

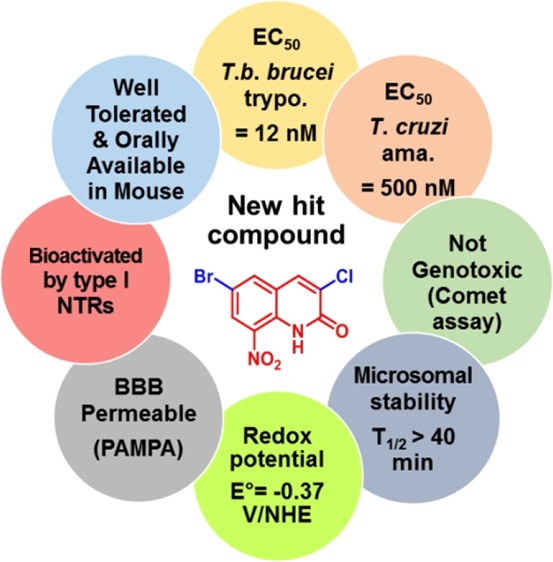
Abstract
An antikinetoplastid pharmacomodulation study was conducted at position 6 of the 8-nitroquinolin-2(1H)-one pharmacophore. Fifteen new derivatives were synthesized and evaluated in vitro against L. infantum, T. brucei brucei, and T. cruzi, in parallel with a cytotoxicity assay on the human HepG2 cell line. A potent and selective 6-bromo-substituted antitrypanosomal derivative 12 was revealed, presenting EC50 values of 12 and 500 nM on T. b. brucei trypomastigotes and T. cruzi amastigotes respectively, in comparison with four reference drugs (30 nM ≤ EC50 ≤ 13 μM). Moreover, compound 12 was not genotoxic in the comet assay and showed high in vitro microsomal stability (half life >40 min) as well as favorable pharmacokinetic behavior in the mouse after oral administration. Finally, molecule 12 (E° = −0.37 V/NHE) was shown to be bioactivated by type 1 nitroreductases, in both Leishmania and Trypanosoma, and appears to be a good candidate to search for novel antitrypanosomal lead compounds.
Keywords: Trypanosoma brucei brucei, Trypanosoma cruzi, 8-nitroquinolin-2(1H)-ones, redox potentials, NTR1
Human African trypanosomiasis (HAT),1 Chagas diseases (CD),2 and visceral leishmaniasis (VL)3 are infectious diseases caused by unicellular flagellated kinetoplastid parasites belonging to the Trypanosoma and Leishmania genera. From an epidemiological point of view, the World Health Organization (WHO) estimates that millions of people are at risk of contracting HAT, CD, and VL. These three diseases are also estimated to be responsible for about 30 000 annual deaths, with the caveat that these numbers may be underestimated due to the difficulty in accessing some rural areas and the unspecific symptoms during the early stage of these infections.4 Although these diseases are lethal if untreated, there are very few efficient and safe drugs available, each of them presenting various drawbacks for the patient (toxicity, emergence of resistances, mode of administration, treatment cost). Thus because of the lack of consideration and investments from the pharmaceutical industry, the WHO classified HAT, CD, and VL among “neglected tropical diseases” (NTDs).5
In this context, nitroaromatic derivatives play a key role in the fight against kinetoplastids (Figure 1). Beyond Nifurtimox and Benznidazole (antitrypanosomal drugs), nitroaromatics are fully part of the research efforts of the Drugs for Neglected Diseases initiative (DNDi). Thus among the few novel chemical entities in clinical trials against these infections,6 the development of fexinidazole (an orally available 5-nitroimidazole derivative that received an agreement from the European Medicines Agency (EMA) in the treatment of HAT in 2018 and that is in phase 2 against CD),7−9 delamanid (a derivative that is marketed against tuberculosis and that was studied against VL),10 and DNDi-0690 (a 2-nitroimidazooxazine derivating from delamanid and that has recently entered phase-I clinical trials against VL) illustrate well the strong potential of nitroaromatic molecules in the search for novel antitrypanosomatids.11
Figure 1.

Structures of nitroaromatic drugs displaying antikinetoplastid activity.
The antiparasitic mechanism of action of these molecules involves parasitic nitroreductases (NTRs). NTRs contain a flavin as a cofactor and catalyze the one- or two-electron reduction of nitrodrugs into cytotoxic electrophilic metabolites such as nitroso and hydroxylamine derivatives, which are able to form covalent adducts with DNA or proteins.12 Two NTRs were found in Leishmania: a mitochondrial NTR1 catalyzing a two-electron reduction13 and a cytosolic NTR2 catalyzing a one-electron reduction.14 In Trypanosoma, only a mitochondrial type 1 NTR is expressed.15,16 It is important to note that these enzymes are absent from mammalian cells, an important feature for developing selective antikinetoplastid molecules. Moreover, depending on the chemical structure of nitrated derivatives, the selectivity for NTRs could be modulated. For instance, fexinidazole is selectively bioactivated by NTR1 in both Leishmania and Trypanosoma; meanwhile, delamanid is only bioactivated by NTR2 in Leishmania.14
In this context, by studying the antiparasitic potential of the 8-nitroquinoline scaffold,17 our research team reported a new antileishmanial pharmacophore: 8-nitroquinolin-2(1H)-one.18 After pharmacomodulation studies at position 4,19,20 an electrochemistry-guided work highlighted the importance of an intramolecular hydrogen bond between the nitro group and the lactam function.21 This work also led to the identification of a new 3-bromosubstituted antikinetoplastid hit compound derivative (Hit 1, Figure 2), which is bioactivated by type 1 NTRs, in both L. donovani and T. brucei brucei (T. b. brucei).21 Recently, by introducing a p-carboxyphenyl functionality at position 3 of the pharmacophore, we reported a new selective antitrypanosomal hit compound (Hit 2, Figure 2).22
Figure 2.

Antitrypanosomatid profile of previously reported 8-nitroquinolin-2(1H)-one hit compounds.
We herein present a medicinal chemistry work that greatly improved the antitrypanosomal activity of the current series via the synthesis of novel derivatives bearing electron-withdrawing groups at position 6 of the quinolinone scaffold.
To increase the oxidant character of the series and facilitate its bioactivation by nitroreductases, 15 derivatives (1–15) were prepared, bearing various electron-withdrawing groups (EWGs) at position 6. As presented in Scheme 1, eight compounds bearing a trifluoromethyl group at position 6 of the quinolinone scaffold were synthesized according to a previously reported protocol.23 In brief, 3-ethoxyacryloyl chloride was synthesized in two steps by the saponification of commercial 3,3′-diethoxyethylpropionate followed by a reaction with thionyl chloride. This acyl chloride was then reacted with p-trifluoromethylaniline to afford, after a cyclization step in H2SO4 (98%), compound 1 in 42% yield (a). Molecule 1 was then either nitrated at position 8, using classical conditions, to afford compound 2 in 89% yield (b) or selectively halogenated at position 3 to generate derivative 3 or 4 in 48 and 54% yield, respectively (c and d). The O-methylation of compound 2 was performed with a protocol using a mixture of methyl iodide and sodium hydride to prepare derivative 5 in 69% yield (e). Nitrated molecule 2 could also undergo chlorination at position 3, affording compound 6 in 88% yield (f), or react with NaBrO3 in refluxing HBr, according to a previously reported protocol,24 to afford halogenated derivative 7 in 94% yield (g). The nitro group of compound 7 was finally reduced to afford the amino-derivative 8, considered as a negative control, using SnCl2, in 68% yield (h).
Scheme 1. Synthesis of Compounds 1–8 from p-Trifluoromethylaniline.

The synthesis pathway used for preparing derivatives bearing a halogen atom at position 6 of the quinolinone ring is presented in Scheme 2. It starts from 8-nitroquinolin-2(1H)-one,21 which was reacted with bromine in refluxing acetic acid, saturated with sodium acetate, leading to a mixture of both 6-bromo and 3,6-dibromo derivatives 9 and 10 in 9 and 33% yield, respectively (a). Dichloro analogue 11 was also prepared from 8-nitroquinolin-2(1H)-one by reacting with 5 equiv of sodium chlorate in refluxing hydrochloric acid for 24 h (b). By using the same reaction, decreasing the amount of sodium chlorate to 3 equiv, and limiting the reaction time to 45 min, 3-chloro-8-nitroquinolin-2(1H)-one was formed (c).21 This compound could be brominated at position 6 by reacting with an excess of NaBrO3 in refluxing HBr, leading to 12 in good yield (d). Taking into account that we previously reported the benefit of introducing a p-carboxyphenyl moiety at position 3 of the scaffold toward antitrypanosomal activity,22 we finally engaged dibromo-derivative 10 into a Suzuki–Miyaura reaction with p-(methoxycarbonyl)phenylboronic acid and then saponified the resulting ester coupling product to afford carboxylic acid 13 (e). Finally, from brominated compound 12, derivatives 14 and 15 were obtained through a Sonogashira coupling reaction (f).
Scheme 2. Synthesis of Compounds 9–15.

Electrochemistry
Because the compounds were expected to be nitroreductase substrates, we studied the influence of the introduction of EWGs at position 6 of the pharmacophore toward redox potentials. Thus redox potentials (couple R-NO2/RNO2•–) of the synthesized compounds were measured by cyclic voltammetry in DMSO (one-electron reversible reduction). Redox potentials ranged between −0.36 and −0.75 V/NHE (Figure 3). In comparison with the original pharmacophore, the introduction of a bromine atom at position 6 (compound 9) led to a significant increase in the E° value from −0.54 to −0.40 V. The same effect was observed when a bromine atom was introduced at position 6 of hit 2 (compound 13), with an increase in the E° value of +0.16 V. It is important to note that introducing a bromine atom has a higher influence on the redox potential at position 6 (compound 9) compared with position 3 (hit 1): +0.14 versus +0.09 V. The influence of the CF3 group toward the redox potential of the scaffold was the same as that of the bromine atom: +0.15 V (comparison between the pharmacophore and compound 2). Then, when two bromine atoms were combined at positions 3 and 6 (compound 10), a slight increase of +0.03 V was observed (no additive effect) in comparison with 6-bromo-derivative 9. Surprisingly, by comparing compounds 7 and 10–12, there was almost no effect of the variation of the EWG at position 3 or 6 toward the redox potential values, with all compounds ranging from −0.36 to −0.37 V. Finally, by comparing compound 2 with its O-methylated derivative 5, it appeared that the redox potential value was drastically lower (−0.36 V shift). This shift is not only due to the donating mesomeric effect of the methoxy group but also due to the disappearance of the intramolecular hydrogen bond between the nitro group and the lactam function.21
Figure 3.

Redox potentials of synthesized nitroaromatic compounds determined by cyclic voltammetry.
Compound Evaluation
The cytotoxicity of all compounds was first assessed on the human HepG2 cell line using doxorubicin as a positive control (Table 1). Compared with this reference drug (CC50 = 0.2 μM), the series showed little (CC50 = 17 μM) to no (CC50 > 100 μM) cytotoxicity. Non-nitrated molecules 1, 3, and 4, O-methylated derivative 5, and 8-amino-derivative 8, considered as negative controls, displayed high CC50 values, >100 μM. The results show that the introduction of a bromine atom or a CF3 group at position 6 of the nitrated scaffold leads to derivatives (2 and 9) that are four to five times more cytotoxic (CC50 = 28–41 μM) than the 8-nitroquinolinone pharmacophore (CC50 = 164 μM). When introducing an additional halogen atom at position 3 of the scaffold (6, 7, 10–12), the cytotoxicity remains close to the ones of 6-monosubstituted derivatives (CC50 = 17–41 μM). Nevertheless, introducing a p-carboxyphenyl functionality at position 3 of the scaffold (compound 13) results in a three-fold decrease in the cytotoxicity (CC50 = 60 μM) in comparison with the 3,6-dibromo derivative 10 (CC50 = 17 μM).
Table 1. Antikinetoplastid Activities and Cytotoxicity of Compounds 1–15.


EC50 or CC50 value was not reached at the highest tested concentration.
Doxorubicin was used as a cytotoxic reference drug.
Amphotericin B, miltefosine, and fexinidazole were used as antileishmanial reference drugs.
Fexinidazole, suramin, and eflornithine were used as anti-Trypanosoma brucei reference drugs.
All synthesized molecules were screened in vitro for their activity against Leishmania infantum axenic amastigotes, and their EC50 values were compared with the ones of three reference drugs (amphotericin B, miltefosine, and fexinidazole) and antikinetoplastid hit 1 (Table 1). Among the tested compounds, only compound 7 (E° = −0.36 V) showed a weak antileishmanial activity, displaying an EC50 value of 3.9 μM in addition to a selectivity index superior to 10. Derivatives of 7 without a nitro group at position 8 of the scaffold (1, 3, 4, and 8) were inactive toward L. infantum as well as the O-methylated analogue 5 (E° = −0.75 V), confirming both the key role of the nitro group toward antiparasitic activity and the importance of the interaction between the nitro group and the lactam function to confer suitable redox potentials for displaying antileishmanial properties in this chemical series.21 Note that, contrary to hit 2, which was not active against L. infantum (EC50 > 100 μM), the 6-bromo derivative 13 was active (EC50 = 8 μM).
All molecules were also screened in vitro for their antitrypanosomal activity against T. b. brucei trypomastigotes, and their EC50 values were compared with the ones of three reference drugs, suramin, eflornithine, and fexinidazole, and with antitrypanosomal hit 2. Among the 15 tested molecules, 7, 9, 10, 11, 12, and 13 displayed both submicromolar activity (12 ≤ EC50 ≤ 200 nM) and good selectivity indices (SI > 200) in comparison with suramin (EC50 = 30 nM), eflornithine (EC50 = 13 μM), fexinidazole (EC50 = 400 nM), and hit 2 (EC50 = 1.5 μM). Molecule 12 was the most active and selective in the series: EC50 = 12 nM and SI = 1508. Compounds without a nitro group at position 8 of the scaffold appeared to be inactive (compounds 1, 3, and 4), and amino-derivative 8 was quite less active (EC50 = 9 μM). It is important to note that the introduction of a bromine atom at position 6 of the scaffold seems crucial to afford highly active antitrypanosomal molecules, with compound 9 being 334 times more active than the 8-nitroquinolin-2(1H)-one pharmacophore and 27 times more active than its 3-bromo-position isomer (hit 1). This key effect of the bromine atom at position 6 of the scaffold also appears when comparing the activities of 3-chloro-6-bromo hit-compound 12 with its 3-chloro-analogue A: 12 is 100 times more active than A. When comparing molecules 2 and 9, it appeared that the CF3 group does not provide the same level of improvement in activity. Introducing an additional halogen atom at position 3 of hit molecule 9, leading to dibromo-derivative 10 and dihalo-derivative 12, afforded the most active compounds in the series (EC50 = 12–50 nM). A comparison between compound 12 and 3,6-dichlorosubstituted derivative 11 showed that the replacement of the bromine atom by a chlorine atom at position 6 was not in favor of antitrypanosomal activity. During pharmacomodulation studies at position 3 of the scaffold, hit 2 was identified as a promising antitrypanosomal hit with an EC50 of 1.5 μM.22 When a bromine atom is introduced at position 6 of hit 2, leading to compound 13, the antitrypanosomal activity is improved, reaching an EC50 value of 200 nM.
Thus the molecule presenting the best antitrypanosomal profile in the series, regarding both the activity and the selectivity, was compound 12. To better evaluate its antitrypanosomal potential, an in vitro evaluation against intracellular T. cruzi amastigotes was carried out using benznidazole and fexinidazole as positive controls (Table 1). Quite interestingly, despite being about 10 times more cytotoxic than reference drugs, molecule 12 displayed a high activity (EC50 = 0.5 μM) against T. cruzi, identical to the one of benznidazole (EC50 = 0.5 μM) and better than the one of fexinidazole (EC50 = 3 μM).
To investigate more deeply the potential of compounds 7, 12, and 13 as the most active antikinetoplastid compounds of the studied series, an in vitro pharmacokinetic evaluation was performed, including a microsomal stability assay (female mouse microsomes), a human albumin binding assay, and a PAMPA assay using a blood–brain barrier (BBB) model (Table 2). All compounds presented good microsomal stability (T1/2 > 40 min) and strong binding to human albumin (≥99%). However, the development of antitrypanosomal agents requires that molecules cross the BBB, as the second stage of HAT is meningoencephalitic. A PAMPA assay on a BBB model was then performed.25,26 Compounds 7 and 12 diffused through the lipid bilayer with Pe values significantly higher than the positive control (Pe = 450.3 and 405.1 nm/s, respectively), whereas compound 13 displayed a low Pe value (Pe = 12.7 nm/s), indicating a limited crossing of the BBB, hindering the further development of 13. This last result is not surprising because electrostatic repulsions between the negatively charged carboxylic acid group of this molecule and the negatively charged membrane constituents are probably responsible for its low permeability across the BBB in the PAMPA model. Moreover, 13 has a molecular weight approaching 400 g/mol, which could also contribute to limiting its diffusion across the BBB.27 Unfortunately, the methylic ester precursor or 13 could not be evaluated in vitro because of a lack of aqueous solubility. The ability of hit-compound 12 to cross the BBB in the PAMPA assay is in accordance with its central nervous system multiparameter optimization (CNS MPO) score28 that has a calculated value of 4.35. Finally, the aqueous solubility of hit-compound 12 was measured with a thermodynamic solubility assay at physiological pH and was determined at 21.7 μM (Table 2).
Table 2. In Vitro Pharmacokinetic, Physicochemical, and Toxicological Properties of Compounds 7, 12, and 13.

Calculated with MarvinSketch (ChemAxon).
To check the antikinetoplastid mechanism of the bioactivation of compounds 7, 12 and 13, their activities were evaluated in vitro toward Leishmania donovani promastigote strains overexpressing leishmanial nitroreductases (NTR1 or NTR2). EC50 values were determined and compared with the ones obtained on a wild-type strain to evaluate the bioactivation of these nitroaromatic compounds (Table 3). As reported for hit 1,21 compounds 7 and 12 are selectively bioactivated by leishmanial NTR1, with EC50 values being 3.5 and 16 times lower on the strain overexpressing the NTR than on the wild type. Contrary to 7 and 12, compound 13 did not demonstrate significant bioactivation by NTR1 or by NTR2 in Leishmania. The same experiments were carried out with compounds 7, 12, and 13 on T. b. brucei strains overexpressing the type 1 trypanosomal NTR, comparing the EC50 values with the ones determined on the wild-type strain (Table 3). All tested compounds were bioactivated by the type 1 nitroreductase of T. b. brucei, being, respectively, 7, 15, and 40 times more active against the strain overexpressing the NTR than against the wild type. It is interesting to note that hit 2 (E° = −0.56 V) did not seem to be bioactivated by T. b. brucei NTR, whereas its 6-brominated derivative 13 (E° = −0.40 V) was. Finally, hit-compound 12 also appeared to be six times more active than nifurtimox against T. b. brucei, underlining the potential of this molecule as an antitrypanosomal candidate.
Table 3. Evaluation of the Bioactivation of Compounds 7, 12, and 13 by Leishmanial and Trypanosomal NTRs.
|
Leishmania donovani promastigotes
EC50 (μM) | |||
|---|---|---|---|
| compound | wild type | NTR1 overexpressing | NTR2 overexpressing |
| Hit 1 | 5.9 ± 0.2 | 0.47 ± 0.02 | 4.6 ± 0.12 |
| 7 | 4.1 ± 0.2 | 1.2 ± 0.05 | 4.9 ± 0.3 |
| 12 | 4.7 ± 0.09 | 0.3 ± 0.01 | 2.9 ± 0.08 |
| 13 | 2.0 ± 0.2 | 1.3 ± 0.1 | 2.1 ± 0.1 |
|
Trypanosoma brucei brucei trypomastigotes EC50 (μM) | ||
|---|---|---|
| compound | wild type | NTR overexpressing |
| Hit 1 | 17.7 ± 1.0 | 3.9 ± 0.1 |
| Hit 2 | 5.4 ± 0.12 | 4.2 ± 0.2 |
| 7 | 6.7 ± 0.4 | 0.9 ± 0.03 |
| 12 | 0.3 ± 0.06 | 0.02 ± 0.008 |
| 13 | 4.0 ± 0.2 | 0.1 ± 0.009 |
| Nifurtimox | 1.9 ± 0.05 | 0.6 ± 0.05 |
Even though many safe nitroaromatic molecules are today on the market (for example, antiprotozoal 5-nitroimidazoles such as metronidazole, several antihypertensive dihydropyridines like nicardipine, some benzodiazepines like loprazolam, anti-Parkinson’s catechol O-methyltransferase (COMT) inhibitors like entacapone, antiandrogens like flutamide, anti-inflammatory nimesulide, immunomodulating azathioprine, anticoagulant acenocoumarol, anthelminthic niclosamide, etc.) or in clinical trials (for instance, delamanid), drugs including a nitroaromatic moiety suffer from a bad reputation as possible mutagenic or genotoxic molecules, which narrows their pharmaceutical development.29 The Ames test is the most commonly used assay to evaluate the mutagenic potential of compounds, using Salmonella typhimurium strains. However, these bacteria possess NTR, making the Ames test generally unsuitable for the evaluation of most of nitroaromatic compounds, with the genotoxicity being attributed to the reduced metabolites that are bioactivated by these bacterial enzymes (absent in human cells).30 Thus it is accepted that the comet assay or the micronucleus assay are more pertinent methods to evaluate the genotoxic potential of nitroaromatic compounds. For example, fexinidazole is positive in the Ames test but negative in the micronucleus assay.31 A comet assay was then performed on the HepG2 cell line with hit compound 12, using methylmethanesulfonate (MMS) as a positive control (see the Supporting Information). Compound 12 was not genotoxic after 2 or 72 h of incubation or at 1 or 10 μM, concentrations chosen to be lower than its CC50 on HepG2 cell line (CC50 = 18 μM).
Regarding tolerance in the mouse, a once daily oral dose of compound 12, at 25 or 50 mg/kg, was administrated to eight mice (four mice in each group). These two doses were well tolerated with just a little apathy during 1 h for all mice receiving the dose of 50 mg/kg. The administration of a repeated dose of 25 mg/kg once daily for 5 days was well tolerated without any adverse effect noted. The no-observed-adverse-effect level (NOAEL)31 in mice was then set at 25 mg/kg/day. After euthanasia, no lesions were found on the different organs (kidney, liver, brain, heart, and lung).
Finally, the in vivo pharmacokinetic parameters were determined in the mouse, after oral administration at 25 mg/kg in a mixture of 5% Tween 80 and 95% of 0.5% carboxymethylcellulose in water, by using the QuEChERS method.32 Monolix Lixoft software was used to analyze the data by a noncompartmental model to fit pharmacokinetics parameters.33 As presented in the Supporting Information (Figure S17), hit-compound 12 showed good pharmacokinetic properties: 12 is highly and rapidly absorbed after oral administration and shows good exposure. Its plasmatic half life (2.9 h) is higher than the one of the drug fexinidazole (0.8 h).31
Conclusions
Via an antitrypanosomal pharmacomodulation study of the 8-nitroquinolin-2(1H)-one scaffold, by introducing an EWG at positions 3 and 6 of the quinolinone ring, a novel potent antitrypanosomal hit-compound (12) was identified, displaying high activities toward T. b. brucei (EC50 = 12 nM) and T. cruzi (EC50 = 0.5 μM) in comparison with eflornithine (EC50 = 13 μM), suramin (EC50 = 30 nM), benznidazole (EC50 = 0.5 μM), and fexinidazole (EC50 = 0.4 and 3.0 μM). Compound 12 displays good in vitro pharmacokinetic parameters (good microsomal stability and BBB permeability), is rapidly absorbed after oral administration, and is well tolerated in the mouse at 25 mg/kg. Nitroaromatic hit compound 12 is selectively bioactivated by type 1 NTR in both Leishmania and Trypanosoma. An electrochemistry study showed the strong influence of the introduction of an EWG in the benzenic part of the pharmacophore toward the redox potentials and the corresponding antitrypanosomal activities. Finally, the lack of genotoxicity of compound 12 in the comet assay is an additional favorable element so as to consider further development. Compound 12 meets all criteria for an antitrypanosomal hit, as defined by Katsuno and coworkers.34In vivo studies on the mice model of trypanosomiasis are now planned to investigate the potential of this compound to become an antitrypanosomal lead.
Acknowledgments
Paul Sabatier University and the Région Occitanie are acknowledged for financial support. A.H.F., S.W., and R.M. are supported by funding from the Wellcome Trust (WT105021). C. Piveteau from Institut Pasteur de Lille, C. Bijani from Laboratoire de Chimie de Coordination de Toulouse, and C. Claparols, N. Martins-Froment, V. Bourdon, and E. Leroy from the Institut de Chimie de Toulouse are also acknowledged for their support. We also thank Dr. J.-B. Woillard for the in vivo pharmacokinetic analysis, Mr. F.-L. Sauvage for his help with the mass spectrometry analysis, and Dr. Ismail Belfquih for his assistance with the culture of Vero cells and T. cruzi.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00566.
Materials and methods, additional analysis of hit compounds 7, 12, and 13 as well as electrochemistry data, in vitro pharmokinetics (PK) data, and in vivo PK data (PDF)
Author Contributions
+ J.P. and C.B. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Büscher P.; Cecchi G.; Jamonneau V.; Priotto G. Human african trypanosomiasis. Lancet 2017, 390, 2397–2405. 10.1016/S0140-6736(17)31510-6. [DOI] [PubMed] [Google Scholar]
- Pérez-Molina J. A.; Molina I. Chagas disease. Lancet 2018, 391, 82–94. 10.1016/S0140-6736(17)31612-4. [DOI] [PubMed] [Google Scholar]
- Burza S.; Croft S. L.; Boelaert M. Leishmaniasis. Lancet 2018, 392, 951–970. 10.1016/S0140-6736(18)31204-2. [DOI] [PubMed] [Google Scholar]
- World Health Organization . Neglected Tropical Diseases. https://www.who.int/neglected_diseases/diseases/en/ (accessed November 1, 2019).
- Molyneux D.; Savioli L.; Engels D. Neglected tropical diseases: progress towards addressing the chronic pandemic. Lancet 2017, 389, 312–325. 10.1016/S0140-6736(16)30171-4. [DOI] [PubMed] [Google Scholar]
- Drugs for Neglected Diseases Initiative (DNDi) . DNDi R&D Portfolio June 2019. https://www.dndi.org/diseases-projects/portfolio/ (accessed November 1, 2019).
- Wyllie S.; Patterson S.; Stojanovski L.; Simeons F. R. C.; Norval S.; Kime R.; Read K. D.; Fairlamb A. H. The anti-trypanosome drug fexinidazole shows potential for treating visceral leishmaniasis. Sci. Transl. Med. 2012, 4, 119re1. 10.1126/scitranslmed.3003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson S.; Fairlamb A. H. Current and future prospects of nitro-compounds as drugs for trypanosomiasis and leishmaniasis. Curr. Med. Chem. 2019, 26, 4454–4475. 10.2174/0929867325666180426164352. [DOI] [PubMed] [Google Scholar]
- Mesu V. K. B. K.; Kalonji W. M.; Bardonneau C.; Mordt O. V.; Blesson S.; Simon F.; Delhomme S.; Bernhard S.; Kuziena W.; Lubaki J. F.; Vuvu S. L.; Ngima P. N.; Mbembo H. M.; Ilunga M.; Bonama A. K.; Heradi J. A.; Solomo J. L. L.; Mandula G.; Badibabi L. K.; Dama F. R.; Lukula P. K.; Tete D. N.; Lumbala C.; Scherrer B.; Strub-Wourgaft N.; Tarral A. Oral fexinidazole for late-stage African Trypanosoma brucei gambiense trypanosomiasis: a pivotal multicentre, randomised, non-inferiority trial. Lancet 2018, 391, 144–154. 10.1016/S0140-6736(17)32758-7. [DOI] [PubMed] [Google Scholar]
- Patterson S.; Wyllie S.; Norval S.; Stojanovski L.; Simeons F. R. C.; Auer J. L.; Osuna-Cabello; Read K. D.; Fairlamb A. H. The anti-tubercular drug delamanid as a potential oral treatment for visceral leishmaniasis. eLife 2016, 5, e09744 10.7554/eLife.09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang C. W.; Jarrad A. M.; Cooper M. A.; Blaskovich M. A. T. Nitroimidazoles: Molecular Fireworks That Combat a Broad Spectrum of Infectious Diseases. J. Med. Chem. 2017, 60, 7636–7657. 10.1021/acs.jmedchem.7b00143. [DOI] [PubMed] [Google Scholar]
- Patterson S.; Wyllie S. Nitro drugs for the treatment of trypanosomatid diseases: past, present, and future prospects. Trends Parasitol. 2014, 30, 289–298. 10.1016/j.pt.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie S.; Patterson S.; Fairlamb A. H. Assessing the essentiality of leishmania donovani nitroreductase and its role in nitro drug activation. Antimicrob. Agents Chemother. 2013, 57, 901–906. 10.1128/AAC.01788-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie S.; Roberts A. J.; Norval S.; Patterson S.; Foth B. J.; Berriman M.; Read K. D.; Fairlamb A. H. Activation of bicyclic nitro-drugs by a novel nitroreductase (NTR2) in Leishmania. PLoS Pathog. 2016, 12, e1005971 10.1371/journal.ppat.1005971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall B. S.; Bot C.; Wilkinson S. R. Nifurtimox activation by trypanosomal type I nitroreductases generates cytotoxic nitrile metabolites. J. Biol. Chem. 2011, 286, 13088–13095. 10.1074/jbc.M111.230847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall B. S.; Wilkinson S. R. Activation of Benznidazole by trypanosomal type I nitroreductases results in glyoxal formation. Antimicrob. Agents Chemother. 2012, 56, 115–123. 10.1128/AAC.05135-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaeghe P.; Rathelot P.; Rault S.; Vanelle P. Convenient preparation of original vinylic chlorides with antiparasitic potential in quinolone series. Lett. Org. Chem. 2006, 3, 891–897. 10.2174/157017806779467997. [DOI] [Google Scholar]
- Paloque L.; Verhaeghe P.; Casanova M.; Castera-Ducros C.; Dumètre A.; Mbatchi L.; Hutter S.; Kraiem-M’Rabet M.; Laget M.; Remusat V.; Rault S.; Rathelot P.; Azas N.; Vanelle P. Discovery of a new antileishmanial hit in 8-nitroquinoline series. Eur. J. Med. Chem. 2012, 54, 75–86. 10.1016/j.ejmech.2012.04.029. [DOI] [PubMed] [Google Scholar]
- Kieffer C.; Cohen A.; Verhaeghe P.; Hutter S.; Castera-Ducros C.; Laget M.; Remusat V.; M’Rabet M. K.; Rault S.; Rathelot P.; Azas N.; Vanelle P. Looking for new antileishmanial derivatives in 8-nitroquinolin-2(1H)-one series. Eur. J. Med. Chem. 2015, 92, 282–294. 10.1016/j.ejmech.2014.12.056. [DOI] [PubMed] [Google Scholar]
- Kieffer C.; Cohen A.; Verhaeghe P.; Paloque L.; Hutter S.; Castera-Ducros C.; Laget M.; Rault S.; Valentin A.; Rathelot P.; Azas N.; Vanelle P. Antileishmanial pharmacomodulation in 8-nitroquinolin-2(1H)-one series. Bioorg. Med. Chem. 2015, 23, 2377–2386. 10.1016/j.bmc.2015.03.064. [DOI] [PubMed] [Google Scholar]
- Pedron J.; Boudot C.; Hutter S.; Bourgeade-Delmas S.; Stigliani J.-L.; Sournia-Saquet A.; Moreau A.; Boutet-Robinet E.; Paloque L.; Mothes E.; Laget M.; Vendier L.; Pratviel G.; Wyllie S.; Fairlamb A. H.; Azas N.; Courtioux B.; Valentin A.; Verhaeghe P. Novel 8-nitroquinolin-2(1H)-ones as NTR-bioactivated antikinetoplastid molecules: synthesis, electrochemical and SAR study. Eur. J. Med. Chem. 2018, 155, 135–152. 10.1016/j.ejmech.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedron J.; Boudot C.; Bourgeade-Delmas S.; Sournia-Saquet A.; Paloque L.; Rastegari M.; Abdoulaye M.; El-Kashef H.; Bonduelle C.; Pratviel G.; Wyllie S.; Fairlamb A. H.; Courtioux B.; Verhaeghe P.; Valentin A. Antitrypanosomal pharmacomodulation at position 3 of the 8-nitroquinolin-2(1H)-one scaffold using pallado-catalyzed cross coupling reactions. ChemMedChem 2018, 13, 2217–2228. 10.1002/cmdc.201800456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaragoza F.; Stephensen H.; Peschke B.; Rimvall K. 2-(4-alkylpiperazin-1-yl)quinolines as a new class of imidazole-free histamine H3 receptor antagonists. J. Med. Chem. 2005, 48, 306–311. 10.1021/jm040873u. [DOI] [PubMed] [Google Scholar]
- O’Brien N. J.; Brzozowski M.; Wilson D. J. D.; Deady L. W.; Abbott B. M. Synthesis and biological evaluation of substituted 3-anilinoquinolin-2(1H)-ones as PDK1 inhibitors. Bioorg. Med. Chem. 2014, 22, 3781–3790. 10.1016/j.bmc.2014.04.037. [DOI] [PubMed] [Google Scholar]
- Di L.; Kerns E. H.; Bezar I. F.; Petusky S. L.; Huang Y. Comparison of blood-brain barrier permeability assays: in situ brain perfusion, MDR1-MDCKII and PAMPA-BBB. J. Pharm. Sci. 2009, 98, 1980–1991. 10.1002/jps.21580. [DOI] [PubMed] [Google Scholar]
- Jourdan J. P.; Since M.; El Kihel L.; Lecoutey C.; Corvaisier S.; Legay R.; Sopkova-de Oliveira Santos J.; Cresteil T.; Malzert-Fréon A.; Rochais C.; Dallemagne P. Benzylphenylpyrrolizinones with anti-amyloid and radical scavenging effects, potentially useful in Alzheimer’s disease treatment. ChemMedChem 2017, 12, 913–916. 10.1002/cmdc.201700102. [DOI] [PubMed] [Google Scholar]
- Van De Waterbeemd H.; Camenisch G.; Folkers G.; Chretien J. R.; Raevsky O. A. Estimation of blood-brain barrier crossing of drugs using molecular size and shape, and H-bonding descriptors. J. Drug Target 1998, 6, 151–165. 10.3109/10611869808997889. [DOI] [PubMed] [Google Scholar]
- Wager T. T.; Hou X.; Verhoest P. R.; Villalobos A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010, 1, 435–449. 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nepali K.; Lee H.-Y.; Liou J.-P. Nitro-Group-Containing Drugs. J. Med. Chem. 2019, 62, 2851–2893. 10.1021/acs.jmedchem.8b00147. [DOI] [PubMed] [Google Scholar]
- Rosenkranz E. J.; McCoy E. C.; Mermelstein R.; Rosenkranz H. S. Evidence for the existence of distinct nitroreductases in Salmonella typhimurium: roles in mutagenesis. Carcinogenesis 1982, 3, 121–123. 10.1093/carcin/3.1.121. [DOI] [PubMed] [Google Scholar]
- Torreele E.; Bourdin Trunz B.; Tweats D.; Kaiser M.; Brun R.; Mazué G.; Bray M. A.; Pécoul B. Fexinidazole - a new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness. PLoS Neglected Trop. Dis. 2010, 4, e923 10.1371/journal.pntd.0000923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westland J.; Dorman F. QuEChERS extraction of benzodiazepines in biological matrices. J. Pharm. Anal. 2013, 3, 509–517. 10.1016/j.jpha.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pkanalix . Data Processing and Calculation Rules. http://pkanalix.lixoft.com/calculation-rules/ (accessed September 15, 2019).
- Katsuno K.; Burrows J. N.; Duncan K.; van Huijsduijnen R. H.; Kaneko T.; Kita K.; Mowbray C. E.; Schmatz D.; Warner P.; Slingsby B. T. Hit and lead criteria in drug discovery for infectious diseases of the developing worlds. Nat. Rev. Drug Discovery 2015, 14, 751–758. 10.1038/nrd4683. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.