their structure-based interaction model into a sequence-based scoring function that is very fast to evaluate.49 The algorithm begins by finding the sequence with the maximum interaction score with the target sequence. A value is then set for the difference between this interaction and the most favorable of the interactions with a set of competitor sequences (see Fig. 8 ). The introduction of competitor sequences is a key concept in protein design, the idea being that it is not enough to simply be stable in the desired fold but that other possible competing folds must be destabilized.
Fig. 8.
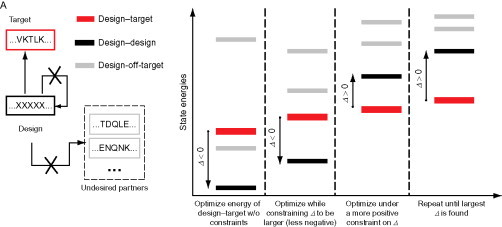
Schematic indicating the operation of the CLASSY algorithm. The design–target interaction is desired, with the design–design and design–off-target interactions being disfavored. Initially, the design–target interaction energy is minimized, and then the energy of the competing interactions is raised until they are higher than that of the design–target interaction. This energy gap is then maximized.
Reproduced with permission from Grigoryan et al.49 copyright Nature Publishing Group (2009).
Once a low-energy sequence is found, it is then mutated until the difference (the specificity) between the energy of the target and of the competitors is maximized. This study demonstrates most vividly the trade-off between stability and specificity of interactions.
Experimentally, 48 peptides designed against 20 natural targets were tested for interaction with 33 representative human bZIP coiled coils and for self-association. Within in this dataset, many designs were found to outcompete the native partners for the targets, and furthermore, several of the designs also exhibited their strongest interaction with their target.
From this work, it was also found that while the designed peptides were somewhat limited in the sequence space they covered: they produced many new interaction profiles indicating that the original bZIP families have only explored a small part of the possible interaction space.
2. Developing a Coiled-Coil Tool Box
With the information required to design sequences that will specifically interact with their target partners in hand, it is possible to address the problem of developing a tool box of synthetic coiled coils. Once such a set of coiled coils exists, their orthogonal interaction profiles can be used to generate more complex systems in which many more self-assembling components can be mixed at will.
Toward this goal, the Woolfson group has used a computer algorithm to find the maximum specificity that can be generated given a set of amino acid choices for positions a, e, and g. 50 This system was limited to coiled coils of only three heptads in length but nevertheless generated a set of six peptides that associated into the three targeted coiled coils preferentially out of all of the 21 possible homo and hetero dimeric possibilities (see Fig. 9 ). The success of this system was tested in two ways. The peptides were all labeled with terminal cysteine residues so that the coiled coils formed in solution could be trapped and examined. First, each of the designed coiled coils was checked for folding preferentially as a parallel dimer, using a combination of analytical ultracentrifugation (under reducing conditions designed to allow nondimeric species to form if desired) and comparing thermal denaturation under both oxidizing and reducing conditions. Second, the mixture of all six peptides was incubated under conditions where exchange of terminal disulfide bonds could occur. The reaction was then quenched, and mass spectrometry was used to identify that only the three desired coiled coils had formed.
Fig. 9.
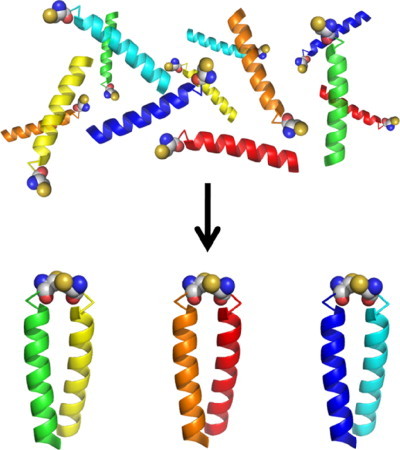
Schematic showing how a mixture of six thiol-labeled peptides assembles into just 3 of the 21 possible disulfide-linked dimers under redox-buffered conditions.
This approach of pulling out selective sets of coiled coils has recently been generalized by Keating's group in a study in which they measured the interactions of 48 designed peptides and 7 natural bZIP proteins with no strong homodimerizing properties.51 Within this dataset, 27 different hetero-specific pairings were found using 26 different peptides, with each peptides being involved in upto 7 different dimers. From this information, 10 different types of subnetworks of interactions were found including orthogonal pairs, orthogonal triplets (as in the previous system), and more complicated hub-type networks in which one peptide interacts with many. The behavior of two sets of four peptides predicted by the interactome to form orthogonal pair systems was experimentally demonstrated as matching the prediction.
One interesting observation from this work was that all of the networks found were sparsely connected, and it was hypothesized that this is due to the peptides having either been designed or selected for a lack of homospecificity. This may, in turn, lead to the desirable outcome that the peptides lack promiscuity, making them ideal for using in synthetic biological applications (Fig. 10 ).
Fig. 10.
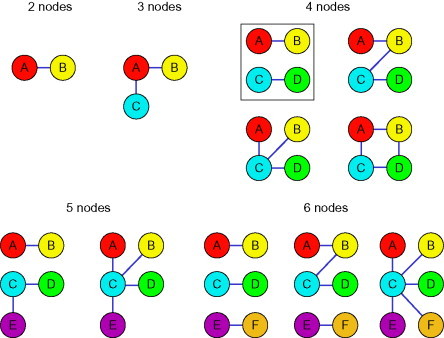
Diagram indicating the subnetworks of interactions found within the combined design and natural bZIP protein interactome. Lines indicate interactions, and all peptides interacting as homodimers were excluded from the analysis. The boxed network corresponds to the system chosen for further characterization.
Adapted with permission from Reinke et al.51 copyright American Chemical Society (2010).
3. Nonnative Amino Acids
One way to extend the specificity available to designers is to incorporate nonnative amino acids. These are simply amino acids with side chains not found in nature, and it is often the case that the interactions between natural and synthetic amino acids show a significant level of specificity. Synthetic amino acids can be incorporated during peptide synthesis and, indeed, commonly used ones may be obtained with all of the protecting groups necessary for routine synthesis.
Several examples of this have come from the Kennan group who have used a variety of nonnatural amino acids to influence coiled-coil assembly (see Fig. 11 ). First, the effect of side-chain length in derivatives of glutamic acid and lysine used at e and g positions was investigated.54 In this study, the number of methylene units separating the amine and carboxylic acid groups from the backbone was varied from 1 to 4. The general trend was a dramatic increase in stability as the total number of methylene units increased.
Fig. 11.
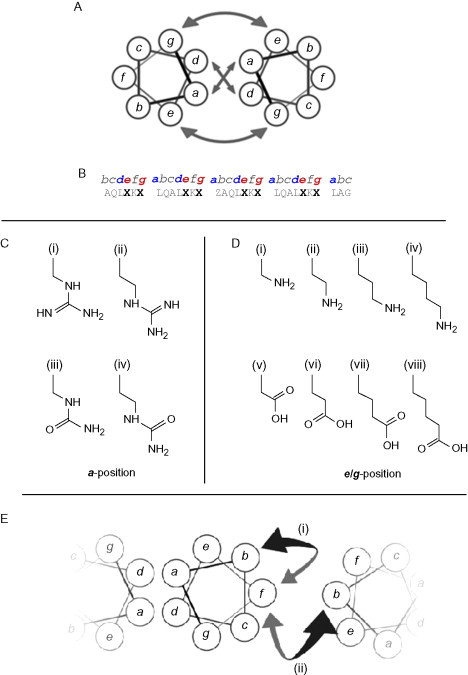
Coiled-coil designs that incorporate nonnative amino acids. (A) Helical wheel illustrating the main interactions available to designers of coiled-coil structures; the core and flanking salt-bridge interactions. (B) A “typical” base sequence for the investigation of nonnative amino-acid interactions in coiled coils.52., 53., 54. Residues in positions marked Z were substituted with amino acids with side chains as shown in C, while those marked X have been substituted with amino acids with side chains shown in D. (C) Guanidinium- and urea-based side chains with differing lengths, used to investigate core interactions. (i) guanidinylated diaminopropionic acid, (ii) guanidinylated diaminobutyric acid, (iii) pUr, a urea-terminated side chain, and (iv) pUr*, a urea-terminated side chain with an additional methylene group in the “linking” region to those in pUr. (D) Amino- and carboxyl-based side chains used to probe salt-bridge interactions. (i–iv) Positively charged, amine-terminated side chains with increasing length (and thus hydrophobic contact area): (iv) forms the naturally occurring lysine. (v–viii) Negatively charged, carboxyl-terminated side chains with increasing length. (vi) and (vii) form the natural amino-acids aspartic acid and glutamic acid, respectively. (E) Intra- and interhelical cation–π interactions investigated using the nonnative amino-acid norleucine.55
The second example from this group involved using guanidinium-functionalized side chains at a positions.52 Again, the effect of chain length was investigated using arginine derivatives with one or two methylene groups removed and looking at interactions with asparagine, glutamic acid, and aspartic acid. The conclusions were that shorter chains in this core position confer more specificity. The combination of positive interactions between aspartic acid and the guanidinium side chains, combined with the negative interaction of asparagine with aspartic acid, allowed the creation of two dimer pairs that assembled with no cross-reactions. This set of coiled coils was extended to three (Section II.B.2) by the addition of peptides with a positions containing urea-functionalized side chains, specifically citrulline.53
Finally from this group, the nonnatural amino acids discussed above were explored in the context of a trimer-forming system.56 In this work, the trimeric nature of the interactions was favored by the use of isoleucine at all a and d positions other than a single mutated a position. It was found that while urea-based side chains allowed trimer formation, guanidinium-based side chains did not—thus providing another rule in the protein design toolbox.
A final example of the utility of nonnative amino acids is the use of fluorinated side chains. Fluorine is not generally found in natural systems because of a lack of bioavailability; however, it has the property that it is immiscible with both water and most organic solvents. This makes it a target for incorporation into biological molecules, as it can provide another orthogonal interaction for increasing the complexity of designed systems.57
To demonstrate the utility of fluorinated side chains, the group of Kumar (and others) has used trifluoroleucine and trifluorovaline substitutions in the a and d positions of the leucine zipper GCN4.58 They found that the substituted peptide formed a dimeric coiled coil which had a higher melting temperature than the original wild-type GCN4. Following on from this, they produced two variants of this system, one with an all leucine core and the other with an all hexafluoroleucine core.59 These two peptides were shown to be mutually exclusive in their self-assembly, with only homodimers forming from mixed solutions. This is attributed to the vastly higher melting temperature of the hexafluoroleucine homodimers.
More recent work in this area by the Koksch group has focused on heterodimeric systems in which the fluorous side chains are partnered with natural amino acids. From their studies, it is found that in this environment fluorous chains pair best with the same hydrophobic amino acids normally found in the core.60 A second study looking at a variety of fluorous side chains in the core a and d positions has found that, similar to the natural hydrophobic amino acids, the specific geometry of the side-chain packing is important in determining stability.61 This suggests that fluorinated side chains may also be capable of specifying helix orientation and the coiled-coil oligomeric state.
C. Applications of Designs
So far, the rules of self-assembly of coiled-coil tectons have been explored. Next, some selected applications in protein design are examined, which are by no means an exhaustive list but should give a flavor of the possibilities.
1. More Complex Discrete Assemblies
The first application examined is simply the building of more complex architectures, involving peptides with more than one coiled-coil domain and hence more than one coiled-coil interaction.
One of the first examples of this was the “belt and braces” system from the Woolfson lab, where a “belt” peptide, six heptads in length, is paired with two “brace” peptides, each three heptads long.62 Specificity was achieved by having the two brace peptides carry only positively charged e and g positions and the belt carrying only negatively charged e and g positions. The two brace peptides were distinguished by one of them possessing an asparagine at a that matched with an asparagine at a in one half of the belt. The external ends of the peptide braces were functionalized with cysteine to enable interactions with gold. The success of the design was demonstrated by a combination of biophysical techniques and by incubation with gold nanoparticles (Fig. 12 ). In the absence of the belt peptide, the gold particles are not seen to assemble by transmission electron microscopy (TEM); however, once the belt is added, the nanoparticles collect together in groups spaced by the expected 6-nm length of the belt peptide.
Fig. 12.

(A) Schematic showing the “belt and braces” principle: dark and light indicate the presence or absence of the specifying asparagine residues. (B) The sequences for the belt and braces. (C) TEM image of resulting gold nanoparticle arrays. Reproduced with permission from Ryadnov et al.62 copyright American Chemical Society (2003).
This concept was extended by the Woolfson group using the set of six helices discussed previously.50 In this case, two sets of two peptides were linked together using flexible glycine residues to create two different belt peptides. By combining the two belt peptides, which assembled in an offset manner, with the remaining two of the six peptides, a 9-nm long construct with four components was formed. Again, this assembly was demonstrated through biophysical techniques. While this increase in the complexity of the constructs seems modest, it opens the way for more complicated nanostructures to be designed in the future.
2. Vesicle Fusion
One recent application of coiled-coil design is the production of a system capable of fusing liposomes.63 The system is based on a simplified model of the action of SNARE proteins, which are responsible for fusing vesicles in the cell (see Fig. 13 ). In nature, three types of SNARE proteins come together to form a coiled coil. One type of SNARE is associated with a transport vesicle; the second is associated with the destination membrane; and the third is present in solution. Once the three types are in proximity with each other, a stable tetrameric coiled-coil forms which colocalizes the membranes, allowing fusion to proceed.
Fig. 13.
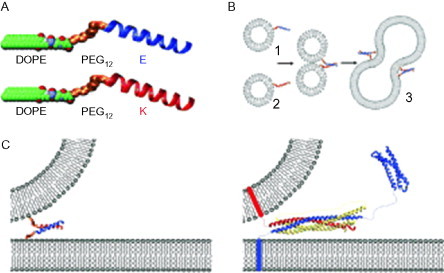
Liposomes labeled with one of two interacting peptides fuse on mixing. (A) The two peptides are linked to DOPE via a PEG linker. (B) The two peptides associate to form a coiled coil which colocalizes the liposomes leading to fusion. (C) Comparison between the reduced SNARE model on the left and the natural process on the right.
Reproduced with permission from Robson Marsden et al.63 copyright Wiley-VCH Verlag GmbH & Co. KGaA (2009).
The designed system uses just two types of peptides, each one anchored to a separate liposome via a PEG linker attached to a lipid domain (DOPE). In this system, a dimeric coiled coil is formed between the two peptides that serves the purpose of colocalizing the membranes to promote fusion. The success of this design has been shown by fusing liposomes that contain one each of a forster resonance energy transfer (FRET) pair of fluorophores. As the contents of the vesicles mix, the FRET becomes measurable, indicating the fusion of oppositely labeled liposomes. Conversely, liposomes labeled with the same peptide do not fuse.
3. Self-Replicating Peptides
Designed coiled coils have also been used to template peptide bond formation, creating a self-replicating system. Ghadiri's group developed a system based on the leucine zipper GCN4 in which the full-length peptide assembles two peptide fragments one of which has been preactivated as a thiobenzyl ester and the other terminated with cysteine.64., 65.
Once these two chemical functionalities are colocalized, they react to produce a peptide bond between the two fragments, making a copy of the full-length peptide (see Fig. 14 ).
Fig. 14.
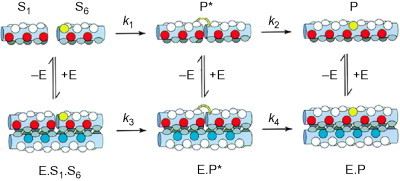
Schematic diagram of the uncatalyzed and catalyzed routes to ligation for a self-replicating peptide. The condensation reaction of the substrate molecules S1 and S6 is greatly facilitated by their colocalization by the catalyst E (rate k3). Residues critical for molecular recognition and ligation are shown colored (red, glutamic acid; blue, lysine and arginine; yellow, cysteine; gray, leucine and valine).
Reproduced with permission from Severin et al.66 copyright Nature Publishing Group (1997).
In this system, a dimeric coiled coil is formed between three components, that is, a full-length peptide and two peptide fragments corresponding to the N- and C-terminal halves of the full-length peptide. Once the peptide bond has formed, the full-length peptide must dissociate in order to leave the catalyst free for the next cycle. The concentration of the full-length peptide has been shown to increase exponentially as expected for this self-catalyzed reaction. This work was extended to using a heterodimeric coiled coil via manipulation of the charged residues at e and g positions.66
The group of Chmielewski has added an interesting design principle to their self-replicating peptide: specifically, the addition of a proline residue close to the ligation site.67 Once ligation has occurred, this proline kinks the resulting full-length helix, destabilizing the coiled coil and leading to an increase in the efficiency of self-replication.
D. Switching and Dynamic Coiled-Coil Systems
As was stated in the introduction, coiled coils are involved in many dynamic processes in the cell and, in many cases, this results from structural switching either between folded and unfolded (in the case of signaling pathways) or more subtly in the case of motor proteins. In this section, some of the ways in which coiled-coil systems have had structural duality introduced is discussed, to bring to bear the function of switching.
1. Solvent Conditions
To begin with, coiled-coil systems that respond to changes in the bulk solvent condition including temperature, pH, and redox potential are discussed.
Some examples for temperature-based switches are peptides that switch from coiled-coil folds to β-sheet-rich folds on heating. Woolfson and colleagues have found that the addition of β-strand favoring threonine at the surface-exposed f position of a dimeric coiled-coil-forming peptide (see Fig. 15 ) produces a switch to β-structure at elevated temperatures.68 This switch is associated with the formation of amyloid fibers and is not therefore reversible. Kammerer also produced a series of peptides that were coiled coils at room temperature but switched to amyloid structures at high temperatures.69 It was found that the coiled-coil structure can tolerate well the addition of several β-strand favoring residues without significant loss of stability.
Fig. 15.
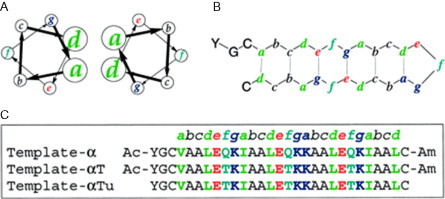
Schematic of design principles for peptides that can adopt both coiled-coil and β-sheet structures. (A) Sequences aligned against the coiled-coil heptad repeat. (B) Sequences aligned against the β-sheet structure.
Proposed structure from Ciani et al.68 (left) and Kammerer et al.69 (right).
An example of a pH-driven switch comes from the Kennan lab in which a heterotrimeric system is employed.70 The assembly of the three different peptides in the trimer is driven by the use of cyclohexylalanine side chains at a positions. At each core a layer, one of the helices supplies a cyclohexylalanine which the other two complement with alanine. Each peptide has either all lysine or all glutamic acid at e and g positions, meaning that one of the e/g interfaces is forced to be mismatched (see Fig. 16 ). At high pH, the system is more tolerant of an all-lysine interface, whereas at low pH, an all glutamic acid interface is preferred. The pH switch was created in the form of a competition between two peptides with the same core pattern but with e and g positions with either all lysine or all glutamic acid. These peptides were shown to exchange with each other when the pH was cycled.
Fig. 16.
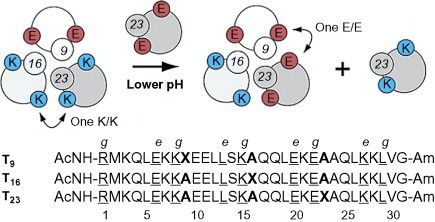
pH-triggered helix exchange. At higher pH, the mismatched lysine interface is favorable and so T23K is incorporated. At lower pH, the mismatched glutamic acid interface becomes favorable and helix exchange occurs. Heptad repeat positions e and g shown only.
Reproduced with permission from Schnarr and Kennan70, copyright American Chemical Society (2003).
This work was extended to apply to a system that switched helices and orientation, between a parallel and antiparallel heterotrimer.71
As an example of a redox-based switch, the Woolfson group produced a system that switched between a monomeric helix–turn–helix hairpin and a dimeric coiled coil.72 The interactions in the monomeric form are those of an antiparallel coiled coil and can be made compatible with the dimeric form by switching the charge pattern at e and g half way along the helix. For the turn region of the monomeric form, a series of sequences were tested, from the flexible to the more rigid. The monomeric form is held together by an intramolecular disulfide bond between the N- and C-termini, and switching to the dimeric form is accomplished by reducing this bond. Although the various design iterations exhibited different stabilities in the two states, the dimeric unstrained state always took precedence once the disulfide bond was reduced, making the switch irreversible.
2. Metal-Driven Switching
In this section, two types of metal-driven switching are discussed: first, switches in which metal binding produces the folded state from the unfolded, and second, reversible switches in which the metal-bound form is different to the unbound but also folded.
As an example of the first type of switch, Tanaka's lab produced a parallel trimeric coiled coil which was adapted to contain metal binding.73 In this design, two core positions were mutated from isoleucine to histidine. This destabilizes the coiled coil until Co(II), Ni(II), or Zn(II) is added, at which point the trimeric coiled-coil forms.
Moving on to the second type of switch, the Ogawa lab has produced a designed dimeric coiled coil which incorporates nonnative metal-binding 4-pyridylalanine side chains in solvent-exposed positions.74 On the addition of a platinum center, this structure rearranges to form a four-helix bundle, and hence this system constitutes an oligomeric state switch.
As a more dramatic structural switch, two groups (Woolfson and Kuhlman) have developed peptides that switch from trimeric coiled coils to a zinc-binding motif known as the “zinc finger” on addition of zinc.75., 76. Although the two states of this switch are similar, the two studies differ in both the approach taken to design and in the details of how the two functionalities are overlaid in the peptide sequence. The Woolfson group switch uses an HX2HX12HX5H zinc-binding motif aligned with the heptad repeat such that none of the core residues of the coiled coil is affected. The sequence was designed manually using knowledge of both structural motifs. In contrast, the Kuhlman sequence uses the CX2CX12HX3H zinc-binding motif in an alignment, which puts the second cysteine residue at a d position in the core of the coiled coil (see Fig. 17 ). This sequence was designed using the rosettadesign program,77 which in this case was used to thread target sequences onto the backbone structures for the two desired states simultaneously. The algorithm then mutates selected side chains to find a minimum-energy sequence for both states. Both these designed sequences produce a structural switch on the addition and removal of zinc.
Fig. 17.
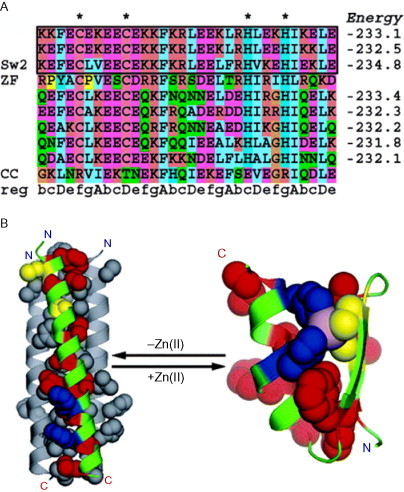
Computational design of a zinc-driven switch. (A) A sequence alignment of the lowest energy sequences from the design process with a zinc finger sequence (residues 3–33 of Zif268) and a coiled-coil sequence (residues 13–44 of hemagglutinin) for comparison. The heptad repeat is also shown along with stars, indicating the zinc-binding residues. (B) The backbone structure of Sw2 (the sequence of the successfully produced switch) threaded onto the two target structures. Hydrophobic residues are colored red, cysteine ligands are colored yellow, histidine ligands are colored blue, and the Zn(II) ion is represented as an exaggerated sphere colored in violet.
Reproduced with permission from Ambroggio and Kuhlman76, copyright American Chemical Society (2006).
As a final example of metal-driven switching, the Koksch lab has produced a metal-driven α- to β-structural switch.78 A peptide has been designed that shares coiled-coil and β-sheet characteristics, which is found to be β-structured under aqueous conditions but will switch to helical with the addition of 40% helix-promoting trifluoroethanol (TFE). A mutation of this peptide, which contains a number of histidine residues, behaves similarly in water and TFE but will switch back to being β-structured on the addition of Zn or Cu. This switch is reversible on the addition of EDTA.
3. Light-Sensitive Switching
Switching using light is desirable because of the difficulty of rapidly changing other solution conditions such as pH or metal ion concentration. One illustration of a possible route to light-controlled conformational switching is the work of Woolley's group using the dye azobenzene as an activating switch within the transcription activator protein-1 system.79 In this work, an azobenzene linker is used to connect two consecutive f positions in a coiled-coil peptide. While the end-to-end distance of the azobenzene linker is compatible with an α-helical fold in the cis form, it is incompatible in the trans form. Switching from the cis to the trans form is achieved through irradiation with light at 460 nm, while switching from the trans back to the cis form requires light at 365 nm.
In order to produce a test system, the linker has been inserted into wFos, the winning peptide partner to wild-type Jun discovered in the work described in Section II.B.1. The idea is that, while the linker is in the trans form, wFos will be unavailable for binding and the native interaction of Fos–Jun will prevail. Once the cis form of the linker is made, wFos will be available to outcompete wild-type Fos and the cellular function of the Fos–Jun dimer will be disrupted. The success of this system was demonstrated via control of DNA-binding activity in cell treated with the modified wFos (Fig. 18 ).
Fig. 18.
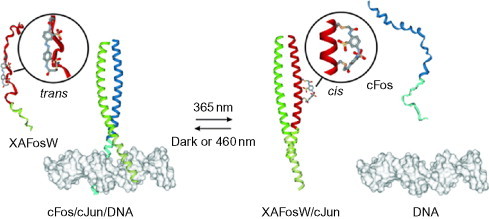
Schematic showing how switching of the azobenzene from trans to cis allows the synthetic peptide to outcompete the natural binding partner of Jun. This in turn prevents binding to DNA.
Reproduced with permission from Zhang et al.79 copyright Wiley-VCH Verlag GmbH & Co. KGaA (2010).
III. Designing Higher-Order Helical Assemblies
Having discussed the way in which coiled coils have been exploited to create new discrete self-assembling systems, the next stage is to look at coiled coils in designed fiber and higher dimensionality assemblies.
A. Lessons from Nature
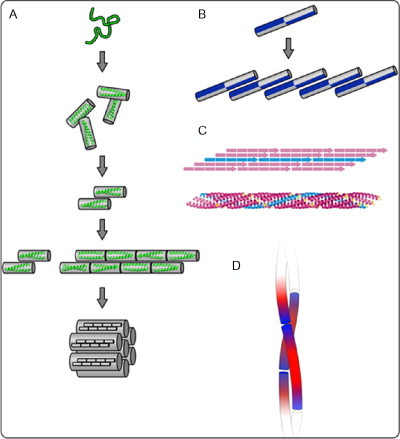
There are many different fibers found in cells performing various functions using a wide variety of structures; however, there are common themes connecting them. Fibers in nature tend to be nucleated; in other words, several monomers must be assembled before a stable growing fiber is formed. This gives the cell spatial and temporal control over the location of fiber growth by disfavoring the spontaneous formation of growing small oligomers in the bulk. Natural fibers are uniform in their width (and sometimes length): that is to say, all fibers formed from the same protein have approximately the same dimensions. This means that physical parameters such as flexibility, persistence length, probability of breakage, and the number of monomers per unit length are controlled. This enables the cell to produce fibers that are fit for purpose and are efficient in their use of protein. Natural fibers are dynamic: they can be switched from assembly to dissociation by changes in chemical potential. Many fibers achieve this property in an active fashion using adenosine triphosphate (ATP) hydrolysis as a method to manipulate the energy landscape; however, others are passively responsive to solvent conditions. Exchange of subunits can usually occur at the free ends of the fiber but can also occur along the length of the fiber. Finally, natural fibers are functional—they possess binding sites on their ends and surfaces that are used to recruit other protein machinery including protein motors.
Examples of naturally occurring coiled-coil fibers are the intermediate filaments, which are a broad class of related proteins containing a rod domain formed from a parallel dimeric coiled coil. The intermediate filaments form one of the primary elements determining the shape of both the nucleus and the cell in general. They have an extraordinarily hierarchical assembly pathway which results in fibers of a determined width and whose mechanical properties allow large deformations.26
The assembly of vimentin is a specific, well-studied example of an intermediate filament,80 in which the first step is association of the parallel dimeric coiled coil (see Fig. 19 ). This leaves non-coiled-coil head and tail domains at either end, which are responsible for the onward assembly of two of these dimers to form a tetrameric species which is antiparallel and sticky-ended (meaning that, while there is a substantial overlap between the dimers, a large fraction of the coiled-coil domains are hanging over either end). Two of these tetramers assemble (via interactions between rod domains) to form an octomer, which can be trapped and examined under specific pH and ionic strength constraints. These octomers will further assemble to form an asymmetric 32-subunit-containing species,81 which represents the unit-length building block of the filaments. Until this species is formed, elongation of the filament is disfavored. Once the elongation has progressed substantially, one final structural rearrangement occurs in which the fiber condenses to a more compact width. The final mature fibers exhibit exchange both at free ends and also in coiled-coil units along the length of the fiber.
Fig. 19.

The structural hierarchy of unit-length vimentin. (A, B) The tetramer state, or antiparallel dimer of parallel dimeric coiled coils. (C, D) The dimer of these tetramers. (E, F) The asymmetric octomer of tetramers.
Reproduced with permission from Parry et al.80 copyright Elsevier (2007).
As such, this example fiber displays nucleation, control of width, and hence mechanical properties and dynamic exchange of subunits along its length. It also displays onward binding motifs for interactions with, amongst other things, cell adhesion proteins.
B. Designed Fiber Assemblies
Armed with a combination of natural examples of fiber assemblies and rules for the design of coiled coils, it is possible to begin to build self-assembling systems that mimic natural fibers.
1. Homotypic Coiled-Coil-Based Systems
The earliest example of a designed coiled-coil fiber was a three-heptad-long homotypic system using the peptide sequence LETLAKA corresponding to heptad positions abcdefg. 82 The use of leucine at a and d positions and alanine at e and g makes this a rather generic coiled-coil sequence in which the oligomer state is not designed in. Indeed, this peptide forms tetrameric bundles under some conditions, but also assemblies into micrometer-long fibrils with widths of 5–10 nm. The morphology of the fibers is found to be dependent on the ionic strength.
The next system to be developed used a similar design principle with just under five-heptad repeats of the sequence QQLAREL at bcdefga.83 This system was designed to assemble into a pentamer initially and then to undergo longitudinal assembly based on the predicted slippage this geometry induces providing staggered ends. This system produced fibrils at pH 6 and below, although this range was later extended by reducing the charge per heptad by replacing glutamic acid with glutamine or serine.
In this design trend, the Hartgerink group has also produced fibers from short, blunt-ended coiled-coil-forming peptides.84 This system has been explored in detail, with control of fiber width being demonstrated via the choice of amino acids used at b, c, and f positions, with higher charge resulting in thinner fibers. This study proposes a general mechanism for this type of assembly in which bundles of helices form first, and then slippage of the alignment allows longitudinal assembly, usually only at higher peptide concentrations (see Fig. 20 ).
Fig. 20.
A variety of possible coiled-coil fiber assembly schemes. (A) Blunt-ended coiled-coil assembly, as demonstrated by Dong et al.84 (B) Coiled-coil assembly using a “staggered” hydrophobic core.85 (C) A five-heptad coiled-coil system with a single heptad overlap.83 (D) A palindromic charge pattern that produces sticky ends and allows fibrilization.55
Both the above systems rely on the slippage between the largely nonspecific hydrophobic cores. It is, however, also possible to promote a staggered assembly (and disfavor the entropically favorable blunt-ended configuration) using the design rules discussed earlier. Namely, this can be done by the insertion of strategically located polar residues into the hydrophobic core and manipulating the charged e and g positions. This was demonstrated in a homotypic system by the Conticello group with a sequence that comprised six heptads.86 Specifying asparagine residues in the third and sixth heptad promote either the blunt-ended dimer or the three-heptad overlapped sticky-ended dimer, while all negative e and g positions in the first three heptads and all positive e and g positions in the last three heptads promote only the sticky-ended dimer. This system indeed forms the α-helical structure which appears as long fibers under TEM.
Another, more recent, implementation of this type is the magic wand system from the Woolfson lab.55 This system demonstrated that the use of charge patterning at e and g positions was sufficient to produce sticky-ended assembly. By removing the polar core residue, this system was able to achieve fibrillogenesis from a shorter four-heptad peptide, with an increase in stability with respect to the previous example. This study also investigated a series of mutations to external coiled-coil positions, and began to highlight the importance of electrostatics and cation–pi interactions as the source of thickening in fiber systems.
A different way to force staggered ends is to break the hydrophobic core. This technique was demonstrated by the Fairman group using a peptide based on the GCN4 leucine zipper but with two key alanine residues inserted into the middle of the sequence.87 This has the effect of locally breaking the heptad repeat and causing the hydrophobic seam of the peptide to be displayed on opposite faces at either end of the helix. This discontinuity of the hydrophobic interface prevents the blunt-ended dimer from forming, and indeed fibers are formed instead. Once again, the morphology of these fibers is dependent on salt concentration.
2. Heterotypic Coiled-Coil-Based Systems
Taking one step further forward in complexity, there are fiber systems made from heterotypic coiled-coil designs. These have the primary advantage over single-peptide systems of being able to control the onset of fibrillogenesis—fibers will only form once the components are mixed.
One of the best studied heterodimeric systems is the SAF system that came out of the Woolfson lab. This system consisted originally of two peptides, each four heptads in length. The design utilizes offset asparagine side chains at a positions and charge patterning to promote staggered heterodimeric assembly. The original peptide design was found to be helical in structure and to produce long, thickened fibers.88
In later work, the stability of the fibers was improved by the inclusion of oppositely charged side chains in surface-exposed positions in the coiled coil.89 For this second iteration of the system, the helices were found to be packed in a helical array with the helix axis parallel to the fiber axis,90 and a mechanism for fiber formation was determined using a combination of kinetic experiments using TEM, circular dichroism (CD), linear dichroism (LD), and nuclear magnetic resonance (NMR).91 The mechanism was found to involve an initial folding in which small oligomers are formed, which are approximately half helical, followed by a nucleation step and by simultaneous thickening and elongation. By adding preformed fiber fragments to fresh peptide solutions (seeding), it was shown that at later times elongation dominates. This method was also used to demonstrate control over the length distribution of the fibers, with high seeding density leading to shorter overall fiber lengths (see Fig. 21 ).
Fig. 21.
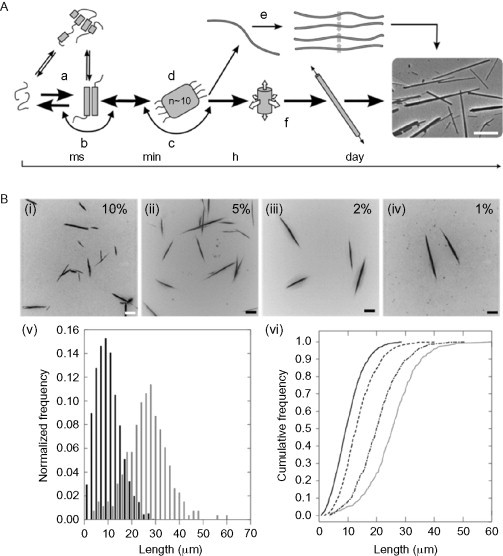
Summary of the model proposed in Bromley et al.50 for the assembly of SAF-type peptide fibers. (A) The proposed assembly mechanism proceeds from an initially unfolded state (a), through dimerization (b), and nucleation (d) steps to form a species that first grows in three dimensions (f) before ending with a species that grows by extension only. Several other possible paths are also shown, but these are found to be not present in this system. (B) The model predicts that control of fiber length should be possible through “seeding”. Figures (i–iv) show representative fluorescence micrographs of SAF samples seeded with varying concentration of seeds. (v) Length distribution of fibers in samples seeded with 10% (black bars) and 1% (gray bars) preformed fibers. (vi) Cumulative length distributions for the four samples imaged, 10% (solid, black), 5% (dashed, black), 2% (dash-dot, black), and 1% (solid, gray).
Reproduced with permission from Bromley et al.50 copyright Elsevier (2009).
A later generation of the SAF system has been made in which the exposed b, c, and f positions have been altered to be alanine and glutamine. These systems produce thinner, more flexible fibers that form system-spanning networks in the form of hydrogels.92 In particular, the alanine-based system has been shown to form a robust gel capable of supporting mammalian cell growth.
C. More Complex and Higher Dimensionality Assemblies
1. Helix–Turn–Helix Motif
A fiber system based on a helix–turn–helix motif forms the first example of a more complex system. The added complexity of the turn region is of interest, especially as the turn regions are notoriously difficult to understand and design. Lazar et al. produced a peptide that comprises two 18-amino-acid long helical segments joined by four different turn regions taken from apolipoprotein A-I.93 Three of these designs, which include a helix-breaking proline in the turn, are found to form fibers, at least some of which have been shown to assemble with the helicies perpendicular to the axis of longitudinal growth. In general, having the helicies running across the fiber axis is considered to be an advantage in terms of being able to functionalize fibers. This is due to the fact that the labels can be added in a position that should be displayed on the outside of the fiber rather than interfering with the end-to-end stacking required by helices running parallel to the fiber axis.
2. Polynanoreactors
The move from one-dimensional fiber systems up to two-dimensional assemblies is made with the creation by Ryadnov of polynanoreactors.94 The key design principle used in this work is the addition of interactions patterned onto the outer surface of the coiled coil. Ryadnov uses arginine in c positions such that an interaction can take place with both the neighboring g positions on the same helix or with an e position on a helix incorporated into a different dimer (see Fig. 22 ). Arginine is chosen for this, as its guanidinium group can form two salt bridges at the same time (though with half of the strength), while all the e and g positions were filled with glutamic acid side chains. This homodimeric system produces very dense mesoscopic assemblies of a hexagonal paracrystalline phase, which was visible by TEM.
Fig. 22.
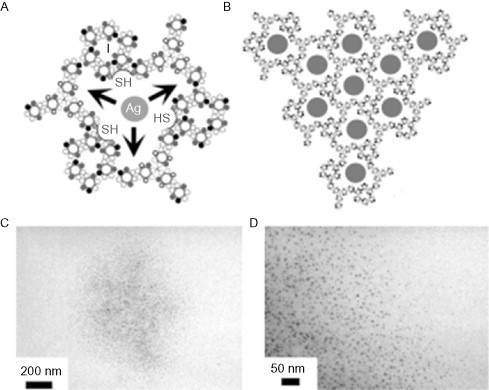
(A) The functionalized nanoreactor design indicating the location of cavities lined with cysteine residues. (B) The network of silver deposits expected to be formed by the design. (C and D) Clusters of colloidal silver as seen by TEM.
Reproduced with permission from Ryadnov94, copyright Wiley-VCH Verlag GmbH & Co. KGaA (2007).
The system was then extended to a heterodimeric format in order to increase the size of the cavities. This was done by the introduction of a second peptide with lysine at all e and g positions and which was covalently triplicated into a starburst molecule. On mixing this peptide with the original, circular dense aggregates are seen by TEM. At higher resolution, these exhibit a surface patterning of rings with an average diameter of around 4.5 nm.
In order to bring function to this system, an f position of the second peptide was mutated to cysteine with the ambition of using the cavities to convert metal ions to colloidal metal. This was demonstrated via the creation of silver nanoparticles, which (once the peptide had been removed) could be seen by TEM to have an average diameter of around 5 nm (see Fig. 22C and D).
3. Peptide Nanoparticles
This example of peptide design links together some of the concepts used earlier in joining together functional tectons, with onwards assembly in three dimensions creating a sphere. Specifically, a peptide designed to form a homopentamer was linked to a homotrimeric sequence via a flexible diglycine linker.95 This system assembles into a sphere containing 60 monomeric units (12 pentamers, 20 trimers) with icosahedral symmetry, via an intermediate with 15 monomeric units (see Fig. 23 ). The final assembly has been shown to contain 60 monomers in solution by analytical ultracentrifugation (AUC) and regular spherical particles by TEM. The particles have a diameter of around 16 nm and an internal cavity of around 6 nm. Both the internal cavity and the free ends of both the trimeric and pentameric peptides are available for functionalization.
Fig. 23.
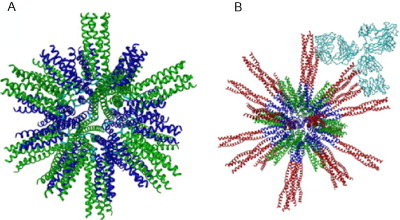
(A) The original peptide nanoparticle structure showing the pentameric and the trimeric coiled coils. (B) The same particle with the trimeric coiled coil extended using the HIV surface protein gp41. Multiple copies are displayed, allowing a strong interaction with antibodies.
Reproduced with permission from Raman et al.95 copyright Elsevier (2006).
One way in which this has been exploited is by appending a trimeric coiled-coil sequence taken from the severe acute respiratory syndrome (SARS) virus to the trimeric domain of the nanoparticle.96 Once the particle assembles, the SARS sequence is displayed multiple times on the surface of the particle in its native trimeric state. As such, the nanoparticles have been shown to be a very promising platform for vaccine design.
Most recently, the nanoparticles have been appended with a tandem repeat of the B cell immunodominant repeat epitope of the malaria parasite Plasmodium berghei circumsporozoite protein.97 The self-assembled nanoparticle was administered as a vaccine to mice and was found to confer a long-lasting, protective immune response.
D. Functionalization of Fiber Assemblies
There are many ways in which one can approach the functionalization of fiber systems.98 In the first approach (coassembly), one may covalently label the peptides before assembly such that they possess both the assembly information and also some other functional domain or moiety. In the second approach, the fibers can be labeled either covalently or noncovalently after assembly. In the third approach (templated assembly), the fibers are used as a scaffold to condense some other functional material.
Each of these cases will be examined with a few examples from the α-helical assembly literature, but it should be noted that the techniques involved have been exploited to a much larger extent in the β-based assembly field and that many of the concepts are transferable. For a recent review of the functionalization of amyloid-based systems, see Ref. 99.
The covalent labeling of peptides has been used extensively in the case of peptides designed for tissue culture scaffolds. Many systems have therefore been modified to include cell recognition motifs including the integrin-dependent cell adhesion motif RGD. Systems modified in this way include the pentameric fibers produced by Potekhin et al.100., 101. Once the fibers are assembled, it is hoped that the RGD motif will be displayed on the surface.
Another form of covalent labeling involves the addition of fluorophores. This was used in the case of the SAF system discussed earlier, with one of the two co-assembling peptides covalently labeled with a fluorescent label.102 Using this technique, it was possible to observe two previously unknown functions of this system. First, polar growth was shown, in that the added labeled peptide associated preferentially to one end of the fibers. This was demonstrated by adding the peptide labeled with rhodamine followed by a peptide labeled with fluorescein, giving fibers with multicolored tips (see Fig. 24 ). Second, a small level of exchange along the length of the fibers was seen, potentially drawing a parallel with the intermediate filament systems. Finally, within the SAF system, various assembling subunits have been covalently linked together, giving systems that produced fibers with branches, waves, and kinks.103., 104.
Fig. 24.

Fibers from the SAF system labeled sequentially with rhodamine (red) and then fluorescein (green) labeled peptide.
Reproduced with permission from Smith et al.101 copyright Wiley-VCH Verlag GmbH & Co. KGaA (2005).
It should be noted that, in many of these systems, it is not desirable, necessary, or even tolerated that every peptide in the assembly carries a function, and that in many cases, only a small amount of functional peptide is doped into the assembly.
Postassembly modification has also been attempted in the SAF system, using biocompatible click chemistry.105 This involves a component of the previous route, in that peptides are labeled with either azide- or alkene-modified side chains. These are small modifications and do not significantly alter the behavior of the peptide system and are incorporated at high levels into the fibers. Once the fibers are assembled, the function bearing click partner is added and labeled fibers are created. This has been demonstrated with gold nanoparticles and fluorphores (Fig. 25A ).
Fig. 25.

Three examples of fiber functionalization applied to the SAF system. (A) Noncovalent attachment of dye to fibers using charged peptides. Scale bar 10 μm. Reproduced with permission from Mahmoud and Woolfson106, copyright Elsevier (2010). (B) In situ functionalization of azide-containing SAFs functionalized with biotin, followed by streptavidin–nanogold. The fibers contained purely azide-labeled P1, scale bar 200 nm. Reproduced with permission from Mahmoud et al.105 copyright Elsevier (2011). (C) TEM images of silica tubes left behind after digestion of the peptide fibers. Scale bar 200 nm.
Reproduced with permission from Holmstrom et al.107 copyright American Chemical Society (2008).
Noncovalent postassembly labeling of the SAF system has also been achieved,106 using short peptide tags with negatively or positively charged or neutral sequences that are incubated with assembled fibers. Given the net positive charge of the SAFs, it is the DEDEDE tag that is found to interact strongly. Again, this has been demonstrated to recruit function in the form of gold nanoparticles and fluorophores to the surface of the fibers (Fig. 25B).
The final route of templated assembly has also been demonstrated in this system, by the deposition of silica onto preformed fibers.107 The fibers themselves can be removed by proteolysis leaving silica tubes, which appear hollow by TEM. Another example of this approach was demonstrated by the Conticello group in which a trimeric coiled-coil-based fiber containing histidine side chains was assembled.108 The peptides could recruit silver, which formed a nanowire within the fiber core (Fig. 25C).
IV. Overview and Future Outlook
As we have seen in the course of this chapter, the design of α-helical tectons for self-assembly is maturing as a science. We have now reached the point where many different coiled-coil topologies can be reliably produced, using design rules that have been extracted from natural systems and validated in synthetic systems. The field is now moving on towards more complex, discrete structures and applications. As we move along, we will encounter increasingly the challenges of creating systems in which multiple self-assembling components must interact. While several of the works discussed here have attempted to address the issues surrounding the orthogonality of tectons for mixed self-assembling systems, to date our success in producing applications using this information has been limited to small proofs of concept. As we move beyond this, we can expect to encounter greater difficulty in maintaining fidelity of interactions—which should in itself provide another rich source of basic research into the way in which natural systems behave.
The level and sophistication of functionalization of discrete, self-assembling structures is also rapidly increasing. In particular, the creation of responsive, structurally switching systems will be of great future interest. The study of natural systems has been moving away from the static and towards the dynamic for some time, and it is encouraging to see that the protein design field is following this trend. With the increase in functional building blocks of this nature, it will not be long before we can realize the ambition of designed protein-based molecular motion.
The story from the perspective of infinite or fiber assemblies is similar: that assemblies can now be designed, modified, and functionalized in a variety of ways. In this case, the future issue is likely to be the control of assembly. Although some control of width and length in fiber systems has emerged, it has mostly been of an observational nature, and developing more complex fiber systems that display the level of width and length control exhibited by nature will indeed be a challenge.
Spatial control of fibrillogenesis will also be a goal for the future, both in terms of controlling the location and direction of nucleation and polymerization, and in the control of labeling such that multiple ordered functionalities can be presented on fibers.
For many of the challenges listed above, it will be necessary to go back to the natural systems to extract more information and to perform more basic science and proof-of-concept experiments. However, the vast increase in the possible applications in both medicine and nanoscience that such research could bring should make the effort well worthwhile.
References
- 1.Szybalski W. In vivo and in vitro initiation of transcription. In: Kohn A., Shatkay A., editors. Control of gene expression. Plenum Press; New York: 1974. [Google Scholar]
- 2.Bromley E.H.C., Channon K., Moutevelis E., Woolfson D.N. Peptide and protein building blocks for synthetic biology: from programming biomolecules to self-organized biomolecular systems. ACS Chem Biol. 2008;3:38–50. doi: 10.1021/cb700249v. [DOI] [PubMed] [Google Scholar]
- 3.Channon K., Bromley E.H.C., Woolfson D.N. Synthetic biology through biomolecular design and engineering. Curr Opin Struct Biol. 2008;18(4):491–498. doi: 10.1016/j.sbi.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 4.Vickers C.E., Blank L.M., Kroemer J.O. Chassis cells for industrial biochemical production. Nat Chem Biol. 2010;6(12):875–877. doi: 10.1038/nchembio.484. [DOI] [PubMed] [Google Scholar]
- 5.Gibson D.G., Glass J.I., Lartigue C., Noskov V.N., Chuang R.Y., Algire M.A. Creation of a bacterial cell controlled by a chemically synthesized genome. Science. 2010;329:52–56. doi: 10.1126/science.1190719. [DOI] [PubMed] [Google Scholar]
- 6.Keasling J.D. Manufacturing molecules through metabolic engineering. Science. 2010;330(6009):1355–1358. doi: 10.1126/science.1193990. [DOI] [PubMed] [Google Scholar]
- 7.Ro D.K., Paradise E.M., Ouellet M., Fisher K.J., Newman K.L., Ndungu J.M. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature. 2006;440:940–943. doi: 10.1038/nature04640. [DOI] [PubMed] [Google Scholar]
- 8.Endy D. Foundations for engineering biology. Nature. 2005;438(7067):449–453. doi: 10.1038/nature04342. [DOI] [PubMed] [Google Scholar]
- 9.Sole R.V., Munteanu A., Rodriguez-Caso C., Marcia J. Synthetic protocell biology: from reproduction to computation. Philos Trans R Soc B. 2007;362(1486):1727–1739. doi: 10.1098/rstb.2007.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luisi P.L. Cambridge University Press; Cambridge: 2006. The emergence of life: from chemical origins to synthetic biology. [Google Scholar]
- 11.Ramussen S., Bedau M.A., Chen L., Deamer D., Krakauer D.C., Packard N.H., editors. Protocells: bridging the nonliving and living matter. The MIT Press; Cambridge and London: 2009. [Google Scholar]
- 12.Andersen E.S., Dong M., Nielsen M.M., Jahn K., Subramani R., Mamdouh W. Self-assembly of a nanoscale DNA box with a controllable lid. Nature. 2009;459:U73–U75. doi: 10.1038/nature07971. [DOI] [PubMed] [Google Scholar]
- 13.Gu H.Z., Chao J., Xiao S.J., Seeman N.C. A proximity-based programmable DNA nanoscale assembly line. Nature. 2010;465:202–206. doi: 10.1038/nature09026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaeger L., Chworos A. The architectonics of programmable RNA and DNA nanostructures. Curr Opin Struct Biol. 2006;16:531–543. doi: 10.1016/j.sbi.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 15.Lund K., Manzo A.J., Dabby N., Michelotti N., Johnson-Buck A., Nangreave J. Molecular robots guided by prescriptive landscapes. Nature. 2010;465:206–210. doi: 10.1038/nature09012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rothemund P.W.K. Folding DNA to create nanoscale shapes and patterns. Nature. 2006;440:297–302. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- 17.Seeman N.C. Nanomaterials based on DNA. Annu Rev Biochem. 2010;79:65–87. doi: 10.1146/annurev-biochem-060308-102244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crick F.H.C. The packing of-helices: simple coiled-coils. Acta Crystallogr. 1953;6:689. [Google Scholar]
- 19.Testa O.D., Moutevelis E., Woolfson D.N. CC plus: a relational database of coiled-coil structures. Nucleic Acids Res. 2009;37(SI):D315–D322. doi: 10.1093/nar/gkn675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moutevelis E., Woolfson D.N. A periodic table of coiled-coil protein structures. J Mol Biol. 2009;385(3):726–732. doi: 10.1016/j.jmb.2008.11.028. [DOI] [PubMed] [Google Scholar]
- 21.Rackham O.J.L., Madera M., Armstrong C.T., Vincent T.L., Woolfson D.N., Gough J. The evolution and structure prediction of coiled coils across all genomes. J Mol Biol. 2010;403(3):480–493. doi: 10.1016/j.jmb.2010.08.032. [DOI] [PubMed] [Google Scholar]
- 22.Hurst H.C. Transcription factors 1: bZIP proteins. Protein Profile. 1995;2(2):101–168. [PubMed] [Google Scholar]
- 23.Rose A., Meier I. Scaffolds, levers, rods and springs: diverse cellular functions of long coiled-coil proteins. Cell Mol Life Sci. 2004;61:1996–2009. doi: 10.1007/s00018-004-4039-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin R.C., Scheller R.H. Mechanisms of synaptic vesicle exocytosis. Annu Rev Cell Dev Biol. 2000;16:19–49. doi: 10.1146/annurev.cellbio.16.1.19. [DOI] [PubMed] [Google Scholar]
- 25.Shu W., Liu J., Ji H., Lu M. Core structure of the outer membrane lipoprotein from Escherichia coli at 1.9 A resolution. J Mol Biol. 2000;299(4):1101–1112. doi: 10.1006/jmbi.2000.3776. [DOI] [PubMed] [Google Scholar]
- 26.Herrmann H., Bar H., Kreplak L., Strelkov S.V., Aebi U. Intermediate filaments: from cell architecture to nanomechanics. Nat Rev Mol Cell Biol. 2007;8(7):562–573. doi: 10.1038/nrm2197. [DOI] [PubMed] [Google Scholar]
- 27.Margolin W. Bacterial shape: concave coiled coils curve caulobacter. Curr Biol. 2004;14(6):R242–R244. doi: 10.1016/j.cub.2004.02.057. [DOI] [PubMed] [Google Scholar]
- 28.Mizuno N., Narita A., Kon T., Sutoh K., Kikkawa M. Three-dimensional structure of cytoplasmic dynein bound to microtubules. Proc Natl Acad Sci USA. 2007;104:20832–20837. doi: 10.1073/pnas.0710406105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vale R.D., Milligan A. The way things move: looking under the hood of molecular motor proteins. Science. 2000;288(5463):88–95. doi: 10.1126/science.288.5463.88. [DOI] [PubMed] [Google Scholar]
- 30.Walshaw J., Woolfson D.N. SOCKET: a program for identifying and analysing coiled-coil motifs within protein structures. J Mol Biol. 2001;307:1427. doi: 10.1006/jmbi.2001.4545. [DOI] [PubMed] [Google Scholar]
- 31.Woolfson D.N. The design of coiled-coil structures and assemblies. Adv Prot Chem. 2005;70:79–112. doi: 10.1016/S0065-3233(05)70004-8. [DOI] [PubMed] [Google Scholar]
- 32.Harbury P.B., Zhang T., Kim P.S., Alber T. A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science. 1993;262:1401. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- 33.Meier M., Stetefeld J., Burkhard P. The many types of interhelical ionic interactions in coiled coils—an overview. J Struct Biol. 2010;170(2):192–201. doi: 10.1016/j.jsb.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 34.Armstrong C.T., Boyle A.L., Bromley E.H.C., Mahmoud Z.N., Smith L., Thomson A.R. Rational design of peptide-based building blocks for nanoscience and synthetic biology. Faraday Discuss. 2009;143:305–317. doi: 10.1039/b901610d. [DOI] [PubMed] [Google Scholar]
- 35.Steinmetz M.O., Stock A., Schulthless T., Landwehr R., Lustig A., Faix J. A distinct 14 residue site triggers coiled-coil formation in cortexillin I. EMBO J. 1998;17(7):1883–1891. doi: 10.1093/emboj/17.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kammerer R.A., Kostrewa D., Progias P., Hannappa S., Avila D., Lustig A. A conserved trimerization motif controls the topology of short coiled coils. Proc Natl Acad Sci USA. 2005;102(39):13891–13896. doi: 10.1073/pnas.0502390102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ciani B., Bjelic S., Honnappa S., Jawhari H., Jaussi R., Payapily A. Molecular basis of coiled-coil oligomerization-state specificity. Proc Natl Acad Sci USA. 2010;107(46):19850–19855. doi: 10.1073/pnas.1008502107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Apgar J.R., Gutwin K.N., Keating A.E. Predicting helix orientation for coiled-coil dimers. Proteins Struct Funct Bioinformatics. 2008;72(3):1048–1065. doi: 10.1002/prot.22118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hadley E.B., Testa O.D., Woolfson D.N., Gellman S.H. Preferred side-chain constellations at antiparallel coiled-coil interfaces. Proc Natl Acad Sci USA. 2008;105:530–535. doi: 10.1073/pnas.0709068105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oakley M.G., Hollenbeck J.J. The design of antiparallel coiled coils. Curr Opin Struct Biol. 2001;11:450–457. doi: 10.1016/s0959-440x(00)00232-3. [DOI] [PubMed] [Google Scholar]
- 41.Newman J.R.S., Keating A.E. Comprehensive identification of human bZIP interactions with coiled-coil arrays. Science. 2003;300(5628):2097–2101. doi: 10.1126/science.1084648. [DOI] [PubMed] [Google Scholar]
- 42.Fong J.H., Keating A.E., Singh M. Predicting specificity in bZIP coiled-coil protein interactions. Genome Biol. 2004;5(2):R11. doi: 10.1186/gb-2004-5-2-r11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grigoryan G., Keating A.E. Structure-based prediction of bZIP partnering specificity. J Mol Biol. 2006;355(5):1125–1142. doi: 10.1016/j.jmb.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 44.Acharya A., Ruvinov S.B., Gal J., Noll J.R., Vinson C. A heterodimerizing leucine zipper coiled coil system for examining the specificity of a position interactions: amino acids I, V, L, N, A, and K. Biochemistry. 2002;41(48):14122–14131. doi: 10.1021/bi020486r. [DOI] [PubMed] [Google Scholar]
- 45.Acharya A., Rishi V., Vinson C. Stability of 100 homo and heterotypic coiled-coil a−a‘ pairs for ten amino acids (A, L, I, V, N, K, S, T, E, and R) Biochemistry. 2006;46(38):11324–11332. doi: 10.1021/bi060822u. [DOI] [PubMed] [Google Scholar]
- 46.Krylov D., Mikhailenko I., Vinson C. A thermodynamic scale for leucine-zipper stability and dimerization specificity: -e and g-interhelical interactions. EMBO J. 1994;13(12):2849–2861. doi: 10.1002/j.1460-2075.1994.tb06579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krylov D., Barchi J., Vinson C. Inter-helical interactions in the leucine zipper coiled coil dimer: ph and salt dependence of coupling energy between charged amino acids. J Mol Biol. 1998;279(4):959–972. doi: 10.1006/jmbi.1998.1762. [DOI] [PubMed] [Google Scholar]
- 48.Mason J.M., Schmitz M.A., Muller K., Arndt K.M. Semirational design of Jun-Fos coiled coils with increased affinity: universal implications for leucine zipper prediction and design. Proc Natl Acad Sci USA. 2006;103(24):8989–8994. doi: 10.1073/pnas.0509880103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grigoryan G., Reinke A.W., Keating A.E. Design of protein-interaction specificity affords selective bZIP-binding peptides. Nature. 2009;458(7240):895. doi: 10.1038/nature07885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bromley E.H.C., Sessions R.B., Thomson A.R., Woolfson D.N. Designed α-helical tectons for constructing multicomponent synthetic biological systems. J Am Chem Soc. 2009;131:928–930. doi: 10.1021/ja804231a. [DOI] [PubMed] [Google Scholar]
- 51.Reinke A.W., Grant R.A., Keating A.E. A synthetic coiled-coil interactome provides heterospecific modules for molecular engineering. J Am Chem Soc. 2010;132(17):6025–6031. doi: 10.1021/ja907617a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Diss M.L., Kennan A.J. Orthogonal recognition in dimeric coiled coils via buried polar-group modulation. J Am Chem Soc. 2008;130(3):1321–1327. doi: 10.1021/ja076265w. [DOI] [PubMed] [Google Scholar]
- 53.Diss M.L., Kennan A.J. Simultaneous directed assembly of three distinct heterodimeric coiled coils. Org Lett. 2008;10(17):3797–3800. doi: 10.1021/ol801461a. [DOI] [PubMed] [Google Scholar]
- 54.Ryan S.J., Kennan A.J. Variable stability heterodimeric coiled-coils from manipulation of electrostatic interface residue chain length. J Am Chem Soc. 2007;129(33):10255–10260. doi: 10.1021/ja073717w. [DOI] [PubMed] [Google Scholar]
- 55.Gribbon C., Channon K.J., Zhang W.J., Banwell E.F., Bromley E.H.C., Chaudhuri J.B. MagicWand: a single, designed peptide that assembles to stable, ordered α-helical fibers. Biochemistry. 2008;47(39):10365–10371. doi: 10.1021/bi801072s. [DOI] [PubMed] [Google Scholar]
- 56.Diss M.L., Kennan A.J. Heterotrimeric coiled coils with core residue urea side chains. J Org Chem. 2008;73(24):9752–9755. doi: 10.1021/jo802379p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Akcay G., Kumar K. A new paradigm for protein design and biological self-assembly. J Fluor Chem. 2009;130(12):1178–1182. doi: 10.1016/j.jfluchem.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bilgicer B., Fichera A., Kumar K. A coiled coil with a fluorous core. J Am Chem Soc. 2001;123(19):4394–4399. doi: 10.1021/ja002961j. [DOI] [PubMed] [Google Scholar]
- 59.Bilgicer B., Xing X., Kumar K. Programmed self-sorting of coiled coils with leucine and hexafluoroleucine cores. J Am Chem Soc. 2001;123(47):11815–11816. doi: 10.1021/ja016767o. [DOI] [PubMed] [Google Scholar]
- 60.Vagt T., Nyakatura E., Salwiczek M., Jackel C., Koksch B. Towards identifying preferred interaction partners of fluorinated amino acids within the hydrophobic environment of a dimeric coiled coil peptide. Org Biomol Chem. 2010;8(6):1382–1386. doi: 10.1039/b917205j. [DOI] [PubMed] [Google Scholar]
- 61.Salwiczek M., Samsonov S., Vagt T., Nyakatura E., Fleige E., Numata J. Position-dependent effects of fluorinated amino acids on the hydrophobic core formation of a heterodimeric coiled coil. Chem A Eur J. 2009;15(31):7628–7636. doi: 10.1002/chem.200802136. [DOI] [PubMed] [Google Scholar]
- 62.Ryadnov M.G., Ceyhan B., Niemeyer C.M., Woolfson D.N. “Belt and braces”: a peptide-based linker system of de novo design. J Am Chem Soc. 2003;125:9388. doi: 10.1021/ja0352045. [DOI] [PubMed] [Google Scholar]
- 63.Robson Marsden H., Elbers N.A., Bomans P.H.H., Sommerdijk N., Kros A. A reduced SNARE model for membrane fusion. Angew Chem Int Ed. 2009;48(13):2330–2333. doi: 10.1002/anie.200804493. [DOI] [PubMed] [Google Scholar]
- 64.Lee D.H., Granja J.R., Martinez J.A., Severin K., Ghadiri M.R. A self-replicating peptide. Nature. 1996;382(6591):525–528. doi: 10.1038/382525a0. [DOI] [PubMed] [Google Scholar]
- 65.Severin K., Lee D.H., Martinez J.A., Ghadiri M.R. Peptide self-replication via template-directed ligation. Chem A Eur J. 1997;3(7):1017–1024. [Google Scholar]
- 66.Severin K., Lee D.H., Kennan A.J., Ghadiri M.R. A synthetic peptide ligase. Nature. 1997;389(6652):706–709. doi: 10.1038/39556. [DOI] [PubMed] [Google Scholar]
- 67.Li X., Chmielewski J. Peptide self-replication enhanced by a proline kink. J Am Chem Soc. 2003;125:11820–11821. doi: 10.1021/ja036569s. [DOI] [PubMed] [Google Scholar]
- 68.Ciani B., Hutchinson E.G., Sessions R.B., Woolfson D.N. A designed system for assessing how sequence affects α to β conformational transitions in proteins. J Biol Chem. 2002;277:10150–10155. doi: 10.1074/jbc.M107663200. [DOI] [PubMed] [Google Scholar]
- 69.Kammerer R.A., Kostrewa D., Zurdo J., Detken A., Garcia Echeverria C., Green J.D. Exploring amyloid formation by a de novo design. Proc Natl Acad Sci USA. 2004;101:4435–4440. doi: 10.1073/pnas.0306786101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schnarr N.A., Kennan A.J. pH-triggered strand exchange in coiled-coil heterotrimers. J Am Chem Soc. 2003;125(21):6364–6365. doi: 10.1021/ja029344a. [DOI] [PubMed] [Google Scholar]
- 71.Schnarr N.A., Kennan A.J. pH-switchable strand orientation in peptide assemblies. Org Lett. 2005;7(3):395–398. doi: 10.1021/ol047720l. [DOI] [PubMed] [Google Scholar]
- 72.Pandya M.J., Cerasoli E., Joseph A., Stoneman R.G., Waite E., Woolfson D.N. Sequence and structural duality: designing peptides to adopt two stable conformations. J Am Chem Soc. 2004;126(51):17016–17024. doi: 10.1021/ja045568c. [DOI] [PubMed] [Google Scholar]
- 73.Suzuki K., Hiroaki H., Kohda D., Nakamura H., Tanaka T. Metal ion induced self-assembly of a designed peptide into a triple-stranded alpha-helical bundle: a novel metal binding site in the hydrophobic core. J Am Chem Soc. 1998;120(50):13008–13015. [Google Scholar]
- 74.Tsurkan M.V., Ogawa M.Y. Metal-mediated peptide assembly: use of metal coordination to change the oligormerization state of an alpha-helical coiled-coil. Inorg Chem. 2007;46:6849. doi: 10.1021/ic700958h. [DOI] [PubMed] [Google Scholar]
- 75.Cerasoli E., Sharpe B.K., Woolfson D.N. ZiCo: a peptide designed to switch folded state upon binding zinc. J Am Chem Soc. 2005;127(43):15008–15009. doi: 10.1021/ja0543604. [DOI] [PubMed] [Google Scholar]
- 76.Ambroggio X.I., Kuhlman B. Computational design of a single amino acid sequence that can switch between two distinct protein folds. J Am Chem Soc. 2006;128(4):1154–1161. doi: 10.1021/ja054718w. [DOI] [PubMed] [Google Scholar]
- 77.Kuhlman B., Baker D. Native protein sequences are close to optimal for their structures. Proc Natl Acad Sci USA. 2000;97(19):10383–10388. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pagel K., Vagt T., Kohadja T., Koksch B. From α-helix to β-sheet—a reversible metal ion induced peptide secondary structure switch. Org Biomol Chem. 2005;3(14):2500–2502. doi: 10.1039/b505979h. [DOI] [PubMed] [Google Scholar]
- 79.Zhang F., Timm K.A., Arndt K.M., Woolley G.A. Photocontrol of coiled-coil proteins in living cells. Angew Chem Int Ed. 2010;49(23):3943–3946. doi: 10.1002/anie.201000909. [DOI] [PubMed] [Google Scholar]
- 80.Parry D.A., Strelkov S.V., Burkhard P., Aebi U., Herrmann H. Towards a molecular description of intermediate filament structure and assembly. Exp Cell Res. 2007;313(10):2204–2216. doi: 10.1016/j.yexcr.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 81.Sokolova A.V., Kreplak L., Wedig T., Mucke N., Svergun D.I., Herrmann H. Monitoring intermediate filament assembly by small-angle x-ray scattering reveals the molecular architecture of assembly intermediates. Proc Natl Acad Sci USA. 2006;103(44):16206–16211. doi: 10.1073/pnas.0603629103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kojima S., Kuriki Y., Yoshida T., Yazaki K., Miura K. Fibril formation by an amphipathic alpha-helix-forming polypeptide produced by gene engineering. Proc Jpn Acad Ser B Phys Biol Sci. 1997;73:7–11. [Google Scholar]
- 83.Potekhin S.A., Melnik T.N., Popov V., Anina N.F., Vazina A.A., Rigler P. De novo design of fibrils made of short alpha-helical coiled coil peptides. Chem Biol. 2001;11(8):1025–1032. doi: 10.1016/s1074-5521(01)00073-4. [DOI] [PubMed] [Google Scholar]
- 84.Dong H., Paramonov S.E., Hartgerink J.D. Self-assembly of α-helical coiled coil nanofibers. J Am Chem Soc. 2008;130(41):13691–13695. doi: 10.1021/ja8037323. [DOI] [PubMed] [Google Scholar]
- 85.Zimenkov Y., Dublin S.N., Ni R., Tu R.S., Breedveld V., Apkarian R.P. Rational design of a reversible pH-responsive switch for peptide self-assembly. J Am Chem Soc. 2006;128:6770. doi: 10.1021/ja0605974. [DOI] [PubMed] [Google Scholar]
- 86.Zimenkov Y., Conticello V.P., Guo L., Thiyagarajan P. Rational design of a nanoscale helical scaffold derived from self-assembly of a dimeric coiled coil motif. Tetrahedron. 2004;60:7237. [Google Scholar]
- 87.Wagner D.E., Phillips C.L., Ali W.M., Nybakken G.E., Crawford E.D., Schwab A.D. Toward the development of peptide nanofilaments and nanoropes as smart materials. Proc Natl Acad Sci USA. 2005;102(36):12656–12661. doi: 10.1073/pnas.0505871102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pandya M.J., Spooner G.M., Sunde M., Thorpe J.R., Rodger A., Woolfson D.N. Sticky-end assembly of a designed peptide fiber provides insight into protein fibrillogenesis. Biochemistry. 2000;39:8728–8734. doi: 10.1021/bi000246g. [DOI] [PubMed] [Google Scholar]
- 89.Smith A.M., Banwell E.F., Edwards W.R., Pandya M.J., Woolfson D.N. Engineering increased stability into self-assembled protein fibers. Adv Funct Mater. 2006;16:1022–1030. [Google Scholar]
- 90.Papapostolou D., Smith A.M., Atkins E.D.T., Oliver S.J., Ryadnov M.G., Serpell L.C. Engineering nanoscale order into a designed protein fiber. Proc Natl Acad Sci USA. 2007;104:10853–10858. doi: 10.1073/pnas.0700801104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bromley E.H.C., Channon K.J., King P.J.S., Mahmoud Z.N., Banwell E.F., Butler M.F. Flow linear dichroism of some prototypical proteins. Biophys J. 2010;98:1668–1676. [Google Scholar]
- 92.Banwell E.F., Aberlardo E.S., Adams D.J., Birchall M.A., Corrigan A., Donald A.M. Rational design and application of responsive alpha-helical peptide hydrogels. Nat Mater. 2009;8:596–600. doi: 10.1038/nmat2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lazar K.L., Miller-Auer H., Getz G.S., Orgel J., Meredisth S.C. Helix-turn-helix peptides that form α-helical fibrils: turn sequences drive fibril structure. Biochemistry. 2005;44:12681. doi: 10.1021/bi0509705. [DOI] [PubMed] [Google Scholar]
- 94.Ryadnov M.G. A self-assembling peptide polynanoreactor. Angew Chem Int Ed. 2007;46(6):969–972. doi: 10.1002/anie.200604014. [DOI] [PubMed] [Google Scholar]
- 95.Raman S., Machaidze G., Lustig A., Aebi U., Burkhard P. Structure-based design of peptides that self-assemble into regular polyhedral nanoparticles. Nanomed Nanotechnol Biol Med. 2006;2:95. doi: 10.1016/j.nano.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 96.Pimentel T.A.P.F., Yan Z., Jeffers S.A., Holmes K.V., Hodges R.S., Burkhard P. Peptide nanoparticles as novel immunogens: design and analysis of a prototypic severe acute respiratory syndrome vaccine. Chem Biol Drug Des. 2009;73(1):53–61. doi: 10.1111/j.1747-0285.2008.00746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kaba S.A., Brando C., Guo Q., Mittelhozer C., Raman S., Tropel D. A nonadjuvanted polypeptide nanoparticle vaccine confers long-lasting protection against rodent malaria. J Immunol. 2009;183(11):7268–7277. doi: 10.4049/jimmunol.0901957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Woolfson D.N., Mahmoud Z.N. More than just bare scaffolds: towards multi-component and decorated fibrous biomaterials. Chem Soc Rev. 2010;39:3464–3479. doi: 10.1039/c0cs00032a. [DOI] [PubMed] [Google Scholar]
- 99.Channon K.J., MacPhee C.E. Possibilities for smart materials exploiting the self-assembly of polypeptides into fibrils. Soft Matter. 2008;4:647–652. doi: 10.1039/b713013a. [DOI] [PubMed] [Google Scholar]
- 100.Melnik T.N., Villard V., Vasiliev V., Corradin G., Kajava A.V., Potehkin S.A. Shift of fibril‐forming ability of the designed α‐helical coiled‐coil peptides into the physiological pH region. Protein Eng. 2003;16:1125. doi: 10.1093/protein/gzg138. [DOI] [PubMed] [Google Scholar]
- 101.Villard V., Kalyuzhniy O., Riccio O., Potehkin S.A., Melnik T.N., Kajava A.V. Synthetic RGD-containing alpha-helical coiled coil peptides promote integrin-dependent cell adhesion. J Pept Sci. 2006;12:206. doi: 10.1002/psc.707. [DOI] [PubMed] [Google Scholar]
- 102.Smith A.M., Acquah S.F.A., Bone N., Kroto H.W., Ryadnov M.G., Stevens M.S.P. Polar assembly in a designed protein fiber. Angew Chem Int Ed. 2005;44:325–328. doi: 10.1002/anie.200461599. [DOI] [PubMed] [Google Scholar]
- 103.Ryadnov M.G., Woolfson D.N. Introducing branches into a self-assembling peptide fiber. Angew Chem Int Ed. 2003;42:3021–3023. doi: 10.1002/anie.200351418. [DOI] [PubMed] [Google Scholar]
- 104.Ryadnov M.G., Woolfson D.N. Engineering the morphology of a self-assembling protein fibre. Nat Mater. 2003;2:329–332. doi: 10.1038/nmat885. [DOI] [PubMed] [Google Scholar]
- 105.Mahmoud Z.N., Gunnoo S.B., Thomson A.R., Fletcher J.M., Woolfson D.N. Bioorthogonal dual functionalization of self-assembling peptide fibers. Biomaterials. 2011;32(15):3712–3720. doi: 10.1016/j.biomaterials.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 106.Mahmoud Z.M., Woolfson D.N. The non-covalent decoration of self-assembling protein fibers. Biomaterials. 2010;31:7468–7474. doi: 10.1016/j.biomaterials.2010.06.041. [DOI] [PubMed] [Google Scholar]
- 107.Holmstrom S.C., King P.J.S., Ryadnov M.G., Butler M.F., Mann S., Woolfson D.N. Templating silica nanostructures on rationally designed self-assembled peptide fibers. Langmuir. 2008;24:11778–11783. doi: 10.1021/la802009t. [DOI] [PubMed] [Google Scholar]
- 108.Dublin S.N., Conticello V.P. Design of a selective metal ion switch for self-assembly of peptide-based fibrils. J Am Chem Soc. 2008;130:49–51. doi: 10.1021/ja0775016. [DOI] [PubMed] [Google Scholar]